

Significance
Here, we address the longstanding question to what extent protein fibrillation behavior as measured under simplified conditions in the test tube is transferrable to protein aggregation disease? Somewhat unexpectedly, we find that superoxide dismutase 1 (SOD1) fibrillation in vitro and growth of pathological SOD1 aggregates in transgenic ALS mice are mechanistically indistinguishable: Both processes reveal exponential kinetics and the typical characteristics of fragment-assisted growth. Although this precise agreement between in vitro and in vivo data opens new possibilities for quantitative examination of the molecular mechanism of neural damage at tissue level, it also moves the question about the medical relevance of our existing experimental tools one step farther: to what extent is protein aggregation in overexpressing mice transferable to late-onset human disease?
Keywords: superoxide dismutase 1, aggregation, transgenic mice, aggregation kinetics
Abstract
A longstanding challenge in studies of neurodegenerative disease has been that the pathologic protein aggregates in live tissue are not amenable to structural and kinetic analysis by conventional methods. The situation is put in focus by the current progress in demarcating protein aggregation in vitro, exposing new mechanistic details that are now calling for quantitative in vivo comparison. In this study, we bridge this gap by presenting a direct comparison of the aggregation kinetics of the ALS-associated protein superoxide dismutase 1 (SOD1) in vitro and in transgenic mice. The results based on tissue sampling by quantitative antibody assays show that the SOD1 fibrillation kinetics in vitro mirror with remarkable accuracy the spinal cord aggregate buildup and disease progression in transgenic mice. This similarity between in vitro and in vivo data suggests that, despite the complexity of live tissue, SOD1 aggregation follows robust and simplistic rules, providing new mechanistic insights into the ALS pathology and organism-level manifestation of protein aggregation phenomena in general.
So far, the difficulty to experimentally measure protein aggregation in live tissue has focused many researchers to infer mechanistic details of neurodegenerative disease from molecular studies in vitro. An important outcome of this in vitro development is the establishment of rational protocols for quantitative assessment of protein aggregation data (1–4), which now start to consolidate our view of what is happening (5). Protein aggregation follows general and simplistic rules dictated by the amino acid sequence. However, the sheer number of competing aggregation sites within a typical protein chain (6) makes the process intrinsically malleable and dependent on experimental conditions (7). The nagging concern is then to what extent these already complex in vitro data are transferable to the even more complex situation in vivo? Here, we shed light on this question by comparing directly in vitro aggregation kinetics with corresponding data from transgenic mice using a recently developed in vivo quantification strategy based on antibodies (8). Our model system is the aggregation of superoxide dismutase 1 (SOD1) associated with the motor neuron disease ALS (8) (Fig. 1). A key feature of this system is that the immature apoSOD1 monomer, which is also implicated as a precursor in human pathology (9–12), needs to be globally unfolded to fibrillate in vitro (7) (Fig. 1). This simplistic behavior presents the experimental advantage that the fibrillation kinetics of apoSOD1 show simple dependence on structural stability (13, 14):
| [1] |
where N is the soluble native structure, and D is the aggregation-competent unfolded state. Accordingly, it has been shown that the in vitro fibrillation of apoSOD1 displays the characteristic fingerprint of fragmentation-assisted growth (15) with a square root dependence on [D] (7), consistent with the requirement of sample agitation to expedite the reaction (1–4, 10). Analogous fibrillation behavior is found for β2-microglobulin (2), yeast prions Sup35 (16) and Ure2p (17), insulin (18), WW domain (19), TI 127 (20), and α-synuclein (21). The main difference between these proteins seems to be that some are intrinsically disordered and constantly aggregation-competent by lacking the ability to hide sticky sequence material by folding. In this study, we see that this simplistic in vitro behavior also translates to the more complex conditions in live tissue: the survival times of ALS mice expressing SOD1 variants of different stabilities are directly correlated with the in vivo levels of globally unfolded protein. Also, spinal cords of mice expressing the human SOD1 mutation G93A show exponential buildup of SOD1 aggregates with a square root dependence on log[D] indistinguishable from the fibrillation kinetics observed in agitated test tubes. The data raise fundamental questions about not only the striking resemblance between mouse and test tube aggregation but also, the apparent differences with human ALS pathology, which seems to have less ordered progression. Clues to the latter, however, are hinted in data from homozygous D90A mice showing two strains of structurally distinct SOD1 aggregates.
Fig. 1.

SOD1 aggregation in vitro and in ALS mice. (A) Aggregation of SOD in test tubes yields fibrillar structures similar to those of other proteins (7). (B) Immunohistochemistry of the ventral horn in the terminal hSOD1G93A mouse showing characteristics of aggresomes (44). (C) Competition between SOD1 folding and fibrillation in vitro, where elongation occurs by unfolded monomers through an encounter complex (7). The question that we ask is how do the in vitro and in vivo aggregations compare mechanistically. (D) Agitation-induced fibrillation in vitro with representative data from an SOD1 mutant in 0 (blue) and 5 M (red) urea with the associated statistics of τ1/2 for repeated measures. To account for this statistical variation, we use the distribution average (Table S1). (E) Log plot of νmax vs. τ1/2 for all individual measures in this study showing uniform behavior of the various SOD1 mutants and a slope of one characteristic for exponential growth (1–4). ALS-associated SOD1 mutations examined in ALS mice (red) (Table S1), other ALS-associated mutations (blue) (Table S1), and SOD1 control mutations (black) (Table S1).
Results
Variability and Statistics of in Vitro Analysis.
A general concern in studies of protein aggregation is that the data show high intrinsic variability and require good statistics to pin down (2, 21). The phenomenon is here illustrated by the aggregation of the SOD1 barrel (SOD1barrel) (22, 23) in pure buffer and 5 M urea (Fig. 1D). Despite a sizable separation of the average times for one-half depletion of the monomer pool (τ1/2) at 3.2 ± 1.0 and 23.0 ± 8.3 h, respectively, the distributions of individual measurements can still overlap. To maximize precision, we, therefore, accumulated experiments for each condition/mutant until statistically acceptable distributions appeared and quantified these by their averages. Unless otherwise stated, individual data points represent the average of 6–198 experiments accumulated over a period of 1–36 mo and typically with different protein batches. To reduce the risk of subjective bias, we included all measurements in the final analysis as well as the clear outliers. Validating the approach, the distribution SDs describe a linear increase with τ1/2 as predicted for stochastic processes (2, 7) (Fig. S1). As a final indicator of systematic behavior, the cumulative data show a 1:1 relation between τ1/2 and the elongation rate (νmax) (7) (Fig. 1E): this correlation signals not only exponential growth but is also the hallmark of kinetic control by secondary nucleation or fragmentation (1–4).
