

Abstract
Alleviating the burden of tuberculosis (TB) requires an understanding of the genetic basis that determines the emergence of drug-resistant mutants. PA-824 (pretomanid) is a bicyclic nitroimidazole class compound presently undergoing the phase III STAND clinical trial, despite lacking identifiable genetic markers for drug-specific resistant Mycobacterium tuberculosis. In the present study, we aimed to characterize the genetic polymorphisms of spontaneously generated PA-824-resistant mutant strains by surveying drug metabolism genes for potential mutations. Of the 183 independently selected PA-824-resistant M. tuberculosis mutants, 83% harbored a single mutation in one of five nonessential genes associated with either PA-824 prodrug activation (ddn, 29%; fgd1, 7%) or the tangential F420 biosynthetic pathway (fbiA, 19%; fbiB, 2%; fbiC, 26%). Crystal structure analysis indicated that identified mutations were specifically located within the protein catalytic domain that would hinder the activity of the enzymes required for prodrug activation. This systematic analysis conducted of genotypes resistant to PA-824 may contribute to future efforts in monitoring clinical strain susceptibility with this new drug therapy.
INTRODUCTION
Tuberculosis (TB) remains a major global health concern, with >8 million new cases and 1.8 million deaths occurring annually (WHO). This pandemic is exacerbated by the pervasive spread of multidrug-resistant (MDR)-TB that challenges clinicians to fight a disease with a limited arsenal of resources. The bicyclic 4-nitroimidazole chemotype has yielded two promising candidates, delamanid (OPC67683) and pretomanid (PA-824), which actively inhibit both nonreplicating and rapidly growing bacilli under aerobic and anaerobic conditions (1). Both drugs are undergoing clinical evaluation and FDA approval is pending for the treatment of MDR-TB. In 2013, delamanid received conditional marketing authorization by the European Medicines Agency (EMA) for use in adult patients deprived of other treatment options (2). PA-824 is in the phase III STAND clinical trial, and at this stage of the development pipeline, it would be beneficial to monitor the genetic basis of resistant clinical strains as they emerge in the wake of future implementation into a treatment protocol.
Bicyclic 4-nitroimidazoles are prodrugs that require metabolic activation by a deazaflavin (cofactor F420)-dependent nitroreductase (Ddn) (3). Ddn (Rv3547) converts the prodrugs into three primary metabolites, a des-nitroimidazole and two unstable by-products (4). Ddn is likely a membrane-bound protein (5) that is involved in a protective mechanism under oxidative stress (6). The major mechanism of action of nitroimidazole in active disease under aerobic conditions is to hinder the formation of mycolic acids, and under anaerobic conditions, the mechanism involves the induction of respiratory poisoning (4, 7). By inhibiting the formation of ketomycolates, a class of mycolic acids, nitroimidazole interferes with Mycobacterium tuberculosis cell wall formation, thus curtailing growth (1). PA-824 also donates nitric oxide (NO), which can accumulate and create toxic conditions within the bacilli that hamper regular electron flow and homeostasis during latency (4). In active aerobic M. tuberculosis, this NO buildup is insufficient to have a significant bactericidal effect (7).
The two-electron transfer cofactor F420 (7,8-didemethyl-8-hydroxy-5-deazaflavin derivative), first reported in mycobacteria (8), plays a role in redox reactions and the methane biosynthesis pathway (9–11). F420 redox cycling requires the NADP-dependent glucose-6-phosphate dehydrogenase (FGD1) to catalyze the oxidation of glucose-6-phosphate to 6-phosphogluconolactone. After the reduction of F420 to the active protonated cofactor, F420-H2, Ddn catalyzes the reverse reaction to oxidize it back to F420. It has been hypothesized that Ddn orients PA-824 so that hydride transfer from F420 can occur and stabilize the transition state during this biochemical reaction (5). In F420 biosynthesis, the FbiC gene encodes a 7,8-didemethyl-8-hydroxy-5-deazariboflavin (FO) synthase, which transfers the hydroxybenzyl group from 4-hydroxy-phenylpyruvate to pyrimidinedione (12). FbiA and FbiB are subsequently involved in the addition of 2-phospho-l-lactate and polymerization of the penta-polyglutamate tail that generates the F420 cofactor (13). FbiA is a 2-phospho-l-lactate transferase responsible for the transfer of the lactyl phosphate moiety of lactyl-2-diphospho-5′-guanosine to FO and is known as CofD in Methanosarcina mazei (14). The biosynthetic contributions of FbiA, FbiB, and FbiC to PA-824 resistance were determined in studies of transposon mutagenesis mutants (13, 15). A susceptible phenotype could be reintroduced by complementation, confirming that F420 depletion is one of the mechanisms of resistance to this compound class (13). The crystal structures and functions of FbiA from M. mazei (14) and FbiB from Archaeoglobus fulgidus (16) were recently deciphered; the functions are depicted in Fig. 1.
FIG 1.

Cofactor F420 biosynthetic pathway. 6P-glucone, 6-phosphogluconolactone; Glu-6-P, glucose 6-phosphate; FO, 7,8-didemethyl-8-hydroxy-5-deazariboflavin (a 5-deazaflavin biosynthetic intermediate of F420); F420-0, FO with side chain phospholactyl; F420, F420-0 with n glutamate moieties (n = 5 or 6); F420-H2, reduced coenzyme F420; P-lactate, 2-phospho-l-lactate; MQ, menaquinone; MQ-H2, menaquinol (reduced menaquinone).
