

Summary
By integrating growth pathways that cancer cells rely on, steroid receptor coactivators (SRC-1, SRC-2, SRC-3) represent emerging targets in cancer therapeutics. High throughput screening for SRC small molecule inhibitors (SMI) uncovered MCB-613 as a potent SRC small molecule ‘stimulator’ (SMS). We demonstrate that MCB-613 can super-stimulate SRCs’ transcriptional activity. Further investigation revealed that MCB-613 increases SRCs’ interactions with other coactivators and markedly induces ER stress coupled to the generation of reactive oxygen species (ROS). Since cancer cells overexpress SRCs and rely on them for growth, we show that we can exploit MCB-613 to induce excessive stress selectively in cancer cells. This suggests that over-stimulating the SRC oncogenic program can be an effective strategy to kill cancer cells.
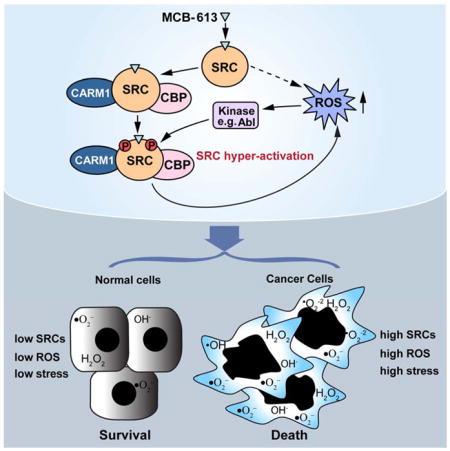
Graphical Abstract
Introduction
Members of the p160 steroid receptor coactivator (SRC) family, SRC-1, SRC-2/TIF2/GRIP1 and SRC-3/AIB1/RAC3/ACTR/pCIP interact with nuclear receptors and other transcription factors to drive target gene expression while also functioning as integrators of upstream cell signaling pathways (Lonard and O’Malley B, 2007). Although they share homology with each other, they have distinct and important roles in multiple physiological processes including growth and development, reproduction and metabolism (Xu et al., 2009; York and O’Malley, 2010). All three proteins also have been found to be broadly involved in different aspects of tumorigenesis. SRC-3 is most well-known for its oncogenic role, whose gene is amplified in 9.5% of breast cancers (Anzick et al., 1997) and whose mRNA has been shown to be overexpressed in different breast cancer cohorts, often at the 50% level or greater (Anzick et al., 1997; Bouras et al., 2001; Glaeser et al., 2001; Zhao et al., 2003). Clinically, SRC-3 overexpression in breast cancer correlates with larger tumor size (Bautista et al., 1998), higher tumor grade (Hudelist et al., 2003) and poor survival rates (Zhao et al., 2003). Direct evidence supporting SRC-3 as a bona fide oncogene comes from a MMTV-SRC-3 transgenic mouse model, in which overexpression of SRC-3 was sufficient to cause spontaneous development of malignant mammary tumors (Torres-Arzayus et al., 2004). SRC-3 overexpression also has been observed in endometrial (Kershah et al., 2004), ovarian (Bautista et al., 1998), prostate (Gnanapragasam et al., 2001), colorectal (Xie et al., 2005), gastric (Sakakura et al., 2000), lung (Cai et al., 2010), pancreatic (Henke et al., 2004) and liver cancers (Wang et al., 2002). Additional in vitro and in vivo studies have bolstered the importance of SRC-3 in tumor initiation, progression, metastasis and drug resistance (Xu et al., 2009). SRC-1 also is overexpressed in about 20% breast cancers and is positively correlated with ERBB2 expression, disease recurrence and poor survival (Fleming et al., 2004; Myers et al., 2004). It has been demonstrated that SRC-1 plays a critical role in cancer cell migration, invasion and metastasis (Qin et al., 2009). Finally, SRC-2 has been proposed as a key oncogene in prostate cancer based on a comprehensive analysis on prostate tumors, cell lines and xenografts, revealing that SRC-2 gene amplification, overexpression and mutations specifically arise to levels of 38% in metastatic prostate tumors (Taylor et al., 2010).
Although tumor formation is a multistage process involving activation of oncogenes and inactivation of tumor suppressors, accumulating evidence indicates that loss of a specific oncogene can frequently reverse the malignant progression of cancer cells, suggesting that cancer cells rely on the continued activation or overexpression of an oncogene (Chin et al., 1999; Felsher and Bishop, 1999; Huettner et al., 2000). This ‘oncogene addiction’ theory, combined with the fact that SRC proteins integrate and promote multiple growth factor signaling pathways crucial for cancer cell growth and survival (Fereshteh et al., 2008; Torres-Arzayus et al., 2004; Torres-Arzayus et al., 2006), highlights the potential value of SRC targeting drugs as future anti-cancer agents. In an initial proof-of-principle study, we identified gossypol as a small molecule inhibitor (SMI) of SRC-1 and SRC-3, which can decrease SRC-1/-3 protein level and cause cell death in various cancer cell lines (Wang et al., 2011). Inspired by this result, a large high throughput compound screening campaign was undertaken against all three SRCs, leading to the identification of improved SRC SMIs including bufalin and verrucarin A (Wang et al., 2014; Yan et al., 2014).
Since cancer cells rely heavily on SRCs to maintain homeostasis, we further hypothesized that the over-stimulation of SRCs through small molecule stimulators, although mechanistically distinct from that of SRC SMIs, might also be able to disrupt the borderline homeostasis of cancer cells, leading to acute stress enhancement and cell death, especially in cancer cells that depend upon SRCs. In this study, we characterize an SRC small molecule stimulator and investigate its biological activities and anti-cancer potential.
Results
MCB-613 is a pan-SRC stimulator
In a series of high throughput screens originally designed to identify SRC SMIs, HEK293 cells transfected with a Gal4 responsive luciferase reporter (pG5-luc) and a construct encoding SRC-1, SRC-2 or SRC-3 fused with the DNA binding domain of Gal4 (pBIND-SRC-1, pBIND-SRC-2 or pBIND-SRC-3) were exposed to treatments with 359,484 compounds from a MLPCN chemical library (PubChem AID: 588362, 652008 and 588354). In addition to inhibitors that were initially sought in these screens, a number of compounds were found to stimulate the activity of SRC-1 and SRC-3. Primary screens indicated that there were 106 and 28 potential SRC-1 and SRC-3 stimulators, respectively, using a 2-fold activation cutoff compared with vehicle controls. Upon further testing and verification, MCB-613 (4-Ethyl-2,6-bis-pyridin-3-ylmethylene-cyclohexanone) (see Figure S1A), which exerted the greatest activation of SRC-1 in the primary screen, was confirmed to be a strong activator of all three SRCs. As shown in Figure 1A, 24-hour treatment with MCB-613 caused an extremely large and unprecedented (maximum 160-fold) induction in the activity of pBIND-SRC-1/-2/-3 in a dose-dependent manner. This stimulation by MCB-613 was selective for SRCs, failing to stimulate a GAL4-PGC-1α coactivator fusion protein (Figure 1B) while minimally increasing the activity of GAL4-VP16 only at higher compound concentrations (Figure S1B). The activation of SRCs by MCB-613 is so strong and rapid that a significant increase in SRC activity was already observed after 4 hours of treatment (Figure S1E); and it represents the strongest stimulation observed to date under any conditions.
Figure 1. MCB-613 selectively activates the intrinsic transcriptional activity of SRCs.

(A) Luciferase assays on HeLa cells co-transfected with pG5-luc and pBIND or pBIND-SRC-1/-2/-3 expression vectors after the treatment of MCB-613 for 24 hours.
(B) Luciferase assays on HeLa cells co-transfected with pG5-luc and Gal4-DBD or Gal4-PGC-1α expression vectors and treated with increasing concentrations of MCB-613 for 24 hours.
(C) Luciferase assays on HeLa cells co-transfected with an SRC-3 expression vector and MMP2-luc or MMP13-luc reporters and treated with MCB-613 for 24 hours.
(D) Luciferase assays on SRC-3 WT or KO HeLa cells transfected with a MMP2-luc reporter plasmid and treated with MCB-613 for 24 hours.
