

Abstract
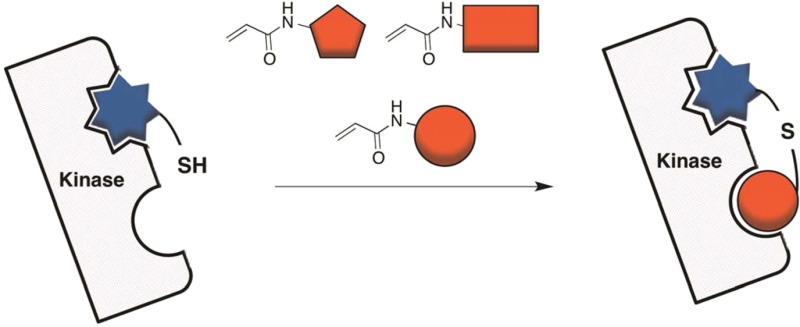
We have employed novel fragment-based screening methodology to discover bivalent kinase inhibitors with improved selectivity. Starting from a low molecular weight promiscuous kinase inhibitor, we appended a thiol for subsequent reaction with a library of acrylamide electrophiles. Enzyme-templated screening was performed to identify acrylamides that assemble into bivalent inhibitors of c-Src kinase. Upon identification of acrylamide fragments that improve the binding affinity of our lead thiol, we characterized the resulting bivalent inhibitors and identified a series of kinase inhibitors with improved potency and selectivity compared to the thiol-containing precursor. Provided that protein can be prepared free of endogenous reactive cysteines, our methodology is general and could be applied to nearly any enzyme of interest.
Keywords: Kinase, selectivity, enzyme-templated
Protein kinases represent one of the largest protein families of the human genome and are responsible for regulating key signaling pathways in eukaryotes.1 Aberrant protein phosphorylation within the cell has been implicated in numerous diseases and has subsequently made kinases attractive targets for medicinal chemistry.2−4 All FDA-approved kinase inhibitors bind to the highly conserved ATP pocket due to the tractable targeting nature of this site.5 Despite successful ATP-competitive inhibitors in the clinic, many inhibitors of this class suffer from poor selectivity profiles and subsequent off-target toxicity.6,7 In an effort to improve inhibitor selectivity, features adjacent to or outside the ATP-binding site have been recently targeted.3,8 The systematic discovery of inhibitors that bind outside the ATP site, however, is challenging and is often serendipitous.9,10
Fragment-based drug discovery (FBDD) has been successful for a number of targets, yet labor-intensive NMR or X-ray crystallography detection methods and ATP-pocket centric design strategies have limited its utility for kinases.11 To apply the principles of FBDD to kinases in an unbiased manner without need for structural information, we developed a fragment elaboration screen where hits can be identified through an activity assay.
Traditional fragment tethering relies on mixed disulfide formation with the protein,12 while our strategy involves a Michael reaction between an ATP-competitive hit compound with a pendent thiol and an acrylamide fragment. We benefit from using a proximity-based, enzyme-templated approach, where increases in local concentration of both reactive components leads to formation of a bivalent inhibitor in situ.13−15 A major benefit of an enzyme-templated approach is the ability to use the same fragment library for diverse targets (as opposed to synthesizing and purifying bivalent inhibitors for each target). Bivalent inhibitors occupy two distinct binding sites connected by a linker. Importantly, bivalent inhibitors often have improved properties such as potency and/or selectivity.
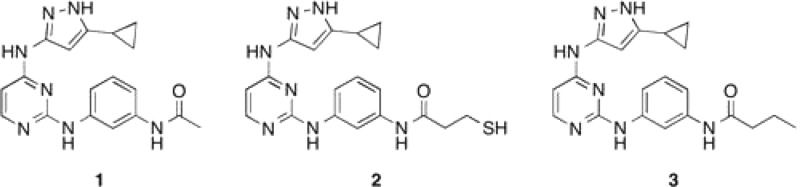
We previously reported compound 1, a highly promiscuous aminopyrazole kinase inhibitor.16 We profiled 1 and found that 1 potently binds 117 of 200 kinases tested.15 To enable enzyme-templated Michael reactions with a library of acrylamides, we synthesized an analogue of 1 that contains a thiol functionality (2) (Chart 1). Compound 3 was synthesized as a control absent a reactive thiol. As the electrophile component, acrylamides are attractive because they have low intrinsic reactivity.16,17 Their relatively low reactivity will ensure that the screen will selectively identify enzyme-templated reactions. Conversely, electrophiles with high intrinsic reactivity (e.g., vinylsulfonamides) could produce conjugates in the absence of enzyme.17
Chart 1. Structure of Promiscuous Kinase Inhibitors 1–3.