Stover et al. (1) selected spontaneous mutants in the presence of PA-824 and found that those strains could not carry out the nitro-reduction required for drug activation, averting bactericidal consequences. Mutant strains containing mutated FGD1, a critical component of F420 activation, conferred PA-824 resistance (1, 13, 15). Choi et al. (13) generated transposon mutants resistant to PA-824, and of those that displayed a negative phenotype for F420 production, insertions were identified in either fbiA or fbiB, for which complementation could restore production. Ddn, FGD1, and FbiA were reported to be nonessential to the optimal growth of M. tuberculosis in vitro, and there are no available data on the essentiality of FbiB and FbiC (17). While findings suggest that the mechanisms of resistance to PA-824 are not essential to proliferation when cells are grown in vitro under aerobic conditions, it is unclear whether they affect the organism while under oxidative stress. Looking forward toward potential implementation in the clinical setting, it is helpful to note that PA-824 exhibits no cross-resistance (18) with other antitubercular drugs, heightening optimism for its use in TB control of multidrug resistant bacilli.
In this study, we used M. tuberculosis strain H37Rv to perform a forward population genetics evaluation of PA-824 resistance. Our aims were to (i) collect spontaneously generated M. tuberculosis strains with a PA-824-resistant phenotype under aerobic conditions, (ii) characterize the genotypes of five genes associated with either PA-824 prodrug activation (ddn and fgd1) or the tangential F420 biosynthetic pathway (fbiA, fbiB, and fbiC), (iii) incorporate mutation and frequency findings into drug target binding models for Ddn and FGD1, and (iv) assess the relative degree of bacterial resistance in representative mutant strains.
MATERIALS AND METHODS
Antibiotics, bacterial strains, and selection of spontaneous mutants.
PA-824 was synthesized and its purity confirmed as previously described (3). M. tuberculosis strain H37Rv (ATCC 29294) was cultured in 7H9 (BD Difco Middlebrook 7H9 broth) liquid medium at 37°C to an optical density at 590 nm (OD590) of 0.6 (∼1 × 108 CFU) for the selection of mutants on agar plates containing either 1 or 5 μM (0.36 or 1.79 μg/ml, respectively) PA-824 under aerobic conditions. The starting inoculum was determined by serial dilution and plating on agar plates in triplicate. As an additional control, two inocula (108 and 109) were plated on 1 μg/ml rifampin yielding 1 to 10 colonies per plate, respectively. After incubation of the seeded plates for 4 weeks at 37°C, resistant colonies were selected and subcultured in 1 ml of 7H9 liquid medium containing an equivalent concentration of the drug used for selection for 12 days. Extracted DNA was used for PCR of the five putative resistance-determining regions, and their corresponding upstream region of 68 to 195 bp were sequenced and analyzed. Sanger sequencing was performed using BigDye Terminator version 3.1 cycle sequencing (1st Base Asia). Five independent selection experiments were performed. All five genes (ddn, fgd1, fbiA, fbiB, and fbiC) were sequenced for the first selection experiment only, comprising 91 samples. For the four subsequent selection experiments, samples were sequenced sequentially, starting with ddn, fgd1, fbiA, fbiB, and fbiC until a mutation was identified. For the four subsequent selection experiments, 107 and 108 bacilli were plated on five plates each at 1 and 5 μM (20 plates per experiment, for 100 plates total), and eight plates for each batch were used to pick three colonies per plate. The primers used for amplification and sequencing are found in Table S1 in the supplemental material.
Determination of MICs.
Transparent flat-bottomed 24-well plates (Nunc) were filled with 1 ml of 7H11 agar containing various drug concentrations of PA-824 (0, 0.5, 1.0, 5.0, and 10.0 μM) and prepared based on previously reported MICs and laboratory observations. A 100-μl culture of M. tuberculosis at an OD590 0.02 (∼1 ×106 CFU) was seeded and incubated at 37°C for 3 to 4 weeks. The MIC99 was assigned at the concentration at which no growth was observed.
RESULTS
Frequency of spontaneous mutations in PA-824.
Spontaneous PA-824-resistant mutants were selected throughout five independent biological experiments, with each experiment distinctly performed on a different date starting from an independent inoculum. The range in mutation rates was determined to be 10−5 to 10−7 CFU in the first selection experiment and found to vary according to the concentrations of PA-824. H37Rv cultures were adjusted to 105, 106, 107, 108, and 109 CFU and plated on 7H11 (BD Difco 7H11) agar plates containing either 1 or 5 μM PA-824 (eight plates each). Only 8 CFU were recovered from the eight plates of 105 CFU at 1 μM and none at 5 μM; consequently, the mutation rate was found to be 1 × 10−5 at 1 μM and undetectable at 5 μM for 1 × 105 CFU. CFU were recovered on plates containing 1 μM PA-824 that had been plated with 106 CFU and on plates containing 5 μM PA-824 at ≥107 CFU. No mutants were selected from plates containing 5 μM PA-824 when plating 106 bacilli (Table 1). In the four subsequent selection experiments, only 107 and 108 CFU were seeded on each plate, from which only three colonies per plate were randomly picked.
TABLE 1.