(E) qRT-PCR analysis of MMP13 in MDA-MB-231 cells treated with MCB-613 for 24 hours.
(F) Immunoblotting of SRCs from HeLa cells treated with MCB-613 for 24 hours.
(G) Surface plasmon resonance (SPR) showing the direct binding of MCB-613 to SRC-3 RID. Upper panel: SPR signal traces of RID in the presence of increasing concentrations of MCB-613; bottom panel: RID’s SPR signals normalized against a reference cell with no immobilized protein exposed to increasing concentrations of MCB-613.
(H) Immunoblotting of CBP and CARM1 in the coimmunoprecipitation (coIP) complex from FLAG or FLAG-SRC-3 overexpressing HeLa cells treated with MCB-613 at the indicated concentrations for 1 hour.
Data are presented as mean ± SEM. ** p<0.01, *** p<0.001. See also Figure S1.
Since SRC-3 has been shown to be preferentially utilized by AP-1 and PEA3 as a coactivator to drive the expression of MMP2 and MMP13 (Yan et al., 2008), we transfected cells with a MMP2 or MMP13 promoter driven luciferase reporter (MMP2-luc or MMP13-luc) and treated these cells with MCB-613. We found that SRC-3’s coactivation of these promoters was greatly enhanced by MCB-613 (Figure 1C). In order to confirm that the MCB-613 effect is SRC-dependent, we performed the same experiment in a SRC-3 KO HeLa cell line, in which both alleles of SRC-3 are knocked out using a zinc finger nuclease (Wang et al., 2014), and found that activation of MMP2-luc by MCB-613 was diminished (Figure 1D). As a further validation, knocking down all three SRCs together by siRNAs significantly impaired the ability of MCB-613 to activate the MMP2 promoter (Figure S1C). Consistent with these findings, endogenous MMP13 expression could be also activated with MCB-613 treatment (Figure 1E).
In order to characterize the underlying mechanism of MCB-613’s activation of SRCs, we initially sought to determine whether MCB-613 increases SRC activity by elevating the concentrations of SRC proteins in the cell. Instead, we found that treatment with MCB-613 resulted in decreases in SRC-1, SRC-2 and SRC-3 protein levels (Figure 1F), indicating that MCB-613 inherently promotes the intrinsic activity of SRCs. The reduction of SRC proteins is not a transcriptional event, since the mRNA levels of SRC-1/-2/-3 were not down-regulated by MCB-613 treatment (Figure S1D). It has been previously demonstrated by our laboratory that SRC-3 activation is coupled to its turnover (Wu et al., 2007). Therefore, the increased SRC protein instability/degradation is probably associated with their hyper-activation. Our time course experiment also supports the idea that there is a correlation between SRC-3’s activity and protein level under the treatment of MCB-613. As shown in Figure S1E, 4 hours was the earliest time point we could see the activation of SRC-3 by MCB-613 in luciferase assays, and it is at this time point that we started to observe a significant decrease in SRC-3 protein level.
Next, we assessed whether MCB-613 physically interacts with SRCs by using the technique of surface plasmon resonance (SPR) (Karlsson and Stahlberg, 1995). A series of concentrations of MCB-613 were flowed over immobilized SRC-3 protein fragments including the receptor interacting domain (RID), CBP interacting domain (CID) and the basic helix-loop-helix (bHLH) domain, with an increase in the SPR signal indicating direct binding. As shown in Figure 1G, MCB-613 binds to the RID of SRC-3 in a reversible manner, as the SPR signal subsided when MCB-613 was removed. The same interaction was confirmed by fluorescence spectroscopy which measured the intrinsic fluorescence of different SRC-3 protein domains in the presence of MCB-613, based on the theory that direct SRC-3 binding with MCB-613 should quench its intrinsic fluorescence emission (Epps et al., 1999; Wang et al., 2011). In Figure S1F, the intrinsic fluorescence of the RID was progressively quenched by increasing concentrations of MCB-613, and the emission maximum was shifted from 340nm to 360nm, again indicating that MCB-613 directly binds to SRC-3 RID. Both these two assays showed that a much higher concentration of MCB-613 is required for the interaction between SRC-3 CID and MCB-613; and due to noisy data, a clear conclusion about the bHLH domain of SRC-3 could not be reached (data not shown). Taken together, these results suggest that MCB-613 selectively and reversibly binds to the RID of SRC-3.
Activated SRC-3 has been shown to recruit other transcriptional coactivators including CBP and CARM1 to form a multi-coactivator complex (Chen et al., 1999; Chen et al., 1997; Foulds et al., 2013). In order to test whether MCB-613 affects the ability of SRC-3 to interact with these coactivator complex members, HeLa cells overexpressing FLAG-SRC-3 were treated with MCB-613 for 1 hour and subjected to coimmunoprecipitation to assess SRC-3-CBP-CARM1 complex formation. As shown in Figure 1H, MCB-613 increased SRC-3’s interaction with CBP and CARM1 robustly in a dose-dependent manner. Longer treatment of 4 or 8 hours with the compound increased the SRC-3-CBP interaction, while no further interaction between SRC-3 and CARM1 was observed (Figure S1G and S1H). These results indicate that MCB-613 binds to SRC-3, where it then promotes coactivator complex formation, consistent with induced coactivator transcriptional activity.
MCB-613 selectively kills cancer cells
While studying the activation of SRCs by MCB-613, we noticed that MCB-613 is cytotoxic. It can efficiently kill a variety of human cancer cell lines (Figure 2A), including MCF-7 (breast), PC-3 (prostate), H1299 (lung) and HepG2 (liver) cells. Intriguingly, although highly toxic to cancer cells, MCB-613 can spare normal cells at these concentrations, as mouse primary hepatocytes and mouse embryonic fibroblasts (MEF) are resistant to this compound (Figure 2A). In order to rule out the possible species-specific toxicity of MCB-613, we tested the compound on primary human umbilical vein endothelial cells (HUVEC) and found that, at low concentration of MCB-613, HUVEC cells are more resistant than almost all of the human cancer cell lines we have tested (Figure S2A), although cell death still occurs at high doses of the compound, which, in part, may be due to the higher fragility of primary human cells in culture.
Figure 2. MCB-613 induces complex cytotoxicity with characteristics of paraptotic-like cell death.

(A) Viability of the indicated cells treated with MCB-613 for 48 hours.
(B) Extensive cytoplasmic vacuolization in the indicated cancer cell lines treated with MCB-613. Scale bar: 20μm.
(C) Immunofluorescence of calnexin in HeLa cells treated with MCB-613 for 24 hours. Scale bar: 20μm.
(D) ROS levels in MCB-613 treated HeLa cells as demonstrated by CM-H2DCFDA, a general ROS indicator. Scale bar: 20μm.
(E) Immunoblotting of ubiquitin from HeLa cells treated with MCB-613 for 5 hours.
(F) Immunoblotting of the indicated UPR markers from HeLa cells treated with MCB-613 for 4 hours.
(G) Viability of HeLa cells treated with MCB-613 for 24 hours in the presence or absence of NAC or CHX.
(H) Morphology of HeLa cells treated as in G. Scale bar: 40μm.
Data are presented as mean ± SD. *** p<0.001. See also Figure S2.
One prominent phenomenon we observed is that MCB-613 treatment induced massive cytoplasmic vacuolization (Figure 2B). To better understand how MCB-613 causes cell death, we first examined if apoptosis or autophagy was activated, especially considering that autophagy is the major cell death mechanism linked to vacuolization. MCB-613 treatment led to the induction of LC3BII and caspase-3 cleavage, markers for autophagy and apoptosis, respectively (Figure S2B). However, autophagy inhibitors, such as chloroquine and 3-methyladenine (3-MA), cannot rescue cell death caused by MCB-613 (Figure S2C), suggesting that autophagy is not involved in the cytotoxicity and that it is probably induced by the cells as a futile survival response. Although a caspase inhibitor, z-VAD-fmk, can inhibit MCB-613 mediated cell death to a significant extent (Figure S2C), MCB-613, unlike other known apoptosis inducers such as etoposide and sodium nitroprusside (SNP), does not cause inter-nucleosomal DNA fragmentation, a classic nuclear characteristic of apoptosis (Figure S2E), implying that apoptosis might not be the primary form of cell death. In addition, neither apoptosis nor autophagy was directly linked to the activation of SRCs, as z-VAD-fmk, chloroquine or 3-MA did not inhibit the activation of SRC-3 by MCB-613 (Figure S2D).
