

Abstract
Qualitative alterations or abnormal expression of microRNAs (miRNAs) in colorectal cancer has mainly been demonstrated in primary tumors. The miRNA expression profiles in 78 samples from 46 patients were analyzed to identify changes in miRNA expression level among normal colon mucosa, primary tumor and liver metastasis samples. Using this dataset, we describe the interplay of miRNA groups in regulating pathways that are important for tumor development. Here we describe in details the contents and quality controls for the miRNA expression and clinical data associated with the study published by Pizzini and colleagues in the BMC Genomics in 2013 (Pizzini et al., 2013). Data are deposited in GEO database as GSE35834 series.
Keywords: microRNA, Microarray, Colorectal cancer, Metastasis
| Specifications | |
|---|---|
| Organism/cell line/tissue | Homo sapiens |
| Strain(s) | Patient's normal mucosa, colorectal tumors and liver metastasis |
| Sequencer or array type | Affymetrix GeneChip miRNA Array 2.0 |
| Data format | Raw data: CEL files, normalized data: SOFT, MINIML, TXT and RData |
| Experimental factors | Liver metastases vs primary colorectal tumor vs normal mucosa |
| Experimental features | We performed a microarray experiment. After quality control and normalization of data, we identified DEMs (differentially expressed miRNAs) involved in colorectal cancer progression. |
| Consent | All patients gave their written informed consent before study entry. |
| Sample source location | Surgery Unit, Department of Surgery, Oncology and Gastroenterology – University of Padova (PD) – ITALY |
Direct link to deposited data
Experimental design, materials and methods
Study population and clinical data
For this study [1], 46 patients with sporadic colorectal adenocarcinomas (CRC), who underwent surgery at the University of Padova (Surgery Unit, Department of Surgery, Oncology and Gastroenterology) between March 1994 and September 2008, were selected from the institutional CRC database. Patients with a known history of a hereditary colorectal cancer syndrome were excluded. The Ethics Committee of the University Hospital of Padova approved the study. All patients provided written informed consent. Enrolled patients did not receive any neo-adjuvant treatment. Table 1 lists the main patient and tumor characteristics. Normal mucosa samples were taken at a minimum distance of 10 cm from the tumor site. All samples were immediately snap-frozen in liquid nitrogen and stored at − 80° until use.
Table 1.
Patients data.
| Characteristics | |
|---|---|
| No of patients (n) | 46 |
| Age (years, mean ± s.d.) | 60,7 ± 10,2 |
| Sex | |
| Female | 17 |
| Male | 29 |
| Tumor site | |
| Cecum, colon ascending, transverse colon | 13 |
| Splenic [left] flexure, colon descending, sigmoid colon | 20 |
| Rectum | 13 |
| TNM stage | IV |
| Liver metastasis | |
| Synchronous | 39 |
| Metachronous | 7 |
miRNA expression data
We selected 78 samples comprising 23 normal colon mucosa (N), 31 primary tumors (T) and 24 liver metastases (M). This dataset included 24 samples belonging to 8 patients with three matched samples (T, N and M from the same patient) as detailed in Table 2.
Table 2.
Sample set description for miRNA array dataset. Column two indicates the number of patients for which we obtained paired data for different tissue types combinations; last column reports the total number of samples for each tissue type.
| Match type | Number of patients | Tissue type | Number of samples |
|---|---|---|---|
| N-T-M | 8 | N | 23 |
| N-T | 7 | ||
| T-M | 8 | T | 31 |
| M-N | 2 | ||
| N | 6 | M | 24 |
| T | 8 | ||
| M | 6 | Total | 78 |
| Total | 45 |
7 μm sections from each tissue sample were prepared using a Leica CM 1950 cryostat (Leica Microsystems, Wetzlar, Germany) and hematoxylin and eosin stained; sections of each specimen were prepared and re-evaluated by one experienced pathologist; only samples with more than 80% of vital tumor tissue were considered for RNA extraction in toto. Laser microdissection was performed on a few frozen samples of primary tumors and metastases with a proportion of neoplastic cells lower than 80% using LMD-6000 Laser Microdissection System (Leica Microsystems, Wetzlar, Germany). Total RNA from samples was isolated using Trizol (Life Technology Corp, Carlsbad, CA, USA) according to manufacturer's instructions. RNA concentration was quantified on a NanoDrop 1000 spectrophotometer (NanoDrop Technologies, Waltham, MA, USA) and RNA quality was evaluated by RNA 6000 Nano LabChip (Agilent Technologies, Santa Clara, CA, USA) on an Agilent 2100 Bioanalyzer. Samples with RNA integrity number (RIN) < 6 were excluded.
miRNA microarray hybridization was performed from total RNA with the Affymetrix GeneChip miRNA Array 2.0 (Affymetrix Santa Clara, CA, USA). Briefly, 100 ng of total RNA from each sample was labeled with the FlashTag Biotin RNA Labeling Kit (Genisphere, Hatfield, PA, USA), hybridized according to manufacturer's instructions, and then scanned with an Affymetrix GCS 3000 7G scanner.