Effective Concentration of Aggregation-Competent Material.
Unlike disordered peptides, globular proteins, like SOD1, need to unfold to aggregate (7). Thus, at any given moment, only a fraction (f) of the protein molecules are aggregation-competent, whereas the rest (1 − f) remain folded and soluble (Fig. 1). Because f further varies with mutation and experimental conditions, this parameter needs to be accounted for in comparison with data. Based on the observation that apoSOD1 aggregates from unfolded monomers (7) (Eq. 1), the concentration of aggregation-competent molecules can be written
| [2] |
where ctotal is total [SOD1], and ΔGD−N is protein stability (Table S1). Accordingly, the concentration of aggregation-competent D can be varied in two ways: by changing (i) ΔGD−N (e.g., by mutation or urea titration) or (ii) ctotal (e.g., by adding more protein to the test tube or increasing expression levels in transgenic mice). However, there is more to it when it comes to the influence of point mutations on the aggregation kinetics. Mutations not only alter [D] (Eq. 2) but also, the intrinsic stickiness of D itself, because the aggregation propensity (p) of any peptide segment depends on amino acid composition (5, 6). To account for such p changes, we introduce here the correction factor pmutant/pwt, which can be measured directly from under standard conditions where the proteins are fully unfolded (i.e., at f = 1) (SI Text, Fig. S2, and Tables S1 and S2). Following the work by Knowles and coworkers (4), this treatment allows the use of a general expression for the aggregation rate constant () in the limit of secondary nucleation/fragmentation (SI Text):
| [3] |
where , [monomer] in the simplest case equals [D] (Eq. 2), and k+ and k− are the elongation and fragmentation rate constants, respectively. The exponent γ determines the [monomer] dependence of , where γ = −0.5 for completely fragmentation-based aggregation and γ = −1.5 for secondary nucleation-driven aggregation assuming no saturation effects (24). As shown below, the implementation of Eq. 3 provides a simple means for not only rationalizing mutational effects in vitro but also, delineating the kinetics of SOD1 aggregation in transgenic mice.
Aggregation of apoSOD1 Monomers in Vitro.
As a benchmark for interpreting in vivo aggregation data, we determined the effect of mutational perturbation on the SOD1 aggregation behavior under simplified conditions in vitro. To achieve as broad as possible a range of [D] = fctotal for defining the protein concentration dependence on (Eq. 3), we devised five sets of SOD1 mutations: (i) ALS-associated mutations of apoSOD1 WT (SOD1wt), (ii) ALS mutations of the apoSOD1 monomer C6A/C111A/F50E/G51E (SOD1pwt), (iii) non-ALS mutations in the SOD1pwt core, (iv) ALS mutations of the loop-truncated SOD1 barrel (SODbarrel), and (v) non-ALS mutations in the SODbarrel core (Fig. 2A). Mutants along with their characteristic parameters for τ1/2, νmax, and KD−N in the reduced apo state at pH 6.3 and 37 °C (Eqs. 1 and S5) are listed in Table S1, and controls of pH effects are in Fig. S3. Now, on plotting log vs. log[D] = logfctotal for these mutants (SI Text), we yield a linear relation with a slope of γin vitro = 0.49 ± 0.03 that spans a concentration range of three orders of magnitude (Fig. 2B). The result matches within error data from urea destabilization (7). Altogether, these observations show that the aggregation mechanism of SOD1 in vitro is robust and largely independent of how the protein is destabilized. ALS-associated mutations do not stand out as special but follow the same trend as any mutation, loop truncation, or even chemical denaturant: the aggregation kinetics depend primarily on [D] that can be varied in many different ways (Fig. 1C). The consistent slope of γin vitro = 0.49 suggests, moreover, that the in vitro fibrillation of unfolded SOD1 is assisted by fragmentation (Eq. 3), which is in full accord with data from other disordered proteins (3). A key feature of such fragmentation is exponential growth followed by monomer depletion (Fig. 1D), where the sigmoidal time courses contain no information about the initial nucleation process (1–4).
Fig. 2.

SOD1 mutations reveal unfolded monomers as aggregation precursors and predict lifetimes of ALS mice. (A) Distribution of analyzed mutations in the apoSOD1 monomer (SOD1pwt; Protein Data Bank ID code 2XJK) and the loop-truncated SOD1 barrel (SOD1barrel; Protein Data Bank ID code 4BCZ) (Tables S1 and S3). (B) Plot of log vs. [D] (Eqs. 2 and 3) for the mutations in Table S1 with γin vitro = 0.49 ± 0.03, consistent with fragmentation-assisted growth (colors are the same as in A). (C) Plot of mice survival times vs. normalized tissue [D], suggesting that apoSOD1 stability (Eq. 2) determines mouse disease (Table S3). The lifespans of G93A mice were around 124 d 10 y ago (25) (blue) and have currently increased to 155 d (red). This drift is a common phenomenon in ALS mouse lines and involves a 12% copy number loss from the transgene insertion (SI Text).
Mouse Survival Times Follow Tissue Levels of Unfolded SOD1 Monomers.
