

Abstract
Introduction
Advancements in epigenetic treatments are not only coming from new drugs but from modifications or encapsulation of the existing drugs into different formulations leading to greater stability and enhanced delivery to the target site. The epigenome is highly regulated and complex; therefore it is important that off-target effects of epigenetic drugs be minimized. The step from in vitro to in vivo treatment of these drugs often requires development of a method of effective delivery for clinical translation.
Areas covered
This review covers epigenetic mechanisms such as DNA methylation, chromatin remodeling and small RNA mediated gene regulation. There is a section in the review with examples of diseases where epigenetic alterations lead to impaired pathways, with an emphasis on cancer. Epigenetic drugs, their targets and clinical status are presented. Advantages of using a delivery method for epigenetic drugs as well as examples of current advancements and challenges are also discussed.
Expert opinion
Epigenetic drugs have the potential to be very effective therapy against a number of diseases, especially cancers and neurological disorders. As with many chemotherapeutics, undesired side effects need to be minimized. Finding a suitable delivery method means reducing side effects and achieving a higher therapeutic index. Each drug may require a unique delivery method exploiting the drug's chemistry or other physical characteristic requiring interdisciplinary participation and would benefit from a better understanding of the mechanisms of action.
Keywords: Epigenetics, Epidrugs, Drug delivery, Cancer, Nanoparticles, Prodrugs, RNA
1. Introduction
Epigenetic mechanisms are known to play a critical role in cancer initiation and development and are implicated in other diseases as well such as multiple sclerosis, neurological disorders, asthma, and depression [1-4]. By exploiting epigenetic regulation, the expression of genes can be controlled or regulated without the risks associated with genetic changes in the DNA sequence itself. There are several epigenetic drugs approved or in clinic trials including 5-azacytidine, 5-aza-2′-deoxycytidine (decitabine), Suberoylanilide hydroxamic acid (SAHA), valproic acid, and entinostat [5-14].
Several methods of enhancing delivery include a delivery system such as different nanocarriers, altering the chemical structure of the drug (pro-drugs), and administration in combination with another drug are aimed at increasing effectiveness of these drugs [15]. Choosing a form of delivery vector includes taking into account properties of the drug or agent to be encapsulated such as the polarity of the drug (some popular epigenetic drugs such as decitabine and 5-azacytidine are hydrophilic), molecular weight of the drug, pharmacokinetics and biodistribution, and effective dose and time of treatment needed at the diseased site [16-18]. Other factors come into play as well such as the target area's pH, mode of cellular uptake, and the desired intracellular release location [18]. Surface modifications can be used to alter or enhance the intracellular interactions or interactions occurring prior to cellular uptake possibly with the blood when given intravenously or gut tissue and fluids when given orally [19].
This review focuses on epigenetic targets where there are drugs in the clinical or preclinical phase targeting their action (i.e. DNMT and HDAC) and the methods of delivering these drugs including delivery systems, altering the chemical structure of the drugs themselves (prodrugs) and using combination therapies to increase efficacy.
2. Epigenetics
Epigenetics is the study of changes in gene activity that do not involve changes in the nucleotide sequence of the gene. Instead of changing nucleotide sequence, gene expression is controlled by modifying nucleotide residues, chromatin structure or degrading the translated messenger ribonucleic acid (RNA). Three distinct mechanisms for epigenetic regulation have been identified. These include DNA methylation, chromatin remodeling (histone modification), small RNA (siRNA) and micro RNA (miRNA) mediated gene regulation [20-22].
Methylation of cytosine residues on DNA is a reversible or irreversible epigenetic mark that regulates several biological processes including gene silencing, imprinting and X-chromosome inactivation [23]. In mammals, DNA methylation is mediated by DNA methyl transferases (DNMT) which add methyl groups to nucleotides in CG context mostly as clusters near gene promoters (termed CpG islands) where they control the expression of the genes they are associated with [24, 25].
The remodeling of chromatin involves post-translational modifications to nucleosomes, particularly to the histone core proteins which allow or restrict access of transcription machinery to the bound DNA. Post-translational modifications of histones generally include acetylation and methylation although modifications such as ubiquitination can also occur [26]. In this section, the most common histone modifications, acetylation and methylation will be discussed in further detail.
Acetylation of histones by histone acetyltransferases (HAT) using acetyl co-A as acetyl donor decreases the positive charge on histones which decreases their affinity to DNA double helix in the nucleosome. This action makes the DNA accessible to transcription factors for gene expression. The added acetyl groups can be removed by histone deacetylases (HDAC) which leads to transcription repression due to chromatin compaction [27].
Methylation of histones occurs on the basic amino acids arginine, lysine and histidine with each experiencing different degrees of methylation [21, 28]. Methyl transferases, which use S-adenosyl methionine as methyl donor, methylate histones to generate methylation profiles that can encourage or prevent gene expression. The methylases are substrate specific and some are sensitive to the degree of methylation [23-25]. Histone methylation can be stable or removed by demethylases.
Unlike the other epigenetic mechanisms that act on just the chromatin, small RNA mediated gene regulation occurs in both the nucleus and cytoplasm. The most common example of RNA mediated gene regulation in the nucleus is the X-inactive specific transcript (Xist) mediated X-chromosome inactivation [29] which results in the random inactivation of one X-chromosome in each cell in females [30]. In the cytoplasm, gene regulation is through the activity of miRNA which are small non-coding single stranded RNAs about 20 to 25 nucleotides in length that regulate a wide range of cellular processes including cellular proliferation, cell death, and developmental processes [31, 32]. Only a small section of the human genome encodes for miRNAs but they may regulate as many as 60% of human genes due to the nature of miRNA interaction with RNA, which enable each miRNA to have several potential targets [33].
3. Diseases with epigenetic mechanisms
The different epigenetic mechanisms modulate high order DNA structure and ultimately gene expression. Stable changes in the epigenetic status of a gene or its associated histones without genetic changes or mutations can lead to diseases. In most diseases however, etiology of a disease is a combination of both epigenetic changes and genetic mutations [34]. Several diseases are implicated where malfunction in epigenetic mechanisms occur (Table 1). In this review we discuss in more detail the role of epigenetic mechanisms in cancer.
Table 1.
Epigenetic diseases and impaired pathways.*
| Disease | Epigenetic alteration | Impaired pathway |
|---|---|---|
|
| ||
| Alpha-Thalassemia X-Linked Intellectual Disability (ATRX) Syndrome | Hypomethylation | Chromatin and transcriptional deregulation [100]. |
|
| ||
| Immunodeficiency, centromere instability and Facial anomalies (ICF) syndrome | Hypomethylation | Loss of DNMT3b activity at multiple immunological and developmental genes [101]. |
|
| ||
| Multiple sclerosis | Hypermethylation | Silencing of FOXP3 [2]. |
| Hypomethylation | Increased expression of PAD2 alters myelin processing [3]. | |
|
| ||
| Asthma | Histone acetylation | Decreased inhibition of NF-κB via histone acetylation [4]. |
|
| ||
| Depression | Hypermethylation | Decreased expression of glucocorticoids[1]. |
|
| ||
| Beckwith–Wiedemann syndrome | Hypomethylation | Increased activity of KCNQ1OT1, a long non-translated RNA [102]. |
Examples of some diseases where an epigenetic malfunction is implicated. Abbreviations: DNMT3b, DNA-methyltransferase 3 beta; FOXP3, scurfin; PAD2, peptidylarginine deiminase 2; NF-kB, nuclear factor kappa-light-chain-enhancer of activated B cells.
3.1 Cancer
Changes in different epigenetic mechanisms have been associated with cancer. Of these, the methylation status of cancer is the most studied. In general, there is an alteration in the methylation status of the promoters of several tumor suppressor genes [35, 36]. Hypermethylation associated with cancer development occurs in the CpG islands of tumor suppressor genes such as retinoblastoma tumor-suppressor gene (Rb) and breast cancer type 1 susceptibility protein (BRCA1), and in genes involved in regulation of the cell cycle, DNA repair and induction of apoptosis. On the other hand, genes that prevent apoptosis and enhance cell survival are hypomethylated [37, 38]. Aberrant changes in activities of HAT and HDAC can lead to cancer.
