

Abstract
Helicases have major roles in genome maintenance by unwinding structured nucleic acids. Their prominence is marked by various cancers and genetic disorders that are linked to helicase defects. Although considerable effort has been made to understand the functions of DNA helicases that are important for genomic stability and cellular homeostasis, the complexity of the DNA damage response leaves us with unanswered questions regarding how helicase-dependent DNA repair pathways are regulated and coordinated with cell cycle checkpoints. Further studies may open the door to targeting helicases in order to improve cancer treatments based on DNA-damaging chemotherapy or radiation.
Helicases are classically defined as molecular motors that are able to couple nucleoside triphosphate (NTP) hydrolysis (typically of ATP) to the unwinding of polynucleic acids1 (FIG. 1). In doing so, the helicase translocates in a directionally specific manner (3′ to 5′ or 5′ to 3′) along the strand it predominantly interacts with. DNA helicases are classified according to their amino acid sequence homology in the ATPase/helicase core domain into two larger superfamilies, superfamily 1 (SF1) and SF2, and four smaller superfamilies1. Although DNA helicases are conventionally known to unwind B-form duplex DNA, some can unwind alternative DNA structures or have specialized functions (FIG. 1). There are an estimated 95 helicases or putative helicases (31 DNA helicases and 64 RNA helicases) encoded by the human genome2. Helicases are ubiquitous in nature and their functions depend on various factors including cell lineage, environmental stress, cell cycle stage and genetic background. Since the discovery of the first DNA helicase in 1976 (REF. 3), researchers have characterized ATP-dependent DNA-unwinding enzymes from all kingdoms of life, and from bacteriophages and eukaryotic viruses, thus providing an immense wealth of information about their mechanistic roles.
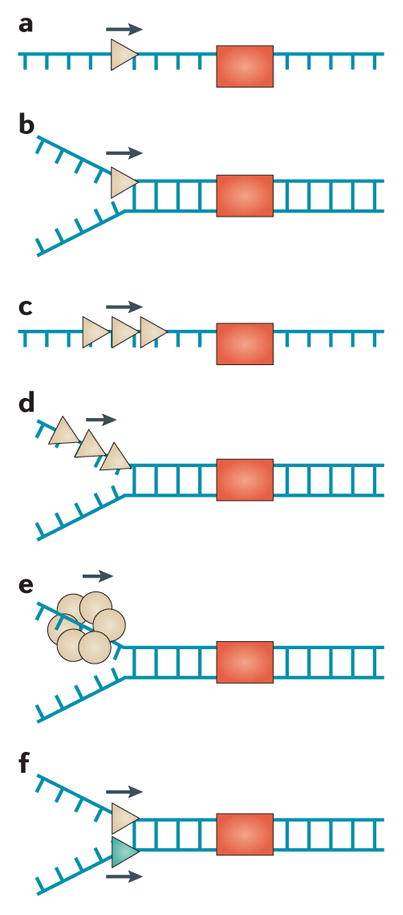
Figure 1. Molecular functions of DNA helicases.

DNA helicases (beige triangles) catalytically disrupt base pairs between complementary strands in an ATP-dependent manner (a), and may have specialized functions. For example, Fanconi anaemia group J protein (FANCJ), the Werner syndrome helicase (WRN), the Bloom syndrome helicase (BLM), and PIF1 disrupt G-quadruplex (G4) DNA structures (b). RECQL5 and FANCJ strip off proteins (for example, RAD51) that are bound to DNA (c). Some helicases (for example, RECQL1, RECQL4, RECQL5, WRN and BLM) carry out strand annealing by promoting base pairing212 (d). Strand annealing directionality by a DNA helicase has not been demonstrated. ATP inhibits strand annealing and promotes duplex unwinding by inducing a conformational change in the helicase protein (for example, RECQL1 (REF. 213)). Some helicases (for example, BLM and regulator of telomere elongation helicase 1 (RTEL1)) suppress homologous recombination (HR)-mediated repair by unwinding displacement loop (D-loop) intermediates (e). Branch-migration of three- or four-stranded joint DNA molecules by a DNA helicase (for example, BLM, WRN or RECQL1) (f) can suppress or promote the formation of Holliday Junction (HJ) structures that can be resolved by specialized endonucleases to create crossover products that are responsible for loss of heterozygosity and cancer predisposition214. The BLM helicase, together with topoisomerase 3α (TOP3α) and RecQ-mediated genome instability 1 (RMI1) and RMI2, dissolves double HJ structures (g) during HR or at converging replication forks to generate non-crossover DNA molecules215. See the main text for details.
Chemical damage to DNA can perturb cellular replication and transcription, and is implicated in mutagenesis, cell lethality, carcinogenesis, ageing and neurological disorders. Helicase-dependent DNA repair systems and DNA damage tolerance mechanisms exist to preserve the informational content and integrity of the genome and to permit timely and efficient replication. Advances in understanding mechanistic and structural aspects of helicase function (BOX 1) suggest new avenues of research for helicase-targeted drugs to combat cancer and other diseases. The importance of DNA helicases in virtually all aspects of nucleic acid metabolism cannot be overestimated and places them at the forefront of biomedical research into genetic disorders, ageing and cancer biology. An important concept that governs all of helicase biology is the crucial role of protein–protein interactions in helicase function to preserve genomic stability, and understanding these interactions is a central area for future research.
Box 1. Advances in understanding helicase mechanisms.
In certain cases, helicase monomers efficiently translocate along single-stranded DNA (ssDNA) and/or unwind double-stranded DNA (dsDNA) (see the figure, parts a and b), whereas multimerization or functional cooperation between monomers promotes dsDNA unwinding or the displacement of proteins bound to DNA, represented by red boxes196 (see the figure, parts c and d). The prototypical Escherichia coli RecQ helicase operates by an inchworm mechanism involving ATP-driven movements of two DNA-binding domains remaining in the same relative orientation along the DNA lattice197,198. By contrast, the replicative bacterial (DnaB) or eukaryotic (minichromosome maintenance (MCM)) helicase forms a hexameric ring-like structure that unwinds dsDNA by steric exclusion199 (see the figure, part e). Coordinate action of helicases with opposite polarities provides a unique dual-motor mechanism89,200 (see the figure, part f). A rolling model for unwinding by a helicase homodimer has also been proposed201. The efficiency of DNA translocation versus unwinding may be affected by mutations that uncouple catalytic activities202,203 or perturb oligomerization204. Certain helicases use their motor ATPase to disrupt protein DNA interactions to enable smooth replication progression or to regulate homologous recombination (HR) by stripping RAD51 from DNA205.
DNA helicases may act by repetitive movements on DNA206,207. The Bloom syndrome helicase (BLM) was shown to unwind a single DNA duplex molecule, re-anneal it by strand switching, and re-initiate unwinding in successive cycles in a replication protein A (RPA)-dependent manner208. Helicase repetitive action may be important for remodelling stalled replication forks, displacing DNA-bound proteins, or preventing illegitimate recombination. This mechanism of action is quite distinct from a hexameric ring-shaped helicase that potentially regulates DNA unwinding during replication by ATP-induced slippage209. It remains to be shown if strand switching, repetitive unwinding, or slippage by a helicase occur in vivo.
Structural analyses of DNA helicases and their accessory domains (for example, human RECQ1 (REF. 210) and Thermoplasma acidophilum XPD211) have provided molecular insights to their unwinding mechanisms and disease pathology. Analysis of disease-causing missense mutations in the context of domain structure has helped to explain the molecular basis for helicase dysfunction in DNA repair.