A non-apoptotic form of cell death, paraptosis, is characterized by extensive cytoplasmic vacuolization caused by the dilation of endoplasmic reticulum (ER) and mitochondria and it does not rely upon hallmarks of apoptosis such as DNA fragmentation and caspase activation (Sperandio et al., 2000). Paraptotic-like cell death has been observed in embryo development (Clarke, 1990) and neuronal degeneration (Pilar and Landmesser, 1976), in several artificial or natural cellular models (Abraham et al., 2007; Chen et al., 2002; Sperandio et al., 2000), and in response to the treatment with some anti-cancer agents (Kar et al., 2009; Yoon et al., 2010). Although the molecular details underlying paraptosis are still relatively understudied, there is evidence suggesting that the vacuolization results from perturbations in ER and proteasome functions (Denoyelle et al., 2006; Mimnaugh et al., 2006; Ustundag et al., 2007). In addition to proteasome dysfunction and ER stress, paraptosis requires de novo protein synthesis and is linked to cellular redox homeostasis (Sperandio et al., 2000; Yoon et al., 2010).
Intrigued by the possibility that paraptosis might be involved in MCB-613’s effect, we used transmission electron microscopy (TEM) to examine in detail the morphology and ultrastructure of the cells after the treatment with the compound. As shown in Figure S2F, compared with vehicle treated cells which have normal mitochondria and ER, cells treated with MCB-613 for 8 hours have vacuoles of various sizes throughout their cytoplasm, consistent with paraptosis. In addition, these cells show no signs of chromatin condensation, nuclear fragmentation or cellular blebbing, again suggesting that they are not going primarily through apoptosis. Moreover, those vacuoles show no resemblance to autophagosomes, which are double-membraned vesicles usually containing proteins or organelles destined for degradation. However, we do see autophagosomes in MCB-613 treated cells by TEM (data not shown), which is in agreement with the induction of LC3BII. In order to further investigate if the cytotoxicity induced by MCB-613 exhibits paraptotic features, we confirmed the ER origin of the MCB-613 induced vacuoles, since they stained positive for calnexin (Figure 2C), an ER-specific marker. Next, by using CM-H2DCFDA, a general ROS indicator, we found that MCB-613 can induce a rapid and marked increase in intracellular ROS levels (Figure 2D). MCB-613 also leads to proteasome dysfunction and ER stress, confirmed by the accumulation of polyubiquitinated proteins (Figure 2E) and the induction of the markers for unfolded protein response (UPR) (Figure 2F), including the phosphorylation of eIF2α and IRE1α as well as the induction of ATF4 protein expression. Since MCB-613 increases ROS levels and paraptosis requires protein synthesis, we next examined whether an antioxidant or a protein synthesis inhibitor can block cell death caused by MCB-613. As shown in Figure 2G, the antioxidant N-Acetyl cysteine (NAC) and the protein synthesis inhibitor cycloheximide (CHX) protected cells from MCB-613 mediated cytotoxicity. We further confirmed that NAC can efficiently eradicate the high level of ROS in the cells treated by MCB-613 (Figure S2G). Consistently, another antioxidant, MnTBAP, a superoxide dismutase (SOD) mimetic, can retard MCB-613 induced cell death, although not as effectively as NAC (Figure S2H). Importantly, vacuolization induced by MCB-613 also was blocked by co-treatment with NAC or CHX (Figure 2H), pointing to paraptotic-like cell death as the primary process underlying the cytotoxic properties of MCB-613. Taken together, these results indicate that MCB-613 selectively kills cancer cells by inducing complex cytotoxicity with features that are characteristic of paraptosis. The apoptotic effect, we speculate, might be a secondary response to higher concentrations or longer treatment time with MCB-613.
SRC hyper-activation is critical for the paraptotic cytotoxicity induced by MCB-613
One key to understanding the mechanism of MCB-613 is to establish the order of events as to whether SRC activation is a consequence of UPR induction or vice versa. We found that known ER stress inducers, such as tunicamycin, failed to recapitulate the effect of MCB-613 to hyper-activate SRC-3 (Figure S3A). In addition, although MCB-613 seems to share some structural similarity with a recently identified PERK activator (Bai et al., 2013), its ability to induce the splicing of Xbp1, another marker of UPR, remains intact in the absence of PERK expression (Figure S3B). Importantly, activation of SRC-3 by MCB-613 is not impacted when essential UPR players like PERK or IRE1α are knocked down by siRNA (Figure S3C and S3D). All these data strongly support the notion that MCB-613 is distinct from other UPR inducers and that SRC hyper-activation by MCB-613 does not rely on but instead precedes UPR induction. We proceeded to determine if a causal link exists between SRC super-activation by MCB-613 and paraptotic-like cell death. SRC-3 KO HeLa cell viability was found to be less affected by MCB-613 compared with WT cells (Figure 3A), suggesting that the cell death induced by MCB-613 is at least partially dependent on SRC-3 (SRC-1 and SRC-2 are still expressed in SRC-3 KO cells). We next sought to examine the expression of downstream stress-response genes that are induced by MCB-613 in a SRC-3 dependent manner, using the human Stress and Toxicity PathwayFinder qPCR array on SRC-3 WT or KO HeLa cells treated with MCB-613. The array focuses on genes involved in oxidative stress, hypoxia, DNA damage and UPR. As shown in Figure 3B, multiple UPR regulators or downstream targets, such as ATF6B, CHOP, DNAJC3 and TNFRSF10B, were either significantly down-regulated or up-regulated upon MCB-613 treatment in WT cells. However, these changes were all blunted in SRC-3 KO cells, again suggesting that SRC-3 stimulation partly underlies the stress response pathways activated by MCB-613 treatment.
Figure 3. SRC hyper-activation is critical for the paraptotic-like cytotoxicity induced by MCB-613.

(A) Viability of SRC-3 WT or KO HeLa cells treated with MCB-613 for 24 hours.
(B) qRT-PCR analysis of the indicated UPR factors in SRC-3 WT and KO HeLa cells treated with MCB-613 for 24 hours.
(C) qRT-PCR analysis of LDHA and GSR from SRC-3 WT and KO HeLa cells treated as in B.
(D) Immunoblotting of spliced Xbp1 and SRCs from HeLa cells transfected with control siRNAs or siRNAs targeting all three SRCs and treated with MCB-613 for 24 hours. Numbers indicate the quantification of the bands for spliced Xbp1.
(E) Morphology of HeLa cells treated with MCB-613 for 24 hours in the presence or absence of bufalin. Scale bar: 40μm.
(F) Immunoblotting of ATF4, cleaved caspase-3 and SRCs from HeLa cells treated as in E.
Data are presented as mean ± SD (MTS assay) or mean ± SEM (qRT-PCR). ** p<0.01, *** p<0.001. See also Figure S3.
We also found that oxidative stress related genes are differentially regulated in MCB-613 treated SRC-3 WT and KO cells (Figure 3C). Lactate dehydrogenase A (LDHA) is an enzyme that converts L-lactate and NAD+ to pyruvate and NADH in the final step of anaerobic glycolysis, whose inhibition has been shown to increase oxidative stress and to interfere with tumor progression (Le et al., 2010). Glutathione reductase (GSR) is a central enzyme responsible for cellular antioxidant defense. MCB-613 treatment resulted in decreased LDHA expression and an increase in GSR expression in WT HeLa cells, consistent with the notion that the cells are attempting to mount a response to the ROS generating effects of MCB-613. Meanwhile, in SRC-3 KO cells treated with MCB-613, there was an attenuated decrease in LDHA expression and a stronger increase in GSR expression compared with WT cells, implying that the KO cells are more resistant to MCB-613 and suffered less oxidative stress due to the absence of SRC-3.
