

Abstract
Nucleic acids are under constant assault from endogenous and environmental agents that alter their physical and chemical properties. O6-methylation of guanosine (m6G) is particularly notable for its high mutagenicity, pairing with T, during DNA replication. Yet, while m6G accumulates in both DNA and RNA, little is known about its effects on RNA. Here, we investigate the effects of m6G on the decoding process, using a reconstituted bacterial translation system. m6G at the first and third position of the codon decreases the accuracy of tRNA selection. The ribosome readily incorporates near-cognate aminoacyl-tRNAs (aa-tRNAs) by forming m6G-uridine codon–anticodon pairs. Surprisingly, the introduction of m6G to the second position of the codon does not promote miscoding, but instead slows the observed rates of peptide-bond formation by >1000-fold for cognate aa-tRNAs without altering the rates for near-cognate aa-tRNAs. These in vitro observations were recapitulated in eukaryotic extracts and HEK293 cells. Interestingly, the analogous modification N6-methyladenosine (m6A) at the second position has only a minimal effect on tRNA selection, suggesting that the effects on tRNA selection seen with m6G are due to altered geometry of the base pair. Given that the m6G:U base pair is predicted to be nearly indistinguishable from a Watson-Crick base pair, our data suggest that the decoding center of the ribosome is extremely sensitive to changes at the second position. Our data, apart from highlighting the deleterious effects that these adducts pose to cellular fitness, shed new insight into decoding and the process by which the ribosome recognizes codon–anticodon pairs.
Keywords: decoding, O6-methylguanosine, RNA damage, ribosome, translation
INTRODUCTION
Cellular nucleic acids are exposed to numerous chemical and environmental insults including ultraviolet radiation, reactive oxygen species, and alkylating agents. Endogenous and exogenous alkylating agents are known to react with and modify the nitrogen and oxygen atoms of all nucleobases (for review, see Wurtmann and Wolin 2009). RNA appears to be more vulnerable to damage presumably due to its prevalent single-stranded nature exposing the Watson-Crick face of the nucleobases (Parsa et al. 1987; Hofer et al. 2005). Unlike programmed methylation of specific nucleotides in rRNA and tRNA, aberrant methylation has the potential to deleteriously alter an RNA's function. Depending on the position, methylation can prevent base-pairing or alter the nucleotide's base-pairing preferences (Ougland et al. 2004). The existence of mechanisms that repair RNA methylation suggests that these modifications are recognized by cells as problematic. The bacterial oxidative demethylase AlkB and its human homolog hABH3 demethylate 1-methyladenine (m1A) and 3-methylcytosine (m3C) in RNA (Aas et al. 2003; Ougland et al. 2004). In DNA, 1-methyladenine can base pair with thymine but stalls the DNA polymerase whereas 3-methylcytosine cannot base pair with guanine.
The methylated adduct O6-methylguanosine (m6G) is particularly notable for its toxicity due to its high rate of mutagenicity during DNA replication. Upon encountering m6G, DNA polymerases preferentially incorporate thymidine, resulting in GC to AT transitions (Fig. 1; Eadie et al. 1984). This mutagenicity has long been exploited by cancer chemotherapeutics, including the glioblastoma therapy temozolomide (O'Reilly et al. 1993). Moreover, organisms from bacteria to man have evolved specific mechanisms to mediate m6G lesions in DNA, underscoring the risk of its accumulation (Sedgwick et al. 2007). And yet, despite having known for a number of years that m6G also accumulates in RNA (Parsa et al. 1987), almost nothing is known about its effects on mRNA and specifically how it is decoded by the ribosome.
FIGURE 1.
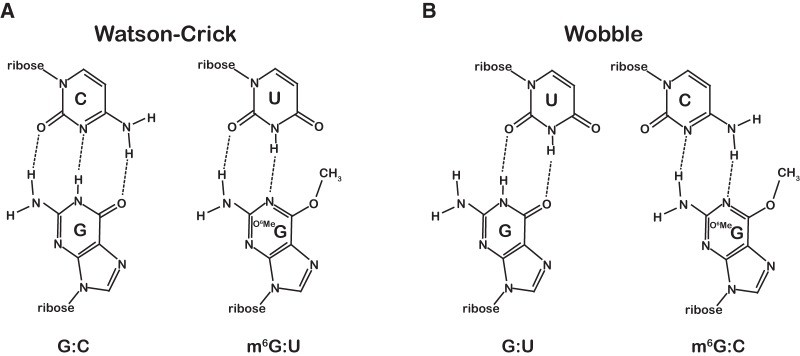
m6G:U base pair adopts a conformation similar to a Watson-Crick base pair. (A) Chemical structure of a normal Watson-Crick G:C base pair compared to the mutagenic m6G:U base pair structure. (B) Chemical structures of the G:U and m6G:C wobble base pairs.
During elongation, ribosomes successfully identify and incorporate the appropriate cognate aminoacylated tRNA (aa-tRNA) from among a large pool of competing aa-tRNAs, release factors, and other A-site binding proteins with remarkable accuracy (error rate of 10−3–10−6) (for review, see Zaher and Green 2009a; Voorhees and Ramakrishnan 2013). To discriminate effectively, the ribosome utilizes both thermodynamic and kinetic strategies to achieve this level of fidelity (Pape et al. 1999; Ogle et al. 2002; Gromadski and Rodnina 2004; Nierhaus 2006; Ninio 2006; Johansson et al. 2008). The tRNA selection process is divided into two main phases, initial selection and proofreading, which are separated by the nearly irreversible step of GTP hydrolysis by elongation factor Tu (EF-Tu). In the initial phase of selection, the incoming aa-tRNA is delivered to the ribosome in a ternary complex with EF-Tu and GTP. Upon recognition of the codon–anticodon interaction, EF-Tu is activated and GTP is rapidly hydrolyzed. The second phase of selection commences with the release of inorganic phosphate followed by conformational rearrangement in the elongation factor resulting in fast dissociation of EF-Tu. In this proofreading phase, naked ribosome bound aa-tRNAs either “accommodate” into the active site of the ribosome and participate in peptidyl transfer, or dissociate from the ribosome. During both phases of the selection process, near-cognate aa-tRNAs are destabilized through increased dissociation rates. In addition, the key forward rates of GTPase activation and accommodation are significantly accelerated for cognate aa-tRNAs, allowing translation to proceed rapidly and accurately (Pape et al. 1999; Ogle et al. 2002; Gromadski and Rodnina 2004).
High-resolution crystal structures of the ribosome have provided important molecular details about this induced-fit mechanism. Binding of cognate aa-tRNA in the A site is accompanied by conformational changes in the decoding center. The universally conserved residues A1493 and A1492 of the 16S rRNA rearrange to interact with the minor groove of the first- and second-position base pairs, respectively (Wimberly et al. 2000; Carter et al. 2001; Ogle et al. 2002). The second position is also inspected through additional interactions with G530 of the 16S rRNA and S50 of ribosomal protein S12. These interactions ensure that only Watson-Crick base pairs are allowed at the first and second position of the codon. Recognition of the third position is less stringent, allowing certain wobble base pairs in the codon anticodon helix. These interactions in the decoding center are thought to initiate conformational changes in the small subunit that ultimately trigger GTP hydrolysis by EF-Tu and subsequent accommodation of the aa-tRNA into the active site of the ribosome (Schmeing et al. 2009).
While much has been learned about how the ribosome distinguishes cognate aa-tRNAs from near-cognate and non-cognate aa-tRNAs, relatively little study has been devoted to understanding how the ribosome decodes aberrant mRNAs. However, it is clear from the limited studies that the ribosomal response to damaged mRNA does not always recapitulate the responses of DNA polymerases to damaged DNA. For instance, our laboratory has recently shown that the oxidized base 8-oxoguanosine, which mispairs with A during DNA replication, instead stalls the translation machinery (Simms et al. 2014).