DNA helicases in genomic stability and cancer
Helicase-dependent mechanisms help cells to cope with endogenous or exogenous stress to prevent chromosomal instability and to maintain cellular homeostasis. Numerous genetic diseases that confer a predisposition to cancer are linked to mutations in genes encoding DNA-helicase-like proteins4 (TABLE 1). The expression of many DNA helicases is upregulated in transformed or neoplastic cells and tissues5–8 and is required for cancer cell proliferation or resistance to DNA damage imposed by chemotherapy (TABLE 2). Continuous upregulation of helicase gene expression in cancer reflects the need for an elevated DNA damage response to deal with replicative lesions that arise in highly proliferative states. Conversely, chromosomal instability from loss of helicase functions in hereditary helicase disorders promotes carcinogenesis, thus implicating DNA helicases as a prominent class of genome caretakers.
Table 1.
DNA helicase gene defects linked to cancer
| Gene | Disease | Cancer type | Genome metabolic pathway | Biochemical function |
|---|---|---|---|---|
| WRN | Werner syndrome | Thyroid neoplasms, melanomas, meningiomas, sarcomas, haematological and lymphoid neoplasms, and osteosarcomas | DSB repair and replication stress response | 3′–5′ helicase, 3′–5′ exonuclease, HJ branch migration, G4 resolution, replication fork regression and strand annealing |
| BLM | Bloom syndrome | Adult epithelial tumours, leukaemias, lymphomas and rare paediatric tumours | DSB repair and repair of replication-associated DNA damage | 3′–5′ helicase, HJ branch migration, G4 resolution, replication fork regression and strand annealing |
| RECQL4 | RTS, BGS and RAPADILINO syndrome | Osteogenic sarcomas, and lymphomas | Replication, mitochondrial genome stability and repair of endogenous base damage | 3′–5′ helicase and strand annealing |
| RECQL1 | Pancreatic cancer (polymorphisms) | Replication and oxidative DNA damage response | 3′–5′ helicase, HJ branch migration and strand annealing | |
| FANCJ | FA | Acute myeloid leukaemia and breast cancer (heterozygotes) | DSB repair and ICL repair | 5′–3′ helicase and G4 resolution |
| XPD | Xeroderma pigmentosum, xeroderma pigmentosum with Cockayne syndrome, TTD and COFS | Skin cancer | NER and transcription | 5′–3′ helicase |
| XPB | Xeroderma pigmentosum, xeroderma pigmentosum with Cockayne syndrome, and TTD | Skin cancer | NER and transcription | 3′–5′ helicase |
| RTEL1 | Dyskeratosis congenita | Adult glioma (polymorphisms) | Telomere maintenance and homologous recombination | 5′–3′ helicase, and disassembly of D-loops and T-loops |
| PIF1 | Breast cancer predisposition | Replication fork progression, telomere maintenance and mitochondrial DNA metabolism | 5′–3′ helicase and G4 resolution |
BGS, Baller–Gerold syndrome; COFS, cerebro-oculo-facial-skeletal syndrome; D-loop, displacement loop; DSB, double-strand break; FA, Fanconi anaemia; G4, G-quadruplex; HJ, Holliday junction; ICL, interstrand crosslink; NER, nucleotide-excision repair; RAPADILINO, syndrome involving radial hypoplasia/aplasia, patellae hypoplasia/aplasia and cleft or highly arched palate, diarrhoea and dislocated joints, little size (height at least 2 standard deviations smaller than average height) and limb malformation, nose slender and normal intelligence; RTS, Rothmund–Thomson syndrome; T-loop, telomeric displacement loop; TTD, trichothiodystrophy.
Table 2.
Effect of helicase depletion on cancer cell proliferation, tumour growth and sensitivity to chemotherapy or radiation
| Helicase | Growth or proliferation | Radiation or chemotherapy sensitivity |
|---|---|---|
| RECQL1 | RECQL1-targeted siRNA decreased proliferation, induced DNA damage and elevated SCE of endocervical carcinoma HeLa cells21, and induced mitotic cell death in lung, prostate, bladder, colon and liver cancer cells26 and HCC cells5 in vitro. RECQL1-targeted siRNA also suppressed tumour growth in mouse xenograft lung, liver, pancreatic and colorectal cancer models27, liver cancer with transplanted human HCC5, and nude mice carrying FaDu or D-562 hypopharyngeal carcinoma xenografts25. RECQL1-targeted siRNA decreased proliferation of human hypopharyngeal carcinoma25 and glioblastoma cells in vitro28. | RECQL1-targeted siRNA decreased proliferation of HeLa cells exposed to camptothecin or ionizing radiation21, and caused a camptothecin-induced replication restart defect in U2OS osteosarcoma cells in vitro129. RECQL1-targeted siRNA enhanced an antitumour effect of cisplatin in nude mice carrying FaDu hypopharyngeal carcinoma xenografts, and co-treatment with cisplatin increased DNA damage, apoptosis and mitotic catastrophe25. RECQL1-targeted siRNA and RECQL1-targeted shRNA sensitized glioblastoma28 and HeLa and U2OS cells24, respectively, to hydroxyurea in vitro. RECQL1-targeted siRNA sensitized glioblastoma cells to TMZ in vitro28. RECQL1-targeted shRNA sensitized HeLa and U2OS cells to 8-MOPS in vitro24. |
| WRN | WRN-targeted siRNA decreased proliferation of human hypopharyngeal carcinoma cells in vitro and suppressed tumour growth in nude mice carrying FaDu or D-562 hypopharyngeal carcinoma xenografts25. WRN-targeted shRNA decreased tumour establishment and tumour growth in MYC overexpressing non-small-cell lung cancer mouse xenografts184. | WRN-targeted siRNA enhanced the antitumour effect of cisplatin in nude mice carrying FaDu hypopharyngeal carcinoma xenografts; this combination increased DNA damage and induced apoptosis and mitotic catastrophe25. |
| BLM | BLM-targeted siRNA caused a 2.5-fold increase in SCE in HeLa cells 223. | NR |
| WRN and BLM | WRN-targeted shRNA or BLM-targeted shRNA decreased proliferation of U2OS cells in vitro with BLM depletion having a greater effect224. Cells co-depleted for WRN and BLM showed reduced proliferation that was comparable to BLM-depleted cells. | BLM-targeted or WRN-targeted shRNA sensitized U2OS cells to camptothecin, cisplatin or 5-FU. BLM-targeted shRNA sensitized U2OS cells to hydroxyurea. Co-depletion did not further sensitize cells to any tested chemotherapies224. |
| RECQL4 | RECQL4-targeted siRNA inhibited proliferation and increased DNA damage and apoptosis of prostate cancer cells in vitro. RECQL4-targeted shRNA caused metastatic cancer cells to display reduced cell invasiveness and suppressed tumour growth in nude mice7. | NR |
| RECQL5 | RECQL5-targeted shRNA decreased proliferation of HeLa cells in vitro29. | RECQL5-targeted shRNA increased 53BP1 foci in HeLa cells225. |
| FANCJ | FANCJ-targeted siRNA caused HeLa cells to display delayed G1/S progression and G1 accumulation132. | FANCJ-targeted siRNA decreased hydroxyurea-induced CHK1 phosphorylation in U2OS cells133 and proliferation of HeLa cells89 in vitro. FANCJ-targeted siRNA reduced the proliferation of MMC-exposed MCF7 breast cancer cells37 or HeLa cells89. FANCJ-targeted shRNA reduced the growth of MCF7 cells following exposure to ionizing radiation226. |
| DDX11 | DDX11-targeted siRNA caused mitotic failure and sister-chromatid cohesion defects in HeLa cells227,228; it also reduced proliferation and elevated chromosome segregation, telomere defects and apoptosis in melanoma cells229. | NR |
| PIF1 | PIF1-targeted siRNA reduced colony formation, increased sub-G1 DNA, and prolonged S phase in colon cancer cells (HCT116)55. | PIF1-targeted siRNA reduced survival and increased apoptosis of HCT116 cells exposed to thymidine, hydroxyurea or gemcitabine. Elevated apoptosis was observed in PIF1-depleted TP53−/− SW480 colon cancer and TP53+/+ MCF7 breast cancer cells55. |
| FBH1 | NR | FBH1-depleted U2OS cells exposed to hydroxyurea show reduced DSBs and apoptosis131. |
| RTEL | NR | RTEL-targeted siRNA decreased MMC resistance in HeLa cells49. |
5-FU, 5-fluorouracil; 8-MOPS, 8-methoxypsoralen; 53BP1, p53-binding protein 1; CHK1, checkpoint kinase 1; DSB, double-strand break; HCC, hepatocellular carcinoma; MMC, mitomycin C; NR, not reported; SCE, sister-chromatid exchange; shRNA, short hairpin RNA; siRNA, small interfering RNA; TMZ, temozolomide.