As a further demonstration that SRC activation drives the paraptotic-like cytotoxicity of MCB-613, siRNA mediated knock down of all three SRCs simultaneously led to a significant impairment of both the splicing of Xbp1 and the induction of ATF4 caused by MCB-613 treatment (Figure 3D and S3E). Along the same lines, cytoplasmic vacuolization and the induction of ATF4 were effectively inhibited by the co-treatment of MCB-613 with the SRC SMI bufalin (Figure 3E and 3F), which has been shown to inhibit all three SRCs (Wang et al., 2014). Instead, bufalin treatment induced caspase-3 cleavage (Figure 3F), consistent with prior findings that loss of SRC-3 can lead to apoptosis (List et al., 2001). All together, these multiple lines of evidence lay down a strong foundation supporting that the UPR induction by MCB-613 is SRC dependent and that hyper-activation of SRCs is responsible for the paraptotic-like cell death response induced by this compound.
One response the cells engage to counteract ER stress is to induce molecular chaperones, such as heat shock proteins, in order to enhance the folding capacity of ER and alleviate the accumulation of misfolded/unfolded proteins. Indeed, many heat shock proteins were significantly induced by treatment with MCB-613 (Figure S3F), suggesting that the cells are striving to battle the enormous stress imposed by the compound. Based on this observation, we speculated that, if excessive ER stress is the primary cause of cell death induced by MCB-613, inhibiting heat shock protein function should further aggravate the deleterious effect of this compound. As expected, geldanamycin, an HSP90 inhibitor, synergizes with MCB-613 to induce ER stress and enhances MCB-613 mediated cell death (Figure S3G and S3H). Consistently, knocking down HSF1 (heat shock factor 1), a major transcription factor for heat shock proteins, also exacerbates the cell death caused by MCB-613 (Figure S3I). Interestingly, the synergism between MCB-613 and geldanamycin also applied to the activation of SRC-3 (Figure S3J), further implying that super-activation of SRCs is integral to the effect of MCB-613 on ER stress and cell viability.
More support for the close relationship between SRCs and paraptotic-like cell death is evidenced by our observations that additional agents, including curcumin, 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2) and MG132, can robustly enhance the intrinsic transcriptional activities of SRCs (Figure S3K and S3L). All these agents have been shown to cause cell death with cytoplasmic vacuolization and devastating stress responses (Ding et al., 2007; Kar et al., 2009; Yoon et al., 2010), suggesting that SRC hyper-stimulation is closely coupled to cellular stress pathways connected to paraptosis.
Oxidative stress induced by MCB-613 is SRC-dependent and contributes to SRC hyper-activation
The rapid increase of intracellular ROS levels in response to MCB-613 treatment led us to examine the relationship between oxidative stress and the activation of SRCs. We found that simultaneous knock down of all three SRCs significantly inhibits the production of ROS by MCB-613 (Figure 4A), suggesting that SRCs are indispensible for MCB-613 induced oxidative stress. Interestingly, co-treatment with the antioxidant NAC was able to abrogate the stimulatory effects of MCB-613 on SRC transcriptional activity (Figure 4B and Figure S4A). At the same time, the decrease of SRC protein levels normally seen in MCB-613 treated cells was blocked by NAC (Figure S4B), which again confirms that there is a correlation between SRC activity and protein level.
Figure 4. Oxidative stress induced by MCB-613 depends on SRCs and drives further SRC hyper-activation via the Abl kinase signaling pathway.

(A) ROS levels as indicated by CM-H2DCFDA in HeLa cells which were transfected with control siRNAs or siRNAs targeting all three SRCs and treated with MCB-613. A representative picture from multiple fields is shown for each treatment (left panel). Fluorescence signals are quantified (middle panel) and knock down efficiency is shown by immunoblotting (right panel). Scale bar: 200μm.
(B) Luciferase assays on HeLa cells co-transfected with pG5-luc and pBIND or pBIND-SRC-3 expression vectors and treated with increasing concentrations of MCB-613 for 24 hours in the presence or absence of NAC.
(C) The immunoprecipitate from FLAG or FLAG-SRC-3 overexpressing HeLa cells treated with MCB-613 at the indicated concentrations for 1 hour was resolved on a 5% SDS-PAGE gel containing 20 μM Phos-tag and immunoblotted with anti-FLAG antibody.
(D) Immunoblotting of phosphorylated CrkL from HeLa cells treated with MCB-613 for 1 hour or 4 hours.
(E) Immunoblotting of Abl in the coIP complex from FLAG or FLAG-SRC-3 overexpressing HeLa cells treated with MCB-613 at the indicated concentrations for 1 hour.
(F) Luciferase assays on HeLa cells co-transfected with pG5-luc and pBIND or pBIND-SRC-3 and treated with MCB-613 for 24 hours in the presence or absence of AT9283 or PHA739358.
(G) HeLa cells transfected with control siRNA or siAbl1 were transfected with pBIND or pBIND-SRC-3 and pG5-luc, followed by treatment with increasing concentrations of MCB-613 for 24 hours and luciferase assays. Knockdown efficiency of Abl is shown in the right panel.
Data are presented as mean ± SEM. * p<0.05, ** p<0.01, *** p<0.001. See also Figure S4.
ROS has been shown to activate multiple kinase signaling pathways (Ray et al., 2012) and because SRCs are phospho-proteins that function as conduits for growth factor kinase cascades (Wu et al., 2004), we wanted to explore the impact of MCB-613 treatment on SRC-3 phosphorylation. Using a phos-tag SDS-PAGE system that allows phosphorylated proteins to be characterized by differences in their migration rate (Kinoshita et al., 2004), we found that MCB-613 treatment resulted in a SRC-3 species with reduced mobility in the gel (Figure 4C) which disappeared after lambda phosphatase treatment (data not shown), indicative of a phosphorylated form of SRC-3. In order to investigate which kinase(s) are responsible for MCB-613 induced phosphorylation, we screened a kinase inhibitor library from Selleck Chemicals, which contained a collection of 141 kinase inhibitors, and found that a number of Abl kinase inhibitors were able to inhibit the activation of pBIND-SRC-3 by MCB-613 (Figure 4F and data not shown).
Abl is a non-receptor tyrosine kinase present in both the cytoplasm and nucleus that has been implicated in a variety of cellular processes such as growth, differentiation and stress response (Greuber et al., 2013). Consequently, Abl is a promising candidate kinase involved in MCB-613 mediated SRC hyper-stimulation because 1) acute hyper-activation of the oncogenic Bcr-Abl fusion protein has been shown to induce severe cytoplasmic vacuolization and ER stress (Dengler et al., 2011); 2) Abl has been shown to phosphorylate and activate SRC-3 (Oh et al., 2008); 3) Abl is activated in response to oxidative stress (Sun et al., 2000). We first tested if Abl is activated by MCB-613 treatment by assaying CrkL (Y207) phosphorylation as a marker for Abl activation (de Jong et al., 1997). As shown in Figure 4D, activation of Abl could be observed as early as one hour after MCB-613 treatment, and reached a high level after four hours of treatment. Moreover, CrkL phosphorylation caused by MCB-613 is significantly attenuated in the presence of NAC (Figure S4C), further proving that Abl activation is dependent on ROS induced by MCB-613. Subsequent co-immunoprecipitation analysis revealed that MCB-613 increases the interaction between SRC-3 and Abl (Figure 4E). Two Abl kinase inhibitors, AT9283 and PHA739358, as well as siRNAs targeting Abl, significantly inhibited the activation of SRC-3 by MCB-613 (Figure 4F and 4G), confirming that oxidative stress induced by MCB-613 contributes to SRC activation via the Abl kinase signaling pathway.