As an organism-level indicator of the in vivo aggregation mechanism, we used the survival times of our six ALS mouse models expressing hSOD1G93A, hSOD1G93A,50%, hSOD1G85R, hSOD1D90A, hSOD1127X, hSOD1127X,50%, and hSOD1wt (Table S3). To allow quantitative comparison with the in vitro data in Figs. 1 and 2, the steady-state concentration of soluble SOD1 molecules in spinal cord (ctotal) was measured from mRNA levels (25) (SI Text and Table S3). For all mutants, this measure X corresponds to the synthesis rate of nascent SOD1 monomers. Under the assumption that X is also proportional to the tissue level of apoSOD1 monomers (X ∝ ctotal), we can further estimate the mutant-induced changes of intracellular [D] from , where the fraction f is mutant-specific and measured from in vitro stability under reducing conditions at 37 °C (Eq. 2), and Xref is the reference mRNA level determined for hSOD1G93A mouse (Table S3). The result of this minimalist treatment is a linear correlation between mouse survival times and tissue [D] with R2 = 0.92 (Fig. 2C). In terms of in vivo mechanism, this scaling with intracellular concentration of unfolded SOD1 has two notable implications. First, it suggests that the in vitro and in vivo aggregation pathways are similar by relying on globally unfolded monomers for growth (Fig. 1C). Second, it suggests that the aggregation mechanism is uniform across the different mouse models (i.e., the process responds only to changes in [D] and is otherwise independent of mutant identity). Now, elucidation of the features of this mechanism requires the ability to follow directly the development of SOD1 aggregates in spinal cord tissue.
Quantification of Aggregate Levels in Spinal Cord.
An inherent challenge in molecular studies of neurodegenerative disease is that the amounts of pathologic aggregates in live tissue are too low for structural and kinetic analysis by conventional methods. To circumvent this problem, we use a newly developed method for quantifying the buildup of SOD1 aggregates in small tissue samples based on antibody mapping (8) (Fig. 3A). In essence, SOD1 aggregates in whole-spinal cord homogenates are captured on a cellulose–acetate filter in a dot-blot apparatus and washed free from any adhered protein components. Samples are then exposed to an array of antibodies recognizing individual segments along the SOD1 sequence in their disordered state, and binding is quantified similar to a Western immunoblot (SI Text). The results yield a binary fingerprint of the in vivo aggregate structure, where binding shows the SOD1 sequence regions protruding from the aggregate surface (i.e., flexible segments that are free to adapt to the antigen binding sites) and no binding shows the sequence regions that are either engaged in ordered aggregate structure or otherwise hidden (Fig. 3A). For quantification of in vivo aggregate growth, we choose antibodies 57–72, which give the strongest signal (Fig. 3A, SI Text, and Fig. S4).
Fig. 3.
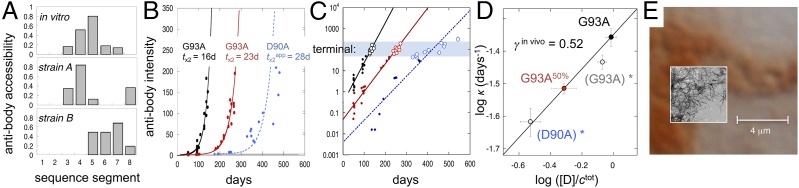
SOD1 aggregate structure and kinetics in ALS mice. (A) Structural fingerprints of in vivo and in vitro aggregates revealing shared fibrillar characteristics. Binding shows SOD1 sequence regions protruding disordered from the aggregate surface, and lack of binding indicates regions hidden in the aggregate core. Modified from ref. 8. (B) Single exponential growth of strain A aggregates in hSOD1G93A mice at two expression levels: 50% and 100% (SI Text). The strain A growth in hSOD1D90A mice occurs in parallel with strain B growth (SI Text) and displays the complexity of a second exponential phase. (C) Log plots of the same data showing the high dynamic range of the antibody detection and the common level of aggregates in terminal mice (blue shading). (D) Log plot of the aggregation rate constant (κ) vs. normalized tissue [D] (Eq. 4) for the hSOD1G93A and hSOD1G93A,50% mice yields a slope of γin vivo = 0.52 ± 0.05, indistinguishable from that in vitro (Fig. 2B). For comparison, we show also log κ from the hSOD1G93A mice (Fig. S5) with 12% expression loss (G93Adrifted)* and hSOD1D90A mice forced to single exponential fit in B (D90A)*; inclusion of these data points yields γin vivo = 0.45 ± 0.06 (R2 = 0.96). (E) Comparison of the dimensions of SOD1 in vivo inclusions and (Inset) in vitro fibrils (EM) illustrating the spatial restrictions that would follow from packing of SOD1 fibrils into cellular aggresomes.
In Vivo Aggregation Reveals Exponential Kinetics and Fragmentation-Assisted Growth.
To target the aggregation kinetics in spinal cord tissue, we used transgenic mice expressing the ALS mutation G93A at two different cellular concentrations: hSOD1G93A and hSOD1G93A,50% (25) (Table S3). The aggregate development in the two mouse lines was followed by (i) euthanizing mice at different time points along the disease progression, (ii) quantifying their spinal cord contents of SOD1 aggregates with the antibody assay (SI Text), and (iii) plotting this content (I) vs. age (Fig. 3 B and C). In hSOD1G93A mice, aggregates are first detected at ∼14 d, and then, they grow exponentially along the lifespan, with a doubling time of tx2 = 16 ± 1 d (Fig. 3 B and C). With the lower expression rate in hSOD1G93A,50%, tx2 increases to 23 ± 1 d (Fig. 3B). Thus, with respect to exponential kinetics, the in vitro and in vivo aggregation processes are indistinguishable. The different shape of raw data (i.e., sigmoidal vs. exponential) is simply caused by the in vitro time courses leveling off with monomer depletion (Fig. 1D), whereas the in vivo time courses remain exponential because of a constant supply on new monomers (Fig. 3B). This simplifying feature of the in vivo kinetics allows us now to take the comparison with in vitro data one step further. To examine how the in vivo aggregation depends on SOD1 concentration, we plotted κ = ln2/tx2 vs. expression level of [D] (SI Text). The result yields γin vivo = 0.52 ± 0.05 (Fig. 3D) within experimental error of γin vitro = 0.49 ± 0.03 observed under fragment-assisted growth (Fig. 2B). A clue to this mechanistic similarity is hinted at the level of aggregate structure. Despite the amorphous look of the SOD1 deposits in vivo, the purified and washed material reveals clear repetitive order when analyzed at sequence level with conformation-sensitive antibodies (Fig. 3A). The cellular aggregates show overall structural resemblance with the in vitro fibrils, albeit that the recruited strands are partly different (Fig. 3A). This similarity suggests that the hSOD1G93A inclusions are, at the molecular level, fibrillar, providing an explanation for in vitro-like, fragment-assisted growth. Notably, such fragmentation-assisted kinetics do not primarily depend on the amino acid identity of the fibrillar structure per se but on its susceptibility to break up in smaller fragments on mechanical deformation (i.e., the fragility stems from the crystalline cross-β packing of the fibrillar backbone rather than from its specific side-chain composition). In the same way the sequencewise disparate proteins SOD1, β2-microglobulin (2), yeast prions (16, 17), insulin (18), WW domain (19), TI 127 (20), and α-synuclein (21) still display similar fibrillation behavior in vitro: the exponential increase of growing fibrillar ends on steady fragmentation comes to dominate the kinetics (3). From Fig. 3, it is evident that this intrinsic kinetic efficiency of fragmentation-assisted growth also persists in live tissue (26).