In this study, we explore the effects of the methylation adduct m6G on the decoding process. We characterize its effects on the key steps of aa-tRNA selection, including accommodation and GTPase activation, as well as on the process of release-factor mediated termination. We then compare these parameters to those measured in the presence of the related modification m6A. Our data show that m6G has differential effects on decoding depending on its position within the codon. At the first and third position, m6G results in efficient miscoding where it pairs with U in the anticodon; at the second position, m6G stalls the ribosome. These observations demonstrate that the alkylation lesion m6G is detrimental to both ribosome speed and fidelity. Our results also highlight the unanticipated distinctions in how the ribosome monitors the first and second positions of the codon–anticodon interface.
RESULTS
Effects of m6G on the fidelity of tRNA selection
To gain a better understanding of the consequences of m6G on the decoding process, we used our bacterial reconstituted translation system to program ribosomes with a single adduct at the first, second, or third position of the A-site codon. Briefly, initiation complexes were generated by loading ribosomes with the initiator tRNA [35S]-fMet-tRNAfMet in the P site in the presence of the appropriate mRNA, initiation factors, and GTP followed by purification over a sucrose cushion. To begin our studies, we set out to explore the overall effects of the adduct on the accuracy of tRNA selection. We utilized a surveying approach wherein the initiation complexes were reacted with individual ternary complexes comprising all of the 20 canonical aa-tRNA isoacceptors. Dipeptide products were then resolved by electrophoretic TLC and the overall efficiency of peptide-bond formation assessed by quantifying the amount of dipeptide products relative to unreacted fMet (Youngman et al. 2004). This approach has been recently used by our laboratory to monitor the efficiency of incorporation of every single amino acid and, importantly, recapitulates to a large extent the level of fidelity measured in vivo (Simms et al. 2014).
We began our surveys by comparing the reactivity of a pair of initiation complexes displaying the intact GUG or the alkylated m6GUG codon in the A site (Fig. 2A; Supplemental Fig. S1). The GUG codon is decoded by Val-tRNAValVAC. The m6GUG codon, if m6G pairs with U in the anticodon of the tRNA, is expected to be decoded by Met-tRNAMetCAU. As predicted, for the intact GUG we observed dipeptide formation only in the presence of the cognate Val-tRNAVal ternary complex (Fig. 2A). In contrast, for m6GUG we observed no accumulation of dipeptide in the presence of Val-tRNAVal ternary complex but instead found robust fMet-Met production (Fig. 2A; Supplemental Fig. S1). This corroborated what has been seen previously for DNA polymerases (Snow et al. 1984). It is worth noting that beyond this switch in reactivity, the modification had no obvious effect on reactivity with other ternary complexes (Supplemental Fig. S1). To ensure that these effects were not specific to the GUG Val codon, we examined two additional mRNAs with m6G in the first position (Fig. 2A; Supplemental Figs. S2, S3). Consistent with our earlier results, complexes displaying the intact Glu GAA and Gly GGC codons in the A site reacted efficiently only with the corresponding Glu-tRNAGluUUC and Gly-tRNAGly ternary complexes, respectively; complexes displaying m6GAA and m6GGC codons in the A site reacted efficiently with the near-cognate Lys-tRNALysUUU and Ser-tRNASerGCU ternary complexes, respectively (Supplemental Figs. S2, S3).
FIGURE 2.
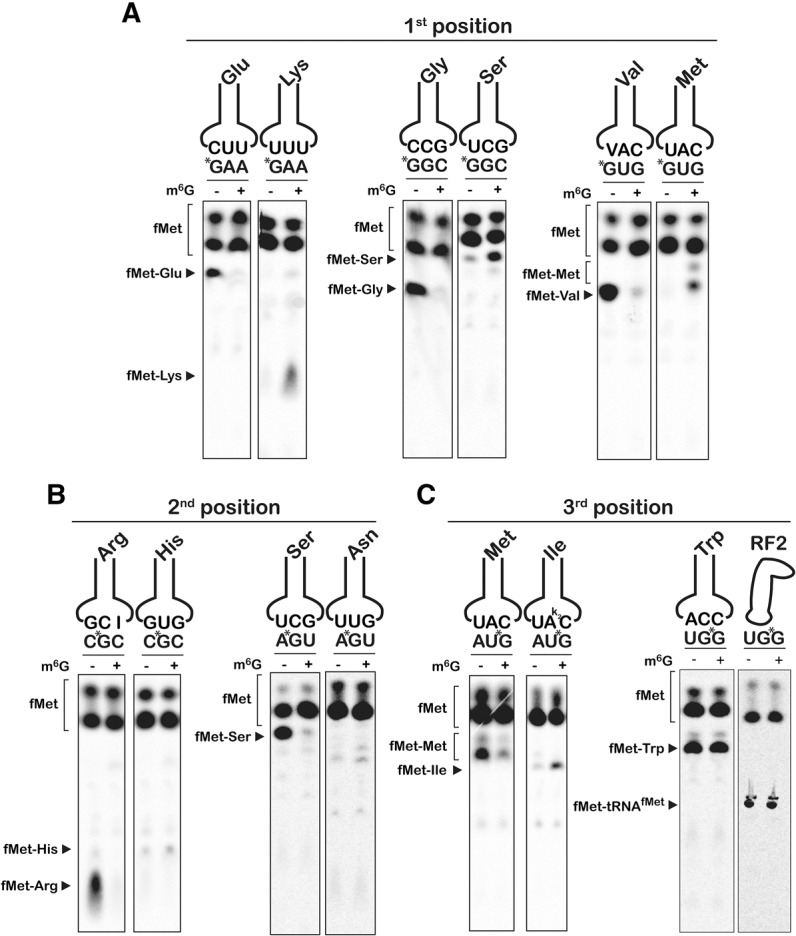
m6G affects the accuracy and speed of the ribosome. (A–C) Phosphorimager scans of electrophoretic TLCs used to follow dipeptide formation in the presence of the indicated initiation and ternary complexes.
Although the first and second positions of the codon are both decoded by monitoring strict Watson-Crick base-pairing interactions, the ribosome utilizes different strategies to inspect the minor groove at each position (Ogle et al. 2001; Demeshkina et al. 2012). To determine whether m6G imparts codon–anticodon mispairing, we generated two pairs of initiation complexes with modifications to the second position. The first pair displayed either the intact CGC Arg codon or the equivalent methylated Cm6GC codon in the A site; the second pair displayed the AGU Ser codon or the equivalent methylated Am6GU codon (Supplemental Figs. S4, S5). For both pairs, the unmodified complexes (CGC and AGU) reacted efficiently only with the corresponding cognate ternary complexes (Arg-tRNAArg and Ser-tRNASer, respectively), but both second-position m6G initiation complexes displayed little detectable reactivity with any ternary complexes, including cognates (Fig. 2B; Supplemental Figs. S4, S5). Thus, even though modification of the second position is predicted to allow m6G:U pairing, the effects on tRNA selection and translation were markedly different than the effects of m6G in the first position.
In contrast to the first and second positions, wobble pairing for most codons permits either A or G in the third position to be read by the same aa-tRNA, preventing us from testing the majority of sense codons for the effects of third-position modification on aa-tRNA selection errors. However, both Met and Trp are each encoded by only one codon (AUG and UGG, respectively). Here, the presence of m6G in the third position would result in codons (AUA and UGA) that are decoded by Ile-tRNAIlek2CAU and release factor 2 (RF2), respectively. As before, we generated two pairs of initiation complexes, intact and m6G-damaged, and tested their reactivities with ternary complexes and release factors (Fig. 2C; Supplemental Figs. S6, S7). For both intact AUG and UGG complexes, we observed robust dipeptide formation only in the presence of cognate Met-tRNAMetCAU and Trp-tRNATrpCCA ternary complexes and not with any near- or non-cognates. Surprisingly, the damaged AUm6G and UGm6G complexes reacted efficiently with both the cognates and the m6G:U near-cognates (Ile-tRNAIlek2CAU and RF2, respectively) (Fig. 2C; Supplemental Figs. S6, S7). Thus, at the wobble position, the ribosome tolerates both m6G:U and m6G:C base pairs.