The RecQ family
The five RecQ family DNA helicases (RECQL1, BLM, WRN, RECQL4 and RECQL5) are highly conserved and are required for genomic stability9,10. Three are implicated in genetic diseases that predispose to cancer: BLM in Bloom syndrome11, WRN in Werner syndrome12 and RECQL4 in Rothmund–Thomson syndrome (RTS). In addition, RECQL4 mutations are genetically linked to RAPADILINO syndrome and Baller–Gerold syndrome (BGS)13–15 (TABLE 1). Individuals affected by Bloom syndrome are diagnosed with a wide variety of cancer types earlier in life than the normal population. Patients with Werner syndrome display many features of premature ageing and frequently develop a broad range of neoplasias including thyroid epithelial tumours, malignant melanomas, meningiomas, soft-tissue sarcomas, haematological and lymphoid neoplasms, and osteosarcoma16. The three genetic disorders involving RECQL4 mutations show overlapping clinical features and a broad range of severity, which is probably influenced by genetic or environmental factors. Individuals with RTS display growth retardation and photosensitivity with poikiloderma, hair thinning and loss, juvenile cataracts and a predisposition to osteogenic sarcomas. The range of tumour types in patients with RECQL4 mutations includes lymphomas, predominantly in patients with RAPADILINO syndrome17. RECQL4 has a crucial role in prostate carcinogenesis7. RECQL4 expression increases as a function of tumour grade; furthermore, RECQL4 depletion was shown to dramatically reduce the growth, survival and invasiveness of metastatic prostate cancer cells in vitro, and to decrease tumorigenicity in a mouse model7.
Although no genetic disorders have been linked to RECQL1 or RECQL5 mutations, these helicases are also likely to have important roles in cancer. RECQL1 polymorphisms are associated with the survival of patients with pancreatic cancer18,19. Despite the absence of phenotypes in unstressed Recql1-knockout mice, primary embryonic fibroblasts from these mice display chromosomal instability20. RECQL1-depleted human cells display increased chromosomal instability21 and sensitivity to ionizing radiation21 — which is consistent with involvement in double-strand break (DSB) repair22 — or agents that induce oxidative damage23 or replication stress24 (TABLE 2). RECQL1 is highly expressed in various cancers, and its depletion reduces tumour cell proliferation5,25–28. RECQL5 is also required for cancer cell proliferation29. Moreover, Recql5-deficient mice are highly cancer-prone; approximately half of these cancers are lymphomas and the rest are solid tumours of different tissue origins30. RECQL5 has roles in transcription through its interaction with RNA polymerase II and in DNA repair by regulating homologous recombination (HR)30,31 and base-excision repair (BER)32.
The Fe-S family
Conserved iron–sulphur (Fe–S) clusters are found in metalloproteins that are implicated in DNA replication and repair33. Of the four human Fe-S DNA helicases (XPD, FANCJ (also known as BRIP1 and BACH1), RTEL1 and DDX11), all are implicated in autosomal recessive genetic diseases: XPD in xeroderma pigmentosum, Cockayne syndrome, trichothiodystrophy (TTD) and cerebro-oculo-facio-skeletal (COFS) syndrome34; FANCJ in Fanconi anaemia (FA)35–37; DDX11 in Warsaw breakage syndrome (WABS)38,39; and regulator of telomere elongation helicase 1 (RTEL1) in dyskeratosis congenita40–42 (TABLE 1). XPD, which was originally discovered through its linkage to xeroderma pigmentosum43,44, encodes a helicase subunit of the transcription factor II H (TFIIH) complex that is implicated in basal transcription and nucleotide-excision repair (NER); recessive mutations in XPD result in diseases with potentially related or overlapping clinical features34. The hallmark of xeroderma pigmentosum is severe sun sensitivity and predisposition to skin cancer. Although less common, individuals with XPD mutations may display xeroderma pigmentosum combined with Cockayne syndrome, characterized by developmental and neurological abnormalities as well as ultraviolet light sensitivity, but not skin cancer.
FA, arising from autosomal recessive mutations in FANCJ or in any of at least 14 other genes, is characterized by progressive bone marrow failure, acute myeloid leukaemia and other cancers45. In some cases, individuals with FA display congenital defects or developmental abnormalities that affect organ systems. All FA-mutant cell lines are hypersensitive to DNA-crosslinking chemotherapies. FANCJ mutations are also associated with breast cancer, which is consistent with the physical interaction between FANCJ and BRCA1 and with the role of FANCJ as a tumour suppressor46.
The Fe-S helicase RTEL1 is essential and has a dominant role in setting telomere length in mice47. Studies with Rtel1-deficient mouse embryonic stem cells demonstrated that the helicase is required for DNA damage resistance and acts as an antirecombinase in HR or counteracts telomeric G-quadruplex (G4) DNA structures48. RTEL1 knockdown in human cells renders them sensitive to the DNA-crosslinking agent mitomycin C (MMC)49. Genome-wide association studies have identified RTEL1 as a susceptibility locus for human glioma50–52. Recently, germline mutations in RTEL1 were linked to dyskeratosis congenita40–42, which is characterized by inherited bone marrow failure and cancer predisposition.
Other human DNA helicases that are important for genomic stability
Other human DNA helicases have genome caretaker roles (TABLE 1). XPB, which was first discovered by its genetic linkage to combined xeroderma pigmentosum and Cockayne syndrome53,54, is a TFIIH component (like XPD) and is also implicated in xeroderma pigmentosum with neurological abnormalities and in TTD34. Another DNA helicase, PIF1, suppresses apoptosis in human tumour cells55, and mutations are associated with breast cancer susceptibility56. Mutations in senataxin, which is a putative RNA/DNA helicase that is involved in transcription termination at RNA polymerase pause sites, are genetically linked to the neurodegenerative disorder ataxia with oculomotor apraxia 2 (AOA-2)57,58. DNA2, which is a helicase-nuclease that is involved in Okazaki fragment processing59, has prominent roles in DNA end resection during HR-mediated repair and in mitochondrial DNA maintenance60–62. Mutations in the mitochondrial DNA helicase twinkle (also known as PEO1) co-segregate with several neuromuscular degenerative disorders63. The number of DNA helicases implicated in cancer and other diseases is likely to grow.
