

Abstract
Background
Bardet–Biedl Syndrome (BBS) is a genetically heterogeneous ciliopathy with clinical cardinal features including retinal degeneration, obesity and renal dysfunction. To date, 20 BBS genes have been identified with BBS10 being a major BBS gene found to be mutated in almost 20 percent of all BBS patients worldwide. It codes for the BBS10 protein which forms part of a chaperone complex localized at the basal body of the primary cilium. Renal dysfunction in BBS patients is one of the major causes of morbidity in human patients and is associated initially with urinary concentration defects related to water reabsorption impairment in renal epithelial cells. The aim of this study was to study and compare the impact of a total Bbs10 inactivation (Bbs10−/−) with that of a specific renal epithelial cells inactivation (Bbs10 fl/fl; Cdh16-Cre+/−).
Results
We generated the Bbs10−/−and Bbs10 fl/fl; Cadh16-Cre+/− mouse model and characterized them. Bbs10−/− mice developed obesity, retinal degeneration, structural defects in the glomeruli, polyuria associated with high circulating arginine vasopressin (AVP) concentrations, and vacuolated, yet ciliated, renal epithelial cells. On the other hand, the Bbs10 fl/fl; Cadh16-Cre+/−mice displayed no detectable impairment.
Conclusions
These data highlight the importance of a systemic Bbs10 inactivation to trigger averted renal dysfunction whereas a targeted absence of BBS10 in the renal epithelium is seemingly non-deleterious.
Electronic supplementary material
The online version of this article (doi:10.1186/s13630-015-0019-8) contains supplementary material, which is available to authorized users.
Keywords: Bardet–Biedl Syndrome, Bbs10, Total knockout, Retinal degeneration, Obesity, Renal epithelial-specific KO
Background
Bardet–Biedl syndrome (BBS) [1] is an autosomal recessive ciliopathy for which 20 genes (BBS1-20) have been identified to date. It is clinically characterized, among features, by early-onset retinitis pigmentosa (RP), obesity and renal dysfunction as well as polydactyly, RP is a rod–cone dystrophy [2], leading to progressive visual loss and blindness [3]. The connecting cilium of photoreceptors, a highly specialized primary cilium [3, 4], plays a pivotal role in intraciliary transport (ICT) between the inner and outer segments [5]. The established mechanism behind this phenotype is an excessive accumulation of protein in the inner segment (IS) of the photoreceptor due to defect in intracellular transport (ICT) [6, 7] triggering an unfolded protein response-mediated apoptosis of the photoreceptors [8–10]. In parallel, obesity develops together with the RP [2] and is associated with hyperleptinemia. The high circulating concentrations of Leptin observed both in patients [11] and mouse models [12, 13] have historically been linked to a hypothalamic defect in the regulation of food intake leading to hyperphagia and decreased energy expenditure [12]. More recently, BBS proteins have been shown to play a key role in the adipose tissue maturation and function [14] which strongly suggest that multiple pathways could be driving the BBS-obese phenotype.
Compared to RP and obesity, BBS-induced renal impairment after birth tends to display a late-onset profile as it is generally detected in the late childhood. This major cause of morbidity and mortality for the BBS patients is characterized by an initial urinary concentrating defects associated with polyuria–polydypsia [15]. Concomitantly, scores of structural abnormalities like small kidneys with normal or irregular contours and reduced parenchyma thickness, calyceal distortion, clubbing and blunting, caliectasis, presence of medullary cysts communicating or not with mildly dilated calyceal system and occasional presence of cortical cysts have been reported and they represent the full clinical spectrum of the BBS renal phenotype [16]. To explain these BBS renal phenotypes several defective signalling pathways have been incriminated such as a defective WNT signalling cascade during development [17, 18] as well as the lack of ciliary targeting of the antidiuretic hormone arginine vasopressin (AVP) isoform 2 receptor (AVPR2). In combination to these intrinsic effects of BBS inactivation, it has been reported that the occurrence of obesity was a leading cause for the presence of renal cysts in mice; an effect which was reverted when they went through food restriction [18]. Although the results highlight the difficulty in identifying the exact pathological mechanism leading to renal dysfunction in the BBS, in an attempt to understand the precise impact of BBS inactivation in the ciliated renal epithelium, we generated the Bbs10−/− mice and the Bbs10 fl/fl; Cadh16-Cre+/− and studied the resulting phenotypes.
Methods
Generation of Bbs10 total knockout mice and Bbs10 renal epithelial-specific knockout mice
All experimental procedures were approved by the local ethical committee of Strasbourg. See Additional file 1: Supporting information for supplementary methods. The inactivation of the Bbs10 gene was achieved by the deletion of the exon 2. Bbs10 fl/fl mice were obtained by DNA recombination, using LoxP sites flanking the 5′ and 3′ regions of the exon 2 in Bbs10 gene in embryonic stem cells line 129OLA using a neomycin cassette as a selectable marker (Additional file 2: Figure S1). These mice were bred with Cadh16-CreDeleter mice (B6.Cg-Tg (Cadh16-Cre) 91Igr/J, stock number 012237 from Jackson). Homozygous knockout mice Bbs10−/− and the control littermates Bbs10+/+ were obtained by mating of heterozygous Bbs10+/−mice. Chimeric mice were obtained on a C57/BL6 genetic background. Bbs10 fl/fl mice were bred with the Ksp1.3/Cre transgenic mouse lines expressing heterozygous Cre recombinase gene under the control of a tissue-specific promoter: Ksp-Cadherin (Cadherin16), exclusively expressed in renal tubular epithelial cells [19]. The resulting Bbs10 fl/fl; Cadh16-Cre+/− together with the Bbs10+/+; Cadh16-Cre+/− mice were generated. All the genotypes were checked by PCR using KAPA Mouse Genotyping Kit (#KK7302, Kapa Biosystems, Woburn, Massachusetts, USA). Primer sequences used for Bbs10 genotyping were 5′-ACA AAT ACA ATT GAT CAT CGA TGT G-3′ (forward primer) and 5′-GTT GCC TGG CTT GGG TGG CA-3′ (reverse primer), and 5′-ACC TCC CCA CTT GAA CGA GGT CT-3′ (forward) and 5′-GTT GCC TGG CTT GGG TGG CA-3′ (reverse) for WT and floxed mice. For Cadh16 Cre genotyping, the primer sequences used were 5′-GCA GAT CTG GCT CTC CAA AG-3′ as sense primer and 5′-AGG CAA ATT TTG GTG TAC GG-3′ as antisense primer, and 5′-CAA ATG TTG CTT GTC TGG TG-3′ (forward) and 5′-GTC AGT CGA GTG CAC AGT TT-3′ (reverse) for the internal positive control. Genotyping at the ROSA26 locus of the RosaTomato/eGFP (Gt(ROSA)26Sortm4(ACTB-tdTomato,−EGFP)Luo/J, Jackson Laboratories Stock # 007576) mice was performed using the following three-primer set: oIMR7318: 5′-CTC TGC TGC CTC CTG GCT TCT-3′; oIMR7319: 5′-CGA GGC GGA TCA CAA GCA ATA-3′; oIMR7320: 5′-TCA ATG GGC GGG GGT CGT T-3′. See Additional file 1: Supporting information for supplementary methods.