Kinetics of peptide-bond formation and proofreading
Because the end-point assays provide little information about the comparative rates of control and m6G aa-tRNA selection, we used pre-steady state quench-flow kinetics to compare the observed rates of cognate and near-cognate dipeptide formation for intact and m6G-containing mRNAs. This observed rate of peptide-bond formation (kpep) reports on the combined rates of aa-tRNA accommodation (k5) (rate-limiting step for peptidyl transfer) and rejection (k7) during the proofreading phase of the selection, whereas the end point of the reaction reports on the effectiveness of proofreading (Fp), i.e., k5 relative to kpep (Pape et al. 1999). As expected, under our experimental conditions (see Materials and Methods) complexes displaying intact codons in the A site reacted rapidly with their cognate ternary complexes with little to no rejection of aa-tRNAs. We measured kpep and Fp values of 10–40 sec−1 and 0.4–0.7, respectively (Figs. 3, 4A; Supplemental Fig. S8). With near-cognate aa-tRNAs, the intact complexes exhibited much slower observed rates of peptide-bond formation (0.01–0.05 sec−1) and higher rates of rejection (0.14–0.34 Fp). Notably, and consistent with our end-point analysis, the first-position m6G complexes also reacted slowly (0.007–0.08 sec−1) and with high rates of rejection (0.04–0.17 Fp) with cognate ternary complexes (Fig. 4A; Supplemental Fig. S8A). In contrast, the m6G complexes reacted much faster (0.2–3.0 sec−1) with near-cognate ternary complexes that preserve m6G:U base pairs between the damaged base at the first position of the codon and the third position of the anticodon (Fig. 4A; Supplemental Fig. S8A). These reactions, compared to the intact complexes, also displayed reduced rates of rejection with Fp values from 0.14 to 0.55 for all three complexes (Supplemental Fig. S8A). These findings indicate that m6G at the first position of the codon is efficiently recognized as an A, highlighting the deleterious effects of the adduct on translational fidelity.
FIGURE 3.
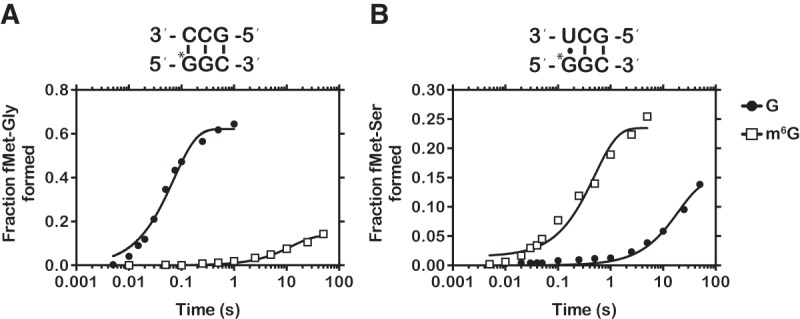
Representative time courses of cognate and near-cognate dipeptide formation. (A) Time course of peptide-bond formation between the native GGC (closed circle), methylated m6GGC (closed square), and cognate Gly-tRNAGly ternary complex. (B) Time course of peptide-bond formation between the native GGC (closed circle), methylated m6GGC (closed square), and near-cognate Ser-tRNASer ternary complex.
FIGURE 4.

m6G affects the decoding process in a position-dependent manner. (A) Bar graphs of the observed rates of peptide-bond formation for complexes carrying m6G at the first position of the codon relative to their native counterparts. For each pair of complexes, rates measured in the presence of cognate ternary complexes are plotted in the left two bars in each graph, whereas those measured in the presence of the near-cognate ternary complexes (U:G at the first position) are plotted in the right two bars. The codon–anticodon interactions are depicted below the x-axis with the corresponding dipeptide depicted above the bars. (B) Same as A, but with initiation complexes harboring m6G at the second position of the A-site codon. (C) Same as A, but with initiation complexes harboring m6G at the third position of the A-site codon. Note that the graph for the UGG and UGm6G complexes was split into two. Error bars represent the standard error of curve fitting from a single representative time course. Twenty-three of the 28 time courses were performed in duplicates with at least <10% variability between samples.
In contrast to the first position, our end-point surveys indicated that initiation complexes programmed with m6G at the second position of the codon react with neither cognate nor near-cognate ternary complexes (Fig. 2B). In full agreement with these initial observations, we measured much slower rates of dipeptide formation for both cognate and near-cognate aa-tRNAs and reduced Fp values typical of increased rates of aa-tRNA rejection (Fig. 4B; Supplemental Fig. S8B). The observed rates of peptide-bond formation between the alkylated Arg Cm6GC and Ser Am6GU complexes and the corresponding cognate ternary complexes were determined to be ∼1000-fold slower (∼0.02 sec−1) than those measured for the analogous intact complexes (Fig. 4B). Furthermore, we observed increased rates of rejection for the methylated complexes as evidenced by the lower Fp values of ∼0.2, versus ∼0.8 for the intact complexes (Supplemental Fig. S8B). As for reactions with the near-cognate ternary complexes, the apparent rates of peptide-bond formation remained slow (0.003–0.01 sec−1, Fig. 4B) in the presence of m6G. Thus, it appears that the addition of m6G at the second position stalls the translational machinery and does not promote miscoding.
At the third position, m6G had limited effects on the rate of cognate aa-tRNA incorporation but substantially increased reactivity with near-cognate aa-tRNAs. AUm6G Met initiation complexes reacted only approximately twofold slower with the cognate Met-tRNAMet ternary complex (28 versus 13 sec−1) and had limited effect on the end point of the reaction (Fig. 4C; Supplemental Fig. S8C). However, the AUm6G complex reacted significantly faster with the near-cognate Ile-tRNAIle (0.07 versus 1.7 sec−1) relative to the control complex and with decreased rejection (0.04 versus 0.15 Fp). Similarly, the rate of peptide-bond formation between the UGm6G initiation complex and Trp-tRNATrp was only fivefold slower than the intact UGG complex (1 versus 5 sec−1), again with little effect on the rate of rejection (Fig. 4C; Supplemental Fig. S8C). Furthermore, the maximal rate of RF2-mediated release was fivefold faster for the methylated complex than its intact control, suggesting that decoding m6G as A is not limited to RNA–RNA interactions but also includes RNA–protein interactions (Fig. 4C). Collectively, these observations are consistent with what we have known for decades about the promiscuous nature of the process by which the ribosome recognizes the third position of the codon.
m6G modulates GTP hydrolysis to promote miscoding and stalling
So far our analysis of m6G's effects on decoding has focused on the end result of the tRNA selection process, and any effects on steps preceding the proofreading phase could have been missed. To address this, we measured the rates of the key step of GTP hydrolysis by EF-Tu (kGTP), which reports on activation of EF-Tu by the decoding center (Pape et al. 1998). Similar to the peptide-bond formation analysis, observed rates of GTP hydrolysis were determined for control and m6G-containing initiation complexes with cognate and near-cognate aa-tRNAs. We found that the trends in the rates of GTP hydrolysis are similar to those observed for the rates of peptide-bond formation. More specifically, for complexes with m6G in the first position, m6G slows GTP hydrolysis of cognate aa-tRNAs ∼300-fold (30 versus 0.1 sec−1) while increasing kGTP for near-cognates ∼30-fold (0.1 versus 3 sec−1) (Fig. 5). Likewise, mRNAs harboring m6G at the second position reflect an almost identical pattern as their corresponding kpep rates, with kGTP rates for near-cognate and cognate both at least 60-fold slower than for intact mRNAs. As well, m6G at the third position does not have a significant effect on cognate selection kGTP (11 versus 13 sec−1) but does increase kGTP for near-cognate selection 10-fold (0.3 versus 3 sec−1). Given the effects we see on proofreading and GTPase activation, m6G appears to alter both phases of the tRNA selection process. This is not unexpected as both phases are well documented to depend critically on proper base-pairing geometry between the codon and anticodon (Gromadski et al. 2006), and m6G is highly likely to disturb this geometry.