There are widespread changes in the expression of miRNA in cancer compared to adjacent normal tissues with miRNA expression profiling of tumors being used in the diagnosis, staging and monitoring of progression and response to treatments [39, 40]. MicroRNAs that have been shown to be down regulated in cancer include the let-7 family of miRNAs which target RAS oncogenes [41, 42]. The reduced expression of these miRNAs results in increased tumor growth and decreased survival [43, 44].
4. Drugs with epigenetic mechanisms
Several classes of drugs that target epigenetic mechanisms are in preclinical or clinical trials (Table 2). Most of these drugs target methylation (inhibit DNMT) or deacetylation (inhibit HDAC). Listed below are the activities of some of the common epigenetic inhibitors.
Table 2.
Epigenetic drugs, target, and clinical status.*
| Drug class | Example | Target | Clinical status |
|---|---|---|---|
| Aza-nucleosides | 5-Azacytidine | DNMT |
|
| 5-Aza-2′-deoxycytidine (Decitabine) | DNMT | ||
| Hydroxamic acids | Suberoylanilide hydroxamic acid (SAHA) | HDAC | |
| Belinostat | HDAC | ||
| Panobinostat | HDAC | ||
| Depsipeptides | Romidepsin | HDAC | |
| Short-chain fatty acids | Valproic acid | HDAC |
|
| Benzamide | Entinostat | HDAC |
Drugs with epigenetic mechanisms including their drug class, target, and clinical status. Abbreviations: DNMT, DNA-methyltransferase; HDAC, histone deacetylase.
4.1 Demethylating agents
5-azacytidine (also known as azacytidine) is an analog to the nucleoside cytidine. Azacytidine is incorporated into DNA and RNA and reversibly inhibits DNA methyl transferase. Because azacytidine is incorporated into both DNA and RNA, it inhibits synthesis of DNA, RNA and proteins [45]. 5-Aza-2′-deoxycytidine (decitabine, DAC)) is another analog of cytidine. Decitabine is incorporated only into DNA, unlike 5-azacytidine which is incorporated into both DNA and RNA. Decitabine incorporated into DNA irreversibly bind to DNMTs leading to rapid depletion of the enzyme and subsequent hypomethylation of DNA [46].
Despite the success of epigenetic drugs such as 5-Aza-2′-deoxycytidine in clinical trials against hematologic malignancies and its antiproliferative effect in vitro, its efficacy in solid tumors has been disappointing [47]. This is due to the difference in the pharmacokinetics and pharmacodynamics of 5-Aza-2′-deoxycytidine in between solid and hematologic malignancies. 5-Aza-2′-deoxycytidine has a very high clearance rate in vivo (the half-life is 10-35 min) [48]. In addition, 5-Aza-2′-deoxycytidine rapidly degrades in acidic conditions, which most solid tumors exhibit [48, 49]. The discrepancy between the in vitro efficacy of epigenetic drugs, including 5-Aza-2′-deoxycytidine, and their in vivo efficacy in solid tumors has been attributed to the slowly dividing nature of cancer cells in vivo versus in vitro [50, 51]. Since 5-Aza-2′-deoxycytidine is S-phase specific, its activity in the tumor, when injected intravenously, may not last long enough for all the tumor cells to pass through the S-phase and thus for 5-Aza-2′-deoxycytidine to be effective. It is estimated that it takes approximately 5-15 days, depending on the tumor type and growth rate, for all the cancer cells in solid tumors to pass through S-phase [51-53]. Since 5-Aza-2′-deoxycytidine targets rapidly dividing cells such as bone marrow stem cells, its repeated high dosing to achieve therapeutic levels in solid tumors was found to have serious side effects, such as chronic myelosuppression and leukopenia [50, 54]. The toxicity of 5-Aza-2′-deoxycytidine at higher doses is due to its DNA-damaging activity rather than DNA-demethylation activity [55].
4.2 HDAC inhibitors
Inhibitors of HDAC arrest growth and induce differentiation of cancer cells. These inhibitors have also been reported to induce apoptosis in cancer cells by changing the expression of genes associated with this process. Common HDAC inhibitors include valproic acid and vorinostat. HDAC inhibitors alter the expression profiles of 2-5% of genes [27, 56, 57].
Valproic acid is an 8-carbon fatty acid that has previously been used in treating epilepsy and bipolar disorder. Valproic acid inhibits HDAC at physiological concentrations used for treating these neurological conditions. As an inhibitor of HDAC, valproic acid acts as a competitive inhibitor to acetyl groups on histone N-terminal tails by binding to the catalytic site of the enzyme [58].
Vorinostat, also known as suberoylanilide hydroxamic acid (SAHA), is an inhibitor of class I and II HDACs [59]. SAHA inhibits cell growth and is known to be better tolerated by normal cells than cancer cells [60]. As a chelator, SAHA acts by binding to the co-factor zinc at the active site of the enzyme making it unavailable for catalysis [61]. Entinostat, also known as MS-275 or SNDX-275, is a synthetic benzamide derivative that inhibits class I HDACs [62, 63]. It is being evaluated in phase I/II clinical trials for Hodgkin's lymphoma and phase III clinical trials for metastatic lung cancer [13, 14]. Entinostat has also shown promise as a pretreatment that could be used to resensitize resistant cells to chemotherapies [64].
5. Delivery of epigenetic drugs
A method of delivering these epigenetic drugs is often necessary due to fast degradation by enzymes in vivo. For instance, decitabine is rapidly degraded by cytidine deaminase which is present in high amount in tissues like liver, gut, etc [48, 65]. In addition, since it does not bind to proteins, its excretion is quite rapid, thus requiring drug to be continuously administered as an infusion [66]. Because of this, systemic drug levels drops rapidly; reaching to almost undetectable levels within 30 min once the infusion is stopped. Additional advantages of nanoparticle-based drug delivery systems may include the ability to target cancer (active, passive, and enhanced permeability and retention [EPR] effect), greater drug stability and lower drug concentrations during administration. All of these advantages have been found to result in lower systemic toxicity and a higher therapeutic index [15, 67, 68]. Targeting has been exploited in cancer therapies and can be passive or active. Passive targeting takes advantage of the EPR effect. This property of the blood vessel walls is caused by abnormal angiogenesis around a primary tumor. The abnormal and rapid formation of the vessels causes them to be ‘leaky’. Nanoparticles, liposomes, and other delivery vectors take advantage of this property and the poor lymphatic drainage characteristic of tumors to achieve greater accumulation in tumor tissue than elsewhere in the body [69, 70]. Active targeting seeks to exploit an extracellular or intracellular mechanism or the increased acidity of a tumor environment (due to increased levels of lactic acid as a product of glycolysis) [15]. Certain proteins or receptors are overexpressed in cancer cells and these can be exploited by conjugating a ligand to the nanoparticle or other delivery vector [71]. Cellular uptake of the delivery system could be by several receptor-mediated endocytotic pathways including clathrin-dependent, clathrin-independent macropinocytosis, and caveole-meditaed [15, 18]. The mechanism of uptake will depend on the size, shape, and surface characteristics such as charge and hydrophobicity of the vector [72].
There are several nanocarriers for drug delivery including polymeric nanoparticles, liposomes, dendrimers, nanogels, and biological vectors (Fig 1). Other ways to enhance the delivery of epigenetic drugs are prodrugs and using a drug combination for synergistic effect [15].
Fig. 1. Example of delivery of epigenetic therapeutics using nanoparticles.

Schematic showing two possible mechanisms of epigenetic drugs being delivered via nanoparticles. The nanoparticles are taken into the cell by pinocytosis and enter an endosome. After endosomal escape two different paths are illustrated. The nanoparticle loaded with pre-miR releases its contents in the cytoplasm and is processed by Dicer. Next, it interacts with RISC to either degrade the target mRNA or inhibit its translation. The nanoparticle loaded with HDAC inhibitor drug enters the nucleus and releases the drug. There it blocks the action of HDAC causing an increase in transcription. Abbreviations: pre-miR, precursor miRNA; RISC, RNA-induced silencing complex; HDAC, histone deacetylase inhibitor; TFC, transcription factor complex.