Helicase functions in response to DNA damage
The DNA damage response mediated by the intra-S-phase checkpoint and DNA repair pathways help cells to cope with a variety of chromosomal lesions that are major contributing factors to various forms of cancer. Helicases have unique roles in these pathways through fork remodelling, DNA damage recognition, damaged strand removal, and recombination-based strategies to restore genomic integrity. The requirement for helicases to carry out roles in DNA repair through their catalytic functions or interactions with other proteins continues to be an important area of study.
Consequences of DNA lesions on helicase functions
Recognition of DNA damage among billions of normal base pairs in the human genome is formidable. Many helicases (for example, WRN64) preferentially bind and unwind forked DNA structures; therefore, they may arrive early at blocked replication forks to facilitate DNA damage recognition. Generally speaking, bulky covalent adducts inhibit helicase-catalysed DNA unwinding in a strand-specific manner; however, the type of adduct and/or helicase under investigation and its assembly in a protein complex is relevant65. Fe-S cluster DNA helicases (for example, XPD) may collaborate with other DNA repair proteins that are able to carry out DNA charge transport to search for DNA damage66. Helicase partnerships with the heterotrimer replication protein A (RPA)67 or other single-stranded DNA (ssDNA)-binding proteins (for example, POT1)68 enable them to unwind past certain DNA lesions. In some cases, replicative helicases (and their associated replisomes) may bypass or hop over template lesions, thus providing the opportunity for ssDNA signalling mechanisms to mediate DNA-damage-induced intra-S-phase checkpoints69. XPD bypasses DNA-bound proteins in vitro70; however, the physiological importance remains unclear.
DNA damage recognition by TFIIH helicases implicated in NER
NER relies on faithful recognition of bulky helix-distorting lesions and subsequent incisions upstream and downstream in the lesion-containing strand71 (FIG. 2a). In order for this to occur, TFIIH must be recruited to the site of DNA damage. The multi-subunit TFIIH complex possesses two opposite-polarity DNA helicases, XPD (5′ to 3′) and XPB (3′ to 5′), which orchestrate the opening of duplex DNA around the lesion and verify the distorting DNA damage72. Considerable efforts have been made to understand the precise roles of XPB, XPD and their protein partners in the various steps that are necessary for NER: TFIIH recruitment, duplex opening and damage verification. XPB ATPase (but not helicase) activity is required for TFIIH recruitment to DNA damage72. Damage recognition in NER involves a scanning mechanism that is dependent on XPD helicase activity73. The ability of XPD to sense a DNA lesion during unidirectional ATP-dependent translocation is required for DNA damage verification, which is necessary for NER74.
Figure 2. Involvement of helicases in nucleotide excision and interstrand crosslink DNA repair mechanisms.

Basic steps of nucleotide-excision repair (NER) (a) and interstrand crosslink (ICL) DNA repair (b) are shown, highlighting the roles of helicases. a | After DNA damage recognition by proteins including xeroderma pigmentosum complementation group C (XPC), XPE and RAD23B, the XPB and XPD helicases act with XPA to create a single-stranded bubble coated by replication protein A (RPA) that is processed by nucleases (the XPF–ERCC1 complex and XPG) to remove the damaged strand in NER72,75. b | Multiple helicases are implicated in crosslink resistance pathways that intersect with the Fanconi anaemia (FA) pathway77. After lesion detection, nuclease-catalysed incisions on each side of the crosslink remove (unhook) it from one strand, leaving a small gap that converts the fork to a double-strand break (DSB). Parallel pathways resolve the lesion. For simplicity, the double replication fork model216 is not shown. HR, homologous recombination; Pol, DNA polymerase; ssDNA, single-stranded DNA.
Lesion verification also involves XPB. One model depicts XPB melting a modest 5 nucleotide (nt) gap and XPD unwinding 22 base pairs (bp) by 5′ to 3′ translocation up to the site of the lesion, thereby defining the characteristic size of the ssDNA bubble to be excised75. Further studies are required to understand how DNA damage recognition by TFIIH-associated helicases is coordinated with subsequent steps in NER. Furthermore, the effects of mutations or polymorphisms in the NER helicases on their catalytic functions, protein interactions, disease pathology and cancer predisposition are of considerable interest4,76.
DNA helicases implicated in crosslink resistance and DSB repair
A DNA interstrand crosslink (ICL) is highly toxic and poses a strong block to cellular replication and transcription77. Reactive oxygen species from cellular biochemical processes may cause peroxidation of lipids, which in turn damage DNA to result in ICLs and other forms of oxidative DNA damage. Moreover, several DNA-damaging agents (for example, MMC and cisplatin) that are used for anticancer therapy induce DNA ICLs77. Thus, a comprehensive understanding of the response to ICLs may provide diagnostic and therapeutic opportunities to combat cancer.
The FA pathway is a highly integrated protein network that provides cellular resistance to DNA ICLs and other forms of replication stress45. Furthermore, it protects haematopoietic stem cells from endogenous aldehydes that produce DNA or protein crosslinks, thus ultimately suppressing bone marrow failure and leukaemia78,79. The currently identified 15 FA gene products collaborate with other proteins that are involved in HR, NER and translesion synthesis to tolerate and repair DNA ICLs77 (FIG. 2b). The FA pathway helps to channel DSBs through HR in order to prevent an inappropriate engagement of breaks by error-prone non-homologous end-joining (NHEJ)80,81. FANCM and FANCJ operate at distinct steps of ICL repair. FANCM shares sequence homology with DNA helicases and can translocate along, but not unwind, duplex DNA82. It is a component of the FA core complex that is important for early ICL recognition, for stabilizing and remodelling the blocked replication fork through interaction with the histone-fold-containing protein complex MHF1–MHF2 (REF. 83), and for signalling from the ICL damage by mono-ubiquitylation of FANCD2 and FANCI, which is a key activation step of the pathway. By contrast, FANCJ operates downstream of FANCD2 and FANCI mono-ubiquitylation to facilitate recombinational repair of a DNA structure (presumably a DSB) that arises from the ‘unhooking’ process, which leaves crosslinked nucleotides covalently attached to the complementary strand. FANCJ helicase activity and interaction with the mismatch repair (MMR) protein MLH1 is required for ICL resistance84, raising the interesting possibility of targeting this protein interaction to sensitize cancer cells to chemotherapeutic agents that induce DNA ICLs.