In order to better understand the phosphorylation pattern of SRC-3 induced by MCB-613 treatment, we generated pBIND-SRC-3 constructs with mutations to some of its known phosphorylation sites, including S505, S543, and S857 (Wu et al., 2004), as well as Y1357 which has been shown to be targeted by Abl (Oh et al., 2008), and tested them alongside the WT pBIND-SRC-3. As shown in Figure S4D, all of these mutants were hyper-activated by MCB-613 in a similar fashion as the WT. Since mutating these sites one by one cannot abrogate the effect of MCB-613, this result suggests that MCB-613 treatment either induces phosphorylation on more than one of these known sites or induces phosphorylation on novel sites.
Despite its necessity in the activation process of SRCs by MCB-613, elevated ROS level alone is not sufficient to achieve a robust stimulatory effect on SRCs, as the ROS inducer H2O2 cannot hyper-activate SRCs, even though it is capable of activating Abl kinase (Figure S4E and S4F). This result also implies that while Abl contributes to SRC hyper-stimulation, it is not a driver of this phenomenon.
MCB-613 inhibits tumor growth in vivo
In order to further evaluate the anti-cancer potential of MCB-613, a MCF-7 breast cancer mouse xenograft model was employed to assess the tumor suppressive effects of MCB-613 in vivo. Tumors were established in athymic nude mice by injecting MCF-7 cells into mammary fat pads. An MCB-613 treated group (n=13) received i.p. injection of the compound (20 mg/kg) three times a week for 7 weeks, while a control group (n=14) were injected with a saline vehicle. As shown in Figure 5A, MCB-613 significantly and dramatically stalled the growth of the tumor compared with the control group while causing no obvious animal toxicity, since the body weights between control and treated groups were not statistically different (Figure 5B).
Figure 5. MCB-613 inhibits tumor growth in a MCF-7 xenograft model.

(A) Tumor volume measurements of the mice treated with vehicle (n=14) or MCB-613 (n=13) for 7 weeks after tumor initiation. Data are presented as mean ± SEM. * p<0.05, ** p<0.01.
(B) Body weights of vehicle or MCB-613 treated mice throughout the treatment.
Discussion
Despite the key roles of SRCs in a broad range of biological processes pertinent to cancer cells, they had been considered by many as potentially undruggable targets in cancer therapeutics due to the lack of a structurally defined enzymatic activation domain or a high affinity ligand binding domain. However, our recent identification of gossypol as a SRC-1/SRC-3 inhibitor (Wang et al., 2011) established the proof-of-concept that it is possible to target these coactivators with small molecule compounds.
Compared with SRC inhibitors, using SRC stimulators in cancer therapy initially appears counterintuitive given the oncogenic actions of SRCs. However, even cancer cells that overexpress SRCs need to delicately orchestrate a wide range of cellular events to execute accelerated SRC-mediated cell proliferation. The demands placed upon cancer cells to cope with cell stress are even more acute than normal cells as they are already forced to deal with much higher rates of protein synthesis/folding and elevated ROS levels (Grek and Tew, 2010). We posit that elevated SRC activity beyond the already high levels present in cancer cells can further pressure their already maximized stress response system and selectively kill them. In fact, we have noted repeatedly that cancer cells overexpressing SRCs cannot be induced to further express increased SRC levels without deleterious effects. Consistent with this phenomenon, forced overexpression of oncoproteins, such as c-Myc, has been shown to cause cell death under certain conditions (Meyer and Penn, 2008), although the mechanisms involved may be distinct from what we observe due to SRC hyper-activation.
A mechanistic model is proposed in Figure 6 to delineate the actions of MCB-613 on SRCs. By directly binding to SRCs, MCB-613 dramatically activates these coactivators and promotes the formation of a coactivator complex with SRC, CBP and CARM1. Meanwhile, the resultant rapid elevation of intracellular ROS activates kinases such as Abl. These two events work in concert to further hyper-activate SRCs. The deregulation of cellular functions and homeostasis downstream of SRCs leads to severe ER stress and UPR, processes known to produce more ROS. Ultimately, this forms a destructive positive feedback loop that overwhelms the cancer cell, resulting in vacuolization and cell death. It is known that, depending on the cellular microenvironment and the nature of the stress, the ER stress-induced UPR program can be either cytoprotective or cytotoxic (Wang and Kaufman, 2012). Excessive ER stress has been proposed as the cause of aberrant vacuolization (Mimnaugh et al., 2006), and our data points to SRC hyper-stimulation as playing a key role in triggering ER stress that drives this phenomenon. We recognize that dramatic increase in ROS levels will result in profound changes in a broad spectrum of cellular processes. For instance, it has been published recently that ROS induced by the metabolic switch in cancer cells treated with a PPARγ agonist can activate PP2A, leading to the dephosphorylation of Rb and the inhibition of cancer cell proliferation (Srivastava et al., 2014). We postulate that the same effect on Rb also might happen as a result of MCB-613 mediated elevation of ROS levels and contribute to the final deleterious effect of MCB-613 on cancer cells, although not necessarily as a direct consequence of SRC hyper-activation.
Figure 6. Mechanistic model of SRC hyper-activation by MCB-613.

By directly binding to SRCs, MCB-613 increases the interaction between SRCs and other coactivators such as CBP and CARM1. Meanwhile, the resultant elevated ROS activates Abl kinase which phosphorylates and further hyper-activates SRCs. The deregulation of cellular functions and homeostasis downstream of SRCs hyper-activation strongly induces ER stress and UPR, producing more ROS and forming a positive feedback loop. The resultant excessive ER and oxidative stress overwhelms cancer cells, leading to vacuolization and cell death.
Because neither Abl kinase inhibitors nor siRNA-mediated Abl knockdown can completely block the activation of SRCs by MCB-613, we believe that Abl may not be the sole stress-related kinase or factor involved in SRC hyper-activation, and that the actions of MCB-613 extend beyond the activation of Abl. Studies using SRC-3 phospho-mutants led us to the conclusion that MCB-613 treatment probably leads to a distinct phosphorylation pattern on SRC-3. Although we have focused on phosphorylation, we haven’t ruled out the possibility that MCB-613 might induce other post-translational modifications on SRCs, which will be interesting to pursue in future studies.
We noted that MCB-613 is part of the chalcone class of compounds. Chalcones are widely produced in plants as intermediates in the biosynthesis of flavonoids. The common 1, 3-diphenyl, propenone template of these compounds allows for easy chemical modification which results in significant and distinct alterations in their molecular targets and biological functions. Various molecular targets of chalcones have been reported, including EGFR, tubulin, NRF2, NF-κB and others, many of which are critical players in carcinogenesis (Karthikeyan et al., 2015). Therefore, the anti-cancer activity of chalcones has been the subject of intensive research and represents a promising class of compounds for therapeutic development. This study establishes the major biological activity of MCB-613 as a compound that can induce catastrophic stress and death in cancer cells primarily by targeting SRC family proteins.
The selective cancer cell cytotoxicity of MCB-613 is similar to that seen with another small molecule compound, piperlongumine, that was identified from a high throughput chemical screen for compounds able to exacerbate cellular stressors that are already fully engaged in cancer cells (Raj et al., 2011). This initial study has led to the development of piperlongumine derivatives with improved drug-like properties (Adams et al., 2013). While these compounds are thought to target enzymes involved in redox maintenance such as glutathione synthetase and glutathionylation (Adams et al., 2012), MCB-613 can be distinguished by the fact that it functions by selectively targeting and stimulating a family of oncogenes.
Virtually all targeted therapies for key cancer oncogenes are designed to inhibit their activities. Here, we argue that while seemingly counterintuitive, oncogene hyper-stimulation warrants consideration as well. Some evidence that supports this idea comes from studies where oncogene activation in an inappropriate context can inhibit cancer growth. For instance, induction of oncogenic Ras in glioblastomas and gastric cancers is very rare and its experimental activation in the cell culture models of these cancer types can lead to cell death (Chi et al., 1999). Just as we have found through our re-analysis of high throughput screens originally designed to identify SRC SMIs, an opportunity to reevaluate compounds identified as oncogene activators in other screens also exists. Based upon our characterization of MCB-613, we anticipate that in the future, other SMSs that target oncogenes can be exploited as anti-cancer agents as well.
Experimental procedures
More procedures can be found in the Supplemental Experimental Procedures.