5.1 Nanoparticles
Nanoparticles (NPs) for drug delivery applications are usually less than 300 nm in diameter [68]. They can be prepared using natural or synthetic polymers. Polymers commonly used in NP formulations include Polyethyleneglycol (PEG), Polylactic acid (PLA), Polyglycolic acid (PGA), Poly(lactic-co-glycolic acid) (PLGA), dextran, and chitosan. NPs are often broken down into monomers or small molecular weight fragments which are removed metabolically through preexisting biological pathways or excreted. For example, PLGA is broken down into lactic acid and glycolic acid. Lactic acid goes through the tricarboxylic acid cycle and is eliminated as carbon dioxide and water [73]. Glycolic acid can be excreted through the kidney or may go through the tricarboxylic acid cycle and be eliminated as carbon dioxide and water [74]. By altering the formulation and manufacturing parameters, tight control is possible over the properties influencing degradation and release kinetics such as molecular weight, composition (lactide to glycolide ratio), hydrophobicity, and crystallinity. The release kinetics may be a first-order release or a triggered release [68]. NPs can be prone to phagocytosis resulting in decreased circulation time and decreased tumor accumulation [75]. Surface modifications (commonly PEG) and/or targeting ligands can be engineered onto the nanoparticles to increase cellular uptake, blood circulation half-life, and impact biodistribution [68].
5.2 Liposomes
Liposomes are self-assembling bilayers of lipids with an aqueous core. Their characteristics such as surface charge, functionality, and size can be controlled by altering the type of lipid or the ratio of the lipids [68]. They are often surface-modified with polyethylene glycol (PEG) to avoid their uptake and clearance by the reticuloendothelial system (RES) and to improve stability in vivo [76]. One advantage of liposomes is that they can be used to deliver both hydrophilic and hydrophobic drugs. The hydrophilic drugs get encapsulated in the inner space of the liposomes, while the hydrophobic drugs are enclosed between the two layers of lipids. The loading for liposomes tends to be less than that of other delivery methods. They also have shown instability in the bloodstream and have a rapid burst release. LiPlasome Pharma has developed non-targeted liposomes with lipids that can be degraded by phospholipase A2 (PLA2) which is up-regulated in the tumor environment. There has also been research into using amphoteric liposomes to deliver nucleic acids [68]. This could include delivering nucleic acids such as RNA sequences which work through epigenetic mechanisms. A novel formulation involving enriched liposomal carriers with encapsulated anacardic acid in the liposomal bilayer with a vitamin C gradient, loaded with mitoxantrone has been developed [77]. The epigenetic agent is the anacardic acid which has been shown to have anticancer properties. This novel formulation has multiple facets. The cytotoxicity of mitoxantrone was shown to be enhanced by the anacardic acid and the vitamin C when used on human melanoma cell lines A375 and Hs294T. However, these same two compounds – anacardic acid and vitamin C – were cytoprotective when used on a normal human fibroblast line [77].
5.3 Dendrimers
Dendrimers are globular macromolecules, about 5-10 nm across. By controlling their properties, the polymer chains can be designed to be degraded for tailored drug release and lengthened circulation time. A dendrimer is built around a core of functional monomers and layers of multifunctional monomeric units are added outwards in a stepwise fashion [78]. Amphiphilic dendrimers can self-assemble to form micelles with hydrophilic surface groups. Delivery of many therapeutics, imaging agents, and targeting agents is possible by using different functionalized monomers [68]. Dendrimers can aid in the delivery of drugs, diagnostic agents, and targeting molecules [68]. CALAA-01, a targeted transferrin-cyclodextrin-siRNA nanoparticle, targets the M2 subunit of ribonucleotide reductase. This product may be effective against solid tumors and is now in phase 1 clinical trials [79]. More studies could be done to elucidate dendrimer biodistribution so that tissue localization can be predicted and improved. The steric hinderance associated with the structure of dendrimers could be a disadvantage [80]. But, it also provides a mechanism for a cascade release, which could be useful in some circumstances such as sequential treatment with an epigenetic drug followed by a chemotherapeutic.
5.4 Nanogels
Nanogels can be formulated so that they release their encapsulated drug under specific conditions or a stimulus such as temperature, pH, magnetic fields, or biomolecule recognition [81]. Vijayaraghavalu and Labhasetwar in their study developed N-isopropylacrylamide (NIPAM)-based biodegradable nanogels which can be loaded with epigenetic drugs (Fig 2). It is known that DNA Methyltransferase 1 (DNMT1) promotes DNA methylation to maintain cancer drug resistance. Decitabine is a potent hypomethylating agent, but its effect is transient because of its short half-life in vivo (10-35 min), and accelerated degradation in acidic conditions such as those found at a tumor site [48].
Fig. 2.

Hydrodynamic diameter and particle size distribution, and transmission electron microscopic analysis of the decitabine-loaded nanogels formulation. Nanogels are synthesized using a combination of N-isopropylacrylamide (NIPAM), vinyl pyrrolidone (VP), and PEG–maleic anhydride (PEG-MA). NG-70 contains 70% NIPAM, 20% VP and 10% PEG-MA. The ratios of these three polymers were varied and nanogels formed were tested for their physical characteristics and drug loading. Composition of other nanogels is given in original publication. Figure reproduced with permission from Elsevier through RightsLink Copyright Clarence Center [82].
The efficacy of decitabine -loaded nanogels was shown in doxorubicin-resistant breast cancer cells (Fig 3), decitabine-resistant melanoma cells, and leukemia cells. The data demonstrated that decitabine in nanogel sustained DNMT1 depletion, prolonged cell arrest in the G2/M cell-cycle phase, and significantly enhanced antiproliferative effect of decitabine [82]. Decitabine-loaded nanogels potentially could be explored for treating solid-tumor because of better drug effect than with decitabine in solution which is cleared rapidly from the circulation following intravenous administration.
Fig. 3.
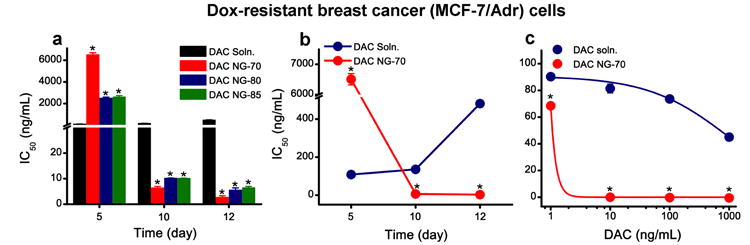
Antiproliferative effect of decitabine-loaded nanogel in doxorubicin-resistant (MCF-7/Adr) breast cancer cells. a) Comparison of IC50 of decitabine in solution vs. in different nanogel formulations over time. Cells treated with decitabine-nanogel formulations demonstrated sustained antiproliferative effect compared with cells treated with decitabine in solution. Efficacy of DAC nanogel depends on the nanogel composition, with NG-70 showing a more sustained antiproliferative effect than other formulations of nanogels. b) Comparison of IC50 of decitabine in solution vs. decitabine loaded in nanogel (NG-70), depicting the transient antiproliferative effect of decitabine in solution vs. the sustained effect with DAC nanogel. c) Dose-response curves showing the difference in efficacy of decitabine in solution and decitabine in nanogel (NG-70) at 12 d post treatment. Data are expressed as mean ± s.e.m, n = 6 *p ≤ 0.0005, #p ≤ 0.005 DAC solution vs. DAC nanogel. Figure reproduced with permission from Elsevier through RightsLink Copyright Clarence Center [82].
5.5 Biological
Biological methods of delivery include the “Nanocell” – a bacteria (∼300nm) void of DNA thereby preventing mutations and replication. This has been loaded with molecules of different solubility and charge such as doxorubicin, paclitaxel, and siRNAs. There is a potential for an immune response due to the lipopolysaccharides [83].
5.6 Prodrugs
Prodrugs are drugs that have been modified so they are inactive until some event in vivo which activates them. Epigentic drugs may be modified to increase their stability leading to slower degradation in vivo and/or the modification can better target the drug to the delivery site (intracellular or extracellular). This is often done by converting the parent drug to an ester, or manipulating the pH [15]. By converting to an ester, the variable expression of esterases in vivo can be used to cleave the prodrug activating the drug. The pH can be manipulated so that the drug is activated in the acidic conditions of tumor tissue [15]. In some cancers, folic acid receptors are overexpressed. In these cases, attaching a folic acid to the drug can enhance cellular uptake in the target cells [15, 84].