Results
Molecular characterisation of the Bbs10−/− mice
Using a PCR-based approach, we generated a conditional floxed mice (Bbs10 fl/fl) in which we flanked the exon2 on the 3′- and 5′-regions with LoxP sites (Fig. 1a and Additional file 2: Figure S1). Total Bbs10 knockout mice (Bbs10−/−) were then produced by breeding the Bbs10 fl/fl mice with a Deleter-Cre mouse line. Following Cre-mediated excision of the Bbs10 allele, the Cre allele was removed through breeding the resulting heterozygous Bbs10 mice Bbs10+/−. The Bbs10+/− mice were then used for breeding to generate the total Bbs10 knockout mice Bbs10−/−, and were subsequently genotyped by PCR (Fig. 1b). Bbs10−/− mice display no Bbs10 expression level in target tissues like kidney and the eyes (Fig. 1c). Bbs10+/+ and Bbs10 fl/fl had no differences in Bbs10 mRNA expression as can be seen in Additional file 3: Figure S2D.
Fig. 1.
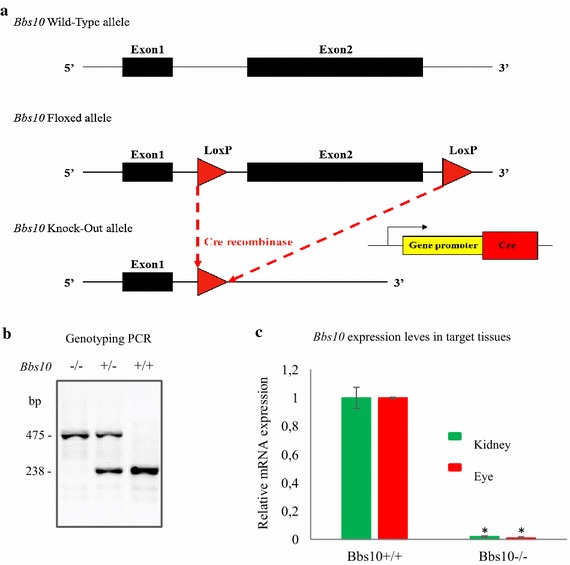
Generating the Bbs10 −/− mouse model. a Schematic representation of the targeting strategy for Bbs10 allele. b Genotyping PCR results for homozygous knock out (Bbs10 −/−), heterozygous (Bbs10 +/−), wild-type (Bbs10 +/+) mice. PCR bands at 475 and 238 bp correspond to the excised allele and to the natural allele, respectively. (bp base pairs). c Relative mRNA expression levels of Bbs10 in kidneys and the eyes of Bbs10 +/+ and Bbs10 −/−. Reference gene Gapdh (n = 4 per genotype).
Bbs10−/− mice display typical BBS-obese phenotype
In the perinatal period, Bbs10−/− mice exhibited a runting phenotype but recovered equivalent body weight with the control Bbs10+/+ littermates by 8 weeks of age. As from 8 weeks onwards, Bbs10−/− mice gained more weight than the Bbs10+/+ (Fig. 2a) and at 3 months, the Bbs10−/− mice were overweight (Fig. 2b). Food intake measurements revealed that Bbs10−/− mice only develop hyperphagia at 8 weeks postnatal (Fig. 2c) and presented severe hyperleptinemia at 3 months with circulating leptin concentration of 125 ng/mL (Fig. 2d). Despite the obese phenotype, subsequent analysis examination of the visceral adipocytes showed no significant difference in cellular diameter between Bbs10−/− and Bbs10+/+ mice (Fig. 2e). Normal glycaemic levels were observed in fasted, unchallenged animals although a delay in the rate of decrease of glucose levels was observed following glucose tolerance test (GTT) (Fig. 2f). Furthermore, insulin sensitivity was maintained in Bbs10−/− as demonstrated by the insulin tolerance test (ITT) results; glucose level patterns were similar between the Bbs10−/− and Bbs10+/+mice (Fig. 2g).
Fig. 2.
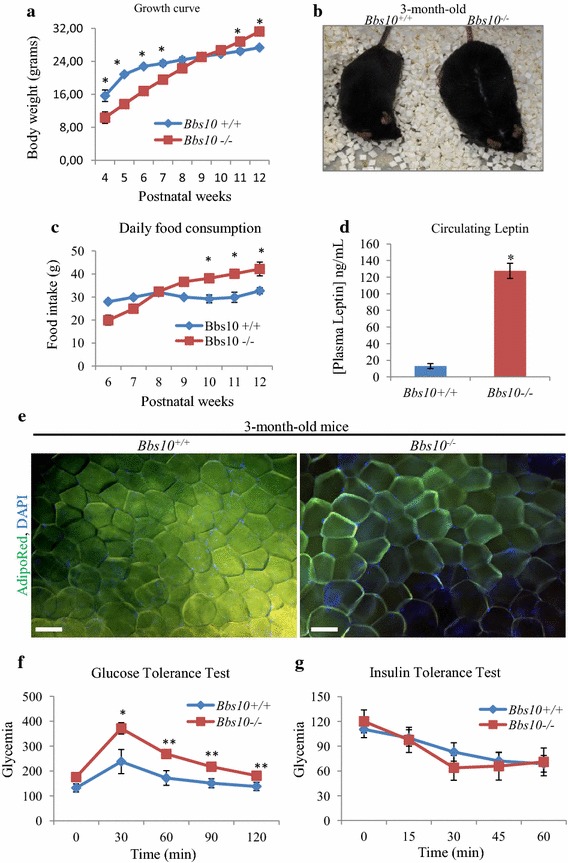
Obesity-associated phenotype in Bbs10 −/− mice. a Growth curve of Bbs10 +/+ and Bbs10 −/− (n = 8). b Photograph of 3-month-old Bbs10 +/+ and Bbs10 −/− mice. c Food intake measurement of Bbs10 +/+ and Bbs10 −/− mice (n = 8 per genotype). d Measurement of the circulating concentrations of leptin in Bbs10 +/+ and Bbs10 −/− (n = 6 per group). e Pictures of AdipoRed-stained visceral adipocytes from Bbs10 +/+ and Bbs10 −/−. Scale bar 50 µm. Data are expressed as mean ± SEM. f Glucose tolerance test (GTT) and g insulin tolerance test (ITT) for the same mice (n = 7 per group). Values are expressed as mean ± SEM. *p < 0.05.