Higher-Order Effects.
Principal component analysis of antibody assay data indicates that the hSOD1G93A mice contain only a single and structurally quite homogenous type of SOD1 aggregates (SI Text), which is referred to as strain A (8). To examine the more complex situation where two types of aggregate strains coexist, we analyzed the mouse model hSOD1D90A. In addition to the common strain A, these mice also contain the structurally distinct strain B. This strain recruits only the N-terminal one-half of the SOD1 sequence (Fig. 3A), is structurally more fragile than strain A (8), and dominates in the spinal cord of hSOD1D90A mice with rapid disease progression (8). For consistency, we used antibodies 57–72 targeting selectively strain A growth (SI Text). The result shows that the presence of strain B adds higher-order detail to strain A kinetics without altering the overall exponential nature of the aggregate buildup (Fig. 3). This deviation from monoexponential kinetics indicates that the growth of the strains A and B aggregates is, at some level, coupled in vivo, although the aggregates themselves show no indication of structural mixing (SI Text). Neglecting the deviation from monoexponential growth, strain A in hSOD1D90A yields an ad hoc doubling time of tx2 = 28 d that falls on the γin vivo = 0.52 line of hSOD1G93A and hSOD1G93A,50%. Another indication that hSOD1D90A mice retain an overall predictable disease progression is given by the survival time that follows the stability trend of pure strain A mice (Fig. 2C).
Aggregate Load in Terminal Mice.
With respect to neural damage, it is easy to envisage that an exponential growth of intracellular SOD1 deposits will eventually end up in a situation where the cells are no longer able to function normally, even if the aggregate structures per se are well-enclosed and molecularly benign. To examine this possibility, we compared aggregate levels in terminal animals across the different mouse models. The results show that the aggregate load at the time of death is similar for all mouse individuals, regardless of expression levels and SOD1 construct (Fig. 3C). Moreover, the span of survival times clearly decreases with increased κ, showing that individual variability is confined to a narrow interval of SOD1 aggregate levels (Fig. 3C). In measures of antibody staining (logIagg), this interval has an SD of ±0.4 on more than three orders of magnitude signal. Such consistency between aggregate level and death questions the need to invoke additional toxic species (27) in the mechanism of neural damage in ALS mice. Much simpler, the neural damage can be directly assigned to stress from critical load. From spatial considerations alone, such critical load of intracellular deposits must, after all, exist, and with exponential growth, it is bound to strike rather suddenly. Intriguingly, we also estimate that the aggregate levels in terminal mice are so high that the sheer amounts of monomers required for sustained exponential growth start to exceed the SOD1 production capacity of motor neurons (SI Text). This possible overrun suggests that glial cells and perhaps, other types of neurons gradually become involved in the aggregation, consistent with their role in modifying disease in the transgenic models (28). Regardless of what mechanistically causes cell death, the asymptomatic lag phase of ALS mice is not linked to delayed nucleation of SOD1 aggregation but arises instead from silent, exponential growth below the pathological threshold (Fig. 3B).
Discussion
Involvement of D.
Unlike other precursors of neurodegenerative disease (21, 29, 30), the SOD1 monomer is not at all times disordered but can hide its sticky sequence material by folding up into a well-ordered and perfectly soluble β-barrel structure (7) (Fig. 1C). This two-state behavior presents a key advantage when it comes to comparing the aggregation mechanism in vitro and in ALS mice: by gradually changing the SOD1 stability, we can gradually change the concentration of species along the folding pathway to identify which state causes the problem (Eq. 1). The result from such tuning shows that SOD1 fibrillation in vitro takes place from D (i.e., the globally unfolded state) (Fig. 2B). Conformationwise, the precursor of SOD1 fibrillation seems, thus, equivalent to those of intrinsically disordered proteins, like α-synuclein (21), polyQ (30), and the Aβ-peptide (29, 31). Changing by mutation the SOD1 stability in ALS mice yields similar results, manifested in an R2 = 0.92 correlation between survival times and tissue concentration of unfolded SOD1 (Fig. 2C). Altering instead [D] by lowering expression level increases survival times along the same correlation (Fig. 2C). Taken together, this close agreement between in vitro fibrillation and mouse pathology suggests that both processes are controlled by [D], where the accelerating effect of mutation is simply caused by an increase of this concentration through decreased protein stability (i.e., ) (Eq. 1 and Fig. 1C).
Aggregate Structure and Growth.
The second agreement between in vitro and in vivo data is found at the level of aggregate structure: although the pathological SOD1 inclusions appear amorphous by tissue staining (Fig. 1B), they reveal characteristic fibrillar structure at the molecular level (Fig. 3A). Also, the register and detailed core composition of these fibrils vary and give rise to different structural strains (8), consistent with disordered SOD1 having multiple sequence segments competing for intermolecular association (32, 33). Strain A fibrils in hSOD1G93A mice are different from those in vitro and clearly distinct from the conspicuous strain B fibrils in D90A mice (Fig. 3A). The third and perhaps, most intriguing similarity between in vitro and in vivo data: the SOD1 aggregation in ALS mice displays exponential growth with γin vivo = 0.52 ± 0.05 (Fig. 3D), consistent with fragmentation control (1–4). Although this precise translation of in vitro behavior to in vivo conditions may be surprising, it shows that the fibrillation process is robust and follows simple rules. As long as the extending fibrils will eventually break, the exponential multiplication of growing ends will come to dominate the kinetics (3). Growth seems, thus, controlled by the generic mechanical properties of the fibrils themselves rather than by their precise structural composition or environmental location. Fragmentation control is accordingly observed as a general feature on stirring, shaking, or sonication in vitro and for a variety of proteins (2, 16, 17, 19, 20) and peptides (18, 21, 29, 30) with little or no sequence similarities. The corresponding source of mechanical stress in vivo (26) could be vascular pounding, cytoskeleton dynamics, spatial restriction of linear growth, or even chaperones in the disaggregation machinery (34). Then, why does SOD1 fibrillation not show any indication of secondary nucleation (i.e., γ = −1.5) (Eq. 3), which was observed for the shorter Aβ-peptide in vitro (35)? The simple answer could be that the leftover parts of the SOD1 sequence that protrude disordered from the fibrillar surface effectively obstruct such sideways templating, leaving fragmentation as the main source of multiplication.