S110, a dinucleotide containing decitabine, has been shown to have decreased depletion by cytidine deaminase [65]. This is attributed to the specificity of cytidine deaminase. The S110 and decitabine were similar in their ability to inhibit DNA methylation, induce expression of the p16 tumor suppressor gene, inhibit tumor cell growth, stability in aqueous solution, and cytotoxicity. The decreased deamination by cytidine deaminase could increase bioavailability. A lower dose would lead to fewer side effects [65].
CP-4200, an elaidic acid derivative of azacytidine has been used in mouse models to demonstrate better therapeutic efficacy than azacytidine alone. This is partly due to this chemical modification causing less dependence on nucleoside transporters [85]. Rather than encapsulating the drug in a liposome, a lipophilic element such as a cholesterol or fatty acid can be attached to the drug to aid in cellular uptake.
HDAC inhibitors – psammaplin A and FK228 (Depsipeptide) – are naturally occurring prodrugs. They work epigenetically as antitumor agents and their disulfide bond is cleaved by glutathione to form an active thiol [86, 87].
5.7 Drug combinations
Enhancing the delivery could also be done by using a combination of drugs. For instance, using tetrahydrouridine (THU) in combination with decitabine to treat β-thalassemia. In this case, THU acts as a competitive inhibitor of cytidine deaminase, which is normally responsible for the rapid metabolization of decitabine in the in the intestines and liver [88]. Low doses of decitabine have been shown to reactivate γ-globin gene (HBG) expression to treat β-thalassemia [89, 90]. Vijayaraghavalu et al have shown that sequential treatment of decitabine and doxorubicin is highly synergistic in inducing cytotoxic effect in drug resistant breast cancer (MCF-7/ADR) cells than simultaneous treatment. The combination treatment could provide an effective therapeutic strategy to overcome drug resistance that can potentially minimize doxorubicin-induced cardiotoxicity because lower doses of doxorubicin may be needed to achieve tumor regression [91].
UVI5008 is a novel epigenetic therapy that independently inhibits three epigenetic targets thereby becoming a combination therapy within itself. These targets are DNMTs, HDACs, and sirtuins (a class of NAD+-dependent HDACs) [92]. In vivo experiments have shown UVI5008 is cancer cell-selective. It is also not reliant on p53, Bcl-2 modifying factor, and/or TNF-related apoptosis-inducing ligand (TRAIL), which increases the range of cancers it could be used to treat [92, 93]. Using one drug to inhibit multiple targets, rather than two or three, is beneficial as it increases ease of manufacturing and research into ADMET profiles, and conceivably reduces costs.
5.8 Other delivery methods of note
Belinostat, a histone deacetylase inhibitor, has been used in clinical trials both intravenously and through oral administration. Oral administration of a drug may be suitable to strengthen the effects on the target and to increase patient convenience. The results show that the oral route is well tolerated for high doses but further studies are necessary to determine dosing and scheduling [94].
To test the potency of an epigenetic drug there is now a cell-based assay called EPISSAY. This uses a non-malignant human breast cancer cell line – MCF10A – with a silenced triple-mutated bacterial nitroreductase (TMnfsB) fused with Red-fluorescent Protein (RFP). The RFP expression can be easily observed and is indicative of the expression of RFP-TMnfsB. The EPISSAY was used to test the potency of decitabine with and without PEGylated liposomal encapsulation. A 50% higher potency was observed when the decitabine was encapsulated in the PEGylated liposomes. The EPISSAY was also shown to be able to indicate the potency of HDAC inhibitors such as Vorinostat [95]. The promoter used in this system – a CMV promoter – had no major changes in methylation before and after treatment with decitabine. Though decitabine is usually solely referred to as a demethylating agent, there have been studies that show it can also work as an HDAC inhibitor [96-98].
6. Conclusions
This is an overview of the current epigenetic drugs and delivery methods being used in coordination. Delivery methods have been broadly defined as carriers such as nanoparticles, liposomes, dendrimers, and nanogels as well as prodrugs and drug combinations. Both epigenetic drugs and delivery methods are in relatively early stages of development and the research with them together is limited. Targeting and sustaining the effect of epigenetic drugs in combination with other drugs could be an effective strategy. There are many epigenetic targets and enzymes not mentioned in this review because no drug has been developed to target them [15]. Efforts are being made to identify these targets and to develop inhibitors/activators [99].
7. Expert Opinion
Nanoparticles, liposomes, prodrugs and other delivery methods hold the key to increasing the efficacy and lowering systemic toxicities of drugs, notably chemotherapeutics. Exploitation of target characteristics such as an acidic microenvironment and/or the altered expression of receptors on the cell surface to influence biodistribution thereby reducing off-target toxicities should. Sometimes the target is an overexpressed receptor. Ideally, in a clinical setting, the presence of these overexpressed receptors in a particular patient would be confirmed. A formulation that could successfully target an overexpressed receptor could theoretically treat metastasis as it could seek out the cancer without the aid of the EPR effect.
Successful targeting has to be paired with an increased retention time for improvement in the efficacy of the drug. For instance, a chemotherapeutic epigenetic drug needs to reach the cancer cells, be taken up by the cells, be released from their encapsulation (if there is one), and get to their intracellular target possibly by a method of endosomal escape.
The majority of epigenetic drugs are hydrophilic and therefore more difficult than other drugs to load into nanoparticles which are mostly hydrophobic. Thus it would require modification of the original method or developing totally new method depending on the original techniques to encapsulate epigenetic drugs. Other factor to consider while developing formulations is their stability in an aqueous environment.
A better understanding of epigenetic mechanisms would provide a larger base for finding novel epigenetic therapeutics. Currently, there are far more delivery methods than there are epigenetic drugs. Epigenetic drugs have potential, especially as a chemotherapy demonstrated by in vitro studies. Some epigenetic drugs are unstable and, like other chemotherapeutics, have a high systemic toxicity. This is why epigenetic drugs in particular could benefit from a delivery system.
Article highlights.
Recent advances in epigenetic treatments are often coming not from new drugs but from modifications to these drugs or encapsulation of the drugs leading to enhanced delivery to the target site.
Epigenetic drugs have the potential to be a powerful tool against a number of diseases, especially cancers. The epigenetic target may have different roles and expression throughout the body. This is why a delivery method which decreases side effects by targeting and therefore increases the therapeutic index is beneficial.
Each drug may require a delivery method exploiting the drug's chemistry or another characteristic requiring interdisciplinary collaboration and would benefit from a better understanding of the mechanisms of action.
Acknowledgments
This study described here from author's laboratory is funded by grant R01 CA149359 (to VL) from the National Cancer Institute of the National Institutes of Health.
