

Abstract
Amplification of multiple unique genetic targets using the polymerase chain reaction (PCR) is commonly required in molecular biology laboratories. Such reactions are typically performed either serially or by multiplex PCR. Serial reactions are time consuming, and multiplex PCR, while powerful and widely used, can be prone to amplification bias, PCR drift, and primer-primer interactions. We present a new thermocycling method, termed thermal multiplexing, in which a single heat source is uniformly distributed and selectively modulated for independent temperature control of an array of PCR reactions. Thermal multiplexing allows amplification of multiple targets simultaneously—each reaction segregated and performed at optimal conditions. We demonstrate the method using a microfluidic system consisting of an infrared laser thermocycler, a polymer microchip featuring 1 μl, oil-encapsulated reactions, and closed-loop pulse-width modulation control. Heat transfer modeling is used to characterize thermal performance limitations of the system. We validate the model and perform two reactions simultaneously with widely varying annealing temperatures (48 °C and 68 °C), demonstrating excellent amplification. In addition, to demonstrate microfluidic infrared PCR using clinical specimens, we successfully amplified and detected both influenza A and B from human nasopharyngeal swabs. Thermal multiplexing is scalable and applicable to challenges such as pathogen detection where patients presenting non-specific symptoms need to be efficiently screened across a viral or bacterial panel.
INTRODUCTION
In 1983, Mullis revolutionized the field of molecular biology by introducing a technique known as the polymerase chain reaction (PCR), a highly specific and sensitive method for exponentially amplifying a DNA sequence from as little as a single copy by thermocycling a biochemical cocktail.1 PCR has become a standard tool for countless applications requiring genetic analysis, including forensics, personalized medicine, and pathogen detection for routine clinical care as well as outbreak response and surveillance.2 Alternatively, infections can be detected by growing a culture from patient samples, which in some cases can be done in less than 24 h but in other cases can require up to 30 days. Culturing has been shown to be less sensitive than PCR in clinical trials.3,4 Other detection techniques, such as immunoassays and direct fluorescent antigen testing, are being replaced in favor of the improved sensitivity and shorter turnaround time of PCR.5 Still, PCR remains relatively expensive due to high reagent costs, and conventional PCR instruments, such as thermoelectric and convective thermocyclers, are limited by large reaction volumes (>5 μl) and thermal inaccuracies.6–8 Further, despite the dozens to hundreds of sample capacities of modern PCR instruments, operation is typically limited to uniform thermal conditions and average reaction durations on the order of 1 h. Yet, different genetic targets have corresponding optimal annealing temperatures that depend on the length and GC content of their primers. Consequently, conventional instruments typically only allow a single type of reaction or multiple reactions that can work at the same conditions, which cannot always be easily accommodated. Exceptions to this trend include the Cepheid SmartCycler, which offers independently controlled sample wells to perform a multitude of thermal cycling profiles simultaneously and asynchronously. Some thermoelectric instruments can generate a temperature gradient across a set of samples but the method is slow, not independently selectable, and as such it has only penetrated niche applications such as primer annealing temperature optimization.
Microfluidic approaches to PCR have become effective in addressing the volume, cost, and speed limitations of conventional instruments. Many microfluidic PCR devices have been reported that require less volume and provide faster turnaround time, as detailed in review articles.9,10 Other advantages of microfluidics include potential for integration with upstream and downstream sample processing11 and portability for point-of-care (POC) applications.12,13 For PCR, the low thermal mass of the reaction volume on a microfluidic device can also help increase specificity due to reduced transition times between PCR steps.14 Similar to commercially available PCR instruments, conductive heating using thermoelectric, or Peltier, elements is common for microfluidic thermocyclers.10 Resistive15 and convective16,17 heating have also been implemented. Radiative heating has been applied to microfluidic PCR with both broadband and focused, monochromatic sources and offers the unique advantage of direct, non-contact heating for greater efficiency and ramping rates as compared with resistive and convective heating instruments. Broadband sources such as a tungsten filament lamp can provide fast thermocycling of small volumes for rapid PCR.11,18–20 Laser-mediated radiative heating offers similar advantages and has been demonstrated for rapidly thermocycling droplets.21–23 The smaller spatial scales and faster heating and cooling rates of microfluidic PCR systems can complicate temperature sensing and control. Techniques used in the previous work include reference chambers with thermocouples,19 fluorescent dyes,22 pyrometers,24 resistance measurements,15 and open-loop control.25,26
Most PCR technologies, especially within the field of microfluidics, have advanced the speed and throughput for screening many samples or patients for a single genetic target. For multiple targets, a biochemical technique called multiplex PCR was developed in the 1980s (Ref. 27) and has since become commonplace. In multiplex PCR, one can amplify multiple gene targets simultaneously using multiple primer sets designed for a common annealing temperature in a single PCR mixture. Challenges that others have reported include amplification bias due to efficiency variations, PCR drift, and the high likelihood of primer-primer interactions.28,29 In addition, skilled primer design and finely tuned reagent concentrations are required and cannot always accommodate all desired target sequences.30
We propose an alternative to traditional multiplex PCR for targeting multiple gene sequences by using a method termed thermal multiplexing, in which the reaction temperatures for an array of samples are controlled independently from a common energy source to achieve multiple, distinct thermal profiles for multiple, optimal amplifications. The end goal is the same as the biochemical approach of multiplex PCR: by targeting multiple sequences at once, additional information may be gained from a single amplification run that otherwise would require more time and reagents. Unlike traditional multiplex PCR, thermally multiplexed PCR does not require specialized reaction design and allows the consolidation of any set of pre-existing reactions into a single instrument run.