Bbs10−/− mice exhibit retinal degeneration
Bbs10−/− mice, like all reported BBS mouse models [3], exhibit severe retinal degeneration. 3-month-old Bbs10−/− mice showed retinal thinning observed in the Optical Coherence Tomography (OCT) (Fig. 3a) and on histological toluidine-stained sections (Fig. 3b). Immunostained cryosections of the corresponding retinas (Fig. 3c) for rhodopsin showed correct localisation in the outer segment (in green) albeit a markedly clear reduction of rhodopsin content in the Bbs10−/− retinas. Transmission electron microscopy (T.E.M.) analysis revealed that the retinal phenotype was primarily due to the loss of the inner and outer segment (IS/OS) of the photoreceptors and the outer nuclear layer (ONL); although the connecting cilium could still be detected, next to the centriole (Fig. 3d, red arrows). This gradual loss of the photoreceptors in the Bbs10−/− retinas correlated with a significant decrease of the a-wave (Fig. 3e, red arrows) and b-wave magnitudes of the scotopic electroretinograms (ERGs) in the Bbs10−/− mice. We also found in the retinas of Bbs10−/− mice an elevated number of TUNEL-positive nuclei indicating an ongoing process of apoptosis in the Bbs10−/− retinas (Additional file 4: Figure S3A, B).
Fig. 3.
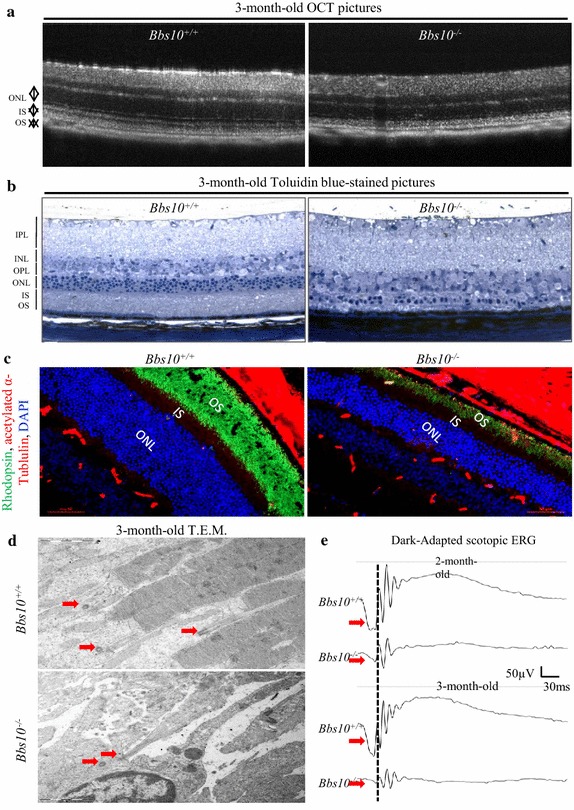
Retinal phenotype in Bbs10 −/− mice. a Optical coherence tomography (OCT) pictures of retinas from Bbs10 +/+ and Bbs10 −/− mice. (OS outer segment, IS inner segment, ONL outer nuclear layer) (n = 3 mice per genotype). b Toluidine blue-stained pictures of 7-µm-thick retinal sections from Bbs10 +/+ and Bbs10 −/− mice (OS outer segment, IS inner segment, ONL outer nuclear layer, OPL outer plexiform layer, INL inner nuclear layer, IPL inner plexiform layer) Scale bar 20 µm. c Immunofluorescence of 7-µm-thick retinal sections from Bbs10 +/+ and Bbs10 −/− mice. Nuclei were counterstained with DAPI (blue), rhodopsin (green), acetylated a-tubulin (red). d Transmission electron microscopy (T.E.M.) pictures of retinal sections in Bbs10 +/+ and Bbs10 −/− mice (ROS retinal outer segment, RIS retinal inner segment, RPE retinal pigment epithelium). Red arrows indicate the centrioles and the connecting cilium. e Scotopic electroretinograms (ERG) recordings of 2- and 3-month-old Bbs10 +/+ and Bbs10 −/− mice. Red arrows indicate a-waves on the ERG recordings (n = 6–8 per genotype).
Structural renal abnormalities associated with functional impairment are observed in Bbs10−/− mice
We next investigated the impact of Bbs10 inactivation on the kidneys. In the glomerular region, T.E.M. analysis revealed substantial decrease of the glomerular basement membrane thickness combined with an absence of primary and secondary podocyte structures (Fig. 4a, b). This structural defect correlated with significant increase in albuminuria (Fig. 4c) although creatinine clearance was maintained in the Bbs10−/− mice (Fig. 4d). As it was reported that BBS protein inactivation could impair ciliogenesis [14, 20, 21], this prompted us to verify the ciliated status of the tubular epithelial cells. Ciliated epithelial cells were readily detected in the Bbs10−/− tubular epithelium as depicted by T.E.M analysis (Fig. 4e). Further analysis showed correctly polarized epithelial cells for both proximal tubular and distal convoluted epithelial cells in the Bbs10−/− kidneys but revealed large intracytoplasmic inclusions in the Bbs10−/− epithelial cells (Fig. 4f and Additional file 5: Figure S4). No detectable cystic lesion were detected and correct targeting of the Aquaporin 2 (AQP2) to the apical side of the epithelial cells was found in the Bbs10−/− epithelial cells (Fig. 4g).
Fig. 4.