Other helicases are involved in resistance to ICLs or other forms of replication stress. BLM resides in a multi-protein nuclear complex with FA core complex proteins to recognize the DNA lesion and stabilize the stalled replication fork85. Together with the RecQ-mediated genome instability (RMI) sub-complex (RMI1 and RMI2) and topoisomerase 3α, BLM interacts with FANCM to help manage the repair of stalled replication forks through its ability to dissolve a double Holliday Junction (HJ) and related DNA structures86; such repair is necessary to avoid loss of heterozygosity and to suppress the cancer that is observed in patients with Bloom syndrome87,88. BLM also interacts with FANCJ to maintain chromosomal stability89. Therefore, BLM probably has upstream and downstream roles in ICL repair, including recognition of the ICL and strand resection of a DSB that arises from an unhooked ICL. FANCJ may also have a role in strand resection through its interaction with BLM and the MRN protein complex, which consists of MRE11–RAD50–Nijmegen breakage syndrome protein 1 (NBS1) and acts as a sensor of DSBs90. Cellular studies suggest that the WRN helicase, in conjunction with BRCA1, has a role in processing DNA ICLs91. WRN is required for ataxia telangiectasia mutated (ATM) activation and the intra-S-phase checkpoint in response to ICL-induced DSBs92 and other forms of fork arrest93. WABS-causing DDX11 mutations render cells sensitive to the chemotherapeutics MMC or camptothecin39, suggesting that they disrupt ICL repair. Depletion of HELQ (also known as HEL308) in human endocervical cancer cells rendered them MMC sensitive and defective in HR94. HELQ was observed to disrupt RAD51–double-stranded DNA filaments. However, this filament-disrupting activity was not dependent on ATP hydrolysis by HELQ95, leaving doubt as to the importance of HELQ catalytic activity in downstream events of HR repair. Human HELQ was shown to localize to damaged replication forks and to preferentially unwind the lagging strand of synthetic replication fork structures96, suggesting a role in fork remodelling or replication restart.
It is difficult to comprehend how so many helicases coordinate their roles in ICL repair and possibly render certain tumours resistant to crosslinking drugs. A key nexus is BLM, which interacts with FA proteins and DNA repair factors to repair the damage that is associated with ICLs and other forms of replication stress. For example, BLM collaborates with the FA pathway during S phase to prevent, and during mitosis to resolve, sister-chromatid bridging at fragile sites, ultimately to avoid chromosomal breakage and aneuploidy97,98. BLM and other helicases are involved in checkpoint signalling, strand resection and resolution of recombination intermediates during DSB repair of processed ICL DNA intermediates99. RECQL5 efficiently displaces RAD51 bound to DNA100, whereas BLM does so poorly101 and requires RAD51 inactivation to disrupt joint D-loop molecules102, thus suggesting that RECQL5 has a more prominent role than BLM in regulating HR by RAD51 filament disassembly. Further studies should delineate specialized functions of DNA helicases in response to drugs that induce ICLs and other forms of replication stress and that are widely used to treat leukaemia and other cancers77.
Processing of ICLs can lead to DSBs, which also arise at broken replication forks as a consequence of topoisomerase inhibitors that trap topoisomerase–DNA complexes103. Ionizing radiation or DNA-damaging agents can also lead to DNA breaks. The RecQ helicases, like numerous tumour suppressors, are implicated in HR-mediated repair of DSBs during the S or G2 phases of the cell cycle9,99 (FIG. 3a). HR is a high-fidelity repair mechanism, whereas NHEJ, which is the other major pathway of DSB repair and which occurs throughout the cell cycle, is error-prone. WRN interacts with the Ku protein complex104 and DNA ligase IV105, which are both implicated in NHEJ, but its precise role as a DNA helicase or exonuclease to trim DNA ends is still poorly understood. It may be that WRN facilitates an alternative form of NHEJ that is mediated by short (5–25 bp) microhomologous DNA sequences to align broken DNA ends, which contributes to the repair of DSBs in chronic myeloid leukaemia cells106. Studies in cell lines suggest that both helicase and exonuclease functions of WRN are necessary for optimal recombinational repair107–109.
Figure 3. Involvement of helicases in homologous recombination and base excision DNA repair mechanisms.

Basic steps of homologous recombination (HR)-mediated DNA repair (a) and base-excision repair (BER) (b) are shown, highlighting the roles of helicases. a | Helicases participate in various steps of conservative HR-mediated repair in somatic cells99. Suppression of crossover and rearrangement events can occur pre- and post-synaptically. Specific roles for helicases are being identified. Helicase disruption of displacement loops (D-loops) promotes synthesis-dependent strand annealing by decreasing double Holliday Junction (HJ) formation. b | The Werner syndrome helicase (WRN) participates in long-patch BER by unwinding 5′ flaps and interacting with BER proteins114, including flap endonuclease 1 (FEN1) (REF. 217). APE1, apurinic/apyrimidinic endonuclease 1; DNA2, DNA replication helicase/nuclease 2; EXO1, exonuclease 1; FANCJ, Fanconi anaemia complementation group J protein; NBS1, Nijmegen breakage syndrome protein 1; OGG1, 8-oxoguanine DNA glycosylase; POLβ, DNA polymerase-β; RTEL1, regulator of telomere elongation helicase 1.
ICL-inducing DNA-damaging agents are among the most widely used classes of chemotherapies77. Understanding the cellular response to ICL-induced DNA damage is a high priority in order to improve chemotherapeutic efficacy. Certain sporadic head and neck, lung, ovarian, cervical and haematological cancers are characterized by epigenetic silencing of wild-type FA gene expression110. It is estimated that 15% of all cancers harbour defects in the FA pathway, and loss of heterozygosity in FA carriers may increase cancer risk later in life45. Therefore, a synthetic lethality approach to target DNA damage response proteins in tumours that are characterized by a defective FA pathway should be evaluated. If FA-deficient tumours become reliant on WRN or other helicases to cope with ICL-induced strand breaks, it will be worthwhile to explore helicase inhibitors as a new class of anticancer therapy (discussed further below).
Helicases help cells to cope with endogenous DNA damage
WRN111, RECQL4 (REF. 112) and RECQL1 (REF. 23) interact with poly(ADP-ribose) polymerase 1 (PARP1), which is a sensor of DNA breaks and is implicated in BER. In mice, WRN and PARP1 collaborate to preserve chromosomal stability and to suppress the early development of neoplasms113. In addition to PARP1, WRN interacts with other BER proteins, notably DNA polymerase-β, apurinic/apyrimidinic endonuclease 1 (APE1), and flap endonuclease 1 (FEN1) to promote long-patch BER114 (FIG. 3b). In addition, RECQL5-depleted cells are sensitive to oxidative agents and accumulate endogenous damage, including strand breaks and 8-oxoguanine32, which is consistent with elevated cancer incidence in Recql5-deficient mice30.
Mutations in MMR genes are linked to colorectal, endometrial, gastric and urothelial cancers115; however, no DNA helicase has been implicated in eukaryotic MMR, unlike the established role of the UvrD helicase in Escherichia coli MMR116. MMR proteins interact with WRN, BLM, RECQL1 and FANCJ, which may regulate the roles of these helicases in recombinational repair117. Interestingly, exonuclease 1 (EXO1), the nuclease that is implicated in eukaryotic MMR, interacts with WRN118, RECQL1 (REF. 119) and BLM61, and the BLM–EXO1 interaction is important for DSB resection during HR-mediated repair.
DNA helicases at replication forks
DNA helicase proteins, which were previously thought to operate strictly in DNA repair or replication, are now known to have interwoven roles in both processes. Maintaining replication fork integrity in vivo is a challenging task that necessitates versatile strategies to cope with fork obstacles (FIG. 4). The importance of helicase-dependent pathways to deal with replication stress is becoming evident from cellular and biochemical studies.
Figure 4. Roles of DNA helicases during replication stress.