In this work, we present a system designed for thermally multiplexed PCR comprising a polymer microfluidic chip, an infrared laser thermocycler that allows for direct heating of the samples, and a closed-loop temperature control method. We demonstrate the ability of this system to simultaneously generate different annealing temperatures in a microchip using a single radiative heat source. We model, characterize, and apply this system to amplifications for a variety of DNA and RNA targets, from both purified templates to clinical specimens.
MATERIALS AND METHODS
To perform thermally multiplexed PCR, a single energy source is uniformly distributed, independently and arbitrarily modulated, and directed to an array of reaction chambers as depicted in Fig. 1(a). The source delivers a constant energy in time, or power, q, divided equally amongst n chambers as q/n. Each attenuator (e.g., shutter, valve, filter, variable resistor, LCD, acousto-optic deflector) modulates the power q/n according to a unique programmed function (e.g., pulse width modulated square wave), fi(t) for 1 < i < n and 0 ≤ fi(t) ≤ 1. Each chamber is thus heated to temperature profile Ti(t), shown in Fig. 1(b), by the power profile qi(t) = (q/n)fi(t).
FIG. 1.

(a) Thermal multiplexing can be generalized to a uniformly distributed and independently attenuated heat source delivered to an array of reaction chambers. (b) Theoretical temperature profiles for n reactions illustrating the capability of maintaining a set of distinct annealing temperatures simultaneously for optimal amplification of unique DNA targets.
Microfluidic device
Polymer microchips were designed and fabricated, as illustrated in Fig. 2, comprising a pair of bonded poly(methyl methacrylate) (PMMA) layers similar to those previously described.26 Each microchip features two 1.6 mm diameter alignment holes for repeatable positioning when installing it in the laser thermocycler and two 1 μl reaction chambers (500 μm wide, 750 μm deep, 2.5 mm average length) accessed via 700 μm diameter fill ports and 250 μm wide × 190 μm deep channels. The chamber length was tapered such that the feature is trapezoidal to minimize bubble trapping. A 1.5 mm substrate thickness was chosen to withstand PCR temperatures (95 °C) and pressures (40 psi/275 kPa).
FIG. 2.
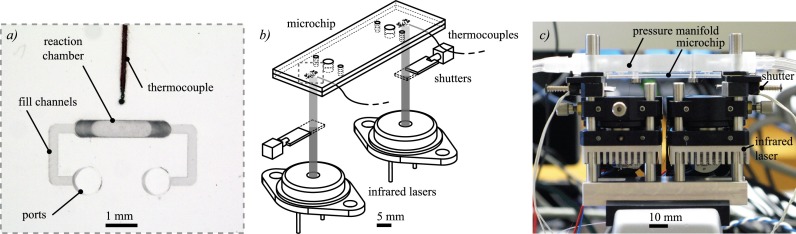
Infrared laser thermocycler comprising two infrared laser sources controlled via a common power supply, two solenoid shutters, and a polymer microchip featuring two 1 μl reaction chambers. (a) Close-up photograph of one of the reaction chambers shows the ports, used for sample loading and pressurization, fill channels, 1 μl reaction chamber, and the tip of the embedded thermocouple bonded between the layers of the microchip. (b) Simplified schematic and (c) photograph show the basic components of the thermocycler assembly.
Chamber spacing on the microchip is a critical design parameter, balancing high chamber density for scaling with manufacturability and thermal crosstalk limiting adjacent reaction temperature differences. To test manufacturability and thermal crosstalk empirically to compare with modeling, microchips featuring chamber spacings of 1 and 40 mm were fabricated.
As described in the previous work,25,31,32 the microchips were fabricated from PMMA substrates using a 3-axis vertical milling center (Haas) at a rate of two chips per minute. The 60 mm × 20 mm substrates were laser cut from sheets of 1.5 mm thick cast PMMA. After milling, microchips were cleaned in a 3-stage process that consisted of rinsing with isopropyl alcohol, rinsing with deionized water, and drying with compressed nitrogen.
Prior to bonding the microchip layers, thermocouples were aligned to be in close proximity (e.g., <1 mm) to the reaction chambers without directly contacting them (see Fig. 2(a)), since the sensors could inhibit PCR by adsorbing reagents such as polymerase33,34 or cause inaccurate temperature measurement due to direct irradiation of the thermocouple tip. Once the thermocouples were accurately positioned, the two microchip layers—one featuring alignment holes, chambers, fill ports, and fill channels and the other featuring only alignment holes—were bonded between clamped, polished aluminum plates featuring alignment pins. The plates were polished to a mirror finish to maintain the optical clarity of the PMMA, useful for minimizing radiation scattering for both inspection and infrared heating. For bonding, the plates were placed on a room-temperature hotplate (Corning) and the clamping bolts were tightened to 0.339 ± 0.014 N m with a torque screwdriver (Seekonk Precision Tools). The hotplate was then heated to 170 °C and held for 30 min, after which the heating was terminated and the microchip was allowed to cool for an additional hour before it was cleaned and used. The temperatures, forces, and times for the bonding process were determined empirically to ensure a strong bond between the two layers without deforming the milled features.
To load the microchip with a PCR sample, a technique analogous to droplet microfluidics was developed in which mineral oil was used as a passivation agent to encapsulate the 1 μl sample and minimize inhibition caused by surface adsorption of critical reagents.33 Specifically, 1.5 μl of dedicated, unused mineral oil was first loaded into a pipette tip using an adjustable micropipette. The pipette volume was increased to 2.5 μl and, after carefully bringing the oil interface to the end of the pipette tip, 1 μl of PCR solution was loaded. The pipette volume was then increased to 4 μl and the remaining section of the pipette tip was filled with another 1.5 μl plug of mineral oil. This entire volume is loaded into the microchip by directly interfacing the pipette tip with a fill port, applying gentle force manually to maintain a seal, and injecting the tip content, making sure to align the PCR solution within the reaction chamber. The channels' U-shape enables facile delivery of pneumatic pressure to both ports of a chamber from a single pressure connection. The microchips are single-use and disposable. After each amplification, thermocouples were recovered from each microchip by delaminating the device.