Frequency of mutations associated with PA-824 resistance at 1 and 5 μM
| PA-824 concn | No. of colonies/plate for: |
||
|---|---|---|---|
| 105 CFU plated | 106 CFU plated | 107 CFU plated | |
| 1 µM | 3, 2, 2, 1, 0, 0, 0, 0 | 28, 28, 25, 18, 17, 17, 14, 6 | >40a |
| Frequency | 1 × 10−5 | 1.9 × 10−5 | |
| 5 µM | None | None | 16, 8, 7, 5, 4, 4, 4, 3 |
| Frequency | 6.38 × 10−7 | ||
Exact number was not recorded.
Distribution of genetic polymorphisms, structural analysis, and homology modeling.
Out of 203 PA-824-resistant M. tuberculosis colonies that were selected and subcultured, 20 were eliminated from the study due to either failure of growth or reconfirmation of resistance. Of the 183 isolated strains that were subjected to target sequencing using PCR, lesions in ddn were most prevalent, accounting for 29% (n = 53), followed by 26% in fbiC (n = 47), 19% in fbiA (n = 35), 7% in fgd1 (n = 12) and 2% in fbiB (n = 4), and the remaining 17% (n = 32) harbored no mutations in the five genes examined (Fig. 2). Insertions and deletions accounted for 36 mutant isolates with lesions on ddn (n = 6), fgd1 (n = 3), fbiA (n = 13), fbiB (n = 1), and fbiC (n = 13), and 40 samples had substitutions leading to an early termination codon ddn (n = 34), fgd1 (n = 2), fbiA (n = 2), fbiB (n = 1), and fbiC (n = 1) (Fig. 2).
FIG 2.
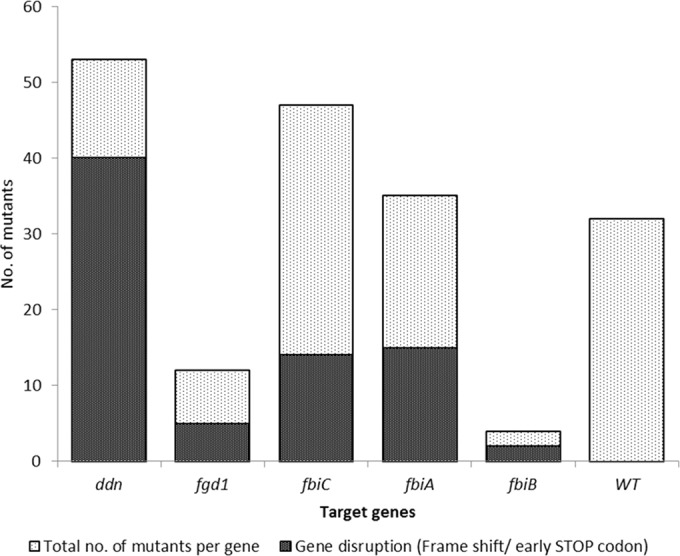
Distribution of mutation frequencies among the five target genes. The relative number of genes encoding early STOP codons and out-of-frame insertions and deletions is shown in gray.
Of the ddn mutants, 58% (31/53) had mutations leading to a 11Ser→STOP substitution, resulting in an early stop codon (Table 2). Eighteen other polymorphisms, including 15 distinct mutations were identified, with three clusters comprising three 133Tyr→Asp and two 88Trp→Arg mutations and two insertions of 55Arg. There were three single and one dinucleotide deletions and two insertions of one and seven amino acids. All insertions were in-frame, and all deletions were out-of-frame. The mutation of 22Ser→Leu mirrored previously reported single nucleotide polymorphisms (SNPs). 22Ser→Ala and 22Ser→Val correlated to strong decreased enzymatic activity in vitro (6). Modeling analysis based on existing crystal structures was completed for the Ddn protein (PDB code 3R5R) (5) (Fig. 3). The 86Pro→Leu and 88Trp→Arg mutations lost contact with F420 and lost a side chain H-bond with F420, respectively (see Fig. S1 in the supplemental material). The two insertions of one and seven amino acids were both found in a loop interacting with the polyglutamate tail of F420 (see Fig. S1) (5), and 133Tyr→Asp mutation localizes in the PA-824 putative binding site.
TABLE 2.