Bibliography
- 1.McGowan PO, Sasaki A, D'Alessio AC, Dymov S, Labonté B, Szyf M, et al. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat Neurosci. 2009;12(3):342–48. doi: 10.1038/nn.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huehn J, Polansky JK, Hamann A. Epigenetic control of FOXP3 expression: the key to a stable regulatory T-cell lineage? Nat Rev Immunol. 2009;9(2):83–89. doi: 10.1038/nri2474. 02//print. [DOI] [PubMed] [Google Scholar]
- 3.Mastronardi FG, Noor A, Wood DD, Paton T, Moscarello MA. Peptidyl argininedeiminase 2 CpG island in multiple sclerosis white matter is hypomethylated. J Neurosci Res. 2007;85(9):2006–16. doi: 10.1002/jnr.21329. [DOI] [PubMed] [Google Scholar]
- 4.Adcock IM, Ito K, Barnes PJ. Histone deacetylation: an important mechanism in inflammatory lung diseases. COPD: Journal of Chronic Obstructive Pulmonary Disease. 2005;2(4):445–55. doi: 10.1080/15412550500346683. [DOI] [PubMed] [Google Scholar]
- 5.Buckstein R, Yee K, Wells RA. 5-Azacytidine in myelodysplastic syndromes: A clinical practice guideline. Cancer Treat Rev. 2011;37(2):160–67. doi: 10.1016/j.ctrv.2010.05.006. 4// [DOI] [PubMed] [Google Scholar]
- 6.Ravandi F, Alattar ML, Grunwald MR, Rudek MA, Rajkhowa T, Richie MA, et al. Phase 2 study of azacytidine plus sorafenib in patients with acute myeloid leukemia and FLT-3 internal tandem duplication mutation. Blood. 2013;121(23):4655–62. doi: 10.1182/blood-2013-01-480228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Greenberg PL, Garcia-Manero G, Moore M, Damon L, Roboz G, Hu K, et al. A randomized controlled trial of romiplostim in patients with low-or intermediate-risk myelodysplastic syndrome receiving decitabine. Leuk Lymphoma. 2013;54(2):321–28. doi: 10.3109/10428194.2012.713477. [DOI] [PubMed] [Google Scholar]
- 8.Thomas X, Dmoszynska A, Wierzbowska A, Kuliczkowski K, Mayer J, Shelekhova T, et al. Results from a randomized phase III trial of decitabine versus supportive care or low-dose cytarabine for the treatment of older patients with newly diagnosed AML. J Clin Oncol. 2011;29(15 Suppl):6504. [Google Scholar]
- 9.Rangwala S, Duvic M, Zhang C. Trends in the treatment of cutaneous T-cell Lymphoma—critical evaluation and perspectives on vorinostat. Blood Lymph Cancer Targets Ther. 2012;2:17–27. [Google Scholar]
- 10.Friday BB, Anderson SK, Buckner J, Yu C, Giannini C, Geoffroy F, et al. Phase II trial of vorinostat in combination with bortezomib in recurrent glioblastoma: a north central cancer treatment group study. Neuro-oncology. 2011:nor198. doi: 10.1093/neuonc/nor198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yoo C, Ryu MH, Na YS, Ryoo BY, Lee CW, Maeng J, et al. Phase I and pharmacodynamic study of vorinostat combined with capecitabine and cisplatin as first-line chemotherapy in advanced gastric cancer. Invest New Drugs. 2014;32(2):271–78. doi: 10.1007/s10637-013-9983-2. [DOI] [PubMed] [Google Scholar]
- 12.Braiteh F, Soriano AO, Garcia-Manero G, Hong D, Johnson MM, Silva LDP, et al. Phase I study of epigenetic modulation with 5-azacytidine and valproic acid in patients with advanced cancers. Clin Cancer Res. 2008;14(19):6296–301. doi: 10.1158/1078-0432.CCR-08-1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jona A, Younes A. Novel treatment strategies for patients with relapsed classical Hodgkin lymphoma. Blood Rev. 2010;24(6):233–38. doi: 10.1016/j.blre.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Witta SE, Jotte RM, Konduri K, Neubauer MA, Spira AI, Ruxer RL, et al. Randomized phase II trial of erlotinib with and without entinostat in patients with advanced non–small-cell lung cancer who progressed on prior chemotherapy. J Clin Oncol. 2012;30(18):2248–55. doi: 10.1200/JCO.2011.38.9411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15**.el Bahhaj F, Dekker FJ, Martinet N, Bertrand P. Delivery of epidrugs. Drug Discov Today September. 2014;19(9):1337–52. doi: 10.1016/j.drudis.2014.03.017. Overview of epigenetic targets, drugs, combined therapies, and some delivery mechanisms. [DOI] [PubMed] [Google Scholar]
- 16.Zhao M, Rudek MA, He P, Hartke C, Gore S, Carducci MA, et al. Quantification of 5-azacytidine in plasma by electrospray tandem mass spectrometry coupled with high-performance liquid chromatography. J Chromatogr B. 2004;813(1):81–88. doi: 10.1016/j.jchromb.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 17.Hua W, Ieradi T, Lesslie M, Hoffman BT, Mulvana D. Development and validation of a HILIC-MS/MS method for quantification of decitabine in human plasma by using lithium adduct detection. J Chromatogr B. 2014 doi: 10.1016/j.jchromb.2014.08.012. [DOI] [PubMed] [Google Scholar]
- 18**.Adjei IM, Sharma B, Labhasetwar V. Nanomaterial. Springer; 2014. Nanoparticles: Cellular Uptake and Cytotoxicity; pp. 73–91. Chapter on the interactions of nanoparticles with cells and cellular uptake. [DOI] [PubMed] [Google Scholar]
- 19*.Brannon-Peppas L, Blanchette JO. Nanoparticle and targeted systems for cancer therapy. Adv Drug Del Rev. 2012;64:206–12. doi: 10.1016/j.addr.2004.02.014. A review of cancer treatment methods and delivery with sections on targeting with degradable and nondegradable polymer nanoparticles. [DOI] [PubMed] [Google Scholar]
- 20*.Bird A. Perceptions of epigenetics. Nature. 2007;447(7143):396–98. doi: 10.1038/nature05913. 05/24/print. Insights into the definition of epigenetics and the potential of epigenetic regulation. [DOI] [PubMed] [Google Scholar]
- 21.Nakao M. Epigenetics: interaction of DNA methylation and chromatin. Gene. 2001 Oct 31;278(1–2):25–31. doi: 10.1016/s0378-1119(01)00721-1. [DOI] [PubMed] [Google Scholar]
- 22.Goldberg AD, Allis CD, Bernstein E. Epigenetics: A Landscape Takes Shape. Cell. 2007 Feb 23;128(4):635–38. doi: 10.1016/j.cell.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 23.Jones PA, Takai D. The Role of DNA Methylation in Mammalian Epigenetics. Science 2001. 2001 Aug 10;293(5532):1068–70. doi: 10.1126/science.1063852. [DOI] [PubMed] [Google Scholar]
- 24.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. 2012 Nat Rev Genet;13(7):484–92. doi: 10.1038/nrg3230. 07//print. [DOI] [PubMed] [Google Scholar]
- 25.Moore LD, Le T, Fan G. DNA Methylation and Its Basic Function. Neuropsychopharmacology. 2013;38(1):23–38. doi: 10.1038/npp.2012.112. 01//print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21(3):381–95. doi: 10.1038/cr.2011.22. 03//print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Richon VM, Sandhoff TW, Rifkind RA, Marks PA. Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proceedings of the National Academy of Sciences 2000. 2000 Aug 29;97(18):10014–19. doi: 10.1073/pnas.180316197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ng HH, Adrian B. DNA methylation and chromatin modification. Curr Opin Genet Dev. 1999;9(2):158–63. doi: 10.1016/s0959-437x(99)80024-0. 4// [DOI] [PubMed] [Google Scholar]
- 29.Penny GD, Kay GF, Sheardown SA, Rastan S, Brockdorff N. Requirement for Xist in X chromosome inactivation. Nature. 1996;379(6561):131–37. doi: 10.1038/379131a0. [DOI] [PubMed] [Google Scholar]
- 30.Avner P, Heard E. X-chromosome inactivation: counting, choice and initiation. Nature reviews genetics. 2001;2(1):59–67. doi: 10.1038/35047580. [DOI] [PubMed] [Google Scholar]