We chose c-Src, a nonreceptor tyrosine kinase, as the initial target to develop our methodology. c-Src has been extensively studied over the years and implicated in a variety of diseases including the metastasis of several cancers,18,19 and few selective inhibitors are available to study c-Src biology.5,8 Importantly, compound 1 is a competent inhibitor of c-Src (Ki = 0.36 ± 0.08 μM). To ensure that our assay would identify only acrylamides that react with 2 and not c-Src itself, we made a mutant c-Src kinase with three nonessential, solvent-accessible cysteines mutated to serines on the surface of c-Src (“3M” c-Src: C277S, C483S, S496S). Of note, we found that compound 2 is a time-dependent, irreversible inhibitor of wild-type c-Src, but not of 3M c-Src.
A library of 110 acrylamide fragments (average molecular weight = 235 Da, see Supplemental Table S1 for structures) was incubated with 3M c-Src in the presence and absence of 2 (2 was added at a concentration that approximates its IC20 value). After a 30 min incubation, we used a continuous activity assay to identify fragments that had significant inhibition difference (>2 standard deviations) between screens performed with and without thiol 2. From the 110 acrylamide fragments, we identified four hits that met our criteria (Figure 1, hit rate of 3.6%). Each of the four hits was confirmed in three subsequent enzyme-templated validation screens (see Supporting Information for details and assay controls performed).
Figure 1.
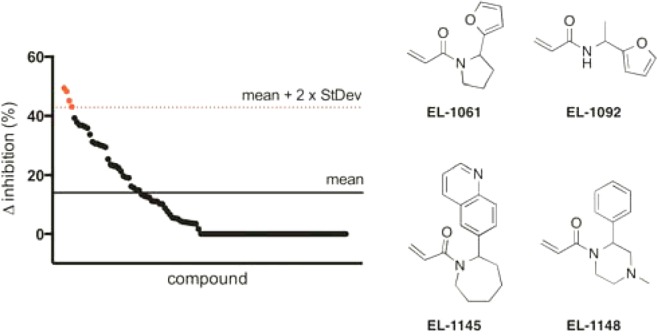
(left) Graph of enzyme-templated screen using c-Src and a library of 110 acrylamides. (right) Structures of acrylamide fragment hits.
To demonstrate that the predicted enzyme-templated Michael additions were occurring, we performed a mass spectrometry analysis of thiol 2 + acrylamide EL-1148 in the presence and absence of c-Src kinase. Only when c-Src was present could we detect a mass for the Michael adduct (4). We next synthesized 4 and found it to have improved binding potency compared to 3 and EL-1148, the individual starting fragments (Chart 2). Because 2 is an irreversible inhibitor of wild-type c-Src, we compared compound 3, where the thiol of 2 is replaced with a carbon. Compound 3 (Ki = 0.40 ± 0.02 μM) was found to be less potent than bivalent inhibitor 4 (Ki = 0.09 ± 0.02 μM) for wild-type c-Src. To enable a comparison of thiol 2 with bivalent inhibitor 4, we tested both for inhibition of 3M c-Src and found 4 to be a more potent inhibitor of this construct (2, 3M c-Src Ki = 0.20 ± 0.06 μM; 4, 3M c-Src Ki = 0.12 ± 0.02 μM).
Chart 2. Structures of Bivalent Kinase Inhibitors.

EL-1148 is a very weak inhibitor of c-Src (3M c-Src, IC50 = 470 ± 110 μM) but was readily identified in our method. In traditional fragment-based screening, it could be difficult to identify weak binding fragments using activity-based assays and even more difficult to discriminate between ATP-competitive and non-ATP-competitive fragments. Using biochemical activity assays, we further demonstrated that EL-1148 is noncompetitive with ATP, suggesting that it is binding in a pocket adjacent to the ATP-pocket (Supplemental Figure S1).