Thermocycler
For proof of concept, we implemented a thermally multiplexed PCR system using infrared laser radiation as the heat source for n = 2 chambers per microchip with mechanical solenoid shutters as attenuators, as shown in Figs. 2(b) and 2(c). Solenoid shutters are low power and digital and can effectively modulate a constant optical energy source. Radiation was supplied by either a single (for chamber spacing of 1 mm) or pair (for chamber spacing of 40 mm) of 1450 nm, 600 mW laser diodes (Hi-Tech Optoelectronics Co.) wired in series and powered by a single constant current laser driver (Wavelength Electronics) for common control and uniform output. The solenoid shutters (Electromechanisms), with 8.9 mm stroke, were driven independently using MOSFETs triggered by 10 Hz pulse width modulated signals. The components were assembled and aligned with a 30 mm cage system (Thorlabs). The laser diodes were mounted on aluminum heat sinks with cooling fans to prevent overheating and to maintain constant optical power output. Collimating lenses (Thorlabs) were mounted on x-y positioners (Thorlabs) to align the lasers with the reaction chambers. The microchip was installed in the optical system by first sandwiching it between a pressure manifold plate and retention plate, with silicone gaskets for sealing and alignment pins for accurate positioning. The pressure manifold plate delivered nitrogen regulated to 275 kPa from a supply tank to the microchip fill ports to prevent the expansion of entrained air bubbles during PCR. This sandwich assembly was mounted on the cage system for repeatable placement. Temperature was measured using 125 μm diameter thermocouples (Physitemp) embedded in the microchip adjacent to each chamber using the calibration and control scheme described below.
To determine the effect of shutter duty cycle on reaction chamber temperature, we measured temperatures in adjacent chambers with varying spacing (from 1 to 40 mm) and varying duty cycle (0.0–1.0). Temperatures were selected in the range useful for PCR, from 48–94 °C.
Closed-loop temperature control
Thermocouples embedded within the microchip were used for feedback control. Since the thermocouple tips were located near the reaction chambers and therefore did not directly measure reaction temperature, calibration was necessary to correlate the measured temperature with the reaction temperature. For this, a calibration microchip was fabricated with thermocouples bonded directly inside the reaction chambers in contact with the top wall of the chamber, furthest away from the laser source. At this location, the measured temperature is close to the true reaction temperature, since <1% of the incident radiation reached the thermocouple to directly heat it. This fraction is determined from the Beer-Lambert Law where PT/P0 = 10−αl, given that α is the absorption coefficient of water at 1450 nm (approximately 30 cm−1), l is the path length (750 μm), PT is the transmitted power, and P0 is the incident power at the relevant wavelength.
In order to use the calibration microchip, the reaction chambers were filled with PCR buffer solution and the microchip was installed in the thermocycler. Starting with the laser turn-on voltage of 0.7 V, the steady state temperature in each chamber was recorded as the voltage was increased to 1.4 V, where the temperature approaches the maximum required temperature of 94 °C for denaturing. This was performed four times to obtain an average steady state chamber temperature as a function of laser driving voltage. Next, this process was repeated using the actual microchip used for PCR, with thermocouples bonded near the reaction chambers. A calibration curve relating the reaction temperature to measured temperature between these microchips was then calculated and subsequently optimized using iterations of PCR thermocycling runs with a test reaction until PCR yield reached expected levels. Calibration was repeated for each experiment to correct for variations in environmental conditions (e.g., room temperature).
A LabVIEW program was developed to operate the laser thermocycler. After setting the desired temperatures and hold times for each stage of PCR (initial denaturing, denaturing, annealing, extension, and final extension), the program was initiated. The temperature for each chamber was measured, the program determined the current stage of PCR, and compared the desired temperature at that stage with the measured temperature. To maintain constant temperatures during denaturation, annealing, and extension, a PID controller modified the duty cycle of the signal driving each solenoid shutter to minimize error between the desired and measured temperature without excessive overshoot or undershoot (>2 °C) at the transitions. If the next stage required a higher temperature, the shutter stayed open until that temperature is reached. Conversely, if the next stage is at a cooler temperature, the shutter closed to completely block the radiation, allowing cooling through natural convection and conduction with the lower temperature ambient environment. When the next desired temperature is reached, the controller again operated the shutters for steady temperature holds. This process was repeated for each stage of PCR for the desired number of PCR cycles. The laser was always operated at 1.4 V, so all temperature control was performed with the shutters.
Heat transfer modeling
Within the temperature range of all possible primer melting temperatures, 48 °C to 76 °C,35 we applied heat transfer modeling to study the effects of thermal crosstalk between adjacent chambers for a range of chamber spacing distances to better understand the limitations of thermal multiplexing. A series of finite element models were built in COMSOL using methods previously reported36 to determine the maximum temperature difference between chambers of various spacing distances when one chamber is heated to steady state using the infrared laser and other is heated solely by conduction through the microchip substrate, just as the temperatures would be achieved in practice using thermal multiplexing in the most extreme scenario. Using this model, we found the minimum spacing necessary to maintain these temperature extremes between adjacent chambers.