High throughput chemical screens
A MLSMR library was provided by Evotec through the NIH’s Roadmap Molecular Libraries Initiative for the high throughput screening for SRC-1/-2/-3 inhibitors. Details about compound selection for this library can be found at http://mli.nih.gov/mli/compound-repository/mlsmr-compounds/.
MCF-7 xenograft tumor model
Animal work was done in accordance with a protocol approved by the Animal Care and Use Committee of Baylor College of Medicine. 6–8 week-old Athymic nude female mice were obtained from Harlan. Two days before cancer cell injection, one estradiol pellet was embedded under the skin for each mouse and a new pellet was added 2 times at a one month interval. 1X 106 MCF-7 cells (50μl) mixed with equal volume of growth factor reduced Matrigel (BD Biosciences) were injected into the fat pad of each of the 2nd pair mammary gland without clearing. The MCB-613 treatment which lasted for seven weeks was started when tumor sizes reached 3–5mm in diameter. Mice of the treatment group (n=13) received MCB-613 (20 mg/kg) in saline by i.p. injection, whereas the control group (n=14) received saline only. The compound was injected three times a week. The mice were weighed and tumors were measured once a week during the treatment period.
Statistics
Statistical significance was determined by 2-tailed Student’s t test. A p value of less than 0.05 was considered statistically significant.
Supplementary Material
Significance.
Due to their essential roles in tumorigenesis, SRC family coactivators have been recognized recently as important targets for future cancer therapeutics. Since SRC proteins integrate and drive key oncogenic pathways that cancer cells use to achieve accelerated growth, invasion, energy metabolism and metastasis, SRC SMIs/SMSs are expected to be effective agents to treat cancers. While a few SRC SMIs have been described, here we report the characterization of an SRC SMS, MCB-613. Our results reveal that acute super-activation of SRC coactivators can paradoxically but effectively kill cancer cells by inducing aberrant cellular stress, suggesting that over-stimulation of the SRC oncogenic program is an alternative approach to selectively kill cancer cells whose cellular stress response pathways already are maximally engaged.
Highlights.
The small molecule compound MCB-613 super-stimulates SRC transcriptional activity.
Acute hyper-activation of SRCs by MCB-613 leads to aberrant cellular stress.
MCB-613 selectively kills cancer cells and inhibits tumor growth in vivo.
Over-activation of SRCs is a potential alternative strategy for cancer therapy.
Acknowledgments
This work was supported by funding from the Susan G. Komen Foundation (PG12221410), the Prostate Cancer Foundation, the Clayton Foundation and the Dunn Foundation, the Department of Defense Breast Cancer Research Program (BC120894), the Cancer Prevention and Research Institute of Texas (RP100348 and RP101251), and from the National Institutes of Health (DK059820) to BWO; and from the National Institutes of Health (HD076596) to DML; and from the Cancer Prevention and Research Institute of Texas (RP120732) and the National Institutes of Health (CA112403) to JX. High throughput screening was supported through the National Institutes of Health Molecular Libraries Program. LW, DML and BWO are co-founders and hold stock in Coregon, Inc. which is developing steroid receptor coactivator stimulators for clinical use.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abraham MC, Lu Y, Shaham S. A morphologically conserved nonapoptotic program promotes linker cell death in Caenorhabditis elegans. Developmental cell. 2007;12:73–86. doi: 10.1016/j.devcel.2006.11.012. [DOI] [PubMed] [Google Scholar]
- Adams DJ, Boskovic ZV, Theriault JR, Wang AJ, Stern AM, Wagner BK, Shamji AF, Schreiber SL. Discovery of small-molecule enhancers of reactive oxygen species that are nontoxic or cause genotype-selective cell death. ACS Chem Biol. 2013;8:923–929. doi: 10.1021/cb300653v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams DJ, Dai M, Pellegrino G, Wagner BK, Stern AM, Shamji AF, Schreiber SL. Synthesis, cellular evaluation, and mechanism of action of piperlongumine analogs. Proc Natl Acad Sci U S A. 2012;109:15115–15120. doi: 10.1073/pnas.1212802109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anzick SL, Kononen J, Walker RL, Azorsa DO, Tanner MM, Guan XY, Sauter G, Kallioniemi OP, Trent JM, Meltzer PS. AIB1, a steroid receptor coactivator amplified in breast and ovarian cancer. Science. 1997;277:965–968. doi: 10.1126/science.277.5328.965. [DOI] [PubMed] [Google Scholar]
- Bai H, Chen T, Ming J, Sun H, Cao P, Fusco DN, Chung RT, Chorev M, Jin Q, Aktas BH. Dual activators of protein kinase R (PKR) and protein kinase R-like kinase PERK identify common and divergent catalytic targets. Chembiochem : a European journal of chemical biology. 2013;14:1255–1262. doi: 10.1002/cbic.201300177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bautista S, Valles H, Walker RL, Anzick S, Zeillinger R, Meltzer P, Theillet C. In breast cancer, amplification of the steroid receptor coactivator gene AIB1 is correlated with estrogen and progesterone receptor positivity. Clinical cancer research : an official journal of the American Association for Cancer Research. 1998;4:2925–2929. [PubMed] [Google Scholar]
- Bouras T, Southey MC, Venter DJ. Overexpression of the steroid receptor coactivator AIB1 in breast cancer correlates with the absence of estrogen and progesterone receptors and positivity for p53 and HER2/neu. Cancer research. 2001;61:903–907. [PubMed] [Google Scholar]
- Cai D, Shames DS, Raso MG, Xie Y, Kim YH, Pollack JR, Girard L, Sullivan JP, Gao B, Peyton M, et al. Steroid receptor coactivator-3 expression in lung cancer and its role in the regulation of cancer cell survival and proliferation. Cancer research. 2010;70:6477–6485. doi: 10.1158/0008-5472.CAN-10-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Ma H, Hong H, Koh SS, Huang SM, Schurter BT, Aswad DW, Stallcup MR. Regulation of transcription by a protein methyltransferase. Science. 1999;284:2174–2177. doi: 10.1126/science.284.5423.2174. [DOI] [PubMed] [Google Scholar]
- Chen H, Lin RJ, Schiltz RL, Chakravarti D, Nash A, Nagy L, Privalsky ML, Nakatani Y, Evans RM. Nuclear receptor coactivator ACTR is a novel histone acetyltransferase and forms a multimeric activation complex with P/CAF and CBP/p300. Cell. 1997;90:569–580. doi: 10.1016/s0092-8674(00)80516-4. [DOI] [PubMed] [Google Scholar]
- Chen Y, Douglass T, Jeffes EW, Xu Q, Williams CC, Arpajirakul N, Delgado C, Kleinman M, Sanchez R, Dan Q, et al. Living T9 glioma cells expressing membrane macrophage colony-stimulating factor produce immediate tumor destruction by polymorphonuclear leukocytes and macrophages via a “paraptosis”-induced pathway that promotes systemic immunity against intracranial T9 gliomas. Blood. 2002;100:1373–1380. doi: 10.1182/blood-2002-01-0174. [DOI] [PubMed] [Google Scholar]
- Chi S, Kitanaka C, Noguchi K, Mochizuki T, Nagashima Y, Shirouzu M, Fujita H, Yoshida M, Chen W, Asai A, et al. Oncogenic Ras triggers cell suicide through the activation of a caspase-independent cell death program in human cancer cells. Oncogene. 1999;18:2281–2290. doi: 10.1038/sj.onc.1202538. [DOI] [PubMed] [Google Scholar]