Because thioethers can undergo retro-Michael reactions in assay buffer conditions (and thus lose inhibitory potency), we synthesized 5, which contains an all-carbon linker between the two fragments (Chart 2). Gratifyingly, 5 was able to inhibit c-Src with identical potency as 4. We then synthesized bivalent inhibitors, again using an all-carbon linker, derived from the two furan-containing fragments (EL-1061 and EL-1092) that were identified in our enzyme-templated screen (bivalent inhibitors 6 and 7, respectively). In addition, because both 6 and 7 are a mixture of enantiomers, we synthesized both R- and S-enantiomers of the furan-containing amine fragments and subsequently produced enantiopure bivalent inhibitors. The amines were synthesized using Ellman sulfinylimine chemistry (see Supporting Information for synthetic details).20,21 Upon characterization of the bivalent inhibitor series, we found that the bivalent inhibitors with R-stereochemistry had improved binding to c-Src (Table 1). A preference for a particular stereochemistry indicates specific binding to c-Src, despite the relatively weak binding observed with the acrylamide fragments.
Table 1. Ki Values for 1 and Enantiomerically Pure Bivalent Kinase Inhibitors.
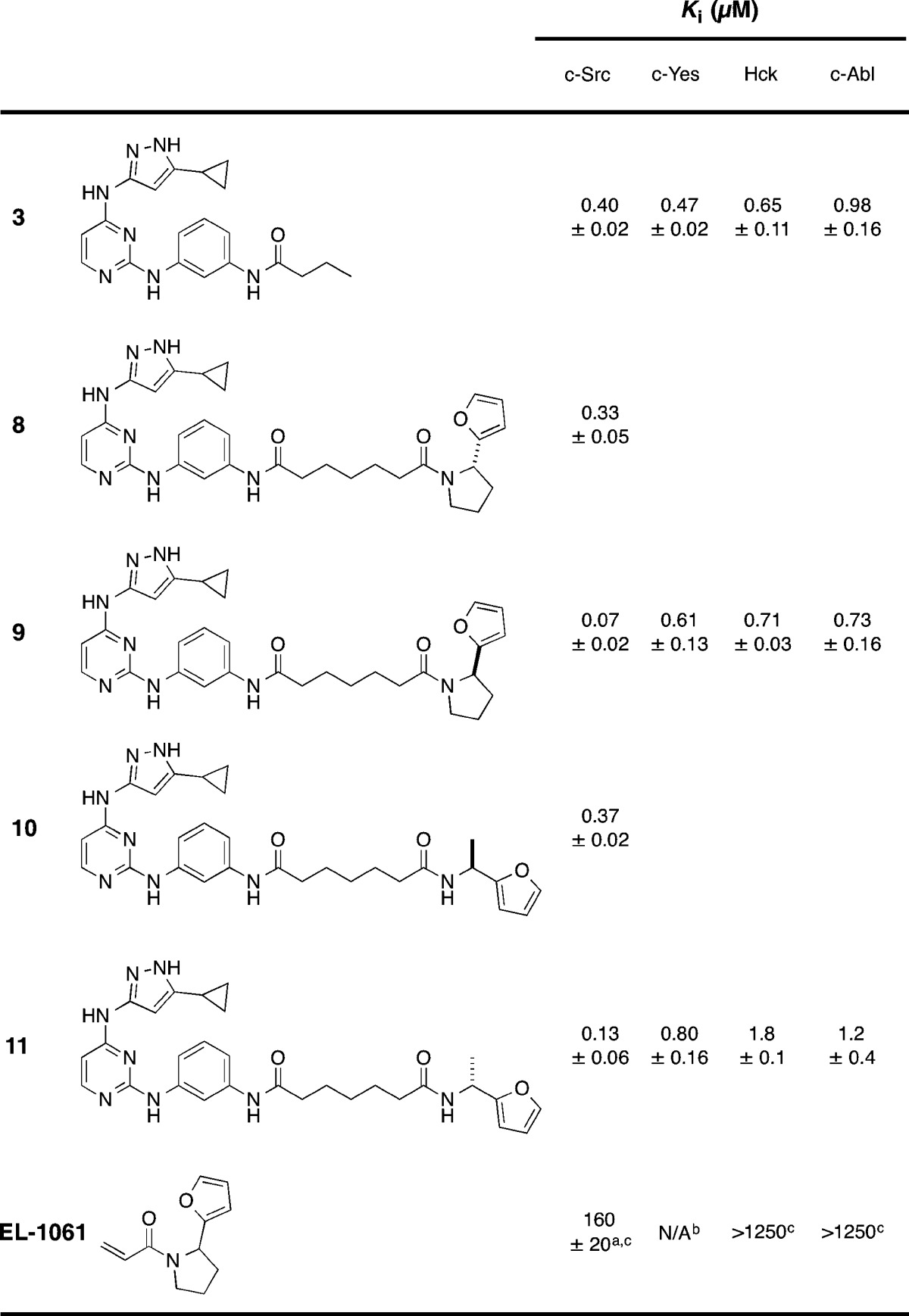
Value is for 3M c-Src.