Exponential Overload.
The only striking difference between the in vivo and in vitro data is in the absolute rates: the aggregate doubling time in mice (weeks) is very much slower than in agitated test tubes (hours). From γin vivo = 0.52 ± 0.05, this ∼103-fold difference in growth rates would require an ∼106-fold difference in monomer concentration to account for the change in aggregation rate (Eq. 3). Considering that steady-state [SOD1] in ALS mice is well in the micromolar range (SI Text), out of which 1–2% is immature apo material (36, 37), this explanation on its own seems insufficient. Most likely, there are also other factors involved stemming from the intracellular environment and how the pathological inclusions are processed. It is reasonable to assume that an aggregate scavenging system with finite capacity cannot handle exponential growth after it has been outpaced. Thus, to maintain balance between clearance and growth (38), the deposits must at all times be kept below a critical threshold. For most proteins, this threshold might never be a problem, because the source of monomers is intrinsically scarce. For proteins with high steady-state levels, however, the situation can rapidly become critical as a result of mosaic copy number distribution (39), stochastic protein expression (40), or reduced capacity of the housekeeping system in aging cells (38). How could the cells stop this from happening? Given the kinetics in Eq. 3, there are two options: reduce expression [i.e., steady-state (D)] or decrease k+k− (i.e., the rates of fibril elongation and fragmentation). However, there is no known mechanism by which a cell can turn down synthesis of a specific protein, because it aggregates. The closest is endoplasmic reticulum (ER) stress and the unfolded protein response that represses synthesis more generally, which indeed seems at work in ALS (41). A drawback is here that unfolded protein response is only temporarily tolerated and can induce apoptosis (42). Also, reduced SOD1 expression comes with loss of native function. The better option could be to target directly elongation and fragmentation. It is, thus, interesting that the foremost apparent in vivo response to protein aggregation involves sequestration, packing, and coating (38, 43, 44), all of which are expected to modulate the fibrils’ growth dynamics.
Implications for Cellular Proteastasis.
Deposition and clearance of protein aggregates in the mammalian cytosol seem to be highly organized processes, acting as buffer to the proteosomal degradation system (38). If the concentration of a certain protein species cannot be controlled by the proteasome, it is sequestered into aggregates, targeted for autophagy (45), and degraded in the lysosome (43). When it comes to the actual handling and molecular environment of the aggregates, this process seems, in turn, to operate at two gears depending on load: either nascent aggregates are cleared directly in the cytosol or if this first line of defense becomes overwhelmed, they are captured in aggresomes for degradation by other more specialized autophagy pathways (43, 44). Although the whole-tissue kinetics in Fig. 3 do not resolve these different operational modes, an indication of their existence is provided by tissue staining, where some cells show overall diffuse cellular distribution SOD1 aggregates, whereas others show granules characteristic for aggresomes (Fig. 3E). Regardless of which pathway the individual fibrils will take, this in vivo processing most likely modulates their behavior by involving association with other cytosolic proteins (30, 46) and spatial restrictions in aggresomes (43) that violate the fibrillar persistence length of >1 μm (47) (Fig. 3E). However, all steps in this regulatory pathway are not necessarily evolved to suppress aggregate growth, because there is evidence that cytosolic chaperone systems, like Hsp110/Hsp70/Hsp40 (48), can also catalyze the sequestration of excess monomers by active fibril fragmentation as observed in yeast (34). On this basis, it is conceivable that stable proteostasis sustains as long as the clearance system maintains the aggregate levels below a safe threshold, where natural fluctuations in the aggregate levels [for example, as a result of stochastic proteome expression (40)] are efficiently buffered by transient overspill into aggresomes and induced autophagy pathways (43, 45) without long-term effects. If the load of monomers is, instead, kept persistently high, like in overexpressing ALS mice, the system is at risk to eventually become outpaced, setting free uncontrolled exponential growth of aggregates. At this point, the only remedy could be to cut the supply of new monomers as already proved to be efficient in G93A mice (49–51). From the data in Fig. 3, it is apparent that proteostasis collapse occurs after just a few weeks in hSOD1G93A mice, leaving steady exponential growth that continues into the terminal stage. We raise here the question if the still relatively slow kinetics of this growth (Table S3) reflect an underlying in vivo suppression of k+k− (Eq. 3) that helps aggregate processing under normal conditions, perhaps through coating or steric interference. It also remains to establish the kinetics of aggregate clearance and where in the aggregate turnover the exponential growth takes place: is it at the level of nascent aggregates, during handling and dynein-mediated transport, or in the aggresomes themselves? An interesting detail is that the total amount of SOD1 aggregates in spinal cord terminal ALS mice (Fig. 3C) corresponds to merely a week of steady-state production (SI Text), indicating an irreversible capture of a small fraction of the total monomer flux, high aggregate turnover, or a combination of both. Extrapolating these data from overexpressing mice to late-onset human pathology is not straightforward. A reductionist view, however, would be that analogous collapse of the human proteostasis occurs as a result of an abnormally age-declined or otherwise compromised housekeeping machinery, leaving SOD1wt as an inherently oversaturated protein (52) to fall through (sporadic or familial SOD1wt ALS). Alternatively, neurons expressing mutant SOD1 with elevated levels of immature apo species or decreased repulsive charge (7, 35) raise the aggregate load to a point where collapse is bound to occur, even with the normally aging housekeeping system (familial SOD1mutant ALS). If so, the SOD1 aggregate levels in the spinal cord should, in both cases, display a 40- to 60-y lag followed by a sudden exponential multiplication, leading to disease onset.
Perspective on Neural Damage and Disease Intervention.