FIGURE 5.

m6G alters the observed rate of GTP hydrolysis by EF-Tu. (A) Observed rates of GTP hydrolysis measured on native and the corresponding first-position modified complexes with cognate (left two bars in each graph) and near-cognate ternary complexes (right two bars in each graph). Labeling scheme of the graphs is identical to the one used in Figure 4. (B,C) Similar to A, but with complexes programmed with m6G at the second position and third position of the codon, respectively. Error bars represent the standard error of curve fit from a single representative time course.
Effects of m6G are recapitulated in eukaryotic extracts and mammalian cell culture
To explore whether the effects that we observed for m6G in our bacterial translation system were conserved in eukaryotes and could be recapitulated in a more in vivo–like setting, we designed model reporter mRNAs encoding an N-terminal HA and C-terminal Flag tag with a single m6G residue (Fig. 6A; Supplemental Fig. S9). In addition to the HA-Stop mRNA, three sets of mRNAs that harbored m6G at the first, second, and third position of a codon were synthesized (HA-m6GAA, HA-Cm6GA, and HA-ACm6G, respectively). The second- and third-position modified reporters were synthesized by inserting 1 and 2 nucleotides (nt), respectively, upstream of the Flag epitope, generating +1 and +2 frameshifted constructs and hence do not generate Flag-tagged protein products (i.e., see Materials and Methods). We then translated these mRNAs together with the corresponding unmodified control reporters in wheat germ extracts that were supplemented with 35S-methionine and separated the peptide products by Bis-Tris PAGE. As predicted, the full-length HA-Flag, HA-GAA, and the first-position modified reporter (HA-m6GAA) mRNAs yielded peptides of identical length, while the control HA-Stop peptide yielded a truncated product (Fig. 6B). The control HA-CGA mRNA yielded a significantly extended peptide as a result of the frameshift removing the stop codon. In contrast, HA-Cm6GA mRNA yielded a shorter product of approximately the same length as the HA-Stop peptide (Fig. 6B). This suggests that the Cm6GA codon stalls translation and results in a prematurely truncated peptide. Finally, m6G at the third position did not appear to affect the production of full-length peptides, corroborating the results of our bacterial ribosome experiments (Fig. 6B).
FIGURE 6.

Effects of m6G on translation in eukaryotic extracts and mammalian cell culture. (A) Schematic of model reporter mRNAs depicting N-terminal HA tag, C-terminal Flag tag, and single m6G residue. (B) Phosphorimage scan of 35S-labeled peptide products after wheat germ extract translation. (C) Immunoblots of wheat germ translated peptides probed with anti-Flag and anti-HA antibodies. (D) Same as in C, but comparing the HA-GAA and HA-m6GAA mRNAs. (E) Immunoblots of HEK293T lysates transfected with m6G reporter mRNAs along with control GFP plasmid and probed with anti-Flag, anti-HA, and anti-GFP antibodies.
To test whether first-position m6G is misread as A, we mutated the central lysine residue (AAA codon) in the Flag epitope to glutamate (GAA codon) and probed the resulting peptide products with the M2 anti-Flag antibody. As predicted, HA-GAA was only recognized by anti-HA antibody, but not with the anti-Flag antibody (Fig. 6C). In direct agreement with our bacterial reconstituted system, the first-position modified reporter HA-m6GAA was efficiently recognized by the anti-Flag antibody, producing a robust signal in the Western blot (Fig. 6D). To provide further support for these observations in a system that closely resembles in vivo conditions, we electroporated the reporters along with a control GFP plasmid into HEK293T cells. Again, we found that only the HA-Flag and HA-m6GAA mRNAs produced peptide products that could be recognized by the anti-Flag antibody (Fig. 6E). We note, however, that we failed to see a signal with the anti-HA antibody, presumably due to the poor yield and the small size of the reporters (Fig. 6E). Nevertheless, because the anti-Flag antibody has no reactivity with the HA-GAA (Glu) reporter product (Fig. 6C), any detectable Flag epitope produced by the HA-m6GAA reporter strongly suggests that m6G at the first position is misread as an A during translation in human cells. Therefore, m6G appears to affect the speed and fidelity of aa-tRNA selection by both bacterial and eukaryotic ribosomes.
m6G does not affect peptide release
Because of the significant effect m6G has on the speed and fidelity of peptide bond formation, especially at the second position, we wondered if m6G might also affect the decoding of stop codons, which utilize protein–mRNA interactions to mediate peptide release. To test this, we programmed initiation complexes with fMet-tRNAfMet in the P site and either control UGA or alkylated Um6GA codons in the A site (Supplemental Fig. S10). We note that RF2 recognizes A and G at the second position of the codon (UGA and UAA are decoded by RF2). In agreement with this, but in contrast to what we saw with second-position modified sense codons, we found that the maximal rate of peptide release was almost completely unaffected by the presence of m6G in the second position (2.6 versus 2.0 sec−1, Fig. 7). This supports the results of the structural and biochemical studies suggesting that peptide release, which is regulated by protein–mRNA and not tRNA–mRNA interactions, utilizes a distinct mechanism to recognize the codon than those used for aa-tRNA selection (Youngman et al. 2006; Korostelev et al. 2008; Laurberg et al. 2008; Weixlbaumer et al. 2008). Moreover, these findings lend further mechanistic insights into the process by which m6G affects decoding of sense codons. In particular, the observation that rate of release is not affected by m6G suggests that the modification does not alter the decoding center significantly on its own, although the decreased fraction of released peptide suggests the occasional formation of a nonproductive complex (Fig. 7). Ultimately, our data suggest that the reduced rates of peptide-bond formation observed with the equivalent sense-codon complexes (Gm6GC and Am6GU) are most likely due to the altered geometries of m6G:C and m6G:U base pairs between the codon and anticodon.
FIGURE 7.
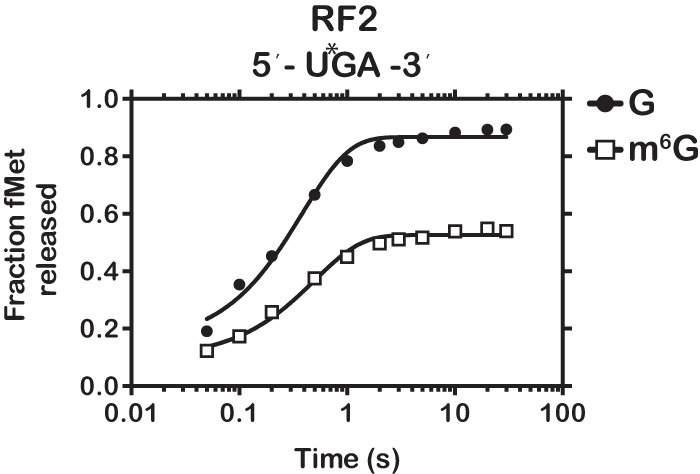
m6G has little effect on peptide release. Time course of RF2-dependent fMet release for initiation complexes programmed with UGA or Um6GA.