EL-1061 can bind irreversibly to c-Yes.
Bivalent inhibitors 9 and 11 have 6- and 3-fold improvements in binding affinity for c-Src, respectively, compared to the control ATP-competitive fragment 3 (Table 1). We next sought to determine whether 9 and 11 have improved selectivity compared to fragment 3. As a measure of selectivity, we assayed for inhibition against a small panel of homologous kinases (c-Yes, 95% similar to c-Src; Hck, 80% similar to c-Src; c-Abl, 69% similar to c-Src). While 3 has virtually no selectivity within this panel (average selectivity = 1.8×), both 9 and 11 have dramatically improved selectivity (average selectivity = 9.5× and 9.4×, respectively). Compounds selective for c-Src over c-Yes have application in pharmacological manipulation of embryonic stem cells,22 and bivalent inhibitor 9 is the most selective c-Src over c-Yes inhibitor reported to date.8,23 In addition, while c-Src has been validated as a target in triple-negative breast cancer (TNBC),8,24 c-Abl has been reported to be a tumor suppressor in TNBC.8,25,26 Thus, compounds that have selectivity for c-Src over these homologous kinases will be useful probe compounds for the biology community.
To explore the role of the non-ATP-competitive fragment in providing selectivity to bivalent inhibitor 9, we determined the selectivity of EL-1061 (the parent fragment of 9) in our homologous kinase panel. EL-1061 is a weak inhibitor of 3M c-Src (IC50 = 160 ± 20 μM); however, it was found not to inhibit Hck and c-Abl at concentrations up to 1250 μM. Because c-Yes has a P-loop cysteine we cannot determine a reversible binding constant for this enzyme. Overall, fragment EL-1061 has a >8× selectivity, which is consistent with the high selectivity obtained from bivalent inhibitor 9 (10× selectivity for c-Src over Hck and c-Abl).
It is worth noting that the potency gained going from 3 to 9 was modest (6-fold) and was at the expense of ligand efficiency (LE = 1.4(−log Ki)/(# heavy atoms)). ATP-competitive inhibitor 3 has a LE = 0.32, while bivalent inhibitor 9 has a LE = 0.24. Despite lowering the overall ligand efficiency, there is a large increase in selectivity due to the addition of the non-ATP-competitive fragment. It is possible that with larger acrylamide fragment libraries (our library is 110 members), a more optimal-binding fragment could be identified.
On the basis of the selectivity data (Table 1), we propose that the non-ATP-competitive fragments are binding in the P-loop (also called the glycine-rich loop) pocket of c-Src (Supplemental Figure S2). This pocket has previously been exploited to improve the selectivity of ATP-competitive kinase inhibitors; however, all previously reported P-loop binding ligands were obtained using structure-based drug design.8,27 One of the key advantages of our methodology is that no structural information is required to identify fragments capable of assembling into bivalent inhibitors.28 Our hypothesis is supported by structural information for analogues of 1 in which the amide functionality would be pointing in the direction of the P-loop pocket (Supplemental Figure S3). The P-loop of c-Src has a unique extended conformation resulting from a three-residue salt bridge between Lys272, Gln275, and Glu280.29 We disrupted this salt bridge by mutating Glu280 to Gly, a mutation previously shown to modulate the P-loop conformation of Lyn, a Src-family kinase.29 We produced E280G c-Src and tested the ability of bivalent inhibitor 9 to inhibit this construct of c-Src. Consistent with our hypothesis, we found that 9 is a 4-fold weaker inhibitor (WT c-Src Ki = 0.07 ± 0.02 μM, E280G c-Src Ki = 0.26 ± 0.03 μM). While our screen identified fragments proposed to bind in the P-loop pocket of c-Src, one could vary the linker length and/or position of the thiol to discover fragments that bind in other locations adjacent to the ATP pocket.