Summary of mutations identified in genes ddn and fgd1
| nt change by gene | aa change | Frequency | Concn used for selection (μM) | Comment(s) |
|---|---|---|---|---|
| Gene Rv3547/ddn | ||||
| 24ΔT | 8Phe→Phe | 1 | 5 | Single-nucleotide deletion→frameshift |
| 32C→A | 11Ser→STOP | 31 | 1/5 | Early termination codon |
| 65C→T | 22Ser→Leu | 1 | 1 | Decrease in enzyme activity (5) |
| 72/73ΔTA | 24Ile→Ile | 1 | 1 | Double-nucleotide deletion→frameshift |
| 73ΔA | 25-Asn→Ile | 1 | 1 | Single-nucleotide deletion→frameshift |
| 87C →A | 29Tyr→STOP | 1 | 5 | Early termination codon |
| 124C→T | 42Gln→STOP | 1 | 5 | Early termination codon |
| 143T→C | 48Leu→Pro | 1 | 5 | Single-nucleotide substitution |
| 163⌂CGC | 55⌂Arg | 2 | 1 | In-frame 1-aa insertion; loop interacting with polyglutamate tail of F420 |
| 163⌂21 bp | 55⌂7 aa | 1 | 5 | In-frame 7-aa insertion; loop interacting with polyglutamate tail of F420 |
| 232T→C | 78Ser→Pro | 1 | 1 | Single-nucleotide substitution |
| 242G→A | 81Gly→Asp | 1 | 1 | Flanking F420-interacting residue |
| 257C→T | 86Pro→Leu | 1 | 5 | Substitution loses contact with F420 cofactor |
| 262T→C | 88Trp→Arg | 2 | 1 | Loss of H-bond with F420 |
| 290ΔA | 97Lys→Arg | 1 | 5 | Single-nucleotide deletion→frameshift |
| 320T→C | 107Leu→Pro | 1 | 1 | Partially accessible β-strand flanking F420-binding site |
| 361G→A | 121Glu→Lys | 1 | 1 | Single-nucleotide substitution |
| 397T→G | 133Tyr→Asp | 3 | 5 | Localize in PA-824 putative binding site; substitutions of 133Tyr previously shown to abolish activity (5) |
| 409C→T | 137Gln→STOP | 1 | 5 | Nucleotide substitution→early termination codon |
| Total no. of ddn mutants | 53 | |||
| Gene Rv0407/fgd1 | ||||
| 128C→G | 43Pro→Arg | 2 | 5 | Putative glucose-6-phosphate-binding site (16) |
| 212G→A | 71Gly→Asp | 1 | 1 | Flanks F420-binding site |
| 317G→T | 106Gly→Val | 1 | 1 | Putative F420-binding site |
| 336C→A | 112Asn→Lys | 1 | 5 | Putative H-bond with F420 cofactor |
| 146ΔGCCATG | 129Gly→Ala, 130ΔH, 131ΔA | 1 | 5 | Out-of-frame 6-nt (2-aa) deletion |
| 428G→A | 143Trp→STOP | 1 | 1 | Nucleotide substitution→early termination codon |
| 429G→A | 143Trp→STOP | 1 | 5 | Nucleotide substitution→early termination codon |
| 498ΔC | 166Pro→Pro | 1 | 5 | Single-nucleotide deletion→frameshift |
| 506G→C | 169Gly→Ala | 1 | 5 | Loop distant from active site, function unknown |
| 678ΔA | 227Asp→Glu | 1 | 5 | Single-nucleotide insertion→frameshift |
| 688G→A | 230Glu→Lys | 1 | 1 | Putative glucose-6-phosphate-binding site (16) |
| Total no. of fgd1 mutants | 12 |
FIG 3.

Ribbon representations of Mycobacterium protein Ddn (PDB code 3R5R). Mutated residues identified are represented on the three-dimensional (3D) protein structures. The F420 is depicted with carbon atoms in yellow. The images were obtained using a consecutive combination of the MolScript (37) and Raster3D (38) programs.
The crystal structure of FGD1 cocrystallized with F420 was used to model the mutations encountered in the present study (PDB code 3B4Y) (Table 2 and Fig. 4A) (19). The 106Gly→Val mutation is located within the F420-binding site. The 112Asn→Lys mutation forms an H-bond with F420, and the 43Pro→Arg and 230Glu→Lys mutations are predicted to bind the substrate glucose-6-phosphate (19). Two of the mutants led to different SNPs on codon 143Trp, resulting in an early STOP codon. In addition, an out-of-frame six-nucleotide deletion responsible for a two-amino acid deletion (130His and 131Ala) and a mutation (129Gly→Ala) were noted.
FIG 4.

Ribbon representation of the crystal structure of Mycobacterium FGD1 (PDB code 3BY4) (A) Mutated residues identified are represented on the 3D protein structures. The F420 is depicted with carbon atoms in yellow. Phylogenetic amino acid substitutions reported by Feuerriegel et al. (35) are shown (B). These residues are found on the protein surface. The image was produced consecutively using the MolScript (37) and Raster3D (38) programs.
Twenty-eight different mutations in 35 independent mutants were identified in fbiA, including 13 frameshift mutations and two early stop codons (Fig. 5; see also Table S2 in the supplemental material). The crystal structure of CofD, an ortholog of FbiA from M. mazei, has been solved in complex with the FO moiety and GDP (PDB code 3C3E) (14). Sequence similarity between M. mazei CofD and M. tuberculosis FbiA proteins suggests that the 63Asp→Gly mutation could be involved in binding of the FO molecule, but this is not sufficient to predict the influence of mutation on FbiA function in the remaining 14 strains.
FIG 5.

Schematic representation of the mutations found in fbiABC.
Genetic alterations in fbiC accounted for 26% (n = 47) of the mutants (Fig. 5; see also Table S2 in the supplemental material). Distinct mutations were observed in 38 isolates, including those leading to one early stop codon and 13 out-of-frame insertions and deletions. The codon leads to the 372Pro→Ser, 387Asp→Tyr, 720Val→Ile mutations, and an insertion of 843C accounted for three, two, four, and four mutants, respectively. Since there is no available crystal structure for FbiC, we could not elucidate a predictable model for PA-824-resistant mutants.