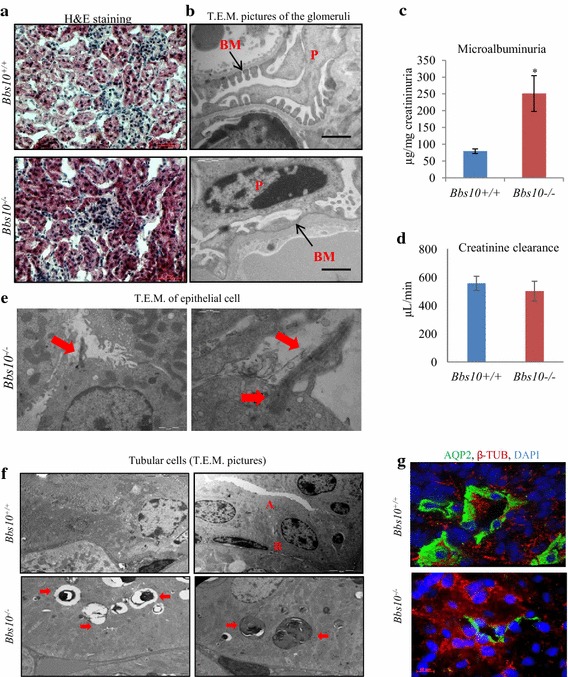
Renal phenotype of the Bbs10 −/− mice and Bbs10 fl/fl ; Cadh16-Cre +/− mice. a Pictures of hematoxylin and eosin-stained kidney sections of 3-month-old Bbs10 +/+ and Bbs10 −/− mice. b Transmission electron microscopy picture of kidney sections of the glomerular region from Bbs10 +/+and Bbs10 −/− mice (P podocyte, BM basement membrane). Scale bars 5 and 2 µm. c Microalbuminuria levels from Bbs10 +/+ and Bbs10 −/− mice. d Creatinine clearance levels (µL/min) and urinary volume under fluid deprivation of Bbs10 +/+ and Bbs10 −/− mice (n = 7–9). Values are expressed as mean ± SEM. e Transmission electron microscopy picture of kidney epithelial cells from Bbs10 +/+, Bbs10 −/− mice. Red arrows indicate the centrioles and the connecting cilium. f Transmission electron microscopy picture of kidney tubular cells from Bbs10 +/+, Bbs10 −/− mice. Red arrows indicate cytoplasmic vacuoles, A apical side, B Basolateral side. g Pictures of immunostained kidney sections for AQP2 (green) and β-tubulin (red) from Bbs10 +/+and Bbs10 −/− mice. Nuclei were counterstained with DAPI (blue). Scale bars 20 µm. Values are expressed as mean ± SEM. *p < 0.05
Bbs10 fl/fl; Cadh16Cre+/− exhibit no detectable impairment in renal morphology or function
Next we generated Bbs10 fl/fl; Cadh16Cre+/− and their controls Bbs10+/+; Cadh16Cre+/− and genotyped those by PCR approach (Fig. 5a). The Bbs10 fl/fl; Cadh16Cre+/− displayed Cre expression in the renal epithelium (Additional file 3: Figure S2A) and the Cre recombinase activity was validated by detecting green fluorescence signal in the renal epithelial cells of the Cadh16Cre+/+; RosaTomatoeGFP+/+ (Additional file 3: Figure S2B). To further establish specific Cre-mediated excision of Bbs10 in the renal epithelial cells, we performed an immunodetection of BBS10 in the tubular renal epithelial cells and found no cells positively stained for BBS10 in the Bbs10 fl/fl; Cadh16Cre+/− compared to control (Additional file 6: Figures S6A, B). As Cadh16-Cre expression targets specifically the renal epithelial cells, no other characteristic BBS phenotype like obesity or retinal degeneration was observed in the Bbs10 fl/fl; Cadh16Cre+/− mice (data not shown). Immunostaining of acetylated-α-tubulin on kidney cryosections showed the presence of ciliated cells in Bbs10 fl/fl; Cadh16Cre+/− mice (Fig. 5c) together with normal apical targeting of AQP2 in the epithelial cells in the Bbs10 fl/fl; Cadh16Cre+/− mice (Fig. 5d). These data demonstrate the correct polarization of the BBS10-deprived renal epithelial cells. Moreover, since we did not target the glomerular region with the Cadh16Cre, no structural defect was observed in any of the podocyte structures of the Bbs10 fl/fl; Cadh16Cre+/− mice (Fig. 5e), an effect correlated with normal microalbuminuria levels (Fig. 5f). Surprisingly, no cytoplasmic inclusion was observed in the epithelial cells in both the proximal and distal tubular regions (Fig. 5g) and creatinine clearance was comparable between the tested groups (Fig. 5h). These results indicate that the sole Bbs10 inactivation in the renal epithelial cells is not enough to trigger massive pathological stress in the renal epithelium.
Fig. 5.
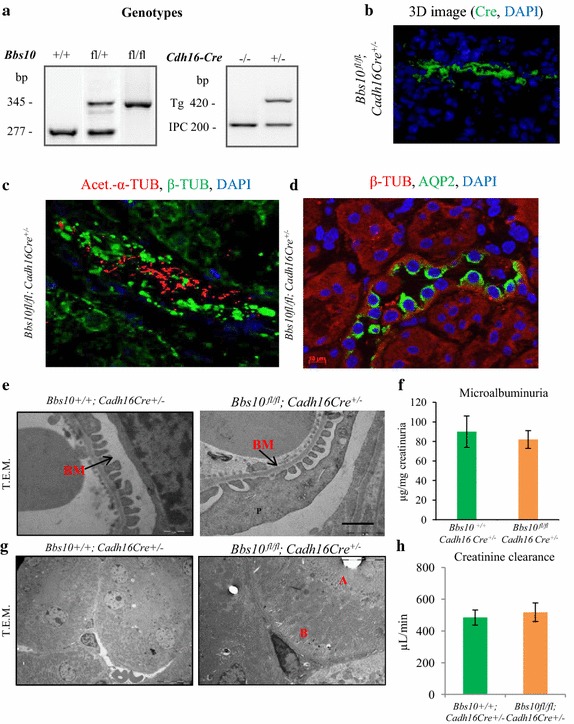
Renal phenotype of the Bbs10 fl/fl ; Cadh16-Cre +/− mice. a PCR genotyping of heterozygous WT (Bbs10 +/+) (Bbs10 fl/+), floxed (Bbs10 fl/fl) mice non-Cre (Cre −/−) and heterozygous (Cre +/−) genotyping (bp base pairs). b 3D image of a kidney section immunostained for Cre (green) and nuclei counterstained with DAPI (blue). c 3D image of a kidney section immunostained for the primary cilia (acetylated α-tubulin in red), cytoskeleton (β-tubulin in green) and nuclei counterstained with DAPI (in blue). d Pictures of immunostained kidney sections for AQP2 (green) and β-tubulin (red) from Bbs10 fl/fl ; Cadh16Cre +/− mice. Nuclei were counterstained with DAPI (blue). Scale bars 20 µm. e T.E.M. analysis of epithelial cells in the proximal tubule from Bbs10 +/+ ; Cadh16Cre +/−and Bbs10 fl/fl ; Cadh16Cre +/− mice. BM basement membrane. f Microalbuminuria levels for of Bbs10 fl/fl ; Cadh16Cre +/− and control Bbs10 +/+ ; Cadh16Cre +/− littermates (n = 6–7 per genotype). g T.E.M. analysis of epithelial cells in the distal tubule from Bbs10 +/+ ; Cadh16Cre +/− and Bbs10 fl/fl ; Cadh16Cre +/− mice. h Creatinine clearance levels (µL/min) and urinary volume under fluid deprivation of Bbs10 fl/fl ; Cadh16Cre +/− and control Bbs10 +/+ ; Cadh16Cre +/− littermates (n = 7–9). Values are expressed as mean ± SEM. *p < 0.05.