- Chin L, Tam A, Pomerantz J, Wong M, Holash J, Bardeesy N, Shen Q, O’Hagan R, Pantginis J, Zhou H, et al. Essential role for oncogenic Ras in tumour maintenance. Nature. 1999;400:468–472. doi: 10.1038/22788. [DOI] [PubMed] [Google Scholar]
- Clarke PG. Developmental cell death: morphological diversity and multiple mechanisms. Anatomy and embryology. 1990;181:195–213. doi: 10.1007/BF00174615. [DOI] [PubMed] [Google Scholar]
- de Jong R, ten Hoeve J, Heisterkamp N, Groffen J. Tyrosine 207 in CRKL is the BCR/ABL phosphorylation site. Oncogene. 1997;14:507–513. doi: 10.1038/sj.onc.1200885. [DOI] [PubMed] [Google Scholar]
- Dengler MA, Staiger AM, Gutekunst M, Hofmann U, Doszczak M, Scheurich P, Schwab M, Aulitzky WE, van der Kuip H. Oncogenic stress induced by acute hyper-activation of Bcr-Abl leads to cell death upon induction of excessive aerobic glycolysis. PloS one. 2011;6:e25139. doi: 10.1371/journal.pone.0025139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denoyelle C, Abou-Rjaily G, Bezrookove V, Verhaegen M, Johnson TM, Fullen DR, Pointer JN, Gruber SB, Su LD, Nikiforov MA, et al. Anti-oncogenic role of the endoplasmic reticulum differentially activated by mutations in the MAPK pathway. Nature cell biology. 2006;8:1053–1063. doi: 10.1038/ncb1471. [DOI] [PubMed] [Google Scholar]
- Ding WX, Ni HM, Yin XM. Absence of Bax switched MG132-induced apoptosis to non-apoptotic cell death that could be suppressed by transcriptional or translational inhibition. Apoptosis : an international journal on programmed cell death. 2007;12:2233–2244. doi: 10.1007/s10495-007-0142-0. [DOI] [PubMed] [Google Scholar]
- Epps DE, Raub TJ, Caiolfa V, Chiari A, Zamai M. Determination of the affinity of drugs toward serum albumin by measurement of the quenching of the intrinsic tryptophan fluorescence of the protein. The Journal of pharmacy and pharmacology. 1999;51:41–48. doi: 10.1211/0022357991772079. [DOI] [PubMed] [Google Scholar]
- Felsher DW, Bishop JM. Reversible tumorigenesis by MYC in hematopoietic lineages. Molecular cell. 1999;4:199–207. doi: 10.1016/s1097-2765(00)80367-6. [DOI] [PubMed] [Google Scholar]
- Fereshteh MP, Tilli MT, Kim SE, Xu J, O’Malley BW, Wellstein A, Furth PA, Riegel AT. The nuclear receptor coactivator amplified in breast cancer-1 is required for Neu (ErbB2/HER2) activation, signaling, and mammary tumorigenesis in mice. Cancer research. 2008;68:3697–3706. doi: 10.1158/0008-5472.CAN-07-6702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming FJ, Myers E, Kelly G, Crotty TB, McDermott EW, O’Higgins NJ, Hill AD, Young LS. Expression of SRC-1, AIB1, and PEA3 in HER2 mediated endocrine resistant breast cancer; a predictive role for SRC-1. Journal of clinical pathology. 2004;57:1069–1074. doi: 10.1136/jcp.2004.016733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foulds CE, Feng Q, Ding C, Bailey S, Hunsaker TL, Malovannaya A, Hamilton RA, Gates LA, Zhang Z, Li C, et al. Proteomic analysis of coregulators bound to ERalpha on DNA and nucleosomes reveals coregulator dynamics. Molecular cell. 2013;51:185–199. doi: 10.1016/j.molcel.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaeser M, Floetotto T, Hanstein B, Beckmann MW, Niederacher D. Gene amplification and expression of the steroid receptor coactivator SRC3 (AIB1) in sporadic breast and endometrial carcinomas. Hormone and metabolic research = Hormon- und Stoffwechselforschung = Hormones et metabolisme. 2001;33:121–126. doi: 10.1055/s-2001-14938. [DOI] [PubMed] [Google Scholar]
- Gnanapragasam VJ, Leung HY, Pulimood AS, Neal DE, Robson CN. Expression of RAC 3, a steroid hormone receptor co-activator in prostate cancer. British journal of cancer. 2001;85:1928–1936. doi: 10.1054/bjoc.2001.2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grek CL, Tew KD. Redox metabolism and malignancy. Current opinion in pharmacology. 2010;10:362–368. doi: 10.1016/j.coph.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greuber EK, Smith-Pearson P, Wang J, Pendergast AM. Role of ABL family kinases in cancer: from leukaemia to solid tumours. Nature reviews Cancer. 2013;13:559–571. doi: 10.1038/nrc3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henke RT, Haddad BR, Kim SE, Rone JD, Mani A, Jessup JM, Wellstein A, Maitra A, Riegel AT. Overexpression of the nuclear receptor coactivator AIB1 (SRC-3) during progression of pancreatic adenocarcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2004;10:6134–6142. doi: 10.1158/1078-0432.CCR-04-0561. [DOI] [PubMed] [Google Scholar]
- Hudelist G, Czerwenka K, Kubista E, Marton E, Pischinger K, Singer CF. Expression of sex steroid receptors and their co-factors in normal and malignant breast tissue: AIB1 is a carcinoma-specific co-activator. Breast cancer research and treatment. 2003;78:193–204. doi: 10.1023/a:1022930710850. [DOI] [PubMed] [Google Scholar]
- Huettner CS, Zhang P, Van Etten RA, Tenen DG. Reversibility of acute B-cell leukaemia induced by BCR-ABL1. Nature genetics. 2000;24:57–60. doi: 10.1038/71691. [DOI] [PubMed] [Google Scholar]
- Kar R, Singha PK, Venkatachalam MA, Saikumar P. A novel role for MAP1 LC3 in nonautophagic cytoplasmic vacuolation death of cancer cells. Oncogene. 2009;28:2556–2568. doi: 10.1038/onc.2009.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson R, Stahlberg R. Surface plasmon resonance detection and multispot sensing for direct monitoring of interactions involving low-molecular-weight analytes and for determination of low affinities. Analytical biochemistry. 1995;228:274–280. doi: 10.1006/abio.1995.1350. [DOI] [PubMed] [Google Scholar]
- Karthikeyan C, Moorthy NS, Ramasamy S, Vanam U, Manivannan E, Karunagaran D, Trivedi P. Advances in chalcones with anticancer activities. Recent patents on anti-cancer drug discovery. 2015;10:97–115. doi: 10.2174/1574892809666140819153902. [DOI] [PubMed] [Google Scholar]
- Kershah SM, Desouki MM, Koterba KL, Rowan BG. Expression of estrogen receptor coregulators in normal and malignant human endometrium. Gynecologic oncology. 2004;92:304–313. doi: 10.1016/j.ygyno.2003.10.007. [DOI] [PubMed] [Google Scholar]
- Kinoshita E, Takahashi M, Takeda H, Shiro M, Koike T. Recognition of phosphate monoester dianion by an alkoxide-bridged dinuclear zinc(II) complex. Dalton transactions. 2004:1189–1193. doi: 10.1039/b400269e. [DOI] [PubMed] [Google Scholar]
- Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM, Royer RE, Vander Jagt DL, Semenza GL, Dang CV. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:2037–2042. doi: 10.1073/pnas.0914433107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- List HJ, Lauritsen KJ, Reiter R, Powers C, Wellstein A, Riegel AT. Ribozyme targeting demonstrates that the nuclear receptor coactivator AIB1 is a rate-limiting factor for estrogen-dependent growth of human MCF-7 breast cancer cells. J Biol Chem. 2001;276:23763–23768. doi: 10.1074/jbc.M102397200. [DOI] [PubMed] [Google Scholar]
- Lonard DM, O’Malley BW. Nuclear receptor coregulators: judges, juries, and executioners of cellular regulation. Mol Cell. 2007;27:691–700. doi: 10.1016/j.molcel.2007.08.012. [DOI] [PubMed] [Google Scholar]
- Meyer N, Penn LZ. Reflecting on 25 years with MYC. Nature reviews Cancer. 2008;8:976–990. doi: 10.1038/nrc2231. [DOI] [PubMed] [Google Scholar]
- Mimnaugh EG, Xu W, Vos M, Yuan X, Neckers L. Endoplasmic reticulum vacuolization and valosin-containing protein relocalization result from simultaneous hsp90 inhibition by geldanamycin and proteasome inhibition by velcade. Molecular cancer research : MCR. 2006;4:667–681. doi: 10.1158/1541-7786.MCR-06-0019. [DOI] [PubMed] [Google Scholar]