a | When the replisome (not shown) encounters a replication-blocking lesion (green oval) or other form of replication stress (for example, nucleotide starvation), certain helicases (for example, the Werner syndrome helicase (WRN) or Bloom syndrome helicase (BLM)126,128), carry out fork regression to create a Holliday Junction (HJ)-like ‘chicken-foot’ DNA structure. Reverse branch-migration of the regressed fork by a helicase (for example, WRN127 or RECQL1 (REF. 129)) potentially allows replication bypass of the lesion in a non-recombinogenic mode. b | The WRN217 or BLM218 helicases can stimulate flap endonuclease 1 (FEN1)-catalyzed cleavage of 5′ flap structures that might arise during lagging-strand synthesis. BLM stimulates FEN1-mediated cleavage on flaps with secondary 5′ flap structure219, and WRN is involved with DNA polymerase-δ in a hairpin repair pathway220. DNA replication helicase/nuclease 2 (DNA2) also facilitates FEN1-mediated cleavage of 5′ flaps221. c | Based on evidence from studies of Escherichia coli replication222, accessory DNA helicases (blue triangle) may coordinate with a eukaryotic minichromosome maintenance (MCM) helicase complex (purple) and replication machinery to displace a protein (orange oval) bound to duplex DNA that impedes fork progression. Replication fork reversal has a role in bacterial replication restart141. Further studies are required to substantiate this model for eukaryotes.
Fork remodelling and checkpoint signalling by helicase proteins
Replication lesions are prominent in rapidly dividing cancer cells120, and persistent replication fork stalling leads to genomic instability. Therefore, understanding how cancer cells deal with such lesions will be informative for developing antitumor strategies. An intra-S-phase checkpoint helps to stabilize proteins at stalled replication forks, to curtail cell cycle progression and to invoke DNA repair pathways. The Saccharomyces cerevisiae RecQ helicase Sgs1, through its interaction with the Rpa70 (also known as Rpa1) subunit of the Rpa heterotrimer121, has a crucial role in checkpoint signalling by recruiting the kinase Rad53 to regions of ssDNA at stalled replication forks and facilitating subsequent Rad53 activation122,123. Conservation of checkpoint activation mechanisms through RecQ protein interactions remains an area of active investigation that will yield new insights into helicase functions at stalled replication forks, the molecular pathology underlying RecQ helicase disorders and targeting DNA damage checkpoints in cancer. For example, the human WRN and BLM RecQ helicases also interact with RPA70 through an amino-terminal acidic domain, which is required for stimulation of helicase activity124. WRN is required for normal replication fork progression after exposure to the DNA-damaging agent methylmethanesulphonate or the replication inhibitor hydroxyurea125 (TABLE 2). Both WRN and BLM may help cells to cope with stalled replication forks by remodelling DNA structures, as demonstrated in vitro126–128 (FIG. 4a,b); however, their precise roles require further investigation. RECQL1 was shown to have a role in the restart of reversed replication forks induced by the topoisomerase 1 inhibitor camptothecin, which is used as an anticancer drug129.
Unlike RecQ helicases, human F-box DNA helicase 1 (FBH1), in cooperation with the MUS81 nuclease130, promotes DNA breakage and apoptosis in response to replication stress, thus suggesting a role for FBH1 during chronic replication stress to suppress oncogenic transformation by eliminating genetically unstable transformed cells that have been overwhelmed by broken replication forks131. Alternatively, FBH1 may promote DNA breakage following replication stress to promote a constitutively high number of DNA ends that can enhance chromosomal rearrangements and promote tumour progression. This would be consistent with the characteristic replicative trauma that transpires during oncogene activation. The role of FBH1 in carcinogenesis requires further study.
FANCJ helicase activity is required for timely progression through S phase132. FANCJ acts with DNA topoisomerase-2-binding protein TOPBP1 in early DNA replication checkpoint control after hydroxyurea-induced replication stress, suggesting that their interaction enables RPA to load onto chromatin, which is a prerequisite for the activation of ataxia telangiectasia and Rad3-related (ATR) and for the replication checkpoint mediated by checkpoint kinase 1 (CHK1)133. The FANCM DNA translocase is recruited to ICL-blocked replication forks, where it suppresses sister-chromatid exchange (SCE) by remodelling DNA134,135. SMARCAL1 (also known as HARP) — which is defective in the autosomal T-cell immunodeficiency disorder Schimke immune-osseous dysplasia — surveys replication forks and catalyses fork regression and HJ branch migration to preserve genome stability136. The ability of SMARCAL1 to anneal complementary strands coated by RPA137 distinguishes it from classic DNA helicases (for example, human RecQ proteins) in which RPA inhibits strand annealing. Helicase-like proteins known as chromatin remodelling enzymes (which are distinct from helicases because they do not separate complementary strands) reconfigure nucleosome interactions by displacing or repositioning histone octamers bound to duplex DNA and by exchanging histone variants138. Chromatin remodelling complexes — for example, the nucleosome remodelling and deacetylase (NuRD) complex, which contains a protein with an ATPase/helicase-like domain — regulate transcriptional events that are important for oncogenesis and cancer progression139.
Collaboration between replicative and accessory helicases
Aside from drug-induced replication stalling, cells must cope with natural barriers to replication, such as stable protein–DNA complexes or a replication fork converging with the transcriptional apparatus. Helicase-dependent mechanisms exist to minimize these collisions140 (FIG. 4c). If the fork collides with transcription complexes, accessory motors help to displace RNA polymerase from DNA to restore fork progression. Genetic evidence from E. coli suggests that replication fork reversal sets the stage for accessory helicases to enable replication restart141. It is unknown whether a similar arrangement exists in eukaryotes.
The atypical helicase-related minichromosome maintenance 8 (MCM8) and MCM9 replication elongation proteins are required for efficient HR-mediated DNA DSB repair142,143. Although MCM9 is dispensable for DNA replication, the characterization of mice lacking Mcm9 demonstrated somatic chromosomal instability, embryonic germ cell depletion in both sexes and hepatocellular carcinoma in males144. It will be important to determine how the recombination functions of MCM paralogues bear on pathways that affect germline stem cell maintenance and proliferation, fertility and cancer suppression.
Accessory DNA helicases facilitate replication fork progression when the forks encounter a protein–DNA complex such as an RNA polymerase, a DNA sequence that is prone to forming an alternative structure (for example, G-quadruplexes (see the next section)), or RNA–DNA hybrids known as R loops145). In Schizosaccharomyces pombe, the Pfh1 helicase is required for replisome fork progression through difficult-to-replicate sequences146, or past protein–DNA obstacles, to ensure chromosomal integrity147. By sequence homology, only one Pfh1 family helicase (PIF1) exists in human cells, raising interest in its role in DNA synthesis at difficult-to-replicate sequences that may be sites of chromosomal fragility and markers or causative factors for cancer or disease.
Helicases at telomeres and G4 DNA
G-quadruplexes, which are composed of planar stacks of four guanines interacting by Hoogsteen hydrogen bonds, have attracted considerable attention, and there is evidence that they form in vivo and affect nucleic acid metabolism148. Certain DNA helicases (for example, WRN, BLM, FANCJ and PIF1) unwind G4 DNA substrates in vitro149. Evidence has emerged that helicase resolution of G4 DNA has important biological outcomes for telomeres (FIG. 5) and other G-rich sequences.
Figure 5. Helicase functions at telomeres and potential for anticancer therapy.