PCR and RT-PCR protocols
To demonstrate thermal multiplexing, we used two unique PCR reactions with relatively high and low annealing temperatures. The high temperature reaction targets a 500 bp amplicon from λ-phage DNA with a 68 °C annealing temperature, while the low temperature reaction targets a 600 bp amplicon from Epstein-Barr virus (EBV) DNA with a 48 °C annealing temperature.37 To demonstrate feasibility of our instrument with clinical samples, we used two RT-PCR reactions for the amplification of influenza A and B.
All PCR reactions for the detection of λ-phage and EBV were prepared from a commercial master mix (Bioneer AccuPower PCR PreMix, 50 μl reaction volume). The premix consists of a lyophilized pellet of 2.5 U Top DNA polymerase, 250 μM dNTPs (dATP, dCTP, dGTP, dTTP), 10 mM Tris-HCl (pH 9.0), 30 mM KCl, 1.5 mM MgCl2, tracking dye, and stabilizer. The master mixes were prepared as follows: 44 μl of nuclease-free water was added to the premix tube, vortexed, and spun down. To this, 1 μl of 20 μM forward and reverse primers were added (see Table I for primer sequences). λ-phage primers target a 500 bp amplicon and EBV primers target a 600 bp amplicon.
TABLE I.
Primer sequences for virus amplifications from purified template and clinical specimens.
| Target | Sequence |
|---|---|
| λ-phagea | Forward: 5′-G(GT)TIGACTTTGCCAGC(TC)T(GC)TACCC-3′ |
| Reverse: 5′-GGGAGTC(AC)GTGTC(GC)CCGTA(GT)ATGA-3′ | |
| EBVb | Forward: 5′-GATGAGTTCGTGTTCGTACAACTGG-3′ |
| Reverse: 5′-GGTTATCGAAATCAGCCACAGCGCC-3′ | |
| Influenza Ac | Forward: 5′-GACCRATCCTGTCACCTCTGAC-3′ |
| Reverse: 5′-AGGGCATTYTGGACAAAKCGTCTA-3′ | |
| Influenza Bc | Forward: 5′-TCCTCAAYTCACTCTTCGAGCG-3′ |
| Reverse: 5′-CGGTGCTCTTGACCAAATTGG-3′ |
From the master mix, reaction mixes were then prepared by adding template DNA. λ-phage template was supplied by Promega and EBV template was supplied by the CDC. For conventional 50 μl control reactions, 5 μl template DNA was added to the master mix. For a 1 μl microchip reaction, the 45 μl master mix was divided into ten 4.5 μl aliquots and 0.5 μl template was added to each tube. From these aliquots, our 1 μl reactions were extracted.
Similarly, all RT-PCR reactions for the detection of influenza A and B were prepared from a commercial master mix (Bioneer, AccuPower one-step RT-PCR PreMix, 50 μl reaction volume). This premix consists of M-MLV Reverse Transcriptase, RNA dependent DNA polymerase, and a thermostable DNA polymerase in a lyophilized mix of dNTPs, reaction buffer, RNase inhibitor, tracking dye, and stabilizer. The master mixes were prepared as follows: 44 μl of nuclease-free water was added to the premix tube, vortexed, and spun down. To this, 1 μl of 20 μM forward and reverse primers were added. Influenza A primers target a 106 bp amplicon and influenza B primers target a 103 bp amplicon.
Reaction mixes were then prepared by adding template RNA. Influenza A and B template was extracted from clinical specimens in the form of nasopharyngeal swabs of known infections collected from pediatric patients at Children's Healthcare of Atlanta (CHOA) Egleston Hospital and initially identified in their clinical laboratory using RT-PCR. These experiments utilized excess swabs that were anonymized in accordance with protocol H13067 approved by the Georgia Institute of Technology Institutional Review Board. Extraction was performed using Qiagen QIAamp Viral RNA Mini Kit. For conventional 50 μl control reactions, 5 μl template RNA was added to the master mix. For a 1 μl microchip reaction, the 45 μl master mix was divided into ten 4.5 μl aliquots and 0.5 μl template RNA was added to each tube. From these aliquots, our 1 μl reactions were extracted.
To show the importance of annealing temperature on final product concentration, the λ-phage and EBV reactions were thermocycled in parallel with four sets of conditions: low annealing temperature (48 °C), high annealing temperature (68 °C), average annealing temperature (58 °C), and ideal annealing temperatures by thermal multiplexing (λ-phage at 68 °C and EBV at 48 °C). A total of 40 cycles were performed, with a 1 min initial denaturation for the first cycle and all subsequent cycles consisted of denaturing at 94 °C for 10 s, annealing for 20 s, and extension at 72 °C for 20 s, as well as a final extension step of 3 min. Both λ-phage and EBV were loaded into the microchip for each experiment. Electrophoretic detection of the PCR products was performed using microchannel electrophoresis (Agilent Bioanalyzer), using a marker set (Agilent DNA 1000 kit) that includes sizing and quantification markers at 15 bp and 1500 bp, with limit of detection of 0.005 ng/μl.
In order to establish repeatable performance and illustrate the utility of our novel multiplexing approach, a set of twelve microchips, each with two chambers, yield twelve pairs of end-point detection trials, t, with λ-phage loaded into chamber 1 and EBV loaded into chamber 2, to test (in triplicate) the four annealing modes described earlier: (1) low, in which both samples were cycled uniformly with an annealing temperature of 48 °C; (2) high, in which both samples were cycled uniformly with an annealing temperature of 68 °C; (3) average, in which both samples were cycled uniformly with an annealing temperature of 58 °C; and (4) thermally multiplexed, in which samples were cycled independently with annealing temperatures of 68 °C and 48 °C for chambers 1 and 2, respectively.