Four mutations within fbiB were identified, including a 17-nucleotide insertion and one leading to a stop codon (Fig. 5; see also Table S2 in the supplemental material). The 361Pro→Ala mutation was located within the FbiB C-terminal domain (portion 245 to 448), whose function remains unknown, and the N-terminal domain encompassed the coenzyme F420-0:γ-glutamyl ligase enzyme. Based on comparative analysis with the structure of a homologous enzyme from A. fulgidus (16), the 153Gly→Val mutation likely localized within the putative active site of the enzyme. To our knowledge, this is the first data reported on mutations in fbiA and fbiB associated with PA-824, two genes associated with bicyclic nitroimidazole resistance. This finding can likely be attributed to the strategy of sequencing all the genes for 91/183 spontaneous mutants associated with the F420 biosynthetic pathway coupled with the large number of isolates analyzed.
The annotated reading frames of fbiA and fbiB overlap by four nucleotides at the C terminus of fbiA. It is possible that the predicted starting codon of fbiB is downstream. Alternatively, the start codon and an additional nucleotide of fbiB might be part of the last codon of fbiA and its termination. This phenomenon is not unusual in plants, whereby two overlapping reading frames can be cotranscribed in a bicistronic mRNA (18).
MIC determination.
The MIC99 was determined for 40 strains on 7H11 agar containing various concentrations of PA-824 (0, 0.5, 1.0, 5.0, and 10.0 μM). The strains used for MIC99 determination were randomly selected prior to knowledge of the sequencing results. Of those mutants exhibiting a high level of resistance, the greatest proportion (56% [n = 15]) led to the 11Ser→STOP mutation in ddn, a single-nucleotide deletion and frameshift at 25Asn in ddn, a single-nucleotide deletion and frameshift at 52Gly in fbiC, a 86Tyr→STOP mutation in fbiC, and a single-nucleotide deletion and resulting frameshift at 71Gly, 74Asp, and 81Gln in fbiA.
DISCUSSION
This study aimed to characterize the genetic polymorphisms of spontaneously generated PA-824-resistant mutant strains by examining drug metabolism genes for potential mutations. The frequency of mutations ranged from 1 × 10−5 to 10−7 in a concentration-dependent manner, which was higher than that previously reported (6.7 × 10−7 to 9.0 × 10−7) (1). Hurdle et al. (20) selected nitrofuranylamide-resistant M. tuberculosis mutants and reported a frequency of 10−5 to 10−7, which was more consistent with our findings and similar to that for isoniazid (21). Concentration dependency was reflected in the increased number of CFU found on plates containing the lowest drug concentration. Noteworthy is the dose of 50 mg/kg of body weight of PA-824 utilized in in vivo combination studies in mouse models, which corresponds to approximately 30 μM, or roughly six times the concentration used to select for mutants in this study (22–25). With the currently recommended 200-mg dose (26) of PA-824 and the corresponding concentration observed in vivo in humans (∼2 to 3 μM [27]), we predict that the mutation frequency will fluctuate around 10−6, although other environmental factors or host immunity may influence this estimate (28). This prediction underscores the critical need for proper drug combination and dosing to avoid the early emergence of drug resistance. The ongoing STAND phase III clinical trial is evaluating a combination of pyrazinamide, PA-824, and moxifloxacin, of which both pyrazinamide and PA-824 have a high frequency of emergent drug resistance, while moxifloxacin shares extensive cross-resistance with other fluoroquinolones (29, 30).
Adding to previous reports of mutations in the ddn, fgd1, and fbiC genes, we found an association between mutations in fbiA and fbiB and PA-824 resistance that supports the transposon mutagenesis work of Choi et al. (13, 15). In assessing the genetic polymorphisms in genes associated with PA-824 prodrug activation (ddn and fgd1) and the tangential F420 biosynthetic pathway (fbiA, fbiB, and fbiC), we hoped to gain a better understanding of the genetic underpinnings that determine the emergence of drug resistance. Within these five genes, we found a diverse set of changes in the majority of the PA-824-resistant mutant strains that were incorporated into a predicted crystal structure analysis of Ddn and FGD1. These structures indicate that identified mutations were specifically located within the protein catalytic domain that would hinder the activity of the enzymes required for prodrug activation.
The greatest diversity in SNP insertions and deletions was identified in fbiC and fbiA. Besides the original transposon mutagenesis reports by Choi et al. (13, 15), no SNPs have been reported in these two genes. The number of early truncations or frameshifts identified in all 5 examined drug target genes corroborate the notion that these enzymes are nonessential for growth under aerobic conditions (17); this finding is similar to those mutations associated with other prodrugs used to treat TB, including ethionamide, pyrazinamide, and para-aminosalicylic acid (29). In contrast, Gurumurthy et al. (6) found fbiC-deficient M. tuberculosis mutants to be hypersensitive to oxidative stress and more susceptible to antitubercular drugs, including isoniazid, moxifloxacin, and clofazimine.
Resistant mutants were organized into three groups: (i) those with complete protein disruption due to a frameshift or early termination, (ii) those with altered critical residues interacting with either the cofactor or substrate, and (iii) those with substitutions for which the function could not be predicted in this study.