Helicases that regulate telomere capping may be targeted for depletion by RNA interference (RNAi) or small-molecule inhibition to selectively kill cancer cells. a | 3′ single-stranded DNA (ssDNA) tail invasion into adjacent telomeric double-stranded DNA (dsDNA) results in displacement of the complementary strand to create a displacement loop (D-loop), the so-called T-loop structure. Human DNA helicases (regulator of telomere elongation helicase 1 (RTEL1)48 and the Werner syndrome helicase (WRN)168; indicated by the beige triangles) resolve T-loops to enable telomere replication or repair. Helicase deficiency by mutation or RNAi-mediated depletion results in the persistence of T-loops which can be nucleolytically cleaved, leading to telomere instability. A helicase inhibitor (purple circle) may prevent T-loop unwinding, thus resulting in telomere instability. b | ssDNA that arises at telomeres or during lagging-strand synthesis can form a G-quadruplex (G4). Certain human helicases with 5′ to 3′ polarity (RTEL, Fanconi anaemia complementation group J protein (FANCJ) and PIF1) or 3′ to 5′ polarity (WRN and the Bloom syndrome helicase (BLM)) may resolve G4 structures to enable replication or repair149. Helicase deficiency by mutation or RNAi-mediated depletion results in telomere fragility. A helicase inhibitor or G4-specific ligand (green circle) may prevent G4 resolution at telomeres or other G-rich genomic sequences. Telomere instability can lead to cellular senescence of rapidly dividing cancer cells. Figure is modified, with permission, from REF. 48 © (2012) Elsevier Science.
Leading versus lagging strand G4 instability
S. cerevisiae Pif1 resolves G4 structures to preserve replication fork progression and suppress chromosomal breakage150. Pif1 inactivation specifically destabilized a G-rich human sub-telomeric CEB1 minisatellite inserted into a yeast chromosome151, and exposure to a G4 ligand exacerbated CEB1 instability152. The CEB1 minisatellite was unstable only when the G-rich strand served as a template for leading-strand replication153. This was not predicted because the lagging-strand template is believed to have more-extensive ssDNA. Genomic instability in Caenorhabditis elegans due to mutation of the Fe-S helicase DOG-1 is thought to be caused by structural blocks to lagging-strand synthesis154. The 5′ to 3′ directionality of Pif1 (or FANCJ155) is well suited for unwinding G4 structures by loading onto downstream ssDNA of the leading-strand template; however, the helicase must be able to ‘turn its shoulder’ as it unwinds G4 to permit the replication machinery to move past the helicase and copy unwound G-rich sequence. Division of labour among G4 helicases may be handled in unique ways depending on the G4 topology or on the type of chromosomal G-rich region (for example, a telomere) that is undergoing replication or repair, or in the case of promoter elements, undergoing transcription.
Roles of vertebrate helicases in telomere maintenance and G4 DNA metabolism
Human cells that are deficient in FANCJ, but not FANCD2 or FANCA, are sensitive to the G4 ligand telomestatin155, which is known to cause telomere instability156. Moreover, patient-derived FANCJ-mutant cells accumulate deletions at genomic sequences that are predicted to form G-quadruplexes157. These findings suggest a model in which FANCJ suppresses genomic instability by resolving G-quadruplexes to enable smooth replication fork progression. A major advance would be obtaining direct evidence of G4-structure accumulation in FANCJ-deficient cells.
The pronounced number of predicted G-quadruplexes in promoter regions suggests their involvement in transcriptional regulation158. Fibroblasts from individuals with Werner syndrome or Bloom syndrome display upregulated expression of genes that are enriched for predicted G4-forming sequences159. In the absence of WRN or BLM, G4 structures may accumulate to exclude nucleosomes or other DNA-binding proteins that repress transcription; however, this hypothesis remains untested.
Continuity of replicative DNA synthesis provides a mechanism for maintaining epigenetic memory by incorporating new histones and depositing parental histones with their characteristic post-translational modifications near their original locations160. Perturbation of DNA synthesis by DNA lesions or structured DNA such as G4 can be overcome by translesion polymerases (for example, REV1)161. FANCJ was shown to maintain epigenetic stability in chicken cells by collaborating independently with REV1 or the RecQ helicases WRN and BLM to replicate G4-forming DNA sequences162. Furthermore, FANCJ counteracts chromatin compaction by enabling smooth fork progression163. Although functional interactions between WRN and DNA polymerases are reported164,165, there is currently no direct evidence that FANCJ interacts with a DNA polymerase. Interaction of FANCJ with BLM might be relevant to the replication or repair of G4 DNA89.
Both FANCJ and BLM are found at telomeres166, where they potentially resolve G4 structures to repress telomere fragility167 (FIG. 5). WRN is also found at telomeres and interacts with shelterin complex proteins that regulate WRN catalytic functions on telomeric D-loops (T-loops)168. The connection of the role of WRN at telomeres to the pathophysiology of Werner syndrome was first made by the observation that late-generation telomerase and WRN-deficient mice (Terc−/− Wrn−/−) have dysfunctional telomeres and chromosomal instability169–171, and exhibit premature ageing and tumours (osteocarcinomas and soft tissue sarcomas) that are typically observed in patients with Werner syndrome169. A role for human WRN in resolving telomeric G4 DNA was suggested by the observation that WRN-deficient cells are defective in replication of the telomeric G-rich lagging-strand template172; however, the WRN–FEN1 interaction may also have a role173,174. Although there is no evidence for a role for FANCJ at telomeres, the FA pathway can influence telomere attrition and telomere recombination175,176. A more-prominent player in telomere metabolism is probably RTEL1, which controls telomere length in mice and is required for telomere stability47 and resistance to telomere fragility induced by the porphyrin G4 ligand TMPyP4 (REF. 48); however, direct evidence that RTEL1 catalytically unwinds G4 DNA substrates to preserve telomere integrity is lacking. Recently, germline mutations in RTEL1 were linked to the telomere and cancer disorder dyskeratosis congenita40–42. Cells from patients with dyskeratosis congenita harbouring biallelic RTEL1 mutations display telomere shortening owing to defective T-loop resolution42.
Helicase-based biomarkers and therapeutics
The elevated expression of DNA helicases in rapidly proliferating cells and tumours suggests they have a role in resistance to DNA-damaging agents and may represent good biomarkers for response to chemotherapies. In general, RecQ and other helicase genes are not mutated or silenced in an appreciable number of tumours, and expression is most often upregulated. Thus, RecQ and perhaps other helicases may be important ‘survival’ factors in many tumours. One reported exception was the epigenetic inactivation of WRN in colorectal cancers177. Studies that deplete helicases using RNA interference reinforce the idea that DNA helicases affect cancer cell proliferation and survival after chemotherapeutic DNA damage (TABLE 2). Recently, a screen to detect predictors of cancer cell response to DNA-damaging agents identified a putative DNA and RNA helicase schlafen 11 (SLFN11) as a potential biomarker178. Inactivation of a single DNA-damage response gene (such as a helicase gene) may serve as a predictive biomarker for personalized cancer therapy179.
Conventional anticancer therapies have relied on the administration of DNA-damaging chemotherapies or radiation; however, these approaches have drawbacks owing to cytotoxic effects on normal cells and the ability of tumours to become resistant180. Personalized medicine to combat cancer is entering an age in which therapies make use of existing tumour deficiencies in the DNA damage response, cell cycle checkpoint signalling or DNA repair pathways179. Synthetic lethality exploits pre-existing DNA repair deficiencies to enable an inhibitor of a given DNA repair pathway to be effective at eliminating those tumours. One potential caveat is therapy-related carcinogenesis that is driven by DNA-damage-induced genomic instability when DNA repair is compromised. Thus, further studies are required to elucidate the precise roles of DNA repair proteins to avoid secondary cancers.