N6 methylation of adenosine at the second position has limited effect on decoding
To further explore our hypothesis that the inhibition of aa-tRNA selection observed with the second-position-modified codon is due to altered base-pairing geometry rather than to a steric effect of a bulky methyl group at the O6 position, we examined the effect of N6 methylation of adenosine (m6A) on the decoding process. It is worth noting that m6A has emerged recently as an abundant and reversible RNA modification (∼1.7 m6A per transcript) (Fu et al. 2014; Meyer and Jaffrey 2014). Interestingly, m6A has been found in both the coding sequence and near the stop codon, suggesting that the modification is likely to be encountered by actively translating ribosomes (Dominissini et al. 2012; Meyer et al. 2012). Important to our studies is the fact that in contrast to m6G, m6A is not predicted to have a drastic effect on the base-pairing properties of the nucleobase (m6A pairs with U). Therefore, any effects we observe for m6A on the decoding process would likely be due to the presence of a hydrophobic methyl group rather than altered base-pairing geometry. We programmed initiation complexes with mRNAs containing the enriched sequence Gm6AC, coding for aspartic acid, and examined its reactivity with all 20 aa-tRNA isoacceptors. Analysis of dipeptide products revealed that m6A had little effect on the fidelity of tRNA selection with cognate fMet-Asp being the major dipeptide product (Supplemental Fig. S11). Moreover, m6A had only a modest effect on the rate of fMet-Asp dipeptide formation (33 versus 7 sec−1, Fig. 8). These results demonstrate that m6A does not decrease fidelity or stall translation as we observed for m6G.
FIGURE 8.
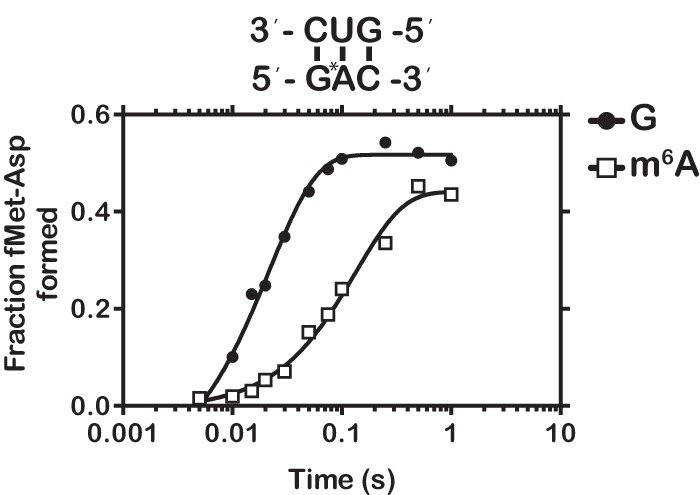
m6A has a marginal effect on the rate of peptide-bond formation. Time course of peptide-bond formation between initiation complexes programmed with GAC or the m6G-related modification Gm6AC and the cognate Asp-tRNAAsp ternary complex.
DISCUSSION
Our study provides important insights into the effects of m6G on tRNA selection by the ribosome. A priori, we predicted that, similar to DNA polymerases, m6G would prefer to pair with U at all codon positions, resulting in the misincorporation of near-cognate aa-tRNAs. Instead, our results demonstrate that m6G affects translation in a position-dependent manner within the codon. When m6G is present at the first position, the ribosome readily decodes m6G as A and incorporates the appropriate near-cognate aa-tRNA. However, m6G at the second position stalls the ribosome, preventing it from reacting efficiently with any of the 20 aa-tRNA isoacceptors. At the third position, m6G had little to no effect on the incorporation of cognate aa-tRNAs, yet significantly increased the incorporation of near-cognate aa-tRNAs. These discrepancies, especially between the first and second position of the codon (both requiring strict Watson-Crick base-pairing), suggest that the ribosome recognizes distinct aspects of the base pair geometries at all three positions and is uniquely affected when the geometries are perturbed. These distinctions will be discussed in greater detail below.
Regardless of its position within the codon, m6G was found to affect both phases of the tRNA selection process. For instance, modification of the first position of the codon was found to significantly decrease the observed rate of GTPase activation and peptide-bond formation for cognate aa-tRNAs, with rates 100- to 3000-fold lower than intact codons (Figs. 4A, 5A). Furthermore, the complexes exhibited higher rates of rejection as evidenced by the decreased end-point values (Supplemental Fig. S8A). On the other hand, the rates of GTPase activation and peptide-bond formation for near-cognate ternary complexes preserving m6G:U base-pairing were 20- to 100-fold faster than control codons (Figs. 4A, 5A). These observations suggest that the geometry of the m6G:U base pair at the first position is similar enough to a Watson-Crick pair to activate the decoding center. Notably, based on studies of DNA polymerases with m6G:T, we would predict that the minor groove of the m6G:U base pair would be nearly identical to that of a G:C base pair (Figs. 1, 9; Leonard et al. 1990; Warren et al. 2006), suggesting that the interaction with A1493 would be maintained. This interaction is central to the transmission of signals required for EF-Tu activation/GTP hydrolysis and aa-tRNA accommodation/peptidyl transfer (Schmeing et al. 2009).
FIGURE 9.

Recognition of base-pairing geometry by the ribosome and a high-fidelity DNA polymerase. (A) Inspection of the minor groove of the codon–anticodon pair by 16S rRNA and rps12 of the small subunit. (PDB IDs: 1XNR and 2WDG) (Murphy and Ramakrishnan 2004; Voorhees et al. 2009). (B) Recognition of stop codons by RF2 at the second position and third position (PDB IDs: 2WH3 and 4V67) (Korostelev et al. 2008; Laurberg et al. 2008; Weixlbaumer et al. 2008). (C) Structural alignments of a dm6G:dC base pair (PDB ID: 2HVH, left) or a dm6G:dT base pair (PDB ID: 2HVW, right) onto a dG:dC base pair (PDB ID: 2HVI) in the active site of a high-fidelity DNA polymerase (Warren et al. 2006). All molecular representations were generated using PyMol (DeLano Scientific).
At the third position we found that m6G increased ribosomal promiscuity during tRNA selection. Our results suggest that both m6G:C (Met-tRNAMet) and m6G:U (Ile-tRNAIle; actually m6G:lysidine) base pairs at the third position are recognized as cognate interactions. While it is easy to explain why the m6G:U base pair is deemed “sufficiently cognate,” since it is very similar to a Watson-Crick base pair, explaining the correct recognition of m6G:C is not as trivial. However, at physiological pH the m6G:C base pair can adopt a geometry similar to a wobble base pair (Figs. 1, 9A; Leonard et al. 1990), which at the third position is recognized as a cognate. Surprisingly, the m6G:C Wobble base pair is even preferred over m6G:lysidine, as peptide bond formation of fMet-Met is ∼10-fold faster than fMet-Ile. However, we cannot rule out the possibility that this effect is due to a steric clash between the bulky lysidine residue and m6G. It would be interesting to determine what effect the lysidine modification may have on the tRNA selection parameters for the cognate A:lysidine base pair compared to those of canonical Watson-Crick base pairs.
We were surprised by the dramatic differences in how m6G affected tRNA selection at the first and second positions. Correct codon–anticodon interactions at the first and second positions are monitored by a “molecular caliper mechanism” wherein the 16S rRNA residues A1493 or A1492/G530 hydrogen bond with the minor groove of the base pair to assess Watson-Crick pairing (Fig. 9A; Ogle et al. 2001). Yet despite their similar mechanisms of recognition, we observed robust miscoding at the first position and stalling at the second. One possible explanation for this is that the residues surrounding the first position of the A site allow greater steric flexibility for a bulky methyl group than does the second position. However, we observed only a modest effect (approximately sixfold) with m6A at the second position (Fig. 8), despite the methyl group of m6A occupying the same steric space as that of m6G. Thus, it seems unlikely that second-position m6G stalls translation because of the inability of the A site to tolerate the methyl group.