Functional redundancy or functional analogs have often been observed in TB, as demonstrated for the 2 isocitrate lyases (31), the 2 thymidylate synthases (32, 33), and the RibD recently identified as a member of the dihydrofolate superfamily (34). The orthologs Rv1261c and Rv1558 may be functional analogs of Ddn (6), while other enzymes (Rv1155 and Rv2991) have been found to be structural analogs of FGD1 (3). In a pool of 65 PA-824-susceptible clinical isolates, Feuerriegel et al. (35) identified five unique phylogenetic or neutral SNPs in fgd1. Accordingly, the FGD1 model elucidated in this study shows that these phylogenetic SNPs are dispersed outside the F420-binding site and are not found in the M. tuberculosis H37Rv resistant mutants selected here (Fig. 4B). Detrimental mutations on the catalase-peroxidase (KatG) required for the activation of the prodrug isoniazid are extremely rare given the associated significant loss of bacterial fitness. In contrast, changes in pyrazinamidase (PncA) associated with pyrazinamide resistance can include complete disruption of the gene with no known associated fitness cost (29). In this study, no visible fitness cost was observed in vitro, as determined aerobically by growth kinetics (data not shown), which is reminiscent of mutants with changes in PncA. It is important to bear in mind that the mutants reported here were identified by analysis of randomly selected mutants aerobically in vitro and may not corroborate to the ratio or targets observed clinically, as previously observed in the cases of ethionamide (P. Bifani and A. Chua, unpublished data) and isoniazid (36).
The wide distribution and diversity of mutations might present a challenge for the development of a molecular diagnostic hybridization-based assay test, such as GeneXpert MTB/RIF or GenoType MTBDRplus. It is possible that the molecular profile for PA-824 resistance in clinical isolates will prove to be more restrictive due to unforeseen fitness costs in the patient; hence, a molecular diagnostic approach could be reconsidered.
The frequency of mutations in PA-824-resistant M. tuberculosis was found to be elevated when selecting at 1 μM and similar to that of isoniazid at 5 μM. Of the resistant strains examined, 83% had single mutations in one of the five genes associated with PA-824 drug metabolism, either directly (ddn and fgd1) or indirectly (fbiA, fbiB, and fbiC). The remaining 17% of the mutants had no identifiable lesions within these genes, and this suggests that other targets might be involved in either the activation pathway or the mechanism of action of the drug. Correlating mutation types and MIC99 is an important next step. The mutant frequency and consequent fitness have yet to be determined in clinical isolates, as nitroimidazoles have not yet been introduced into standard clinical practice. With this systematic analysis of PA-824-resistant M. tuberculosis mutants, we aimed to provide a reference to support the tools used to monitor TB drug resistance.
Supplementary Material
ACKNOWLEDGMENTS
H.L.H. is a fellow of the Joint Master of Science in Infectious Diseases, Vaccinology & Drug Discovery, National University of Singapore (NUS), the Novartis Institute for Tropical Diseases (NITD), the University of Basel (UB), and the Swiss Tropical and Public Health Institute (STPH). R.W. is a research associate at the National Fund for Scientific Research (FNRS-FRS) (Belgium). P.B. is an adjunct staff, Department of Microbiology and Immunology Program, Yong Loo Lin School of Medicine, NUS.
H.L.H., B.M., and P.B. developed the concept; A.C. and P.B. carried-out biosafety level 3 (BSL3) work, including spontaneous-resistant mutant selection; P.G. and H.L.H. sequenced and analyzed the spontaneous mutants; R.W. performed the protein modeling; R.W. H.L.H, A.S., S.B.L., and P.B. analyzed the results; and H.L.H., A.C., B.M., R.W., and P.B. wrote the paper.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.00308-15.
REFERENCES
- 1.Stover CK, Warrener P, VanDevanter DR, Sherman DR, Arain TM, Langhorne MH, Anderson SW, Towell JA, Yuan Y, McMurray DN, Kreiswirth BN, Barry CE, Baker WR. 2000. A small-molecule nitroimidazopyran drug candidate for the treatment of tuberculosis. Nature 405:962–966. doi: 10.1038/35016103. [DOI] [PubMed] [Google Scholar]
- 2.European Medicines Agency. 2013. European Medicines Agency recommends two new treatment options for tuberculosis. EMA/717915/2013. European Medicines Agency, London, United Kingdom: http://www.ema.europa.eu/ema/index.jsp?curl=pages/news_and_events/news/2013/11/news_detail_001972.jsp&mid=WC0b01ac058004d5c1. [Google Scholar]