- 31.Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek SH, et al. MicroRNA genes are transcribed by RNA polymerase II. The EMBO journal. 2004;23(20):4051–60. doi: 10.1038/sj.emboj.7600385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.John B, Enright AJ, Aravin A, Tuschl T, Sander C, Marks DS. Human microRNA targets. PLoS Biol. 2004;2(11):e363. doi: 10.1371/journal.pbio.0020363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brennecke J, Stark A, Russell RB, Cohen SM. Principles of microRNA–target recognition. PLoS Biol. 2005;3(3):e85. doi: 10.1371/journal.pbio.0030085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martin DI, Cropley JE, Suter CM. Epigenetics in disease. Epigenetics. 2011;6(7):843–48. doi: 10.4161/epi.6.7.16498. [DOI] [PubMed] [Google Scholar]
- 35.Esteller M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nature reviews genetics. 2007;8(4):286–98. doi: 10.1038/nrg2005. [DOI] [PubMed] [Google Scholar]
- 36.Flanagan JM, Munoz-Alegre M, Henderson S, Tang T, Sun P, Johnson N, et al. Gene-body hypermethylation of ATM in peripheral blood DNA of bilateral breast cancer patients. Hum Mol Genet. 2009;18(7):1332–42. doi: 10.1093/hmg/ddp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Friedrich MG, Weisenberger DJ, Cheng JC, Chandrasoma S, Siegmund KD, Gonzalgo ML, et al. Detection of methylated apoptosis-associated genes in urine sediments of bladder cancer patients. Clin Cancer Res. 2004;10(22):7457–65. doi: 10.1158/1078-0432.CCR-04-0930. [DOI] [PubMed] [Google Scholar]
- 38.Gopisetty G, Ramachandran K, Singal R. DNA methylation and apoptosis. Mol Immunol. 2006;43(11):1729–40. doi: 10.1016/j.molimm.2005.11.010. 4// [DOI] [PubMed] [Google Scholar]
- 39.Calin GA, Croce CM. MicroRNA-cancer connection: the beginning of a new tale. Cancer Res. 2006;66(15):7390–94. doi: 10.1158/0008-5472.CAN-06-0800. [DOI] [PubMed] [Google Scholar]
- 40.Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A. 2006;103(7):2257–61. doi: 10.1073/pnas.0510565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu F, Yao H, Zhu P, Zhang X, Pan Q, Gong C, et al. < i> let-7</i> Regulates Self Renewal and Tumorigenicity of Breast Cancer Cells. Cell. 2007;131(6):1109–23. doi: 10.1016/j.cell.2007.10.054. [DOI] [PubMed] [Google Scholar]
- 42.Akao Y, Nakagawa Y, Naoe T. let-7 microRNA functions as a potential growth suppressor in human colon cancer cells. Biol Pharm Bull. 2006;29(5):903–06. doi: 10.1248/bpb.29.903. [DOI] [PubMed] [Google Scholar]
- 43.Takamizawa J, Konishi H, Yanagisawa K, Tomida S, Osada H, Endoh H, et al. Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res. 2004;64(11):3753–56. doi: 10.1158/0008-5472.CAN-04-0637. [DOI] [PubMed] [Google Scholar]
- 44.Esquela-Kerscher A, Trang P, Wiggins JF, Patrawala L, Cheng A, Ford L, et al. The let-7 microRNA reduces tumor growth in mouse models of lung cancer. CELL CYCLE-LANDES BIOSCIENCE- 2008;7(6):759. doi: 10.4161/cc.7.6.5834. [DOI] [PubMed] [Google Scholar]
- 45*.Christman JK. 5-Azacytidine and 5-aza-2′-deoxycytidine as inhibitors of DNA methylation: mechanistic studies and their implications for cancer therapy. Oncogene. 2002;21(35):5483–95. doi: 10.1038/sj.onc.1205699. Thorough look at the mechanisms of demethylation by 5-azacytidine and 5-aza-2′-deoxycytidine. [DOI] [PubMed] [Google Scholar]
- 46.Nguyen CT, Weisenberger DJ, Velicescu M, Gonzales FA, Lin JCY, Liang G, et al. Histone H3-Lysine 9 Methylation Is Associated with Aberrant Gene Silencing in Cancer Cells and Is Rapidly Reversed by 5-Aza-2′-deoxycytidine. Cancer Res 2002. 2002 Nov 15;62(22):6456–61. [PubMed] [Google Scholar]
- 47.Cherblanc F, Chapman-Rothe N, Brown R, Fuchter M. Current limitations and future opportunities for epigenetic therapies. Future Med Chem. 2012;4(4):425–46. doi: 10.4155/fmc.12.7. [DOI] [PubMed] [Google Scholar]
- 48*.Jabbour E, Issa JP, Garcia-Manero G, Kantarjian H. Evolution of decitabine development. Cancer. 2008;112(11):2341–51. doi: 10.1002/cncr.23463. Pharmacokinetic aspects of decitabine are covered thoroughly including in vivo molecular effects. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Trédan O, Galmarini CM, Patel K, Tannock IF. Drug resistance and the solid tumor microenvironment. J Natl Cancer Inst. 2007;99(19):1441–54. doi: 10.1093/jnci/djm135. [DOI] [PubMed] [Google Scholar]
- 50.Momparler RL, Bouffard DY, Momparler LF, Dionne J, Bélanger K, Ayoub J. Pilot phase l-ll study on 5-aza-2′-deoxycytidine (Decitabine) in patients with metastatic lung cancer. Anticancer Drugs. 1997;8(4):358–68. doi: 10.1097/00001813-199704000-00008. [DOI] [PubMed] [Google Scholar]
- 51.Brünner N, Bronzert D, Vindeløv LL, Rygaard K, Spang-Thomsen M, Lippman ME. Effect on growth and cell cycle kinetics of estradiol and tamoxifen on MCF-7 human breast cancer cells grown in vitro and in nude mice. Cancer Res. 1989;49(6):1515–20. [PubMed] [Google Scholar]
- 52.Terry N, Meistrich M, Roubein L, Lynch P, Dubrow R, Rich T. Cellular kinetics in rectal cancer. Br J Cancer. 1995;72(2):435. doi: 10.1038/bjc.1995.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Oloumi A, Maidan M, Lock FE, Tearle H, McKinney S, Muller WJ, et al. Cooperative signaling between Wnt1 and integrin-linked kinase induces accelerated breast tumor development. Breast Cancer Research. 2010;12(3):R38. doi: 10.1186/bcr2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hollenbach PW, Nguyen AN, Brady H, Williams M, Ning Y, Richard N, et al. A comparison of azacitidine and decitabine activities in acute myeloid leukemia cell lines. Plos One. 2010;5(2):e9001. doi: 10.1371/journal.pone.0009001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brown R, Plumb JA. Demethylation of DNA by decitabine in cancer chemotherapy. Expert Rev Anticancer Ther. 2004;4(4):501–10. doi: 10.1586/14737140.4.4.501. [DOI] [PubMed] [Google Scholar]
- 56.Butler LM, Zhou X, Xu WS, Scher HI, Rifkind RA, Marks PA, et al. The histone deacetylase inhibitor SAHA arrests cancer cell growth, up-regulates thioredoxin-binding protein-2, and down-regulates thioredoxin. Proceedings of the National Academy of Sciences 2002. 2002 Sep 3;99(18):11700–05. doi: 10.1073/pnas.182372299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huang L, Sowa Y, Sakai T, Pardee AB. Activation of the p21WAF1/CIP1 promoter independent of p53 by the histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA) through the Sp1 sites. Oncogene. 2000;19(50):5712. doi: 10.1038/sj.onc.1203963. [DOI] [PubMed] [Google Scholar]
- 58.Chavez-Blanco A, Segura-Pacheco B, Perez-Cardenas E, Taja-Chayeb L, Cetina L, Candelaria M, et al. Histone acetylation and histone deacetylase activity of magnesium valproate in tumor and peripheral blood of patients with cervical cancer. A phase I study Molecular Cancer. 2005;4(1):22. doi: 10.1186/1476-4598-4-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Marks PA, Miller T, Richon VM. Histone deacetylases. Curr Opin Pharm. 2003;3(4):344–51. doi: 10.1016/s1471-4892(03)00084-5. 8// [DOI] [PubMed] [Google Scholar]
- 60.Qiu L, Kelso MJ, Hansen C, West ML, Fairlie DP, Parsons PG. Anti-tumour activity in vitro and in vivo of selective differentiating agents containing hydroxamate. British journal of cancer. 1999;80(8):1252–58. doi: 10.1038/sj.bjc.6690493. 06//print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Richon VM. Cancer biology: mechanism of antitumour action of vorinostat (suberoylanilide hydroxamic acid), a novel histone deacetylase inhibitor. British journal of cancer. 95(S1):S2–S6. 0000 //print. [Google Scholar]