Template concentrations of 5.4 pg/μl (1 × 105 copies/μl) and 500 fg/μl (1 × 105 copies/μl) were used for λ-phage and EBV, respectively, resulting in 540 fg of λ-phage and 50 fg of EBV. (Note: λ-phage DNA was provided in purified form with a known concentration while EBV template was supplied by the Centers for Disease Control and and Prevention (CDP) in Atlanta, GA, which they use for positive controls during herpesvirus screening, and was quantitated based on optical density, OD, and the known template size of 4485 bp.) These dilutions provided, identically, 10 000 starting copies for each reaction.
To verify that our microfluidic PCR platform was capable of performing clinically relevant reactions of a viral template in a background of human DNA, we performed amplifications of influenza A and influenza B. These reactions were first performed at a 50 μl volume using a conventional thermocycler (Bio-Rad MJ Mini) with a 1 h reverse transcription step at 42 °C, 2 min initial denaturation at 94 °C, then 30 cycles of denaturing at 94 °C for 15 s, annealing at 55 °C (influenza A) and 61 °C (influenza B) for 30 s, and extension at 72 °C for 30 s, then a final extension step for 7 min. Once verified, these reactions were then tested at 1 μl in our microchips using the custom, automated water bath thermocycler described in previous work.32 For this, hold times were modified to accommodate slower sample equilibration. Following the 1 h reverse transcription, samples were cycled with a 1 min denaturation, 3 min annealing, and 3 min extension. Finally, microfluidic reactions were performed with our infrared laser system with a 30 min reverse transcription, 1 min initial denaturation, then 30 cycles of 10 s denaturation, 20 s annealing, 20 s extension, and a final extension step for 3 min. Two clinical specimens of each of our targets, influenza A and B, were screened. All samples were previously tested at CHOA facilities and confirmed positive for influenza infections. Aliquots of extracted viral RNA template were each tested in triplicate and detected by electrophoresis using the Agilent Bioanalyzer to confirm repeatable amplifications.
RESULTS AND DISCUSSION
Heat transfer modeling and empirical validation
We applied our heat transfer model to study the effect of chamber spacing on steady-state temperature difference between adjacent chambers. Our objective in the design of the microfluidic chip is to be able to achieve the full range of possible annealing temperatures, from 48 °C to 76 °C (28 °C range), in adjacent reaction chambers.
For manufacturability, we determined that the closest distance that two chambers can be spaced on a single microchip is 1 mm center-to-center, limited by inadequate bonding causing leaking across the thin wall between chambers. Starting with a chamber spacing of 1 mm, we modeled and measured the temperature difference between adjacent chambers, as shown in Fig. 3(a). The model closely matches the experimentally measured thermal behavior and reveals that at the maximum practical annealing temperature of 76 °C, 1 mm spaced chambers can only achieve temperatures differences of 12 °C, which would greatly limit the range of possible reactions.
FIG. 3.

(a) Finite element model (line) and experimental data (circles) showing temperature difference between two 1 μl chambers as a function of the steady state temperature of the heated chamber in a PMMA microchip with 1 mm chamber spacing. Temperature of adjacent chambers as shutter duty cycle of one is varied for spacings of (b) 1 mm and (c) 40 mm.
Using this validated model, we computed that a minimum chamber spacing of at least 3 mm was necessary to achieve the full range of annealing temperatures from 48 °C to 76 °C in adjacent reaction chambers.
We next measured the effect of shutter duty cycle on temperature for small (1 mm) and large (40 mm) chamber spacings, as shown in Figs. 3(b) and 3(c), where the chamber 1 shutter was held at a constant duty cycle while the chamber 2 shutter was stepwise increased. Results indicate that systematic control of chamber temperatures can be enabled by optical modulation. In addition, 40 mm spacing can be seen to cover the full range of relevant temperatures, as predicted by the model. When implemented experimentally, such large chamber spacing is not only beneficial for steady state temperature difference but also improves transient thermal crosstalk affecting heating and cooling rates (data not shown).
PCR and RT-PCR
In order to simultaneously perform the λ-phage and EBV amplifications, the laser thermocycler with solenoid shutters and thermocouple feedback was used to generate two distinct thermocycling profiles, shown in Fig. 4, in a two-chamber microchip with 40 mm chamber spacing. Total runtime was roughly 110 min to complete 40 cycles. The average cycle duration was 155 s with average heating rates of 2.54 °C/s and cooling rates of 2.53 °C/s (n = 5). Although the total runtime of the system does not match the speed of the fastest reported PCR systems, thermal multiplexing divides the runtime per target by a factor of n chambers, since they would otherwise be performed in a serial fashion. Future design iterations will also incorporate active cooling using a fan for faster cooling rates. Regarding thermal accuracy and repeatability, we measured temperate accuracy as compared to the set point within 0.64 °C (average absolute difference over three consecutive cycles) and standard deviation around the set point of 0.46 °C, both of which were constant over the tested range of 48–94 °C.
FIG. 4.

Independent temperature profiles for λ-phage and EBV amplifications were generated simultaneously with distinct annealing temperatures of 48 °C and 68 °C. This system utilized closed-loop control via thermocouples feedback and shutter-based optical modulation of infrared laser radiation in 1 μl reaction chambers on a polymer microchip.
Following the execution of all 12 microchip runs, PCR products were detected by electrophoresis. The resulting PCR yield (or final amplicon concentration) measurements are shown in Fig. 5.
FIG. 5.

Detection results for λ-phage and EBV amplification using our laser-mediated thermocycler to test four distinct thermal conditions to demonstrate the advantage of thermal multiplexing for optimal performance. Each condition was tested in triplicate end-point detection trials.