- Myers E, Fleming FJ, Crotty TB, Kelly G, McDermott EW, O’Higgins NJ, Hill AD, Young LS. Inverse relationship between ER-beta and SRC-1 predicts outcome in endocrine-resistant breast cancer. British journal of cancer. 2004;91:1687–1693. doi: 10.1038/sj.bjc.6602156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh AS, Lahusen JT, Chien CD, Fereshteh MP, Zhang X, Dakshanamurthy S, Xu J, Kagan BL, Wellstein A, Riegel AT. Tyrosine phosphorylation of the nuclear receptor coactivator AIB1/SRC-3 is enhanced by Abl kinase and is required for its activity in cancer cells. Molecular and cellular biology. 2008;28:6580–6593. doi: 10.1128/MCB.00118-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilar G, Landmesser L. Ultrastructural differences during embryonic cell death in normal and peripherally deprived ciliary ganglia. The Journal of cell biology. 1976;68:339–356. doi: 10.1083/jcb.68.2.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, Liu Z, Chen H, Xu J. The steroid receptor coactivator-1 regulates twist expression and promotes breast cancer metastasis. Cancer research. 2009;69:3819–3827. doi: 10.1158/0008-5472.CAN-08-4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj L, Ide T, Gurkar AU, Foley M, Schenone M, Li X, Tolliday NJ, Golub TR, Carr SA, Shamji AF, et al. Selective killing of cancer cells by a small molecule targeting the stress response to ROS. Nature. 2011;475:231–234. doi: 10.1038/nature10167. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Ray PD, Huang BW, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cellular signalling. 2012;24:981–990. doi: 10.1016/j.cellsig.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakakura C, Hagiwara A, Yasuoka R, Fujita Y, Nakanishi M, Masuda K, Kimura A, Nakamura Y, Inazawa J, Abe T, Yamagishi H. Amplification and over-expression of the AIB1 nuclear receptor co-activator gene in primary gastric cancers. International journal of cancer Journal international du cancer. 2000;89:217–223. [PubMed] [Google Scholar]
- Sperandio S, de Belle I, Bredesen DE. An alternative, nonapoptotic form of programmed cell death. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:14376–14381. doi: 10.1073/pnas.97.26.14376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava N, Kollipara RK, Singh DK, Sudderth J, Hu Z, Nguyen H, Wang S, Humphries CG, Carstens R, Huffman KE, et al. Inhibition of cancer cell proliferation by PPARgamma is mediated by a metabolic switch that increases reactive oxygen species levels. Cell metabolism. 2014;20:650–661. doi: 10.1016/j.cmet.2014.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Majumder P, Shioya H, Wu F, Kumar S, Weichselbaum R, Kharbanda S, Kufe D. Activation of the cytoplasmic c-Abl tyrosine kinase by reactive oxygen species. The Journal of biological chemistry. 2000;275:17237–17240. doi: 10.1074/jbc.C000099200. [DOI] [PubMed] [Google Scholar]
- Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, et al. Integrative genomic profiling of human prostate cancer. Cancer cell. 2010;18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres-Arzayus MI, Font de Mora J, Yuan J, Vazquez F, Bronson R, Rue M, Sellers WR, Brown M. High tumor incidence and activation of the PI3K/AKT pathway in transgenic mice define AIB1 as an oncogene. Cancer cell. 2004;6:263–274. doi: 10.1016/j.ccr.2004.06.027. [DOI] [PubMed] [Google Scholar]
- Torres-Arzayus MI, Yuan J, DellaGatta JL, Lane H, Kung AL, Brown M. Targeting the AIB1 oncogene through mammalian target of rapamycin inhibition in the mammary gland. Cancer research. 2006;66:11381–11388. doi: 10.1158/0008-5472.CAN-06-2316. [DOI] [PubMed] [Google Scholar]
- Ustundag Y, Bronk SF, Gores GJ. Proteasome inhibition-induces endoplasmic reticulum dysfunction and cell death of human cholangiocarcinoma cells. World journal of gastroenterology : WJG. 2007;13:851–857. doi: 10.3748/wjg.v13.i6.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Kaufman RJ. The impact of the unfolded protein response on human disease. The Journal of cell biology. 2012;197:857–867. doi: 10.1083/jcb.201110131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Lonard DM, Yu Y, Chow DC, Palzkill TG, O’Malley BW. Small molecule inhibition of the steroid receptor coactivators, SRC-3 and SRC-1. Molecular endocrinology. 2011;25:2041–2053. doi: 10.1210/me.2011-1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Lonard DM, Yu Y, Chow DC, Palzkill TG, Wang J, Qi R, Matzuk AJ, Song X, Madoux F, et al. Bufalin is a potent small-molecule inhibitor of the steroid receptor coactivators SRC-3 and SRC-1. Cancer research. 2014;74:1506–1517. doi: 10.1158/0008-5472.CAN-13-2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Wu MC, Sham JS, Zhang W, Wu WQ, Guan XY. Prognostic significance of c-myc and AIB1 amplification in hepatocellular carcinoma. A broad survey using high-throughput tissue microarray. Cancer. 2002;95:2346–2352. doi: 10.1002/cncr.10963. [DOI] [PubMed] [Google Scholar]
- Wu RC, Feng Q, Lonard DM, O’Malley BW. SRC-3 coactivator functional lifetime is regulated by a phospho-dependent ubiquitin time clock. Cell. 2007;129:1125–1140. doi: 10.1016/j.cell.2007.04.039. [DOI] [PubMed] [Google Scholar]
- Wu RC, Qin J, Yi P, Wong J, Tsai SY, Tsai MJ, O’Malley BW. Selective phosphorylations of the SRC-3/AIB1 coactivator integrate genomic reponses to multiple cellular signaling pathways. Molecular cell. 2004;15:937–949. doi: 10.1016/j.molcel.2004.08.019. [DOI] [PubMed] [Google Scholar]
- Xie D, Sham JS, Zeng WF, Lin HL, Bi J, Che LH, Hu L, Zeng YX, Guan XY. Correlation of AIB1 overexpression with advanced clinical stage of human colorectal carcinoma. Human pathology. 2005;36:777–783. doi: 10.1016/j.humpath.2005.05.007. [DOI] [PubMed] [Google Scholar]
- Xu J, Wu RC, O’Malley BW. Normal and cancer-related functions of the p160 steroid receptor co-activator (SRC) family. Nature reviews Cancer. 2009;9:615–630. doi: 10.1038/nrc2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan F, Yu Y, Chow DC, Palzkill T, Madoux F, Hodder P, Chase P, Griffin PR, O’Malley BW, Lonard DM. Identification of verrucarin a as a potent and selective steroid receptor coactivator-3 small molecule inhibitor. PLoS One. 2014;9:e95243. doi: 10.1371/journal.pone.0095243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Erdem H, Li R, Cai Y, Ayala G, Ittmann M, Yu-Lee LY, Tsai SY, Tsai MJ. Steroid receptor coactivator-3/AIB1 promotes cell migration and invasiveness through focal adhesion turnover and matrix metalloproteinase expression. Cancer research. 2008;68:5460–5468. doi: 10.1158/0008-5472.CAN-08-0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon MJ, Kim EH, Lim JH, Kwon TK, Choi KS. Superoxide anion and proteasomal dysfunction contribute to curcumin-induced paraptosis of malignant breast cancer cells. Free radical biology & medicine. 2010;48:713–726. doi: 10.1016/j.freeradbiomed.2009.12.016. [DOI] [PubMed] [Google Scholar]
- York B, O’Malley BW. Steroid receptor coactivator (SRC) family: masters of systems biology. The Journal of biological chemistry. 2010;285:38743–38750. doi: 10.1074/jbc.R110.193367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Yasui K, Lee CJ, Kurioka H, Hosokawa Y, Oka T, Inazawa J. Elevated expression levels of NCOA3, TOP1, and TFAP2C in breast tumors as predictors of poor prognosis. Cancer. 2003;98:18–23. doi: 10.1002/cncr.11482. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.