The cause of neural damage in ALS is yet poorly understood, largely because of the inherent challenge of singling out a key decisive factor in the global cellular response to SOD1 aggregation. Qualified suggestions range from erroneous radical production (53), chaperone overload (54, 55), and aggregate sequestration of essential proteins (56) to toxic oligomers analogous to those implicated in other neurodegenerative diseases (37, 57). The results from this study add yet another possibility to put to experimental test: general aggregate overload. Exponential aggregate growth as observed in the ALS mice will inevitably come to a point where the cells can no longer cope with the mass load, although the inclusions themselves are molecularly benign. Although this perspective is admittedly imprecise about the mechanism of neural damage, it has the advantage of moving focus upstream: what triggers the chain of events leading to this criticality? Because innate housekeeping apparently succeeds to keep patients with highly aggressive SOD1 mutations symptom-free for decades (58), it is reasonable to assume that even a subtle help on the way could significantly delay onset. As inferred from the discussion above, a challenge could be that therapeutic intervention directed to, for example, reduce SOD1 expression (49–51) or otherwise aid aggregate turnover (57) needs to be implemented presymptomatically to yield optimal results: after exponential aggregate growth has commenced, any interference with expression or k+k− (Eq. 3) would be limited to square root response on the progression (Fig. 3D). Although the validity of these extrapolations has yet to be confirmed, the results presented in this study show that the field is now in position to put these mechanistic questions and related ones to strict experimental test in vivo. At a more fundamental level, the results also move the question about how simplistic in vitro aggregation behavior translates to in vivo conditions one step farther: how does the test tube behavior of overexpressing mice translate to human disease?
Materials and Methods
Mutagenesis, expression, and purification were the same as in refs. 14 and 22, and experiments were performed at 37 °C in 10 mM MES buffer at pH 6.3 unless otherwise stated. EM pictures were prepared as in ref. 7. Determination of the unfolded protein population was according to refs. 7, 14, and 59–62 and SI Text. Fibrillation was measured at 37 °C in standard 96-well plates in a FLUOstar Omega Microplate Reader (BMG Labtech,) with 3/32-in Teflon beads. All animal use and procedures were approved by the Umeå Regional Ethics Committee for Animal Research.
Supplementary Material
Acknowledgments
Support was from the Swedish Research Council, Hjärnfonden, The Knut and Alice Wallenberg Foundation, The Bertil Hållsten Foundation, and The Magnus Bergwall Foundation.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1503328112/-/DCSupplemental.
References
- 1.Ferrone F. Analysis of protein aggregation kinetics. Methods Enzymol. 1999;309:256–274. doi: 10.1016/s0076-6879(99)09019-9. [DOI] [PubMed] [Google Scholar]
- 2.Xue WF, Homans SW, Radford SE. Systematic analysis of nucleation-dependent polymerization reveals new insights into the mechanism of amyloid self-assembly. Proc Natl Acad Sci USA. 2008;105(26):8926–8931. doi: 10.1073/pnas.0711664105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Knowles TP, et al. An analytical solution to the kinetics of breakable filament assembly. Science. 2009;326(5959):1533–1537. doi: 10.1126/science.1178250. [DOI] [PubMed] [Google Scholar]
- 4.Cohen SI, et al. Nucleated polymerization with secondary pathways. I. Time evolution of the principal moments. J Chem Phys. 2011;135(6):065105. doi: 10.1063/1.3608916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buell AK, Dobson CM, Knowles TP. The physical chemistry of the amyloid phenomenon: Thermodynamics and kinetics of filamentous protein aggregation. Essays Biochem. 2014;56:11–39. doi: 10.1042/bse0560011. [DOI] [PubMed] [Google Scholar]
- 6.Maurer-Stroh S, et al. Exploring the sequence determinants of amyloid structure using position-specific scoring matrices. Nat Methods. 2010;7(3):237–242. doi: 10.1038/nmeth.1432. [DOI] [PubMed] [Google Scholar]
- 7.Lang L, Kurnik M, Danielsson J, Oliveberg M. Fibrillation precursor of superoxide dismutase 1 revealed by gradual tuning of the protein-folding equilibrium. Proc Natl Acad Sci USA. 2012;109(44):17868–17873. doi: 10.1073/pnas.1201795109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bergh J, et al. Structural and kinetic analysis of protein-aggregate strains in vivo using binary epitope mapping. Proc Natl Acad Sci USA. 2015;112(14):4489–4494. doi: 10.1073/pnas.1419228112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Subramaniam JR, et al. Mutant SOD1 causes motor neuron disease independent of copper chaperone-mediated copper loading. Nat Neurosci. 2002;5(4):301–307. doi: 10.1038/nn823. [DOI] [PubMed] [Google Scholar]
- 10.Banci L, et al. Metal-free superoxide dismutase forms soluble oligomers under physiological conditions: A possible general mechanism for familial ALS. Proc Natl Acad Sci USA. 2007;104(27):11263–11267. doi: 10.1073/pnas.0704307104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sheng Y, Chattopadhyay M, Whitelegge J, Valentine JS. SOD1 aggregation and ALS: Role of metallation states and disulfide status. Curr Top Med Chem. 2012;12(22):2560–2572. doi: 10.2174/1568026611212220010. [DOI] [PubMed] [Google Scholar]
- 12.Luchinat E, et al. In-cell NMR reveals potential precursor of toxic species from SOD1 fALS mutants. Nat Commun. 2014;5:5502. doi: 10.1038/ncomms6502. [DOI] [PubMed] [Google Scholar]
- 13.Jackson SE, Fersht AR. Folding of chymotrypsin inhibitor 2. 1. Evidence for a two-state transition. Biochemistry. 1991;30(43):10428–10435. doi: 10.1021/bi00107a010. [DOI] [PubMed] [Google Scholar]