The results show that when microchips were run with identical annealing temperatures, only the reaction operating at its ideal annealing condition was successful while the other failed, with the exception of one of the λ-phage samples that produced a detectable peak when run with the non-ideal low annealing temperature of 48 °C. Due to the low yield, this data point is difficult to discern in Fig. 5 but present in the first trial for the common low temperature condition. This is not surprising considering the tendency for primers to anneal within a large range of temperatures under its melting temperature. A major consequence of operating at an excessively low annealing temperature not captured with this demonstration is the appearance of non-specific products due to non-specific annealing observed when host DNA is present.39 Since only purified template was used for these experiments, 100% specificity was observed as expected (Fig. 6). Average yields at the low annealing condition were 0.04 and 8.71 ng/μl for λ-phage and EBV, respectively. For the high annealing condition, average yield was 14.17 and 0 ng/μl for λ-phage and EBV, respectively.
FIG. 6.

Representative electropherograms show the PCR products for λ-phage (top row) and EBV (bottom row) amplifications run in parallel using four different annealing modes (represented by each column). From left to right, annealing temperature was uniform across the two reaction chambers (1 μl each, 40 mm spacing) at low, high, and average values and then thermally multiplexed to maintain the ideal temperatures for each reaction. Small peaks correspond to sizing and quantification markers (15 bp and 1500 bp).
When the microchips were run at the average of the two optimal annealing temperatures, weak performance was observed as indicated by the relatively low yield for all samples. Although still detectable with 10 000 starting copies, observed yields were just within the detectable range, indicating that more sensitive detections would likely be impossible. Average yields at the average annealing condition were 0.45 and 0.78 ng/μl for λ-phage and EBV, respectively. While not demonstrated in this work, microfluidic PCR has been shown to have sensitivity down to 140 starting copies.32
When thermal multiplexing was utilized for the dual amplification at the ideal conditions for each reaction, electropherograms (Fig 6, right) indicate efficient amplifications. Average yields for the multiplexed annealing conditions were 8.05 and 10.54 ng/μl for λ-phage and EBV, respectively. The same reaction chemistries run using conventional PCR at 50 μl yielded 39.34 ng/μl and 29.38 ng/μl for λ-phage and EBV, respectively. While these yields are somewhat higher, both microfluidic and conventional reactions yielded target concentrations well above the limit of detection of 0.005 ng/μl.
With the data from the twelve trials performed with each respective target, λ-phage and EBV, we examined the standard deviation of the yield for the six run at ideal annealing temperature to assess chip-to-chip repeatability. For λ-phage and EBV, the standard deviations were 6.4 ng/μl and 2.0 ng/μl, respectively. Thus, for both targets, the average yield exceeds the standard deviation and these variations are typical with end-point PCR detection.40
In an effort to validate our microfluidic PCR system for clinically relevant reactions, we performed RT-PCR for amplifications of influenza A and influenza B. Despite observing slightly higher yields using conventional 50 μl reactions (flu A = 4.58 ng/μl and flu B = 7.96 ng/μl), we performed consistent amplifications at 1 μl using our microchip and laser-mediated thermocycling. For two specimens of known influenza A infection tested in triplicate, we observed successful detections in all cases with yields of 1.96, 1.86, 2.95 ng/μl, and 2.68, 3.58, 2.06 ng/μl. Similarly, for two specimens tested in triplicate for influenza B, we achieved yields of 6.48, 7.18, 6.75 ng/μl and 2.23, 2.55, 2.14 ng/μl. Representative electropherograms for these amplifications are shown in Fig. 7. These final amplicon concentrations exhibit expected ranges of sample-to-sample variation resulting from differing viral loads and extraction efficiencies and trial-to-trial variation resulting from the inconsistency inherent to end-point detection. Non-specific products and primer dimers were observed in both the conventional and microfluidic amplifications, but they were observed to be smaller in the microfluidic amplifications. The outermost peaks are sizing and quantification markers (15 bp and 1500 bp). Qualitatively, the microchip exhibits good specificity. In cases where primer design is inflexible to annealing temperature considerations, such as consensus degenerate PCR37,38 and biochemical multiplexing is infeasible, the freedom to perform reactions with independent thermal conditions is advantageous.
FIG. 7.

Representative electropherograms show the RT-PCR products for (a) influenza A with 106 bp amplicon and (b) influenza B with 103 bp amplicon performed with our microfluidic system. Small non-specific peaks were common and can be seen for both samples. The outermost peaks are sizing and quantification markers (15 bp and 1500 bp).
CONCLUSIONS
We report a novel approach to thermocycling, termed thermal multiplexing, to increase the throughput and flexibility of multi-chamber microfluidic PCR while maintaining optimal performance. A system consisting of an infrared laser thermocycler with optical modulation and a polymer microchip was used to perform a multiplexed temperature feedback control scheme. Solenoid shutters enabled effective, multi-chamber control of the reaction temperatures as a function of duty cycle. We performed thermal modeling to determine optimum spacing between chambers and empirically verified the modeling results. Amplifications of λ-phage (at 68 °C) and EBV template (at 48 °C) DNA in 110 min demonstrate the capability of thermal multiplexing near the extremes of PCR primer design temperatures. Further, we were able to achieve the full range of annealing temperatures theoretically necessary (28 °C range between adjacent chambers). We also demonstrate successful amplification of RNA viruses influenza A and B from human nasopharyngeal swabs to show the utility of this instrument for clinical specimens with good specificity. Thermal multiplexing does not appear to have inherent limitations to scaling many reactions, reaction volumes, or adaptations to different heating modalities. In contrast to the Cepheid SmartCycler, this system operates from a single heating source and uses a single integrated microfluidic chip, allowing for smaller sample volumes, higher throughput, and greater portability. For this system to be further developed for POC applications, refinements would be needed, including simplification of the sample loading procedure to reduce the required skill of the user. Further, active cooling, rather than passive, natural convection described in this work, could reduce cycle time and allow the system to operate in extreme environments. For suppressing formation and expansion of bubbles, we explored numerous strategies, including degassing, adjusting surfactant concentration, and varying reaction chamber geometry with limited success. We believe pressurization of the device is effective and viable but could be more elegantly incorporated into a future refinement of the instrument. A POC thermally multiplexed system could enable applications such as pathogen detection, where patients presenting non-specific or inconclusive symptoms need to be screened across an entire viral or bacterial panel, in a few hours.