Quality control and normalization
A first quality control check was performed on miRNAs arrays, with Affymetrix® Expression Console™ software (v.1.0) to determine the success of hybridizations. miRNA expression measure was reconstructed from. cel files by using the Robust Multichip Average (RMA) method implemented in the Bioconductor R package affy [2], [3].
Quality control of samples was carried out with R software. Three descriptive plots (MA-plot, NUSE plot and RLE plot) were generated for each sample using a customized version of the Array Quality Metrics Bioconductor package [4].
MA-plots evaluate the dependency between intensity levels and expression ratios comparing expression data coming from one chip to a pseudo-median chip of the dataset. The shape of the point cloud and the loess curve interpolating the cloud is examined under the hypothesis that the majority of points should stay near the x-axis and loess curve should be straight and parallel to the x-axis. RLE (Relative Log Expression) plots are box plots of values computed for each probeset by comparing the expression value on each array against the median expression value for that probeset across all arrays. Potential outlier chips are identified as boxes not centered on 0 or more spreadout than the others.
NUSE plots (Normalized Unscaled Standard Error) are box plots of normalized standard errors obtained by fitting probe linear models as in RMA. An array with elevated standard errors relative to the other arrays is typically of suspect quality. Samples that were considered of low quality were excluded from further analysis. miRNAs detected in fewer than 20 samples were discarded, without filtering out miRNAs undetected only in one sample class.
Data are available at the GEO database [5] as GSE35834 series.
Basic analysis
Unsupervised clustering
Unsupervised hierarchical cluster analysis was performed on selected miRNAs expression data, using Pearson Correlation-based distance and average clustering. Normal samples clustered together and were relatively well separated from T and M samples. Considerable per-patient pairing of T and M samples was observed in the dendrogram (Fig. 1), in which triplets and pairs of samples from the same patient are shown in the same color. In roughly 25% of patients, the M samples were more similar to the T from which it derives, rather than to the M samples of other patients (20% of per-patient sample pairing, in miRNA-based heatmap).
Fig. 1.

Sample classification and heatmap based on 309 miRNAs expression profile. Color-coding of samples reported in three different lines refers to different information. First line indicates tissue type (N, T and M) as shown in the legend. The two lines below indicate the per-patient matching of samples, separately for triples (upper line) and couples (lower line) of samples from the same patient (i.e. samples from the same patient are in the same color).
Paired vs unpaired comparisons
A Significance Analysis of Microarrays (SAM) [6] with unpaired and paired two-class design was performed in parallel to identify differences in miRNA expression between groups of N, T and M samples.
In the larger unpaired dataset, we identified 62, 63 and 11 DEMs (Differentially Expressed miRNAs) in T vs N, M vs N and M vs T comparisons, respectively (Supplementary Table 1). Several miRNAs were significantly modulated in more than one contrast. Only 5 miRNAs varied merely when unpaired M and T samples were compared (miR-146a, miR-15a, miR-15b, miR-196a, miR-708). Of 53 DEMs shared by at least two unpaired comparisons, 25 were always under- and 26 over-expressed, whereas two miRNAs did not follow the same trend in the various comparisons. In both T vs N and M vs T contrasts, we observed a comparable number of up- and down-modulated DEMs: 29 and 33 were respectively up- and down-modulated in T vs N; as against 5 and 6 in M vs T. Regarding global miRNA expression, when we considered the distribution of all DEM expression levels measured in N, T and M, again we could not find any significant differences between the groups (mean values 6.10, 5.95 and 6.01, respectively; p-value of pairwise mean equality t-test > 0.7).
Differentially expressed miRNA in pairwise group comparisons, involving groups of samples matched per patient (e.g., T vs N samples matched per patient) was calculated with SAM using a two-class paired design. The cut-off for significance (determined by tuning parameter delta) corresponded to a false discovery rate (FDR) < 0.01. Sample subsets matched by patient with miRNA expression data were considered for paired comparisons (e.g., subset of T vs N samples, with T and N from the same patient). We found 34, 38 and 5 DEMs, respectively in T vs N, M vs N and M vs T comparisons. We found hsa-miR-139-5p, hsa-miR-210, hsa-miR-150, hsa-miR-100 and hsa-miR-122 as miRNAs involved in M vs T comparison.