- 3.Manjunatha UH, Boshoff H, Dowd CS, Zhang L, Albert TJ, Norton JE, Daniels L, Dick T, Pang SS, Barry CE III. 2006. Identification of a nitroimidazo-oxazine-specific protein involved in PA-824 resistance in Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 103:431–436. doi: 10.1073/pnas.0508392103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Singh R, Manjunatha U, Boshoff HI, Ha YH, Niyomrattanakit P, Ledwidge R, Dowd CS, Lee IY, Kim P, Zhang L, Kang S, Keller TH, Jiricek J, Barry CE III. 2008. PA-824 kills nonreplicating Mycobacterium tuberculosis by intracellular NO release. Science 322:1392–1395. doi: 10.1126/science.1164571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cellitti SE, Shaffer J, Jones DH, Mukherjee T, Gurumurthy M, Bursulaya B, Boshoff HI, Choi I, Nayyar A, Lee YS, Cherian J, Niyomrattanakit P, Dick T, Manjunatha UH, Barry CE III, Spraggon G, Geierstanger BH. 2012. Structure of Ddn, the deazaflavin-dependent nitroreductase from Mycobacterium tuberculosis involved in bioreductive activation of PA-824. Structure 20:101–112. doi: 10.1016/j.str.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gurumurthy M, Rao M, Mukherjee T, Rao SP, Boshoff HI, Dick T, Barry CE III, Manjunatha UH. 2013. A novel F(420)-dependent anti-oxidant mechanism protects Mycobacterium tuberculosis against oxidative stress and bactericidal agents. Mol Microbiol 87:744–755. doi: 10.1111/mmi.12127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Manjunatha U, Boshoff HI, Barry CE. 2009. The mechanism of action of PA-824: novel insights from transcriptional profiling. Commun Integr Biol 2:215–218. doi: 10.4161/cib.2.3.7926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cousins FB. 1960. The prosthetic group of a chromoprotin from mycobacteria. Biochim Biophys Acta 40:532–534. doi: 10.1016/0006-3002(60)91396-2. [DOI] [PubMed] [Google Scholar]
- 9.Jones WJ, Nagle DP Jr, Whitman WB. 1987. Methanogens and the diversity of archaebacteria. Microbiol Rev 51:135–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bleicher K, Winter J. 1991. Purification and properties of F420- and NADP(+)-dependent alcohol dehydrogenases of Methanogenium liminatans and Methanobacterium palustre, specific for secondary alcohols. Eur J Biochem 200:43–51. doi: 10.1111/j.1432-1033.1991.tb21046.x. [DOI] [PubMed] [Google Scholar]
- 11.Purwantini E, Daniels L. 1996. Purification of a novel coenzyme F420-dependent glucose-6-phosphate dehydrogenase from Mycobacterium smegmatis. J Bacteriol 178:2861–2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li H, Xu H, Graham DE, White RH. 2003. Glutathione synthetase homologs encode alpha-l-glutamate ligases for methanogenic coenzyme F420 and tetrahydrosarcinapterin biosyntheses. Proc Natl Acad Sci U S A 100:9785–9790. doi: 10.1073/pnas.1733391100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choi KP, Bair TB, Bae YM, Daniels L. 2001. Use of transposon Tn5367 mutagenesis and a nitroimidazopyran-based selection system to demonstrate a requirement for fbiA and fbiB in coenzyme F(420) biosynthesis by Mycobacterium bovis BCG. J Bacteriol 183:7058–7066. doi: 10.1128/JB.183.24.7058-7066.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Forouhar F, Abashidze M, Xu H, Grochowski LL, Seetharaman J, Hussain M, Kuzin A, Chen Y, Zhou W, Xiao R, Acton TB, Montelione GT, Galinier A, White RH, Tong L. 2008. Molecular insights into the biosynthesis of the F420 coenzyme. J Biol Chem 283:11832–11840. doi: 10.1074/jbc.M710352200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Choi KP, Kendrick N, Daniels L. 2002. Demonstration that fbiC is required by Mycobacterium bovis BCG for coenzyme F(420) and FO biosynthesis. J Bacteriol 184:2420–2428. doi: 10.1128/JB.184.9.2420-2428.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nocek B, Evdokimova E, Proudfoot M, Kudritska M, Grochowski LL, White RH, Savchenko A, Yakunin AF, Edwards A, Joachimiak A. 2007. Structure of an amide bond forming F(420):gamma-glutamyl ligase from Archaeoglobus fulgidus–a member of a new family of non-ribosomal peptide synthases. J Mol Biol 372:456–469. doi: 10.1016/j.jmb.2007.06.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Griffin JE, Gawronski JD, Dejesus MA, Ioerger TR, Akerley BJ, Sassetti CM. 2011. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog 7:e1002251. doi: 10.1371/journal.ppat.1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hiesel R, Brennicke A. 1985. Overlapping reading frames in Oenothera mitochondria. FEBS Lett 193:164–168. doi: 10.1016/0014-5793(85)80143-5. [DOI] [Google Scholar]
- 19.Bashiri G, Squire CJ, Moreland NJ, Baker EN. 2008. Crystal structures of F420-dependent glucose-6-phosphate dehydrogenase FGD1 involved in the activation of the anti-tuberculosis drug candidate PA-824 reveal the basis of coenzyme and substrate binding. J Biol Chem 283:17531–17541. doi: 10.1074/jbc.M801854200. [DOI] [PubMed] [Google Scholar]
- 20.Hurdle JG, Lee RB, Budha NR, Carson EI, Qi J, Scherman MS, Cho SH, McNeil MR, Lenaerts AJ, Franzblau SG, Meibohm B, Lee RE. 2008. A microbiological assessment of novel nitrofuranylamides as anti-tuberculosis agents. J Antimicrob Chemother 62:1037–1045. doi: 10.1093/jac/dkn307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson R, Streicher EM, Louw GE, Warren RM, van Helden PD, Victor TC. 2006. Drug resistance in Mycobacterium tuberculosis. Curr Issues Mol Biol 8:97–111. [PubMed] [Google Scholar]