ACKNOWLEDGMENTS
C.R.F. acknowledges the NIH BRAIN Initiative Grant (NEI and NIMH 1-U01-MH106027-01), NIH Single Cell Grant 1 R01 EY023173, NSF (EHR 0965945 and CISE 1110947), NIH Computational Neuroscience Training grant (DA032466-02), Georgia Tech Translational Research Institute for Biomedical Engineering & Science (TRIBES) Seed Grant Awards Program, Georgia Tech Fund for Innovation in Research and Education (GT-FIRE), Wallace H. Coulter Translational/Clinical Research Grant Program and support from Georgia Tech through the Institute for Bioengineering and Biosciences Junior Faculty Award, Technology Fee Fund, Invention Studio, and the George W. Woodruff School of Mechanical Engineering. We would like to acknowledge Mark McJunkin and Marty Jacobson for their assistance with fabrication and machine shop training. This research was performed under an appointment to the Department of Homeland Security (DHS) Scholarship and Fellowship Program, administered by the Oak Ridge Institute for Science and Education (ORISE) through an interagency agreement between the U.S. Department of Energy (DOE) and DHS. ORISE is managed by Oak Ridge Associated Universities (ORAU) under DOE Contract No. DE-AC05-06OR23100. All opinions expressed in this paper are the authors and do not necessarily reflect the policies and views of DHS, DOE, or ORAU/ORISE.
References
- 1. Mullis K., Faloona F., Scharf S., Saiki R., Horn G., and Erlich H., Cold Spring Harbor Symp. Quant. Biol. 51, 263–273 (1986). 10.1101/SQB.1986.051.01.032 [DOI] [PubMed] [Google Scholar]
- 2. Henderson D. A., Inglesby T. V., O'Toole T., Fine A., and Layton M., Clin. Infect. Dis. 32, 277–282 (2001). 10.1086/318469 [DOI] [PubMed] [Google Scholar]
- 3. van Elden L. J. R., van Kraaij M. G. J., Nijhuis M., Hendriksen K. A. W., Dekker A. W., Rozenberg-Arska M., and van Loon A. M., Clin. Infect. Dis. 34, 177–183 (2002). 10.1086/338238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Virolainen A., Salo P., Jero J., Karma P., Eskola J., and Leinonen M., J. Clin. Microbiol. 32, 2667–2670 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cao Q., Mahalanabis M., Chang J., Carey B., Hsieh C., Stanley A., Odell C. A., Mitchell P., Feldman J., Pollock N. R., and Klapperich C. M., PLoS ONE 7, e33176 (2012). 10.1371/journal.pone.0033176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wittwer C. T., Ririe K. M., Andrew R. V., David D. A., Gundry R. A., and Balis U. J., BioTechniques 22, 176–181 (1997). [DOI] [PubMed] [Google Scholar]
- 7. Kim Y. H., Yang I., Bae Y.-S., and Park S.-R., BioTechniques 44, 495–496, 498, 500 passim (2008). 10.2144/000112705 [DOI] [PubMed] [Google Scholar]
- 8. Yang I., Kim Y.-H., Byun J.-Y., and Park S.-R., Anal. Biochem. 338, 192–200 (2005). 10.1016/j.ab.2004.09.035 [DOI] [PubMed] [Google Scholar]
- 9. Zhang C. and Xing D., Nucleic Acids Res. 35, 4223–4237 (2007). 10.1093/nar/gkm389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang C., Xu J., Ma W., and Zheng W., Biotechnol. Adv. 24, 243–284 (2006). 10.1016/j.biotechadv.2005.10.002 [DOI] [PubMed] [Google Scholar]
- 11. Easley C. J., Karlinsey J. M., Bienvenue J. M., Legendre L. A., Roper M. G., Feldman S. H., Hughes M. A., Hewlett E. L., Merkel T. J., Ferrance J. P., and Landers J. P., Proc. Natl. Acad. Sci. U.S.A. 103, 19272–19277 (2006). 10.1073/pnas.0604663103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Belgrader P., Young S., Yuan B., Primeau M., Christel L. A., Pourahmadi F., and Northrup M. A., Anal. Chem. 73, 286–289 (2001). 10.1021/ac000905v [DOI] [PubMed] [Google Scholar]
- 13. Liu P., Yeung S. H. I., Crenshaw K. A., Crouse C. A., Scherer J. R., and Mathies R. A., Forensic Sci. Int. Genet. 2, 301–309 (2008). 10.1016/j.fsigen.2008.03.009 [DOI] [PubMed] [Google Scholar]
- 14. Wittwer C. T. and Garling D. J., BioTechniques 10, 76–83 (1991). [PubMed] [Google Scholar]
- 15. Liao C.-S., Lee G.-B., Liu H.-S., Hsieh T.-M., and Luo C.-H., Nucleic Acids Res. 33, e156 (2005). 10.1093/nar/gni157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Agrawal N., Hassan Y. A., and Ugaz V. M., Angew. Chem., Int. Ed. 46, 4316–4319 (2007). 10.1002/anie.200700306 [DOI] [PubMed] [Google Scholar]