The observation that the ribosome is more sensitive to changes at the second position than at the first position of the codon agrees with biochemical and structural studies of the tRNA selection process. Kinetic analysis of ribosomal response to mismatches by the Rodnina group revealed that while the ribosome discriminates exceptionally well against mismatches at all positions, mismatches at the second position result in much slower rates of GTPase activation and accommodation relative to mismatches at the first and third position (Gromadski et al. 2006). Furthermore, whereas the minor groove of the first-position base pair is inspected through A-minor interaction with only one ribosomal residue (A1493), the minor groove of the second-position base pair is closely inspected through interactions with multiple residues, including A1492 and G530 of 16S rRNA. These residues are then themselves stabilized through hydrogen bond interactions with C518 of 16S and S50 of ribosomal protein S12 (Fig. 9A; Ogle et al. 2001). Structures of a high-fidelity DNA polymerase in complex with m6G show that the incoming m6G:T base pair adopts a conformation that is nearly indistinguishable from canonical Watson-Crick base pairs (Warren et al. 2006). Moreover, after replication, the m6G:T base pair is similar enough to G:C that it is not detected by the mismatch-repair machinery (Leonard et al. 1990) but is instead detected by proteins devoted entirely to removing m6G lesions (Demple et al. 1982). How could the m6G:T base pair trick the DNA replication machinery and yet m6G:U at the second codon position of the A site stall the ribosome? One possibility is that the m6G:T base pair deviates slightly from planarity relative to a G:C Watson-Crick base pair (Fig. 9C; Warren et al. 2006). The effect of this twisting is detectable during DNA replication. Specifically, while the kcat/Km for dTMP incorporation across m6G is much higher than that of dCMP, it is significantly lower than the normal incorporation of dCMP across G (Warren et al. 2006). It is tempting to speculate that the ribosome recognizes this slight geometric change through the intricate hydrogen bonding network that forms around the base pair. Alternatively, it is feasible that the change is transmitted though the mRNA structure itself to the adjacent positions. Whatever the mechanism, it is clear that m6G at the second position slows translation at least 1000-fold. Such a strong roadblock would likely engage the ribosomal rescue pathways (tmRNA in bacteria and No-Go Decay in eukaryotes) (Doma and Parker 2006; Moore and Sauer 2007; Tsuboi et al. 2012) as we have observed recently for mRNAs containing the oxidation adduct 8-oxo-G (Simms et al. 2014). Further study will be necessary to delineate how m6G at the second position stalls translation and to assess what effect this would have on cellular fitness.
In addition to aa-tRNA selection, we also examined the effects of m6G on peptide release by the class I release factor RF2. Surprisingly, we found that RF2 recognized UGA and Um6GA equally well. Although this observation on its own is not surprising (RF2 recognizes both UGA and UAA codons), it was unexpected given the striking effect of m6G at the second position during aa-tRNA selection. This finding also supports our hypothesis that m6G stalls aa-tRNA selection due to its distorted base-pairing interactions and not through other independent interactions. Moreover, the data highlight a key difference between the recognition of sense and nonsense codons: release factors engage stop codons through protein–RNA interactions (Korostelev et al. 2008; Laurberg et al. 2008; Weixlbaumer et al. 2008). Specifically, RF2 interacts with the second-position G of the UGA stop codon via hydrogen bonds between T203/S204 and N2 and between S193 and N1 and O6 (Fig. 9B). Importantly, threonine and serine residues can serve as both hydrogen bond donors and acceptors. Thus, during recognition of the UAA stop codon the hydrogen bond directionalities of S193 are reversed, allowing the factor to recognize A (Fig. 9B). Inspection of the 70S ribosome crystal structure in complex with a UGA stop codon and RF2 suggests that m6G at the second position would still permit hydrogen bonding between T203/S204 and N2 and one hydrogen bond between S193 and either O6 or N1 to maintain RF2 stabilization. We also note that the third-position codon UGm6G was decoded by RF2 fivefold faster than control UGG (0.60 versus 0.12 sec−1), although this rate was still fourfold slower than that of an authentic UGA stop codon (2.6 sec−1). At the third position, T203 of RF2 accepts a hydrogen bond from N6 and donates one to N7 of A; R201 also likely donates a hydrogen bond to N1 of the A (Fig. 9B; Weixlbaumer et al. 2008). Methylation of O6 would restore some of these interactions as N1 would no longer be a hydrogen bond donor but instead serve as a hydrogen bond acceptor. However, the hydrogen bond accepting of O6 does not change upon methylation and as a result it is unable to donate to T203, explaining why the rate of release on UGm6G, although faster than on UGG, is slower than on the UGA stop codon.
Our data show that m6G has dramatic effects on mRNA decoding at all positions of the codon. At the second position, m6G essentially stalls translation while at the third and first positions m6G impairs selection fidelity. The efficient incorporation of near-cognate aa-tRNAs by m6G at the first position would rapidly result in the production of potentially deleterious protein products. Moreover, this miscoding would be magnified as each mRNA is translated numerous times over the course of its lifespan. In the case of DNA replication, it is clear that m6G's mutagenic potential poses a significant risk to organismal fitness if not repaired. However, whether cells also detect damaged RNA remains unclear. Several studies have in fact demonstrated that certain RNA modifications including N1-methyladenosine (m1A) and N3-methyladenosine (m3A) are efficiently repaired by the bacterial oxidative demethylase AlkB and its human ortholog ABH3 (Aas et al. 2003; Ougland et al. 2004). In addition, the natural modification m6A is also removed by the demethylase FTO (Jia et al. 2011). Taken together, these studies raise the exciting possibility that cells have evolved pathways to detect, decode, modulate, and repair nucleobase modifications in both DNA and RNA. It will be interesting to determine whether the systems that detect and repair the mutagenic adduct m6G in DNA might also serve to protect the cell from its deleterious effects in RNA.
MATERIALS AND METHODS
Reagents
All experiments were performed in 1× polymix buffer containing 95 mM KCl, 5 mM NH4Cl, 5 mM Mg(OAc)2, 0.5 mM CaCl2, 8 mM putrescine (pH 7.5), 1 mM spermidine (pH 7.5), 10 mM K2HPO4 (pH 7.5), and 1 mM DTT (Jelenc and Kurland 1979).
70S ribosomes were purified from MRE600 Escherichia coli by the double-pelleting technique as described previously (Zaher and Green 2010). E. coli translation factors were overexpressed and purified as described previously (Zaher and Green 2009b).
Control mRNAs were transcribed from dsDNA templates by T7 RNA polymerase and purified by denaturing PAGE (Zaher and Unrau 2004). Experimental mRNAs containing either m6G or m6A were purchased from GE Healthcare and examined before use by denaturing PAGE to ensure purity. The final mRNAs had the following sequence: CAGAGGAGGUAAAAAAAUG (X) UUGUACAAA, where X represents the variable A-site codon.
Control reporter mRNAs for wheat germ and HEK293 experiments were transcribed from dsDNA templates as described above. Reporter mRNAs with m6G were synthesized using RNA ligation as described previously (Simms et al. 2014). Briefly, an upstream RNA encoding a 3′ hammerhead ribozyme was PCR amplified and transcribed with T7 RNA polymerase and then purified by denaturing PAGE. The 2′/3′ phosphate was removed with T4 PNK. The downstream RNA oligo containing the m6G residue was purchased from GE Healthcare, purified by denaturing PAGE, and 5′ phosphorylated using T4 PNK. The two fragments were then combined, along with a 60-mer reverse complement DNA oligo splint, annealed, and ligated using T4 RNA ligase 2 (NEB). For the HEK293 experiments, the reporter mRNAs were capped using the Vaccinia capping system (NEB) and polyadenylated with E. coli poly(A) polymerase (NEB) followed by phenol–chloroform purification.
aa-tRNA charging
Individually purified tRNAs (Glu, Lys, Val, Met, Arg) were purchased from Chemical Block and aminoacylated using purified aa tRNA synthetases from E. coli in 100 mM K-HEPES (pH 7.6), 20 mM MgCl2, 10 mM KCl, 1 mM DTT, and 10 U thermostable inorganic pyrophosphatase (Walker and Fredrick 2008). Additional aa-tRNAs were charged individually using total E. coli MRE600 tRNAs (Roche) and individual amino acids as above. AA-tRNAs were generally charged for 30 min at 37°C followed by phenol/chloroform extraction. AA-tRNAs were resuspended in 20 mM KOAc (pH 5.2), 1 mM DTT and used immediately for peptidyl transfer or GTP hydrolysis assay.