Herein, we have demonstrated the application of a fragment elaboration method using thiol–acrylamide chemistry to generate bivalent enzyme inhibitors. We applied our method to c-Src kinase; however, our methodology could easily be applied to other enzyme systems, including proteases and phosphatases. The assembled bivalent inhibitors possess improved potency and selectivity compared to the starting fragments. Work applying this methodology to additional kinases targeted by 1 is ongoing in our laboratory.
Acknowledgments
We would like to thank J. Kuriyan (UC Berekely) and M. Seeliger (Stony Brook) for providing expression plasmids for c-Src, Hck, and c-Abl.
Glossary
ABBREVIATIONS
- ATP
adenosine triphosphate
- FBDD
fragment-based drug discovery
- TNBC
triple-negative breast cancer
- WT
wild-type
Supporting Information Available
Synthetic procedures, assay details, and compound characterization data. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00167.
Author Contributions
The manuscript was written through contributions of all authors and all authors have given approval to the final version of the manuscript.
F.E.K. was supported in part by a National Institutes of Health Chemistry–Biology Interface Training Grant (T32GM008597) and a University of Michigan Rackham Predoctoral Fellowship. Funding of this research was provided in part by NIH grant R01GM088546 to M.B.S. and the University of Michigan College of Pharmacy.
The authors declare no competing financial interest.
Supplementary Material
References
- Manning G.; Whyte D. B.; Martinez R.; Hunter T.; Sudarsanam S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- Cohen P. Protein kinases – the major drug targets of the twenty-first century?. Nat. Rev. Drug Discovery 2002, 1, 309–315. 10.1038/nrd773. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Yang P. L.; Gray N. S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. 10.1038/nrc2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P.; Alessi D. R. Kinase drug discovery--what’s next in the field?. ACS Chem. Biol. 2013, 8, 96–104. 10.1021/cb300610s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breen M. E.; Steffey M. E.; Lachacz E. J.; Kwarcinski F. E.; Fox C. C.; Soellner M. B. Substrate Activity Screening with Kinases: Discovery of Small-Molecule Substrate-Competitive c-Src Inhibitors. Angew. Chem., Int. Ed. 2014, 53, 7010–7013. 10.1002/anie.201311096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight Z. A.; Shokat K. M. Features of selective kinase inhibitors. Chem. Biol. 2005, 12, 621–637. 10.1016/j.chembiol.2005.04.011. [DOI] [PubMed] [Google Scholar]
- Morphy R. Selectively nonselective kinase inhibition: striking the right balance. J. Med. Chem. 2010, 53, 1413–1437. 10.1021/jm901132v. [DOI] [PubMed] [Google Scholar]
- Brandvold K. R.; Steffey M. E.; Fox C. C.; Soellner M. B. Development of a highly selective c-Src kinase inhibitor. ACS Chem. Biol. 2012, 7, 1393–1398. 10.1021/cb300172e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Z.; Grutter C.; Rauh D. Strategies for the selective regulation of kinases with allosteric modulators: exploiting exclusive structural features. ACS Chem. Biol. 2013, 8, 58–70. 10.1021/cb300663j. [DOI] [PubMed] [Google Scholar]
- Breen M. E.; Soellner M. B. Small molecule substrate phosphorylation site inhibitors of protein kinases: approaches and challenges. ACS Chem. Biol. 2015, 10, 175–189. 10.1021/cb5008376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott D. E.; Coyne A. G.; Hudson S. A.; Abell C. Fragment-based approaches in drug discovery and chemical biology. Biochemistry 2012, 51, 4990–5003. 10.1021/bi3005126. [DOI] [PubMed] [Google Scholar]
- Erlanson D. A.; Braisted A. C.; Raphael D. R.; Randal M.; Stroud R. M.; Gordon E. M.; Wells J. A. Site-directed ligand discovery. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 9367–9372. 10.1073/pnas.97.17.9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasinski A. R.; Radic Z.; Manetsch R.; Raushel J.; Taylor P.; Sharpless K. B.; Kolb H. C. In situ selection of lead compounds by click chemistry: Target-guided optimization of acetylcholinesterase inhibitors. J. Am. Chem. Soc. 2005, 127, 6686–6692. 10.1021/ja043031t. [DOI] [PubMed] [Google Scholar]