Systemic Bbs10 inactivation is a prerequisite for the classical polyuria phenotype in vivo
The most prominent renal phenotype in BBS patients is polyuria; a phenotype which was linked to a defective AVP signalling cascade. To identify the cellular mechanism behind this renal phenotype, we tested the Bbs10−/− mice for fluid retention. The mice underwent 24-h fluid deprivation and the collected urinary volumes were analysed. Bbs10−/− mice suffered of increased diuresis (Fig. 6a). This increased discharge of urine correlated with a decrease in mRNA expression of aquaporin (Aqp2), aquaporin 3 (Aqp3) and Avpr2 (Fig. 6b), nor decrease in protein levels of AQP2 and AVPR2 (Fig. 6c). Circulating AVP levels revealed a similar drastic increase in the Bbs10−/− mice irrespective of fluid intake (Fig. 6d), contrasting with the Bbs10+/+ mice that exhibit the normal physiological response upon the fluid restriction. Next, we performed the same series of tests with the Bbs10 fl/fl; Cadh16Cre+/− mice, to verify whether the specific Bbs10 inactivation could impair AVP signalling in the kidneys. Interestingly, neither the 24-h urinary volumes (Fig. 6e), nor renal mRNA expression levels of AQP2, AQP3 and AVPR2 (Fig. 6f) nor protein expression levels of AQP2 and AVPR2 (Fig. 6g) were impacted in the Bbs10 fl/fl; Cadh16-Cre+/− mice. Compared to controls, normal physiological AVP response to fluid restriction was observed in the Bbs10 fl/fl; Cadh16-Cre+/− mice (Fig. 6h). All together these data demonstrate that the specific inactivation of Bbs10 in the renal epithelial cells does not induce polyuria and the associated high AVP circulating levels.
Fig. 6.
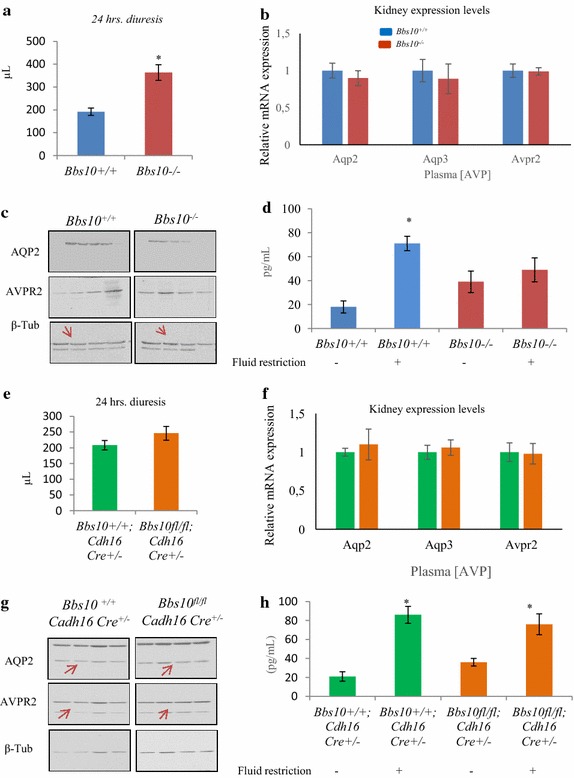
Bbs10−/− mice but not Bbs10 fl/fl ; Cadh16-Cre +/− mice display polyuria associated with increased circulating AVP concentrations. a Urinary volume (µL) under 24-h fluid deprivation of Bbs10 +/+ and Bbs10 −/− mice (n = 6 per genotype). b Relative mRNA expression levels of Aqp2, Aqp3 and Avpr2 in Bbs10 +/+ and Bbs10 −/− kidneys. Gene of reference Gapdh. c Entire photographs of immunoblots for AQP2, AVPR2 and β-tub in kidneys of Bbs10 +/+ and Bbs10 −/−. d AVP plasma levels in Bbs10 +/+ and Bbs10 −/− under 24-h fluid restriction or in normal conditions. (n = 5 per genotype). e Urinary volume (µL) under 24-h fluid deprivation of Bbs10 fl/fl ; Cadh16Cre +/− and control Bbs10 +/+ ; Cadh16Cre +/− littermates (n = 6 per genotype). f Relative mRNA expression levels of Aqp2, Aqp3 and Avpr2 in Bbs10 +/+ ; Cadh16Cre +/− and Bbs10 fl/fl ; Cadh16Cre +/−. g Entire photographs of the immunoblots for AQP2, AVPR2 and β-tubulin (β-tub) in kidneys of Bbs10 +/+ ; Cadh16Cre +/− and Bbs10 fl/fl ; Cadh16Cre +/−; the immunoblots are shown in Additional file 4: Figure S4A, B. h AVP plasma levels in Bbs10 fl/fl ; Cadh16Cre +/− and control Bbs10 +/+ ; Cadh16Cre +/− under 24-h fluid restriction or in normal conditions. (n = 5 per genotype). Values are expressed as mean ± SEM. *p < 0.05.
Discussion
Herein, we describe and compare the phenotypes of a Bbs10−/− versus Bbs10 fl/fl; Cadh16Cre+/− mice. The Bbs10−/− mice recapitulated most clinical features of the human condition, ranging from early-onset obesity, retinal degeneration and renal dysfunction whereas the Bbs10 fl/fl; Cadh16Cre+/− mice show neither detectable renal defect at 3 months nor any other noticeable manifestations. The Cadh16 Cre mouse used in this work to mediate the specific inactivation of Bbs10 in renal epithelial cells was the Cre Ksp1.3/Cre transgenic mouse. This model carries the 1,329 bp fragments of the 5′ flanking region of the Ksp-cadherin promoter [19]. Two Cadh16-Cre mice were developed by the group of Igarashi in 2002 containing different fragments of the 5′ flanking region of the Ksp-cadherin promoter [19], one of the mice carrying the 1,329 bp (Ksp1.3/Cre mouse) and the other carrying the 324 bp fragment (Ksp0.3/Cre mice). Cre expression in Ksp1.3 mice was restricted to the tubular epithelial cells in the mature and developing kidney, while the Ksp0.3 mice produce a variegated expression pattern [19, 22]. In this work we used the Ksp1.3/Cre transgenic mice to specifically target the tubular epithelial cells of the kidney and no other renal cells types. Based on the results presented here and combined with the use of the specified Cre mouse line, it seems that specific Bbs10 inactivation in the renal epithelium on its own, may not be sufficient to be deleterious to renal function up to 3 months. This being said, we cannot entirely exclude especially in absence of a phenotype in the Bbs10 fl/fl; Cadh16Cre+/− that this lack of impact on the renal physiology could be due to a heterogeneous and variable activity of the Cre in vivo; a proof that will require further investigations.