- 14.Lindberg MJ, Normark J, Holmgren A, Oliveberg M. Folding of human superoxide dismutase: Disulfide reduction prevents dimerization and produces marginally stable monomers. Proc Natl Acad Sci USA. 2004;101(45):15893–15898. doi: 10.1073/pnas.0403979101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hwang YM, et al. Nonamyloid aggregates arising from mature copper/zinc superoxide dismutases resemble those observed in amyotrophic lateral sclerosis. J Biol Chem. 2010;285(53):41701–41711. doi: 10.1074/jbc.M110.113696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Collins SR, Douglass A, Vale RD, Weissman JS. Mechanism of prion propagation: Amyloid growth occurs by monomer addition. PLoS Biol. 2004;2(10):e321. doi: 10.1371/journal.pbio.0020321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhu L, Zhang XJ, Wang LY, Zhou JM, Perrett S. Relationship between stability of folding intermediates and amyloid formation for the yeast prion Ure2p: A quantitative analysis of the effects of pH and buffer system. J Mol Biol. 2003;328(1):235–254. doi: 10.1016/s0022-2836(03)00249-3. [DOI] [PubMed] [Google Scholar]
- 18.Foderà V, et al. Thioflavin T hydroxylation at basic pH and its effect on amyloid fibril detection. J Phys Chem B. 2008;112(47):15174–15181. doi: 10.1021/jp805560c. [DOI] [PubMed] [Google Scholar]
- 19.Ferguson N, et al. Rapid amyloid fiber formation from the fast-folding WW domain FBP28. Proc Natl Acad Sci USA. 2003;100(17):9814–9819. doi: 10.1073/pnas.1333907100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wright CF, Teichmann SA, Clarke J, Dobson CM. The importance of sequence diversity in the aggregation and evolution of proteins. Nature. 2005;438(7069):878–881. doi: 10.1038/nature04195. [DOI] [PubMed] [Google Scholar]
- 21.Buell AK, et al. Solution conditions determine the relative importance of nucleation and growth processes in α-synuclein aggregation. Proc Natl Acad Sci USA. 2014;111(21):7671–7676. doi: 10.1073/pnas.1315346111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Danielsson J, Kurnik M, Lang L, Oliveberg M. Cutting off functional loops from homodimeric enzyme superoxide dismutase 1 (SOD1) leaves monomeric β-barrels. J Biol Chem. 2011;286(38):33070–33083. doi: 10.1074/jbc.M111.251223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Danielsson J, et al. Global structural motions from the strain of a single hydrogen bond. Proc Natl Acad Sci USA. 2013;110(10):3829–3834. doi: 10.1073/pnas.1217306110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meisl G, et al. Differences in nucleation behavior underlie the contrasting aggregation kinetics of the Aβ40 and Aβ42 peptides. Proc Natl Acad Sci USA. 2014;111(26):9384–9389. doi: 10.1073/pnas.1401564111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jonsson PA, et al. Disulphide-reduced superoxide dismutase-1 in CNS of transgenic amyotrophic lateral sclerosis models. Brain. 2006;129(Pt 2):451–464. doi: 10.1093/brain/awh704. [DOI] [PubMed] [Google Scholar]
- 26.Xue WF, Hellewell AL, Hewitt EW, Radford SE. Fibril fragmentation in amyloid assembly and cytotoxicity: When size matters. Prion. 2010;4(1):20–25. doi: 10.4161/pri.4.1.11378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baglioni S, et al. Prefibrillar amyloid aggregates could be generic toxins in higher organisms. J Neurosci. 2006;26(31):8160–8167. doi: 10.1523/JNEUROSCI.4809-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ilieva H, Polymenidou M, Cleveland DW. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J Cell Biol. 2009;187(6):761–772. doi: 10.1083/jcb.200908164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cohen SI, et al. Proliferation of amyloid-β42 aggregates occurs through a secondary nucleation mechanism. Proc Natl Acad Sci USA. 2013;110(24):9758–9763. doi: 10.1073/pnas.1218402110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Park SH, et al. PolyQ proteins interfere with nuclear degradation of cytosolic proteins by sequestering the Sis1p chaperone. Cell. 2013;154(1):134–145. doi: 10.1016/j.cell.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 31.Danielsson J, Jarvet J, Damberg P, Gräslund A. The Alzheimer beta-peptide shows temperature-dependent transitions between left-handed 3-helix, beta-strand and random coil secondary structures. FEBS J. 2005;272(15):3938–3949. doi: 10.1111/j.1742-4658.2005.04812.x. [DOI] [PubMed] [Google Scholar]
- 32.Furukawa Y, Kaneko K, Yamanaka K, Nukina N. Mutation-dependent polymorphism of Cu,Zn-superoxide dismutase aggregates in the familial form of amyotrophic lateral sclerosis. J Biol Chem. 2010;285(29):22221–22231. doi: 10.1074/jbc.M110.113597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ivanova MI, et al. Aggregation-triggering segments of SOD1 fibril formation support a common pathway for familial and sporadic ALS. Proc Natl Acad Sci USA. 2014;111(1):197–201. doi: 10.1073/pnas.1320786110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shorter J, Lindquist S. Hsp104 catalyzes formation and elimination of self-replicating Sup35 prion conformers. Science. 2004;304(5678):1793–1797. doi: 10.1126/science.1098007. [DOI] [PubMed] [Google Scholar]
- 35.Sandelin E, Nordlund A, Andersen PM, Marklund SS, Oliveberg M. Amyotrophic lateral sclerosis-associated copper/zinc superoxide dismutase mutations preferentially reduce the repulsive charge of the proteins. J Biol Chem. 2007;282(29):21230–21236. doi: 10.1074/jbc.M700765200. [DOI] [PubMed] [Google Scholar]
- 36.Rhoads TW, et al. Using theoretical protein isotopic distributions to parse small-mass-difference post-translational modifications via mass spectrometry. J Am Soc Mass Spectrom. 2013;24(1):115–124. doi: 10.1007/s13361-012-0500-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zetterström P, et al. Soluble misfolded subfractions of mutant superoxide dismutase-1s are enriched in spinal cords throughout life in murine ALS models. Proc Natl Acad Sci USA. 2007;104(35):14157–14162. doi: 10.1073/pnas.0700477104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tyedmers J, Mogk A, Bukau B. Cellular strategies for controlling protein aggregation. Nat Rev Mol Cell Biol. 2010;11(11):777–788. doi: 10.1038/nrm2993. [DOI] [PubMed] [Google Scholar]