- Namelikonda N. K.; Manetsch R. Sulfo-click reaction via in situ generated thioacids and its application in kinetic target-guided synthesis. Chem. Commun. 2012, 48, 1526–1528. 10.1039/C1CC14724B. [DOI] [PubMed] [Google Scholar]
- Levitsky K.; Ciolli C. J.; Martinez R.; Belshaw P. J. Selective inhibition of engineered receptors via proximity-accelerated alkylation. Org. Lett. 2003, 5, 693–696. 10.1021/ol027448k. [DOI] [PubMed] [Google Scholar]
- Kwarcinski F. E.; Fox C. C.; Steffey M. E.; Soellner M. B. Irreversible inhibitors of c-Src kinase that target a nonconserved cysteine. ACS Chem. Biol. 2012, 7, 1910–1917. 10.1021/cb300337u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kathman S.; Ziyang X.; Statsyuk A. V. A Fragment-based method to discover irreversible covalent inhibitors of cysteine proteases. J. Med. Chem. 2014, 57, 4969–4974. 10.1021/jm500345q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas S. M.; Brugge J. S. Cellular functions regulated by Src family kinases. Annu. Rev. Cell Dev. Biol. 1997, 13, 513–609. 10.1146/annurev.cellbio.13.1.513. [DOI] [PubMed] [Google Scholar]
- Martin G. S. The hunting of the Src. Nat. Rev. Mol. Cell Biol. 2001, 2, 467–475. 10.1038/35073094. [DOI] [PubMed] [Google Scholar]
- Liu G. C.; Cogan D. A.; Owens T. D.; Tang T. P.; Ellman J. A. Synthesis of enantiomerically pure N-tert-butanesulfinyl imines (tert-butanesulfinimines) by the direct condensation of tert-butanesulfinamide with aldehydes and ketones. J. Org. Chem. 1999, 64, 1278–1284. 10.1021/jo982059i. [DOI] [Google Scholar]
- Brinner K. M.; Ellman J. A. A rapid and general method for the asymmetric synthesis of 2-substituted pyrrolidines using tert-butanesulfinamide. Org. Biomol. Chem. 2005, 3, 2109–2113. 10.1039/b502080h. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Meyn M. A.; Smithgall T. E. c-yes tyrosine kinase is a potent suppressor of ES cell differentiation and antagonizes the actions of its closest phylogenetic relative, c-Src. ACS Chem. Biol. 2014, 9, 139–146. 10.1021/cb400249b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M. I.; Hunt J. P.; Herrgard S.; Ciceri P.; Wodicka L. M.; Pallares G.; Hocker M.; Trieber D. K.; Zarrinkar P. P. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. 10.1038/nbt.1990. [DOI] [PubMed] [Google Scholar]
- Finn R. S. Targeting Src in breast cancer. Ann. Oncology 2008, 19, 1379–1386. 10.1093/annonc/mdn291. [DOI] [PubMed] [Google Scholar]
- Allington T. M.; Schiemann W. P. The Cain and Abl of epithelial-mesenchymal transition and transforming growth factor-B in mammary epithelial cells. Cells Tissues Organs 2011, 193, 98–113. 10.1159/000320163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allington T. M.; Galliher-Beckley A. J.; Schiemann W. P. Activated Abl kinase inhibits oncogenic transforming growth factor-beta signaling and tumorigenesis in mammary tumors. FASEB J. 2009, 23, 4231–4243. 10.1096/fj.09-138412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaikuad A.; Tacconi E. M.; Zimmer J.; Liang Y.; Gray N. S.; Tarsounas M.; Knapp S. A unique inhibitor binding site in ERK1/2 is associated with slow binding kinetics. Nat. Chem. Biol. 2014, 10, 853–862. 10.1038/nchembio.1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gower C. M.; Chang M. E.; Maly D. J. Bivalent inhibitors of protein kinases. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 102–115. 10.3109/10409238.2013.875513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barouch-Bentov R.; Che J.; Lee C. C.; Yang Y.; Herman A.; Jia Y.; Velentza A.; Watson A.; Sternberg L.; Kim S.; Ziaee N.; Miller A.; Jackson C.; Fujimoto M.; Young M.; Batalov S.; Liu Y.; Warmuth M.; Wiltshire T.; Cooke M. P.; Sauer K. A conserved salt bridge in the G loop of multiple protein kinases is important for catalysis and for in vivo Lyn function. Mol. Cell 2009, 33, 43–52. 10.1016/j.molcel.2008.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.