On the other hand Bbs10−/− mice developed progressive obesity associated with hyperphagia and hyperleptinemia. The absence of hypertrophic adipose tissue at 3 months in both Bbs10−/− and Bbs12−/− mice does not support the classical leptin resistance theory leading to obesity in the BBS (32). In fact, our results follow the same line of thought that was suggested by another group studying the Bbs4−/− mice. The authors demonstrated that leptin resistance was not a constitutive defect but seemingly an acquired one appearing as the mice were ageing [13]. Besides, we and others have also shown that the inactivation of BBS genes was favouring pre-adipocyte lineage [14, 23] and adipogenesis [24]. These effects could explain the observed hyperplastic adipose tissue as well as the higher circulating levels of leptin observed in Bbs10−/− mice (Fig. 2d); a phenotype observed in all BBS mouse models and in human patients (15, 16, 31, 30). Moreover, the 3-month-old Bbs10−/− mice were not hyperglycaemic but exhibited a significant delay in bringing their glucose levels back to normal following GTT (Fig. 2f). Simultaneously, these mice showed insulin sensitivity, indicating that the delay in glucose handling observed with the GTT was not related to insulin resistance. Interestingly, it has been described that leptin could be as effective as insulin in controlling glucose levels [25]. It is, therefore, conceivable that the delay in glucose handling observed in the Bbs10−/− mice could be due to the hyperleptinemia; a hypothesis that warrants further investigations with targeted tissue BBS inactivation. In this present study, 3-month-old mice were used to clearly demonstrate that in absence of obesity Bbs10 inactivation has no major impact on renal function. Although it might seem interesting to have older mice to investigate the effect of Bbs10 inactivation at later stages, it then becomes difficult to dissect the exact contribution of Bbs inactivation due to the effects of ageing itself.
Coincidently, the Bbs10−/− mice suffered from RP. The retinal degeneration observed in the Bbs10−/− was similar to the other previous Bbs mouse models (33, 34, 29, 27, 19), characterized by the loss of photoreceptors resulting in a thinning of the retina and a progressive flattening of the ERGs. Of note, is the presence of connecting cilium (Fig. 3c). With the complete photoreceptor structure, the Bbs10−/− retina highlights the fact that BBS10, as was the case for BBS12 [26], is not required for ciliogenesis. The observed defect is probably related to an impaired ICT from the inner segment to the outer segment.
BBS renal phenotype is a mix of different defects of the nephron correlating with the variable human renal phenotype. We, therefore, investigated the effect of a total knockout condition versus a specific renal epithelial inactivation. In the Bbs10−/− mice, the glomeruli suffered from the absence of the primary and secondary podocyte structures (Fig. 4b) correlating with elevated albuminuria (Fig. 4c). Accordingly, no such glomerulopathy was present in the Bbs10 fl/fl; Cadh16Cre+/− mice (Fig. 5f, g), as the Cre activity was limited to the renal epithelial regions (Figure S2B). The primary cilium is known to play a role in development of the podocytes and that later in life, the podocytes lose their ciliated status as the flow gets stronger to prevent excessive polycystin-mediated calcium signalling [27]. As the observed structural defects in the Bbs10-deprived podocytes seemingly bear a developmental aspect and not an acquired one, we hypothesized that BBS10 is crucial for proper podocyte development. Surprisingly, human BBS patients do not present overt early-onset glomerulopathy but do exhibit decreasing glomerular function with age [28, 29]. This discrepancy could result from inherent differences between species as exemplified by the fact that rodent podocytes seem to be ciliated only during development [27] whereas human podocytes remain ciliated in fully differentiated and functional podocytes [30, 31].
Moreover, cyst was undetectable in both Bbs10−/− and the Bbs10 fl/fl; Cadh16Cre+/− kidneys (Figs. 4b–f, 5f–h and Additional file 5: Figure S4). This feature shared with other BBS chaperone KO mice, namely the Bbs12−/−, suggests that chaperone BBS inactivation does not impact the planar cell polarization (PCP) of the developing kidney like other ciliopathies. In addition, proper PCP of the Bbs10-deprived renal epithelial cells was further evidenced by the presence of primary cilia on the apical side of the epithelium (Figs. 4e, 5d) combined with the AQP2 apical localisation (Figs. 4h, 5e). Interestingly, only Bbs2−/− and Bbs4−/− mice suffered from late-onset cystogenesis which was secondary and uniquely associated to obesity [32]. Altogether, these data highlight that cyst formation is not, per se, intrinsically correlated to BBS gene inactivation but could nevertheless favour cystogenesis when combined with BBS-induced obesity. Besides cytoplasmic vacuoles, an established parameter for cellular stress of the renal epithelium [33] was observed in the Bbs10−/− renal epithelial cells but not in the Bbs10 fl/fl; Cadh16Cre+/− ones (Figs. 4g, 5h). These highlight the prerequisite of multiple defects to induce detectable renal defects.
Finally, polyuria, the most prominent human renal phenotype in BBS was observed in the Bbs10−/− and not in Bbs10 fl/fl; Cadh16Cre+/− mice. Given that AVPR2 is localized in the primary cilium of renal epithelial cells and that BBS protein inactivation in renal cells impairs their capacity to respond to AVP and activate their luminal AQP2 in vitro [15], we measured the protein levels of AQP2, AQP3 and AVPR2. No difference in expression levels of these proteins were found in both tested models. This indicates that polyuria in the Bbs10−/− mice is not linked to a local decrease in expression levels of these proteins (Fig. 6b, c, f, g). The fact that the AVP levels were always high in the Bbs10−/− mice, irrespective of the fluid intake condition (Fig. 6d), highlights an AVP-resistant status in Bbs10−/− mice; an effect which was absent in the Bbs10 fl/fl; Cadh16Cre+/− mice (Fig. 6h). Based on these findings, we hypothesized that the BBS-induced polyuria is not simply related to the absence of BBS10 in the renal epithelium but is the result of more complex interactions between several pathways being impacted in the Bbs10−/−. These could be linked to the hypothalamic–pituitary axis sensing osmolality and secreting AVP compared to the other ciliated cells.
Conclusions
The data presented herein describe the phenotype of a new mouse model for one of the most commonly mutated genes in BBS human patients, namely BBS10, with special emphasis in the kidney phenotype. Our findings show that deletion of Bbs10 is able to recapitulate most of the clinical BBS features, whereas the Bbs10 fl/fl; Cadh16Cre+/− did not induce any detectable defect. Overall, these results highlight once more the complexity of renal dysfunction characterizing this emblematic ciliopathy and like for the study of the origins of obesity, an integrative approach is required to understand and ultimately cure these genetic disorders.
Authors’ contributions
NC performed the renal phenotyping experiments, the analysis of the data and wrote the paper, MJS performed the metabolic phenotyping, analysed the data and wrote the paper, XY, EH, CO, FC, DL generated the mice, performed the retinal phenotyping and the morphological phenotyping. CS performed the real-time experiments. MJR, BM and HD analysed the data, wrote the manuscript, VM conceived and coordinated the experiments, analysed the data and wrote the manuscript. All authors have been involved in drafting the manuscript and in its revision. All authors read and approved the final manuscript.
Acknowledgements
We thank the Mouse Clinic Institute for generating the Bbs10 fl/fl mouse model. We are grateful to Jean-Louis Mandel for his constant support and Eric Robinet from the Institute of Virology, INSERM_U1110 for all the technical support. This project was supported by the ANR Calls 2006, 2009, UNADEV, Retina France and by the National Institute for Health and Medical Research funding (INSERM).
Compliance with ethical guidelines
Competing interests The authors declare that they have no competing interests.