- 39.McConnell MJ, et al. Mosaic copy number variation in human neurons. Science. 2013;342(6158):632–637. doi: 10.1126/science.1243472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Junker JP, van Oudenaarden A. Every cell is special: Genome-wide studies add a new dimension to single-cell biology. Cell. 2014;157(1):8–11. doi: 10.1016/j.cell.2014.02.010. [DOI] [PubMed] [Google Scholar]
- 41.Atkin JD, et al. Endoplasmic reticulum stress and induction of the unfolded protein response in human sporadic amyotrophic lateral sclerosis. Neurobiol Dis. 2008;30(3):400–407. doi: 10.1016/j.nbd.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 42.Hetz C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13(2):89–102. doi: 10.1038/nrm3270. [DOI] [PubMed] [Google Scholar]
- 43.Chin LS, Olzmann JA, Li L. Parkin-mediated ubiquitin signalling in aggresome formation and autophagy. Biochem Soc Trans. 2010;38(Pt 1):144–149. doi: 10.1042/BST0380144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang H, Ying Z, Wang G. Ataxin-3 regulates aggresome formation of copper-zinc superoxide dismutase (SOD1) by editing K63-linked polyubiquitin chains. J Biol Chem. 2012;287(34):28576–28585. doi: 10.1074/jbc.M111.299990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bandyopadhyay U, Nagy M, Fenton WA, Horwich AL. Absence of lipofuscin in motor neurons of SOD1-linked ALS mice. Proc Natl Acad Sci USA. 2014;111(30):11055–11060. doi: 10.1073/pnas.1409314111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Olzscha H, et al. Amyloid-like aggregates sequester numerous metastable proteins with essential cellular functions. Cell. 2011;144(1):67–78. doi: 10.1016/j.cell.2010.11.050. [DOI] [PubMed] [Google Scholar]
- 47.Smith JF, Knowles TP, Dobson CM, Macphee CE, Welland ME. Characterization of the nanoscale properties of individual amyloid fibrils. Proc Natl Acad Sci USA. 2006;103(43):15806–15811. doi: 10.1073/pnas.0604035103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shorter J. The mammalian disaggregase machinery: Hsp110 synergizes with Hsp70 and Hsp40 to catalyze protein disaggregation and reactivation in a cell-free system. PLoS One. 2011;6(10):e26319. doi: 10.1371/journal.pone.0026319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ralph GS, et al. Silencing mutant SOD1 using RNAi protects against neurodegeneration and extends survival in an ALS model. Nat Med. 2005;11(4):429–433. doi: 10.1038/nm1205. [DOI] [PubMed] [Google Scholar]
- 50.Raoul C, et al. Lentiviral-mediated silencing of SOD1 through RNA interference retards disease onset and progression in a mouse model of ALS. Nat Med. 2005;11(4):423–428. doi: 10.1038/nm1207. [DOI] [PubMed] [Google Scholar]
- 51.Saito Y, et al. Transgenic small interfering RNA halts amyotrophic lateral sclerosis in a mouse model. J Biol Chem. 2005;280(52):42826–42830. doi: 10.1074/jbc.M507685200. [DOI] [PubMed] [Google Scholar]
- 52.Ciryam P, Tartaglia GG, Morimoto RI, Dobson CM, Vendruscolo M. Widespread aggregation and neurodegenerative diseases are associated with supersaturated proteins. Cell Reports. 2013;5(3):781–790. doi: 10.1016/j.celrep.2013.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Beckman JS, Carson M, Smith CD, Koppenol WH. ALS, SOD and peroxynitrite. Nature. 1993;364(6438):584. doi: 10.1038/364584a0. [DOI] [PubMed] [Google Scholar]
- 54.Okado-Matsumoto A, Fridovich I. Amyotrophic lateral sclerosis: A proposed mechanism. Proc Natl Acad Sci USA. 2002;99(13):9010–9014. doi: 10.1073/pnas.132260399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shinder GA, Lacourse MC, Minotti S, Durham HD. Mutant Cu/Zn-superoxide dismutase proteins have altered solubility and interact with heat shock/stress proteins in models of amyotrophic lateral sclerosis. J Biol Chem. 2001;276(16):12791–12796. doi: 10.1074/jbc.M010759200. [DOI] [PubMed] [Google Scholar]
- 56.Yu A, et al. Protein aggregation can inhibit clathrin-mediated endocytosis by chaperone competition. Proc Natl Acad Sci USA. 2014;111(15):E1481–E1490. doi: 10.1073/pnas.1321811111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rotunno MS, Bosco DA. An emerging role for misfolded wild-type SOD1 in sporadic ALS pathogenesis. Front Cell Neurosci. 2013;7:253. doi: 10.3389/fncel.2013.00253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Turner MR, Swash M. The expanding syndrome of amyotrophic lateral sclerosis: A clinical and molecular odyssey. J Neurol Neurosurg Psychiatry. 2015;86(6):667–673. doi: 10.1136/jnnp-2014-308946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fersht AR. Optimization of rates of protein folding: The nucleation-condensation mechanism and its implications. Proc Natl Acad Sci USA. 1995;92(24):10869–10873. doi: 10.1073/pnas.92.24.10869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nordlund A, Oliveberg M. Folding of Cu/Zn superoxide dismutase suggests structural hotspots for gain of neurotoxic function in ALS: Parallels to precursors in amyloid disease. Proc Natl Acad Sci USA. 2006;103(27):10218–10223. doi: 10.1073/pnas.0601696103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Svensson AK, Bilsel O, Kondrashkina E, Zitzewitz JA, Matthews CR. Mapping the folding free energy surface for metal-free human Cu,Zn superoxide dismutase. J Mol Biol. 2006;364(5):1084–1102. doi: 10.1016/j.jmb.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 62.Leinartaite L, Saraboji K, Nordlund A, Logan DT, Oliveberg M. Folding catalysis by transient coordination of Zn2+ to the Cu ligands of the ALS-associated enzyme Cu/Zn superoxide dismutase 1. J Am Chem Soc. 2010;132(38):13495–13504. doi: 10.1021/ja1057136. [DOI] [PubMed] [Google Scholar]
- 63.Price JC, Guan S, Burlingame A, Prusiner SB, Ghaemmaghami S. Analysis of proteome dynamics in the mouse brain. Proc Natl Acad Sci USA. 2010;107(32):14508–14513. doi: 10.1073/pnas.1006551107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Burke R, Marks W. Computational Neuroanatomy. Humana Press; Clifton, NJ: 2002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.