- 22.Tasneen R, Li SY, Peloquin CA, Taylor D, Williams KN, Andries K, Mdluli KE, Nuermberger EL. 2011. Sterilizing activity of novel TMC207- and PA-824-containing regimens in a murine model of tuberculosis. Antimicrob Agents Chemother 55:5485–5492. doi: 10.1128/AAC.05293-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harper J, Skerry C, Davis SL, Tasneen R, Weir M, Kramnik I, Bishai WR, Pomper MG, Nuermberger EL, Jain SK. 2012. Mouse model of necrotic tuberculosis granulomas develops hypoxic lesions. J Infect Dis 205:595–602. doi: 10.1093/infdis/jir786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Williams K, Minkowski A, Amoabeng O, Peloquin CA, Taylor D, Andries K, Wallis RS, Mdluli KE, Nuermberger EL. 2012. Sterilizing activities of novel combinations lacking first- and second-line drugs in a murine model of tuberculosis. Antimicrob Agents Chemother 56:3114–3120. doi: 10.1128/AAC.00384-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lakshminarayana SB, Boshoff HI, Cherian J, Ravindran S, Goh A, Jiricek J, Nanjundappa M, Nayyar A, Gurumurthy M, Singh R, Dick T, Blasco F, Barry CE III, Ho PC, Majunatha UH. 2014. Pharmacokinetics-pharmacodynamics analysis of bicyclic 4-nitroimidazole analogs in a murine model of tuberculosis. PLoS One 9:e105222. doi: 10.1371/journal.pone.0105222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Diacon AH, Dawson R, Hanekom M, Narunsky K, Maritz SJ, Venter A, Donald PR, van Niekerk C, Whitney K, Rouse DJ, Laurenzi MW, Ginsberg AM, Spigelman MK. 2010. Early bactericidal activity and pharmacokinetics of PA-824 in smear-positive tuberculosis patients. Antimicrob Agents Chemother 54:3402–3407. doi: 10.1128/AAC.01354-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ginsberg AM, Laurenzi MW, Rouse DJ, Whitney KD, Spigelman MK. 2009. Safety, tolerability, and pharmacokinetics of PA-824 in healthy subjects. Antimicrob Agents Chemother 53:3720–3725. doi: 10.1128/AAC.00106-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McGrath M, Gey van Pittius NC, van Helden PD, Warren RM, Warner DF. 2014. Mutation rate and the emergence of drug resistance in Mycobacterium tuberculosis. J Antimicrob Chemother 69:292–302. doi: 10.1093/jac/dkt364. [DOI] [PubMed] [Google Scholar]
- 29.Stoffels K, Mathys V, Fauville-Dufaux M, Wintjens R, Bifani P. 2012. Systematic analysis of pyrazinamide-resistant spontaneous mutants and clinical isolates of Mycobacterium tuberculosis. Antimicrob Agents Chemother 56:5186–5193. doi: 10.1128/AAC.05385-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Diacon AH, Dawson R, von Groote-Bidlingmaier F, Symons G, Venter A, Donald PR, van Niekerk C, Everitt D, Winter H, Becker P, Mendel CM, Spigelman MK. 2012. 14-day bactericidal activity of PA-824, bedaquiline, pyrazinamide, and moxifloxacin combinations: a randomised trial. Lancet 380:986–993. doi: 10.1016/S0140-6736(12)61080-0. [DOI] [PubMed] [Google Scholar]
- 31.Muñoz-Elias EJ, McKinney JD. 2005. Mycobacterium tuberculosis isocitrate lyases 1 and 2 are jointly required for in vivo growth and virulence. Nat Med 11:638–644. doi: 10.1038/nm1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mathys V, Wintjens R, Lefevre P, Bertout J, Singhal A, Kiass M, Kurepina N, Wang XM, Mathema B, Baulard A, Kreiswirth BN, Bifani P. 2009. Molecular genetics of para-aminosalicylic acid resistance in clinical isolates and spontaneous mutants of Mycobacterium tuberculosis. Antimicrob Agents Chemother 53:2100–2109. doi: 10.1128/AAC.01197-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sampathkumar P, Turley S, Ulmer JE, Rhie HG, Sibley CH, Hol WG. 2005. Structure of the Mycobacterium tuberculosis flavin dependent thymidylate synthase (MtbThyX) at 2.0Å resolution. J Mol Biol 352:1091–1104. doi: 10.1016/j.jmb.2005.07.071. [DOI] [PubMed] [Google Scholar]
- 34.Zheng J, Rubin EJ, Bifani P, Mathys V, Lim V, Au M, Jang J, Nam J, Dick T, Walker JR, Pethe K, Camacho LR. 2013. para-Aminosalicylic acid is a prodrug targeting dihydrofolate reductase in Mycobacterium tuberculosis. J Biol Chem 288:23447–23456. doi: 10.1074/jbc.M113.475798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Feuerriegel S, Köser CU, Baù D, Rüsch-Gerdes S, Summers DK, Archer JA, Marti-Renom MA, Niemann S. 2011. Impact of Fgd1 and ddn diversity in Mycobacterium tuberculosis complex on in vitro susceptibility to PA-824. Antimicrob Agents Chemother 55:5718–5722. doi: 10.1128/AAC.05500-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bergval IL, Schuitema AR, Klatser PR, Anthony RM. 2009. Resistant mutants of Mycobacterium tuberculosis selected in vitro do not reflect the in vivo mechanism of isoniazid resistance. J Antimicrob Chemother 64:515–523. doi: 10.1093/jac/dkp237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Esnouf RM. 1999. Further additions to MolScript version 1.4, including reading and contouring of electron-density maps. Acta Crystallogr D Biol Crystallogr 55:938–940. doi: 10.1107/S0907444998017363. [DOI] [PubMed] [Google Scholar]
- 38.Merritt EA, Murphy ME. 1994. Raster3D version 2.0. A program for photorealistic molecular graphics. Acta Crystallogr D Biol Crystallogr 50:869–873. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.