Abbreviations
- BBS
Bardet–Biedl syndrome
- BBS10
Bardet–Biedl syndrome gene 10
- KO
knock out
- ERGs
scotopic electroretinograms
- GTT
glucose tolerance test
- ICT
intraciliary transport
- IS
inner segment
- ITT
insulin tolerance test
- ONL
outer nuclear layer
- OS
outer segment
- RP
retinitis pigmentosa
- T.E.M
transmission electron microscopy
- AQP2
aquaporin 2
- AQP3
aquaporin 3
- AVP
arginine vasopressin
- AVPR2
arginine, vasopressin receptor 2
Additional files
Supporting information: Methods; Additional Table 1: list of primers used for RT-qPCR; Additional Table 2: List of antibodies used. Supplementary references.
Figure S1. Schematic representation of the DNA construct for the Knock out Allele.
Figure S2. Bbs10 fl/fl ; Cadh16Cre +/− mice model. (A) Immunostaining of Cre recombinase on kidney sections of WT and Bbs10 fl/fl ; Cadh16Cre +/− mice counterstained with DAPI. (B) Fluorescence images of kidney sections from RosaTomatoeGFP +/− ; Cadh16Cre +/− mice. Cells expressing Cadh16-Cre lineage marker GFP (green) are indicated with arrows. Scale bars: 200 μm (C) Growth curve of Bbs10 fl/fl ; Cadh16Cre +/− and control littermates (n = 8, mean ± SEM). (D) Relative mRNA expression of Bbs10 gene in kidneys of Bbs10 fl/fl and Bbs10 +/+. Reference gene: Gapdh (n = 4, mean ± SEM).
Figure S3. Apoptosis of photoreceptors in Bbs10 −/− mice. Immunofluorescence of 7 µm thick retinal sections from Bbs10 +/+ and Bbs10 −/− mice. Fluorescence signal in green correspond to TUNEL signal and in blue the nuclei counterstained with DAPI.
Figure S4. Transmission Electron Microscopy of kidney tubular cells from Bbs10 +/, Bbs10 −/− and Bbs10 fl/fl ; Cadh16Cre +/− mice. Scale bars: 5 µm and 2 µm.
Figure S5. Immunodetection of BBS10 in the Bbs10 fl/fl ; Cadh16-Cre +/− tubular region. (A) Representative epifluorescence picture of BBS10-immunostained cryosection of a distal tubule in the medullary region of mice with the indicated genotype. (B) 3D images of immunostained distal tubules against BBS10 for the indicated genotype from 3-month-old male mice generated from a Z-stack of epifluorescence pictures including the image showed in (A).
Footnotes
Noëlle Cognard, Maria J Scerbo and Cathy Obringer contributed equally to the manuscript
Contributor Information
Noëlle Cognard, Email: ncognard@unistra.fr.
Maria J Scerbo, Email: jscerbo@unistra.fr.
Cathy Obringer, Email: c.obringer@unistra.fr.
Xiangxiang Yu, Email: xiangxiang.yu@etu.unistra.fr.
Fanny Costa, Email: fcosta@unistra.fr.
Elodie Haser, Email: ehaser@unistra.fr.
Dane Le, Email: dane.le@etu.unistra.fr.
Corinne Stoetzel, Email: cstoetzel@unistra.fr.
Michel J Roux, Email: mjroux@igbmc.fr.
Bruno Moulin, Email: bmoulin@me.com.
Hélène Dollfus, Email: dollfus@unistra.fr.
Vincent Marion, Email: vincent.marion@unistra.fr.
References
- 1.Mueller WM, Gregoire FM, Stanhope KL, Mobbs CV, Mizuno TM, Warden CH, et al. Evidence that glucose metabolism regulates leptin secretion from cultured rat adipocytes. Endocrinology. 1998;139(2):551–558. doi: 10.1210/endo.139.2.5716. [DOI] [PubMed] [Google Scholar]
- 2.Forsythe E, Beales PL. Bardet–Biedl syndrome. Eur J Human Genet EJHG. 2013;21(1):8–13. doi: 10.1038/ejhg.2012.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mockel A, Perdomo Y, Stutzmann F, Letsch J, Marion V, Dollfus H. Retinal dystrophy in Bardet–Biedl syndrome and related syndromic ciliopathies. Prog Retin Eye Res. 2011;30(4):258–274. doi: 10.1016/j.preteyeres.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 4.Badano JL, Mitsuma N, Beales PL, Katsanis N. The ciliopathies: an emerging class of human genetic disorders. Annu Rev Genomics Hum Genet. 2006;7:125–148. doi: 10.1146/annurev.genom.7.080505.115610. [DOI] [PubMed] [Google Scholar]
- 5.Cheung CC, Clifton DK, Steiner RA. Proopiomelanocortin neurons are direct targets for leptin in the hypothalamus. Endocrinology. 1997;138(10):4489–4492. doi: 10.1210/endo.138.10.5570. [DOI] [PubMed] [Google Scholar]
- 6.Nishimura DY, Fath M, Mullins RF, Searby C, Andrews M, Davis R, et al. Bbs2-null mice have neurosensory deficits, a defect in social dominance, and retinopathy associated with mislocalization of rhodopsin. Proc Natl Acad Sci USA. 2004;101(47):16588–16593. doi: 10.1073/pnas.0405496101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abd-El-Barr MM, Sykoudis K, Andrabi S, Eichers ER, Pennesi ME, Tan PL. Impaired photoreceptor protein transport and synaptic transmission in a mouse model of Bardet–Biedl syndrome. Vision Res. 2007;47(27):3394–3407. doi: 10.1016/j.visres.2007.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davis RE, Swiderski RE, Rahmouni K, Nishimura DY, Mullins RF, Agassandian K, et al. A knockin mouse model of the Bardet–Biedl syndrome 1 M390R mutation has cilia defects, ventriculomegaly, retinopathy, and obesity. Proc Natl Acad Sci USA. 2007;104(49):19422–19427. doi: 10.1073/pnas.0708571104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mykytyn K, Mullins RF, Andrews M, Chiang AP, Swiderski RE, Yang B, et al. Bardet–Biedl syndrome type 4 (BBS4)-null mice implicate Bbs4 in flagella formation but not global cilia assembly. Proc Natl Acad Sci USA. 2004;101(23):8664–8669. doi: 10.1073/pnas.0402354101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fath MA, Mullins RF, Searby C, Nishimura DY, Wei J, Rahmouni K, et al. Mkks-null mice have a phenotype resembling Bardet–Biedl syndrome. Hum Mol Genet. 2005;14(9):1109–1118. doi: 10.1093/hmg/ddi123. [DOI] [PubMed] [Google Scholar]