- 17. Wheeler E. K., Benett W., Stratton P., Richards J., Chen A., Christian A., Ness K. D., Ortega J., Li L. G., Weisgraber T. H., Goodson K., and Milanovich F., Anal. Chem. 76, 4011–4016 (2004). 10.1021/ac034941g [DOI] [PubMed] [Google Scholar]
- 18. Giordano B. C., Ferrance J., Swedberg S., Hühmer A. F. R., and Landers J. P., Anal. Biochem. 291, 124–132 (2001). 10.1006/abio.2000.4974 [DOI] [PubMed] [Google Scholar]
- 19. Huhmer A. F. R. and Landers J. P., Anal. Chem. 72, 5507–5512 (2000). 10.1021/ac000423j [DOI] [PubMed] [Google Scholar]
- 20. Oda R. P., Strausbauch M. A., Huhmer A. F. R., Borson N., Jurrens S. R., Craighead J., Wettstein P. J., Eckloff B., Kline B., and Landers J. P., Anal. Chem. 70, 4361–4368 (1998). 10.1021/ac980452i [DOI] [PubMed] [Google Scholar]
- 21. Kim H., Vishniakou S., and Faris G. W., Lab Chip 9, 1230–1235 (2009). 10.1039/b817288a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Slyadnev M. N., Tanaka Y., Tokeshi M., and Kitamori T., Anal. Chem. 73, 4037–4044 (2001). 10.1021/ac010318p [DOI] [PubMed] [Google Scholar]
- 23. Terazono H., Hattori A., Takei H., Takeda K., and Yasuda K., Jpn. J. Appl. Phys., Part 1 47, 5212 (2008). 10.1143/JJAP.47.5212 [DOI] [Google Scholar]
- 24. Roper M. G., Easley C. J., Legendre L. A., Humphrey J. A. C., and Landers J. P., Anal. Chem. 79, 1294–1300 (2007). 10.1021/ac0613277 [DOI] [PubMed] [Google Scholar]
- 25. Pak N., Saunders D. C., Phaneuf C. R., and Forest C. R., Biomed. Microdevices 14, 427–433 (2012). 10.1007/s10544-011-9619-2 [DOI] [PubMed] [Google Scholar]
- 26. Saunders D. C., Holst G. L., Phaneuf C. R., Pak N., Marchese M., Sondej N., McKinnon M., and Forest C. R., Biosens. Bioelectron. 44, 222–228 (2013). 10.1016/j.bios.2013.01.019 [DOI] [PubMed] [Google Scholar]
- 27. Chamberlain J. S., Gibbs R. A., Rainer J. E., Nguyen P. N., and Thomas C., Nucleic Acids Res. 16, 11141–11156 (1988). 10.1093/nar/16.23.11141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Edwards M. C. and Gibbs R. A., Genome Res. 3, S65–S75 (1994). 10.1101/gr.3.4.S65 [DOI] [PubMed] [Google Scholar]
- 29. Markoulatos P., Siafakas N., and Moncany M., J. Clin. Lab. Anal. 16, 47–51 (2002). 10.1002/jcla.2058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Henegariu O., Heerema N. A., Dlouhy S. R., Vance G. H., and Vogt P. H., BioTechniques 23, 504–511 (1997). [DOI] [PubMed] [Google Scholar]
- 31. Phaneuf C. R. and Forest C. R., in Proceedings of the 25th Annual Meeting of the American Society for Precision Engineering, Atlanta, GA (2010), Vol. 50, pp. 345–347. [Google Scholar]
- 32. Phaneuf C. R., Oh K., Pak N., Saunders D. C., Conrardy C., Landers J. P., Tong S., and Forest C. R., Biomed. Microdevices 15, 221–231 (2013). 10.1007/s10544-012-9720-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kodzius R., Xiao K., Wu J., Yi X., Gong X., Foulds I. G., and Wen W., Sens. Actuators, B 161, 349–358 (2012). 10.1016/j.snb.2011.10.044 [DOI] [Google Scholar]
- 34. Scherag F. D., Brandstetter T., and Rühe J., Colloids Surf. B 122, 576–582 (2014). 10.1016/j.colsurfb.2014.07.035 [DOI] [PubMed] [Google Scholar]
- 35. Kibbe W. A., Nucleic Acids Res. 35, W43–W46 (2007). 10.1093/nar/gkm234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Phaneuf C. R., Pak N., and Forest C. R., Sens. Actuators, A 167, 531–536 (2011). 10.1016/j.sna.2011.02.002 [DOI] [Google Scholar]
- 37. Rose T. M., Schultz E. R., Henikoff J. G., Pietrokovski S., McCallum C. M., and Henikoff S., Nucleic Acids Res. 26, 1628–1635 (1998). 10.1093/nar/26.7.1628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rose T. M., Strand K. B., Schultz E. R., Schaefer G., Rankin G. W., Thouless M. E., Tsai C. C., and Bosch M. L., J. Virol. 71, 4138–4144 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. van Pelt-Verkuil E., van Belkum A., and Hays J. P., Principles and Technical Aspects of PCR Amplification ( Springer, 2008). [Google Scholar]
- 40. Arya M., Shergill I. S., Williamson M., Gommersall L., Arya N., and Patel H. R., Expert Rev. Mol. Diagn. 5, 209–219 (2005). 10.1586/14737159.5.2.209 [DOI] [PubMed] [Google Scholar]