Most DEMs identified with the paired test were confirmed with the unpaired test conducted on the larger dataset, as shown in Fig. 2 (top panel) which, for each comparison, gives the numbers of DEM obtained with paired and unpaired designs, and intersections thereof.
Fig. 2.

Venn diagrams of intersections among DEMs obtained with differing contrasts and methods. Top: overlap of DEMs in same contrast (e.g., T vs N) with paired and unpaired tests; bottom: overlap among DEMs obtained by the same test, with differing contrasts.
DEMs between tumor and normal mucosa include ones previously described as members of a “signature” common to various types of solid tumors. Many of them have also been implicated in the molecular and biological processes driving tumorigenesis in CRC.
miRNAs involved in EMT
Of 82 miRNAs modulated during tumor progression, 22 were involved in EMT (Epithelial to Mesenchymal Transition), a critical step which drives tumor metastasis [7]: 19 were differentially expressed in T vs N and one in M vs T, and one was common to both comparisons. We observed that the expression of many DEMs involved in EMT was modulated in tumor development (T vs N comparison) and then remained stable or at a similar level in metastasis.
Fig. 3 shows the expression profiles of 22 miRNAs involved in EMT which were differentially expressed in TN and/or MT comparisons.
Fig. 3.

Expression profiles in considered sample classes of 22 miRNAs reportedly involved in EMT that are differentially expressed in the TN and/or MT comparisons.
miR-10b and potential association with survival
We investigated the association between the expression levels of 26 DEMs (present in reconstructed post-transcriptional regulatory networks) in biopsies obtained from distinct primary [n = 26] or metastatic [n = 20] colorectal cancers and patients' disease-specific survival (interval between diagnosis of primary or metastatic disease and death by disease or last follow-up).
Given the relatively low sample size, only univariate survival analysis was performed, and the Cox proportional hazard regression model was used, assuming a linear functional form of the covariates being assumed. The risk associated with a unit increase in miRNA levels was expressed as hazard ratio (HR) and its 95% confidence interval (CI). With Bonferroni's p-value adjustment for multiple comparisons, the alpha level of significance was set at 0.002. In order to illustrate prognosis associated with different levels of the relevant miRNAs, Kaplan–Meier survival curves were generated after dichotomizing (high vs low categories) originally continuous covariates based on the median values of miRNA expression levels. All analyses were performed with Stata/SE software (version 11.0, StataCorp LP, College Station, TX, USA). The 26 miRNAs included in the T vs N and M vs T networks were considered for survival analysis.
The expression of miR-10b measured in liver metastasis showed a statistically significant association with the survival of patients affected with stage IV CRC (hazard ratio = 1.47, 95% confidence interval = 1.23–1.75; adjusted p-value: 0.00052). The effect of miR-10b on prognosis is given in Fig. 4, which shows that patients with high levels of miR-10b expression in their metastatic disease have a shorter time to event (median survival: 8 months) compared with those with low levels (51 months). In our study, the expression levels of miR-10b measured in primary tumors had no significant impact on prognosis.
Fig. 4.
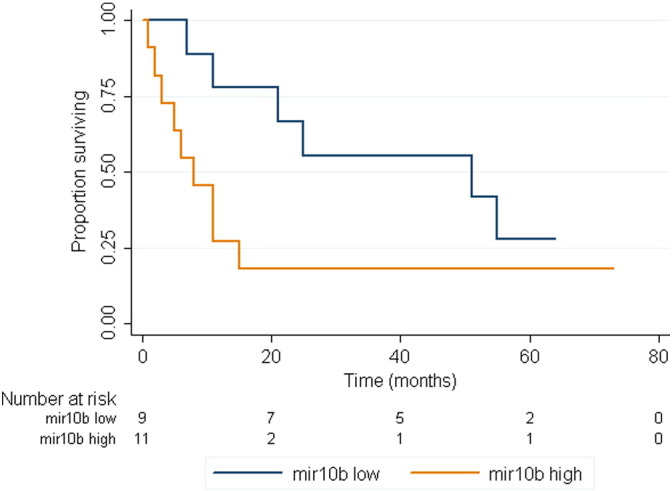
Kaplan–Meier overall survival curve was plotted based on miR-10b expression showing relationship between miR-10b expression and survival in CRC cancer.