- 62*.Zhu S, Denman CJ, Cobanoglu ZS, Kiany S, Lau CC, Gottschalk SM, et al. The narrow-spectrum HDAC inhibitor entinostat enhances NKG2D expression without NK cell toxicity, leading to enhanced recognition of cancer cells. Pharm Res. 2013:1–14. doi: 10.1007/s11095-013-1231-0. A novel dinucleotide containing decitabine showing decreased depletion by cytidine deaminase leading to greater bioavailability. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Duque-Afonso J, Yalcin A, Berg T, Abdelkarim M, Heidenreich O, Lübbert M. The HDAC class I-specific inhibitor entinostat (MS-275) effectively relieves epigenetic silencing of the LAT2 gene mediated by AML1/ETO. Oncogene. 2011;30(27):3062–72. doi: 10.1038/onc.2011.32. [DOI] [PubMed] [Google Scholar]
- 64.Oronsky B, Oronsky N, Scicinski J, Fanger G, Lybeck M, Reid T. Rewriting the Epigenetic Code for Tumor Resensitization: A Review. Translational oncology. 2014;7(5):626–31. doi: 10.1016/j.tranon.2014.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65**.Yoo CB, Jeong S, Egger G, Liang G, Phiasivongsa P, Tang C, et al. Delivery of 5-aza-2′-deoxycytidine to cells using oligodeoxynucleotides. Cancer Res. 2007;67(13):6400–08. doi: 10.1158/0008-5472.CAN-07-0251. An overview of many nanoparticle platforms and their use in cancer therapy. [DOI] [PubMed] [Google Scholar]
- 66.Karahoca M, Momparler RL. Pharmacokinetic and pharmacodynamic analysis of 5-aza-2′-deoxycytidine (decitabine) in the design of its dose-schedule for cancer therapy. Clin Epigenetics. 2013;5(1):3. doi: 10.1186/1868-7083-5-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Maeda H. Tumor-selective delivery of macromolecular drugs via the EPR effect: background and future prospects. Bioconj Chem. 2010;21(5):797–802. doi: 10.1021/bc100070g. [DOI] [PubMed] [Google Scholar]
- 68.Alexis F, Pridgen EM, Langer R, Farokhzad OC. Drug Deliv. Springer; 2010. Nanoparticle technologies for cancer therapy; pp. 55–86. [DOI] [PubMed] [Google Scholar]
- 69.Danhier F, Feron O, Préat V. To exploit the tumor microenvironment: Passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J Control Release. 2010;148(2):135–46. doi: 10.1016/j.jconrel.2010.08.027. [DOI] [PubMed] [Google Scholar]
- 70.Allen TM, Cullis PR. Liposomal drug delivery systems: from concept to clinical applications. Adv Drug Del Rev. 2013;65(1):36–48. doi: 10.1016/j.addr.2012.09.037. [DOI] [PubMed] [Google Scholar]
- 71*.Sharma B, Peetla C, Adjei IM, Labhasetwar V. Selective biophysical interactions of surface modified nanoparticles with cancer cell lipids improve tumor targeting and gene therapy. Cancer Lett. 2013;334(2):228–36. doi: 10.1016/j.canlet.2013.03.011. PLGA used for drug delivery - surface modification with surfactants for tumor localization. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhao F, Zhao Y, Liu Y, Chang X, Chen C, Zhao Y. Cellular uptake, intracellular trafficking, and cytotoxicity of nanomaterials. Small. 2011;7(10):1322–37. doi: 10.1002/smll.201100001. [DOI] [PubMed] [Google Scholar]
- 73.Crotts G, Park TG. Protein delivery from poly (lactic-co-glycolic acid) biodegradable microspheres: release kinetics and stability issues. J Microencaps. 1998;15(6):699–713. doi: 10.3109/02652049809008253. [DOI] [PubMed] [Google Scholar]
- 74.Makadia HK, Siegel SJ. Poly lactic-co-glycolic acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers. 2011;3(3):1377–97. doi: 10.3390/polym3031377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75*.Wang AZ, Langer R, Farokhzad OC. Nanoparticle delivery of cancer drugs. Annu Rev Med. 2012;63:185–98. doi: 10.1146/annurev-med-040210-162544. A study on using nanogel as a decitabine carrier to overcome resistance in cancer cells. [DOI] [PubMed] [Google Scholar]
- 76.Gabizon A, Martin F. Polyethylene glycol-coated (pegylated) liposomal doxorubicin. Drugs. 1997;54(4):15–21. doi: 10.2165/00003495-199700544-00005. [DOI] [PubMed] [Google Scholar]
- 77.Legut M, Lipka D, Filipczak N, Piwoni A, Kozubek A, Gubernator J. Anacardic acid enhances the anticancer activity of liposomal mitoxantrone towards melanoma cell lines–in vitro studies. International journal of nanomedicine. 2014;9:653. doi: 10.2147/IJN.S54911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kaminskas LM, McLeod VM, Porter CJ, Boyd BJ. Association of chemotherapeutic drugs with dendrimer nanocarriers: an assessment of the merits of covalent conjugation compared to noncovalent encapsulation. Mol Pharm. 2012;9(3):355–73. doi: 10.1021/mp2005966. [DOI] [PubMed] [Google Scholar]
- 79.Oh YK, Park TG. siRNA delivery systems for cancer treatment. Adv Drug Del Rev. 2009;61(10):850–62. doi: 10.1016/j.addr.2009.04.018. [DOI] [PubMed] [Google Scholar]
- 80.Gillies ER, Frechet JM. Dendrimers and dendritic polymers in drug delivery. Drug Discov Today. 2005;10(1):35–43. doi: 10.1016/S1359-6446(04)03276-3. [DOI] [PubMed] [Google Scholar]
- 81.Zha L, Banik B, Alexis F. Stimulus responsive nanogels for drug delivery. Soft Matter. 2011;7(13):5908–16. [Google Scholar]
- 82.Vijayaraghavalu S, Labhasetwar V. Efficacy of decitabine-loaded nanogels in overcoming cancer drug resistance is mediated via sustained DNA methyltransferase 1 (DNMT1) depletion. Cancer Lett. 2013;331(1):122–29. doi: 10.1016/j.canlet.2012.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.MacDiarmid JA, Mugridge NB, Weiss JC, Phillips L, Burn AL, Paulin RP, et al. Bacterially derived 400 nm particles for encapsulation and cancer cell targeting of chemotherapeutics. Cancer Cell. 2007;11(5):431–45. doi: 10.1016/j.ccr.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 84.Kapoor M, Siegel RA. Prodrug/Enzyme Based Acceleration of Absorption of Hydrophobic Drugs: An in Vitro Study. Mol Pharm. 2013;10(9):3519–24. doi: 10.1021/mp400272m. [DOI] [PubMed] [Google Scholar]
- 85.Brueckner B, Rius M, Markelova MR, Fichtner I, Hals PA, Sandvold ML, et al. Delivery of 5-azacytidine to human cancer cells by elaidic acid esterification increases therapeutic drug efficacy. Mol Cancer Ther. 2010;9(5):1256–64. doi: 10.1158/1535-7163.MCT-09-1202. [DOI] [PubMed] [Google Scholar]
- 86.García J, Franci G, Pereira R, Benedetti R, Nebbioso A, Rodríguez-Barrios F, et al. Epigenetic profiling of the antitumor natural product psammaplin A and its analogues. Biorg Med Chem. 2011;19(12):3637–49. doi: 10.1016/j.bmc.2010.12.026. [DOI] [PubMed] [Google Scholar]
- 87.Furumai R, Matsuyama A, Kobashi N, Lee KH, Nishiyama M, Nakajima H, et al. FK228 (depsipeptide) as a natural prodrug that inhibits class I histone deacetylases. Cancer Res. 2002;62(17):4916–21. [PubMed] [Google Scholar]
- 88.Lavelle D, Vaitkus K, Ling Y, Ruiz MA, Mahfouz R, Ng KP, et al. Effects of tetrahydrouridine on pharmacokinetics and pharmacodynamics of oral decitabine. Blood. 2012;119(5):1240–47. doi: 10.1182/blood-2011-08-371690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Olivieri NF, Saunthararajah Y, Thayalasuthan V, Kwiatkowski J, Ware RE, Kuypers FA, et al. A pilot study of subcutaneous decitabine in β-thalassemia intermedia. Blood. 2011;118(10):2708–11. doi: 10.1182/blood-2011-03-341909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Saunthararajah Y, DeSimone J. Semin Hematol. Vol. 2004. Elsevier; 2004. Clinical studies with fetal hemoglobin—enhancing agents in sickle cell disease; pp. 11–16. [DOI] [PubMed] [Google Scholar]