Generation of initiation complexes
Initiation complexes (ICs) were prepared by incubating 2 μM 70S ribosomes with 6 μM mRNA, 3 μM [35S]fMet-tRNAfMet, 3 μM of each IF1, IF2, and IF3, and 2 mM GTP in polymix buffer for 15 min at 37°C. The ICs were then layered over 700 μL sucrose cushions (1.1 M sucrose, 20 mM Tris-Cl [pH 7.5], 500 mM NH4Cl, 10 mM MgCl2, and 0.5 mM Na-EDTA [pH 7.5]) and centrifuged in an MLA-130 rotor at 267,000g for 2 h at 4°C. Pelleted ICs were resuspended in polymix buffer at a final concentration of 2 μM and stored at −80°C until use. ICs were stable for at least 1 mo at −80°C. IC formation efficiency was assessed by the fraction of radiolabeled [35S]fMet-tRNAfMet in the ribosome pellet and was typically >75%.
Ternary complexes reactivity survey
Individual ternary complexes were first prepared by incubating 40 μM EF-Tu and 2 mM GTP in polymix buffer for 10 min at 37°C. Individually charged Roche total aa-tRNAs were then added to a final concentration of 2 μM (80 μM total tRNA, reasoning that there are ∼40 equally distributed tRNA isoacceptors) and incubated for another 10 min at 37°C. 4 μL of ICs (2 μM) were then combined with 4 μL of each TC in 96-well plates using a multichannel pipette and reacted for 30 sec at 37°C before being quenched with 2 μL of 1 M KOH. 0.5 μL of each product was spotted at the center of cellulose TLC plates (Merck) and resolved using an electrophoretic TLC system containing PyrAc buffer (3.48 M acetic acid, 62 mM pyridine) and Stoddard's solvent at 1200 V (Youngman et al. 2004). TLC plates were analyzed by phosphorimaging and quantified using Bio-Rad Quantity One software.
Kinetics of peptidyl transfer
To measure rates of peptidyl transfer, ternary complexes (TCs) were prepared as described above. Reactions were initiated by combining equal volumes of TCs and ICs (prepared as above) using a quench-flow instrument (RQF-3, KinTek Corporation) and quenched with 1 M KOH. Reactions were spotted, resolved, and quantified as described above and reaction rates were calculated using GraphPad Prism software.
Kinetics of GTP hydrolysis
To measure GTP hydrolysis, we programmed ICs as above with the exception of using only a trace amount of radiolabeled [35S]fMet-tRNAfMet (just sufficient to quantify charging efficiency). TCs were prepared by incubating 20 μM EF-Tu with 20 μCi (6000 Ci/mmol) [γ-32P]-GTP, and 50 μM cold GTP, 2× polymix in a 20 μL volume at 37°C for 10 min. An equal volume (20 μL) of 40 μM aa-tRNA was then added and further incubated at 37°C for 10 min. TCs were then layered over polymix-equilibrated P-30 gel filtration spin columns (Bio-Rad) and centrifuged at 1000g for 1 min at 4°C. The flow through was then applied to a second spin column. The final flow through was diluted to 400 μL (1 μM EF-Tu final) with polymix buffer kept on ice until use. To measure rates, ICs and TCs were combined at 20°C using a quench-flow instrument and quenched with 2% formic acid. Products were spotted (0.5 μL) on PEI-cellulose TLC plates (Sigma), separated in 0.5 M KH2PO4 (pH 3.5), and analyzed by phosphorimaging. Rate constants were calculated using GraphPad Prism.
Kinetics of peptide release
ICs (2 μM) programmed with a stop codon in the A site were reacted with an equal volume of 20 μM RF2 at 37°C using a quench-flow apparatus and quenched with 50 mM K-EDTA (pH 6.0). 0.5 μL of each reaction was spotted and resolved as above using the electrophoretic TLC system. Rate constants were calculated using GraphPad Prism.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article
Supplementary Material
ACKNOWLEDGMENTS
We thank Carrie Simms for her help in constructing the mRNA reporters and the electroporation experiments. We also thank Doug Chalker and Joe Jez for careful reading of the manuscript and members of the laboratory for useful discussions and comments. This work was supported by a National Institutes of Health grant (R00GM094210) and funding from the Searle Scholars Program to H.S.Z.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.052464.115.
REFERENCES
- Aas PA, Otterlei M, Falnes PO, Vågbø CB, Skorpen F, Akbari M, Sundheim O, Bjørås M, Slupphaug G, Seeberg E, et al. 2003. Human and bacterial oxidative demethylases repair alkylation damage in both RNA and DNA. Nature 421: 859–863. [DOI] [PubMed] [Google Scholar]
- Carter AP, Clemons WM Jr, Brodersen DE, Morgan-Warren RJ, Hartsch T, Wimberly BT, Ramakrishnan V. 2001. Crystal structure of an initiation factor bound to the 30S ribosomal subunit. Science 291: 498–501. [DOI] [PubMed] [Google Scholar]
- Demeshkina N, Jenner L, Westhof E, Yusupov M, Yusupova G. 2012. A new understanding of the decoding principle on the ribosome. Nature 484: 256–259. [DOI] [PubMed] [Google Scholar]
- Demple B, Jacobsson A, Olsson M, Robins P, Lindahl T. 1982. Repair of alkylated DNA in Escherichia coli. Physical properties of O6-methylguanine-DNA methyltransferase. J Biol Chem 257: 13776–13780. [PubMed] [Google Scholar]
- Doma MK, Parker R. 2006. Endonucleolytic cleavage of eukaryotic mRNAs with stalls in translation elongation. Nature 440: 561–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob-Hirsch J, Amariglio N, Kupiec M, et al. 2012. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485: 201–206. [DOI] [PubMed] [Google Scholar]
- Eadie JS, Conrad M, Toorchen D, Topal MD. 1984. Mechanism of mutagenesis by O6-methylguanine. Nature 308: 201–203. [DOI] [PubMed] [Google Scholar]
- Fu Y, Dominissini D, Rechavi G, He C. 2014. Gene expression regulation mediated through reversible m6A RNA methylation. Nat Rev Genet 15: 293–306. [DOI] [PubMed] [Google Scholar]
- Gromadski KB, Rodnina MV. 2004. Kinetic determinants of high-fidelity tRNA discrimination on the ribosome. Mol Cell 13: 191–200. [DOI] [PubMed] [Google Scholar]
- Gromadski KB, Daviter T, Rodnina MV. 2006. A uniform response to mismatches in codon-anticodon complexes ensures ribosomal fidelity. Mol Cell 21: 369–377. [DOI] [PubMed] [Google Scholar]
- Hofer T, Badouard C, Bajak E, Ravanat JL, Mattsson A, Cotgreave IA. 2005. Hydrogen peroxide causes greater oxidation in cellular RNA than in DNA. Biol Chem 386: 333–337. [DOI] [PubMed] [Google Scholar]
- Jelenc PC, Kurland CG. 1979. Nucleoside triphosphate regeneration decreases the frequency of translation errors. Proc Natl Acad Sci 76: 3174–3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, Yi C, Lindahl T, Pan T, Yang YG, et al. 2011. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol 7: 885–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson M, Bouakaz E, Lovmar M, Ehrenberg M. 2008. The kinetics of ribosomal peptidyl transfer revisited. Mol Cell 30: 589–598. [DOI] [PubMed] [Google Scholar]