- 11.Feuillan PP, Ng D, Han JC, Sapp JC, Wetsch K, Spaulding E, et al. Patients with Bardet–Biedl syndrome have hyperleptinemia suggestive of leptin resistance. J Clin Endocrinol Metab. 2011;96(3):E528–E535. doi: 10.1210/jc.2010-2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rahmouni K, Fath MA, Seo S, Thedens DR, Berry CJ, Weiss R, et al. Leptin resistance contributes to obesity and hypertension in mouse models of Bardet–Biedl syndrome. J Clin Investig. 2008;118(4):1458–1467. doi: 10.1172/JCI32357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berbari NF, Pasek RC, Malarkey EB, Yazdi SM, McNair AD, Lewis WR, et al. Leptin resistance is a secondary consequence of the obesity in ciliopathy mutant mice. Proc Natl Acad Sci USA. 2013;110(19):7796–7801. doi: 10.1073/pnas.1210192110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marion V, Stoetzel C, Schlicht D, Messaddeq N, Koch M, Flori E, et al. Transient ciliogenesis involving Bardet–Biedl syndrome proteins is a fundamental characteristic of adipogenic differentiation. Proc Natl Acad Sci USA. 2009;106(6):1820–1825. doi: 10.1073/pnas.0812518106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Imhoff O, Marion V, Stoetzel C, Durand M, Holder M, Sigaudy S, et al. Bardet–Biedl syndrome: a study of the renal and cardiovascular phenotypes in a French cohort. Clin J Am Soc Nephrol. 2011;6(1):22–29. doi: 10.2215/CJN.03320410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Putoux A, Attie-Bitach T, Martinovic J, Gubler MC. Phenotypic variability of Bardet–Biedl syndrome: focusing on the kidney. Pediatric Nephrol. 2012;27(1):7–15. doi: 10.1007/s00467-010-1751-3. [DOI] [PubMed] [Google Scholar]
- 17.Nauli SM, Alenghat FJ, Luo Y, Williams E, Vassilev P, Li X, et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet. 2003;33(2):129–137. doi: 10.1038/ng1076. [DOI] [PubMed] [Google Scholar]
- 18.Gerdes JM, Katsanis N. Ciliary function and Wnt signal modulation. Curr Top Dev Biol. 2008;85:175–195. doi: 10.1016/S0070-2153(08)00807-7. [DOI] [PubMed] [Google Scholar]
- 19.Shao X, Somlo S, Igarashi P. Epithelial-specific Cre/lox recombination in the developing kidney and genitourinary tract. J Am Soc Nephrol JASN. 2002;13(7):1837–1846. doi: 10.1097/01.ASN.0000016444.90348.50. [DOI] [PubMed] [Google Scholar]
- 20.Schroder JM, Larsen J, Komarova Y, Akhmanova A, Thorsteinsson RI, Grigoriev I, et al. EB1 and EB3 promote cilia biogenesis by several centrosome-related mechanisms. J Cell Sci. 2011;124(Pt 15):2539–2551. doi: 10.1242/jcs.085852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cardenas-Rodriguez M, Osborn DP, Irigoin F, Grana M, Romero H, Beales PL, et al. Characterization of CCDC28B reveals its role in ciliogenesis and provides insight to understand its modifier effect on Bardet–Biedl syndrome. Hum Genet. 2013;132(1):91–105. doi: 10.1007/s00439-012-1228-5. [DOI] [PubMed] [Google Scholar]
- 22.Shao X, Johnson JE, Richardson JA, Hiesberger T, Igarashi P. A minimal Ksp-cadherin promoter linked to a green fluorescent protein reporter gene exhibits tissue-specific expression in the developing kidney and genitourinary tract. J Am Soc Nephrol JASN. 2002;13(7):1824–1836. doi: 10.1097/01.ASN.0000016443.50138.CD. [DOI] [PubMed] [Google Scholar]
- 23.Aksanov O, Green P, Birk RZ. BBS4 directly affects proliferation and differentiation of adipocytes. Cell Mol Life Sci CMLS. 2014;71(17):3381–3392. doi: 10.1007/s00018-014-1571-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marion V, Mockel A, De Melo C, Obringer C, Claussmann A, Simon A, et al. BBS-induced ciliary defect enhances adipogenesis, causing paradoxical higher-insulin sensitivity, glucose usage, and decreased inflammatory response. Cell Metab. 2012;16(3):363–377. doi: 10.1016/j.cmet.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 25.Wang MY, Chen L, Clark GO, Lee Y, Stevens RD, Ilkayeva OR, et al. Leptin therapy in insulin-deficient type I diabetes. Proc Natl Acad Sci USA. 2010;107(11):4813–4819. doi: 10.1073/pnas.0909422107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mockel A, Obringer C, Hakvoort TB, Seeliger M, Lamers WH, Stoetzel C, et al. Pharmacological modulation of the retinal unfolded protein response in Bardet–Biedl syndrome reduces apoptosis and preserves light detection ability. J Biol Chem. 2012;287(44):37483–37494. doi: 10.1074/jbc.M112.386821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ichimura K, Kurihara H, Sakai T. Primary cilia disappear in rat podocytes during glomerular development. Cell Tissue Res. 2010;341(1):197–209. doi: 10.1007/s00441-010-0983-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marion V, Schlicht D, Mockel A, Caillard S, Imhoff O, Stoetzel C, et al. Bardet–Biedl syndrome highlights the major role of the primary cilium in efficient water reabsorption. Kidney Int. 2011;79(9):1013–1025. doi: 10.1038/ki.2010.538. [DOI] [PubMed] [Google Scholar]
- 29.Parfrey PS, Davidson WS, Green JS. Clinical and genetic epidemiology of inherited renal disease in Newfoundland. Kidney Int. 2002;61(6):1925–1934. doi: 10.1046/j.1523-1755.2002.00305.x. [DOI] [PubMed] [Google Scholar]
- 30.Saleem MA, O’Hare MJ, Reiser J, Coward RJ, Inward CD, Farren T, et al. A conditionally immortalized human podocyte cell line demonstrating nephrin and podocin expression. J Am Soc Nephrol JASN. 2002;13(3):630–638. doi: 10.1681/ASN.V133630. [DOI] [PubMed] [Google Scholar]
- 31.Coward RJ, Foster RR, Patton D, Ni L, Lennon R, Bates DO, et al. Nephrotic plasma alters slit diaphragm-dependent signaling and translocates nephrin, Podocin, and CD2 associated protein in cultured human podocytes. J Am Soc Nephrol JASN. 2005;16(3):629–637. doi: 10.1681/ASN.2004030172. [DOI] [PubMed] [Google Scholar]
- 32.Guo DF, Beyer AM, Yang B, Nishimura DY, Sheffield VC, Rahmouni K. Inactivation of Bardet–Biedl syndrome genes causes kidney defects. Am J Physiol Renal Physiol. 2011;300(2):F574–F580. doi: 10.1152/ajprenal.00150.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Z, Havasi A, Gall J, Bonegio R, Li Z, Mao H, et al. GSK3beta promotes apoptosis after renal ischemic injury. J Am Soc Nephrol JASN. 2010;21(2):284–294. doi: 10.1681/ASN.2009080828. [DOI] [PMC free article] [PubMed] [Google Scholar]