- 91.Vijayaraghavalu S, Dermawan JK, Cheriyath V, Labhasetwar V. Highly synergistic effect of sequential treatment with epigenetic and anticancer drugs to overcome drug resistance in breast cancer cells is mediated via activation of p21 gene expression leading to G2/M cycle arrest. Mol Pharm. 2012;10(1):337–52. doi: 10.1021/mp3004622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nebbioso A, Pereira R, Khanwalkar H, Matarese F, García-Rodríguez J, Miceli M, et al. Death receptor pathway activation and increase of ROS production by the triple epigenetic inhibitor UVI5008. Mol Cancer Ther. 2011;10(12):2394–404. doi: 10.1158/1535-7163.MCT-11-0525. [DOI] [PubMed] [Google Scholar]
- 93.Carafa V, Nebbioso A, Altucci L. Sirtuins and disease: the road ahead. Frontiers in pharmacology. 2012:3. doi: 10.3389/fphar.2012.00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Steele N, Plumb J, Vidal L, Tjørnelund J, Knoblauch P, Buhl-Jensen P, et al. Pharmacokinetic and pharmacodynamic properties of an oral formulation of the histone deacetylase inhibitor Belinostat (PXD101) Cancer Chemother Pharmacol. 2011;67(6):1273–79. doi: 10.1007/s00280-010-1419-5. [DOI] [PubMed] [Google Scholar]
- 95.Lim SP, Kumar R, Akkamsetty Y, Wang W, Ho K, Neilsen PM, et al. Development of a novel cell-based assay system EPISSAY for screening epigenetic drugs and liposome formulated decitabine. BMC Cancer. 2013;13(1):113. doi: 10.1186/1471-2407-13-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Halaban R, Krauthammer M, Pelizzola M, Cheng E, Kovacs D, Sznol M, et al. Integrative analysis of epigenetic modulation in melanoma cell response to decitabine: clinical implications. Plos One. 2009;4(2):e4563. doi: 10.1371/journal.pone.0004563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Scott SA, Dong WF, Ichinohasama R, Hirsch C, Sheridan D, Sanche SE, et al. 5-Aza-2′-deoxycytidine (decitabine) can relieve p21WAF1 repression in human acute myeloid leukemia by a mechanism involving release of histone deacetylase 1 (HDAC1) without requiring p21WAF1 promoter demethylation. Leuk Res. 2006;30(1):69–76. doi: 10.1016/j.leukres.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 98.Lavelle D, Vaitkus K, Hankewych M, Singh M, DeSimone J. Effect of 5-aza-2′-deoxycytidine (Dacogen) on covalent histone modifications of chromatin associated with the ε-, γ-, and β-globin promoters in< i> Papio anubis</i>. Exp Hematol. 2006;34(3):339–47. doi: 10.1016/j.exphem.2005.12.010. [DOI] [PubMed] [Google Scholar]
- 99.Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M. Epigenetic protein families: a new frontier for drug discovery. Nature reviews Drug discovery. 2012;11(5):384–400. doi: 10.1038/nrd3674. [DOI] [PubMed] [Google Scholar]
- 100.Urdinguio RG, Sanchez-Mut JV, Esteller M. Epigenetic mechanisms in neurological diseases: genes, syndromes, and therapies. The Lancet Neurology. 2009;8(11):1056–72. doi: 10.1016/S1474-4422(09)70262-5. 11// [DOI] [PubMed] [Google Scholar]
- 101.Jin B, Tao Q, Peng J, Soo HM, Wu W, Ying J, et al. DNA methyltransferase 3B (DNMT3B) mutations in ICF syndrome lead to altered epigenetic modifications and aberrant expression of genes regulating development, neurogenesis and immune function. Hum Mol Genet. 2008;17(5):690–709. doi: 10.1093/hmg/ddm341. [DOI] [PubMed] [Google Scholar]
- 102.Weksberg R, Shuman C, Caluseriu O, Smith AC, Fei YL, Nishikawa J, et al. Discordant KCNQ1OT1 imprinting in sets of monozygotic twins discordant for Beckwith–Wiedemann syndrome. Hum Mol Genet. 2002;11(11):1317–25. doi: 10.1093/hmg/11.11.1317. [DOI] [PubMed] [Google Scholar]
- 103.Greco M, Criscuolo M, Fianchi L, Fabiani E, Pagano L, Voso M. 5-azacytidine in chronic myelomonocytic leukemia: case report and review of literature. Mediterranean journal of hematology and infectious diseases. 2010;3(1):e2011011–e11. doi: 10.4084/MJHID.2011.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Raza A, Raza FZ, Galili N. Low-dose decitabine and high-risk MDS. Blood. 2006;108(13):4291–92. doi: 10.1182/blood-2006-08-041145. [DOI] [PubMed] [Google Scholar]
- 105.Kantarjian HM, Thomas XG, Dmoszynska A, Wierzbowska A, Mazur G, Mayer J, et al. Multicenter, randomized, open-label, phase III trial of decitabine versus patient choice, with physician advice, of either supportive care or low-dose cytarabine for the treatment of older patients with newly diagnosed acute myeloid leukemia. J Clin Oncol. 2012;30(21):2670–77. doi: 10.1200/JCO.2011.38.9429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist. 2007;12(10):1247–52. doi: 10.1634/theoncologist.12-10-1247. [DOI] [PubMed] [Google Scholar]
- 107.National Cancer Institute (NCI) ClinicalTrials.gov [Internet] Bethesda (MD): National Library of Medicine (US); 2000. Vorinostat in Treating Patients With Metastatic or Unresectable Melanoma. [cited 2015 January 31]. Available from: http://clinicaltrials.gov/show/NCT00121225 NLM Identifier: NCT 00121225. [Google Scholar]
- 108.Ngan SY, Burmeister B, Fisher RJ, Solomon M, Goldstein D, Joseph D, et al. Randomized trial of short-course radiotherapy versus long-course chemoradiation comparing rates of local recurrence in patients with T3 rectal cancer: Trans-Tasman Radiation Oncology Group trial 01. 04 J Clin Oncol. 2012;30(31):3827–33. doi: 10.1200/JCO.2012.42.9597. [DOI] [PubMed] [Google Scholar]
- 109.TopoTarget A/S. ClinicalTrials.gov [Internet] Bethesda (MD): National Library of Medicine (US); 2000. Study of Oral PXD101 in Patients With Advanced Solid Tumors or Lymphoma. [cited 2015 January 31]. Available from: http://clinicaltrials.gov/show/ NCT00413075 NLM Identifier: NCT00413075. [Google Scholar]
- 110.University of Wisconsin, Madison. ClinicalTrials.gov [Internet] Bethesda (MD): National Library of Medicine (US); 2000. Trial of LBH589 in Metastatic Thyroid Cancer. [cited 2015 January 31]. Available from: http://clinicaltrials.gov/show/NCT01013597 NLM Identifier: NCT01013597. [Google Scholar]
- 111.Novartis Pharmaceuticals. ClinicalTrials.gov [Internet] Bethesda (MD): National Library of Medicine (US); 2000. A Phase III Randomized, Double Blind, Placebo Controlled Multi-center Study of Panobinostat for Maintenance of Response in Patients With Hodgkin's Lymphoma (PATH) [cited 2015 January 31]. Available from: http://clinicaltrials.gov/show/NCT01034163 NLM Identifier: NCT01034163. [Google Scholar]
- 112.West AC, Smyth MJ, Johnstone RW. The anticancer effects of HDAC inhibitors require the immune system. Oncoimmunology. 2014;3(1) doi: 10.4161/onci.27414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.National Cancer Institute (NCI) ClinicalTrials.gov [Internet] Bethesda (MD): National Library of Medicine (US); 2000. FR901228 in Treating Patients With Recurrent High-Grade Gliomas. [cited 2015 January 31]. Available from: http://clinicaltrials.gov/show/ NCT00085540 NLM Identifier: NCT00085540. [Google Scholar]
- 114.National Cancer Institute (NCI) ClinicalTrials.gov [Internet] Bethesda (MD): National Library of Medicine (US); 2000. A Phase II Trial of Valproic Acid in Patients With Advanced Thyroid Cancers of Follicular Cell Origin. [cited 2015 January 31]. Available from: http://clinicaltrials.gov/show/ NCT01182285 NLM Identifier: NCT01182285. [Google Scholar]