- Korostelev A, Asahara H, Lancaster L, Laurberg M, Hirschi A, Zhu J, Trakhanov S, Scott WG, Noller HF. 2008. Crystal structure of a translation termination complex formed with release factor RF2. Proc Natl Acad Sci 105: 19684–19689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurberg M, Asahara H, Korostelev A, Zhu J, Trakhanov S, Noller HF. 2008. Structural basis for translation termination on the 70S ribosome. Nature 454: 852–857. [DOI] [PubMed] [Google Scholar]
- Leonard GA, Thomson J, Watson WP, Brown T. 1990. High-resolution structure of a mutagenic lesion in DNA. Proc Natl Acad Sci 87: 9573–9576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, Jaffrey SR. 2014. The dynamic epitranscriptome: N6-methyladenosine and gene expression control. Nat Rev Mol Cell Biol 15: 313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. 2012. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 149: 1635–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore SD, Sauer RT. 2007. The tmRNA system for translational surveillance and ribosome rescue. Annu Rev Biochem 76: 101–124. [DOI] [PubMed] [Google Scholar]
- Murphy FV IV, Ramakrishnan V. 2004. Structure of a purine-purine wobble base pair in the decoding center of the ribosome. Nat Struct Mol Biol 11: 1251–1252. [DOI] [PubMed] [Google Scholar]
- Nierhaus KH. 2006. Decoding errors and the involvement of the E-site. Biochimie 88: 1013–1019. [DOI] [PubMed] [Google Scholar]
- Ninio J. 2006. Multiple stages in codon-anticodon recognition: double-trigger mechanisms and geometric constraints. Biochimie 88: 963–992. [DOI] [PubMed] [Google Scholar]
- Ogle JM, Brodersen DE, Clemons WM Jr, Tarry MJ, Carter AP, Ramakrishnan V. 2001. Recognition of cognate transfer RNA by the 30S ribosomal subunit. Science 292: 897–902. [DOI] [PubMed] [Google Scholar]
- Ogle JM, Murphy FV, Tarry MJ, Ramakrishnan V. 2002. Selection of tRNA by the ribosome requires a transition from an open to a closed form. Cell 111: 721–732. [DOI] [PubMed] [Google Scholar]
- O'Reilly SM, Newlands ES, Glaser MG, Brampton M, Rice-Edwards JM, Illingworth RD, Richards PG, Kennard C, Colquhoun IR, Lewis P, et al. 1993. Temozolomide: a new oral cytotoxic chemotherapeutic agent with promising activity against primary brain tumours. Eur J Cancer 29A: 940–942. [DOI] [PubMed] [Google Scholar]
- Ougland R, Zhang CM, Liiv A, Johansen RF, Seeberg E, Hou YM, Remme J, Falnes PØ. 2004. AlkB restores the biological function of mRNA and tRNA inactivated by chemical methylation. Mol Cell 16: 107–116. [DOI] [PubMed] [Google Scholar]
- Pape T, Wintermeyer W, Rodnina MV. 1998. Complete kinetic mechanism of elongation factor Tu-dependent binding of aminoacyl-tRNA to the A site of the E. coli ribosome. EMBO J 17: 7490–7497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pape T, Wintermeyer W, Rodnina M. 1999. Induced fit in initial selection and proofreading of aminoacyl-tRNA on the ribosome. EMBO J 18: 3800–3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsa I, Friedman S, Cleary CM. 1987. Visualization of O6-methylguanine in target cell nuclei of dimethylnitrosamine-treated human pancreas by a murine monoclonal antibody. Carcinogenesis 8: 839–846. [DOI] [PubMed] [Google Scholar]
- Schmeing TM, Voorhees RM, Kelley AC, Gao YG, Murphy FV IV, Weir JR, Ramakrishnan V. 2009. The crystal structure of the ribosome bound to EF-Tu and aminoacyl-tRNA. Science 326: 688–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedgwick B, Bates PA, Paik J, Jacobs SC, Lindahl T. 2007. Repair of alkylated DNA: recent advances. DNA Repair (Amst) 6: 429–442. [DOI] [PubMed] [Google Scholar]
- Simms CL, Hudson BH, Mosior JW, Rangwala AS, Zaher HS. 2014. An active role for the ribosome in determining the fate of oxidized mRNA. Cell Rep 9: 1256–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snow ET, Foote RS, Mitra S. 1984. Base-pairing properties of O6-methylguanine in template DNA during in vitro DNA replication. J Biol Chem 259: 8095–8100. [PubMed] [Google Scholar]
- Tsuboi T, Kuroha K, Kudo K, Makino S, Inoue E, Kashima I, Inada T. 2012. Dom34:hbs1 plays a general role in quality-control systems by dissociation of a stalled ribosome at the 3′ end of aberrant mRNA. Mol Cell 46: 518–529. [DOI] [PubMed] [Google Scholar]
- Voorhees RM, Ramakrishnan V. 2013. Structural basis of the translational elongation cycle. Annu Rev Biochem 82: 203–236. [DOI] [PubMed] [Google Scholar]
- Voorhees RM, Weixlbaumer A, Loakes D, Kelley AC, Ramakrishnan V. 2009. Insights into substrate stabilization from snapshots of the peptidyl transferase center of the intact 70S ribosome. Nat Struct Mol Biol 16: 528–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker SE, Fredrick K. 2008. Preparation and evaluation of acylated tRNAs. Methods 44: 81–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren JJ, Forsberg LJ, Beese LS. 2006. The structural basis for the mutagenicity of O6-methyl-guanine lesions. Proc Natl Acad Sci 103: 19701–19706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weixlbaumer A, Jin H, Neubauer C, Voorhees RM, Petry S, Kelley AC, Ramakrishnan V. 2008. Insights into translational termination from the structure of RF2 bound to the ribosome. Science 322: 953–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimberly BT, Brodersen DE, Clemons WM Jr, Morgan-Warren RJ, Carter AP, Vonrhein C, Hartsch T, Ramakrishnan V. 2000. Structure of the 30S ribosomal subunit. Nature 407: 327–339. [DOI] [PubMed] [Google Scholar]
- Wurtmann EJ, Wolin SL. 2009. RNA under attack: cellular handling of RNA damage. Crit Rev Biochem Mol Biol 44: 34–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youngman EM, Brunelle JL, Kochaniak AB, Green R. 2004. The active site of the ribosome is composed of two layers of conserved nucleotides with distinct roles in peptide bond formation and peptide release. Cell 117: 589–599. [DOI] [PubMed] [Google Scholar]
- Youngman EM, Cochella L, Brunelle JL, He S, Green R. 2006. Two distinct conformations of the conserved RNA-rich decoding center of the small ribosomal subunit are recognized by tRNAs and release factors. Cold Spring Harb Symp Quant Biol 71: 545–549. [DOI] [PubMed] [Google Scholar]
- Zaher HS, Green R. 2009a. Fidelity at the molecular level: lessons from protein synthesis. Cell 136: 746–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaher HS, Green R. 2009b. Quality control by the ribosome following peptide bond formation. Nature 457: 161–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaher HS, Green R. 2010. Hyperaccurate and error-prone ribosomes exploit distinct mechanisms during tRNA selection. Mol Cell 39: 110–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaher HS, Unrau PJ. 2004. T7 RNA polymerase mediates fast promoter-independent extension of unstable nucleic acid complexes. Biochemistry 43: 7873–7880. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.