Discussion
Several miRNAs significantly differentially expressed (DEMs) during tumor progression were identified. The whole set of samples with miRNA expression data was considered in unpaired tests (e.g., all T vs all N samples), and other sample subsets matched by patient were considered for paired comparisons (e.g., subset of T vs N samples, with T and N from the same patient). Identification of DEM confirmed that more miRNAs are modulated in N vs T than in T vs M transitions; however, DEMs in metastasis compared with primary tumors may be of great importance since they include key regulators of several processes relative to disease progression. Almost all of the miRNAs which vary their expression in the N vs T transition remain stable after metastasis development; three-quarters of miRNAs modulated in the T vs M transition are invariant in the N vs T transition [8], [9], [10], [11]. Although purely exploratory in nature, due to the relatively low number of subjects analyzed, our data support the prognostic value of miR-10b, in line with available evidence regarding the role played by this microRNA in cancer biology, according to both preclinical and clinical models. miR-10b over-expression has been associated not only with enhanced aggressiveness of malignant cells in a variety of experimental models, but also with worse prognosis in patients with breast and pancreatic carcinoma. To our knowledge, our results suggest the potential involvement of this microRNA in CRC, with special regard to the modulation of the biological behavior of metastatic disease.
The following are the supplementary data related to this article.
miRNA differentially expressed in different contrasts, according to unpaired and paired tests.
Competing interests
The authors declare that they have no competing interests.
Acknowledgments
We would like to thank Marco Agostini and Loris Bertazza for their technical assistance. This work was supported by grants from the Associazione Italiana per la Ricerca sul Cancro (AIRC-Regional Research Program 2008) to PZ; the Fondazione Cassa di Risparmio di Padova e Rovigo (CARIPARO) to PZ and SB; Italian Ministry of Health to PZ; Italian Ministry for Education, Universities and Research and SB (PRIN 2010-11, project number 2010NYKNS7), and Alleanza Contro il Cancro to PZ. MB received a grant from the Fondazione Cassa di Risparmio di Padova e Rovigo, and AB received a grant from the Associazione Italiana per la Ricerca sul Cancro (AIRC-Gruppo Italiano Malattie Mieloproliferative, Special Program Molecular Clinical Oncology 5 × 1000, project number #10005).
References
- 1.Pizzini S., Bisognin A., Mandruzzato S., Biasiolo M., Facciolli A., Perilli L., Rossi E., Esposito G., Rugge M., Pilati P., Mocellin S., Nitti D., Bortoluzzi S., Zanovello P. Impact of microRNAs on regulatory networks and pathways in human colorectal carcinogenesis and development of metastasis. BMC Genomics. 2013 Aug 29;14:589. doi: 10.1186/1471-2164-14-589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Irizarry R.A., Hobbs B., Collin F., Beazer-Barclay Y.D., Antonellis K.J., Scherf U., Speed T.P. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 3.Gentleman R., Carey V., Bates D., Bolstad B., Dettling M., Dudoit S., Ellis B., Gautier L., Ge Y., Gentry J. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kauffmann A., Gentleman R., Huber W. arrayQualityMetrics—a bioconductor package for quality assessment of microarray data. Bioinformatics. Feb 1 2009;25(3):415–416. doi: 10.1093/bioinformatics/btn647. (Epub 2008 Dec 23) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barrett T., Suzek T.O., Troup D.B., Wilhite S.E., Ngau W.C., Ledoux P., Rudnev D., Lash A.E., Fujibuchi W., Edgar R. NCBI GEO: mining millions of expression profiles—database and tools. Nucleic Acids Res. Jan 1 2005;33(Database issue):D562–D566. doi: 10.1093/nar/gki022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tusher, Tibshirani R., Chu G. Significance analysis of microarrays applied to transcriptional responses to ionizing radiation. Proc. Natl. Acad. Sci. U. S. A. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bullock M.D. MicroRNAs: critical regulators of epithelial to mesenchymal (EMT) and mesenchymal to epithelial transition (MET) in cancer progression. Biol. Cell. 2012;104(1):3–12. doi: 10.1111/boc.201100115. [DOI] [PubMed] [Google Scholar]
- 8.Luo X. MicroRNA signatures: novel biomarker for colorectal cancer? Cancer Epidemiol. Biomarkers Prev. 2011;20(7):1272–1286. doi: 10.1158/1055-9965.EPI-11-0035. [DOI] [PubMed] [Google Scholar]
- 9.Agostini M., Chim Acta Clin. miRNAs in colon and rectal cancer: a consensus for their true clinical value. Clin. Chim. Acta. 2010;411(17-18):1181–1186. doi: 10.1016/j.cca.2010.05.002. [DOI] [PubMed] [Google Scholar]
- 10.Liu M., Chen H. The role of microRNAs in colorectal cancer. J. Genet. Genomics. 2010;37(6):347–358. doi: 10.1016/S1673-8527(09)60053-9. [DOI] [PubMed] [Google Scholar]
- 11.Schee K., Fodstad O., Flatmark K. microRNAs as biomarkers in colorectal cancer. Am. J. Pathol. 2010;177(4):1592–1599. doi: 10.2353/ajpath.2010.100024. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
miRNA differentially expressed in different contrasts, according to unpaired and paired tests.