The involvement of DNA helicases in the repair of endogenous DNA damage in rapidly dividing cells has prompted interest in their potential as therapeutic targets, following the path of inhibitors of the single-strand break repair protein PARP1. A proof-of-principle for the synthetic lethal approach was made by the discovery that tumours that are deficient for BRCA1 or BRCA2, which are defective in HR, are highly sensitive to PARP inhibitors181,182. Results from preclinical and clinical trials (Phase I and Phase II) with PARP inhibitors that potentiate the cytotoxicity and antitumor effects of chemotherapy drugs or radiation suggest that tumours that are defective in other DNA repair pathways (such as NER or BER) may also be susceptible179. HR-defective tumours that are characterized by a deficiency of a key DNA repair helicase may also be amenable to DNA-damaging chemotherapies183.
Small-molecule helicase inhibitors
DNA helicase inhibitors may improve the efficacy of anticancer therapies183. Moreover, helicase inhibitors may have value as mono-therapies, as the inactivation of specific DNA helicases promotes synthetic lethality in contexts of specific genetic deficiencies and may impair the replication or division of actively dividing cancer cells. For example, recent work showed that conditional silencing of WRN expression in MYC-overexpressing non-small-cell lung cancer xenografts impaired tumour growth and establishment184. Thus, targeting WRN may be effective to combat MYC-associated cancers.
Screening of chemical libraries for helicase-docking small molecules that perturb unwinding may lead to the development of anticancer drugs. Alternatively, chemical rescue may restore the function of a helicase protein that is misfolded owing to a disease-causing or cancer-promoting missense mutation. Further studies are required to determine the efficacy of these strategies. Recently, a small molecule (NSC 19630) was identified that specifically inhibited WRN helicase activity in vitro185. Exposure of human cancer cells to NSC 19630 dramatically impaired growth and proliferation, and induced apoptosis and DNA damage in a WRN-dependent manner. The dominant-negative effect of the WRN helicase inhibitor is reminiscent of certain clinical PARP inhibitors that exert their poisonous effect by trapping PARP on DNA186. Exposure of cancer cells to a sublethal dose of NSC 19630 sensitized them to telomestatin, a PARP inhibitor or topotecan chemotherapy185. Further investigation, such as a recent study that identified a BLM inhibitor187, is necessary to characterize helicase-specific small molecules, their mechanisms of action and their potential as therapeutic drugs. To achieve synthetic lethality, a helicase inhibitor should be successful in a genetic background that is defective in a key protein that is involved in HR or another repair pathway, checkpoint signalling, DNA replication or the cell cycle.
Targeting G-quadruplexes to combat cancer
In approximately 90% of cancers, progressive shortening of chromosomes by loss of telomeric DNA is counteracted by upregulated telomerase activity188. Inhibiting telomerase may be useful to combat cancer, and a number of telomerase inhibitors are in clinical trials189. There is growing interest in anticancer drugs that stabilize telomeric G-quadruplexes190. Understanding the roles of G4-resolving DNA helicases may lead to new insights for the design of G4 ligands that block G4 helicases or related proteins, thereby interfering with telomere semiconservative replication or impeding telomerase-catalysed elongation in cancer cells and causing them to senesce (FIG. 5).
The G4 ligand telomestatin induces telomere destabilization by perturbing the interaction of the shelterin protein POT1 with telomeres, thus resulting in apoptosis of cancer cells191. Similarly, the synthetic G4-binding ligand pyridostatin inhibits the proliferation of numerous cancer cell lines and uncaps POT1 from telomeric ssDNA in fibrosarcoma cells to trigger a DNA damage response192. Chromatin immunoprecipitation followed by high-throughput sequencing (ChIP–seq) analysis of DNA sequences associated with the DNA damage marker γH2AX was used to search for pyridostatin-stabilized G-quadruplexes in human cells that displayed replication- and transcription-dependent DNA lesions193. Fluorescently labelled pyridostatin, detected mainly at non-telomeric G4 foci, colocalized with human PIF1. Pyridostatin modulated the expression of genes with high G4-forming potential, including the proto-oncogene SRC. The prospect of targeting G-quadruplexes in gene promoters for anticancer therapy prompts a greater understanding of helicases that are involved in gene regulation. G4 ligands that target promoter G-quadruplexes are in Phase I and Phase II clinical trials for cancer158. Pyridostatin analogues also cause telomere dysfunction, resulting in long-term growth arrest and senescence of cancer cells, suggesting a small-molecule approach to target G-quadruplexes for anticancer therapy194 (FIG. 4).
Targeting helicases that resolve unusual DNA structures (such as G-quadruplexes and T-loops) found at telomeres is a possibility (FIG. 5). In addition, RecQ helicases have a role in the alternative lengthening of telomeres (ALT) pathway, which affects proliferation in 5–10% of tumours195. Characterization of small molecules that modulate helicase function in the ALT pathway may be valuable for devising strategies to treat ALT-dependent tumours (for example, osteosarcomas) that are resistant to conventional anticancer therapies.
Concluding remarks
Helicases have crucial roles in DNA metabolism, and their clinical importance is evident from the discovery of links between helicase gene mutations and cancer-predisposing diseases. The prominent role of helicases in the DNA damage response and cell proliferation has prompted researchers to investigate their molecular and cellular functions. However, there is still much to learn about the underlying mechanisms that are responsible for genomic instability when human DNA helicases are defective. This challenge is particularly daunting owing to the complex network of interconnected roles of DNA helicases and their protein interactions, which profoundly influence DNA repair and replication.
Continued interest in DNA repair as a target for anticancer strategies is propelling the field forwards to develop clinical applications for the diagnosis or treatment of individuals that are predisposed to or experiencing cancer. As chemotherapy or radiation frequently introduces DNA damage, helicases that are involved in DNA repair are moving to the forefront of cancer research. To target helicase-dependent processes, a greater understanding of the molecular and cellular functions of helicases will be required. Modulation of helicase-mediated DNA damage response pathways to enhance anticancer efforts will require the development of tumour-targeted gene-silencing strategies and small molecules that bind to helicase proteins or their protein partners to alter catalytic functions and/or protein interactions. Whole-genome RNA interference screens should help to identify synthetic lethal relationships of target helicases that may potentially be exploited to sensitize tumours by personalized medicine. The knowledge gained from mechanistic and structural studies of helicases (for example, BOX 1) and high-throughput screens to identify small molecules that directly bind to and modulate helicase function will hopefully result in translational advances. As we learn from the successes and limitations of the first wave of treatment strategies to target DNA damage response proteins, DNA helicases are now an important focus for understanding both the origins and treatment of human cancer.
Key points.
Helicase-dependent DNA damage response and repair mechanisms help cells to cope with endogenous or exogenous stress to prevent chromosomal instability and maintain cellular homeostasis.
Inactivating mutations in DNA helicase genes are linked to genetic disorders that are frequently associated with various cancers. However, the expression of many DNA helicases is upregulated in transformed or neoplastic cells and tissues and is required for cancer cell proliferation or resistance to DNA damage imposed by chemotherapies.
The RecQ and iron-sulphur (Fe-S) families of DNA helicases have prominent roles in the maintenance of genomic stability through their catalytic functions and protein interactions in telomere maintenance and DNA repair pathways including nucleotide-excision repair (NER), homologous recombination (HR)-mediated repair of double-strand breaks (DSBs), interstrand crosslink (ICL) repair, and base-excision repair (BER).
Specialized DNA helicases efficiently unwind G-quadruplex (G4) DNA and other forms of alternative DNA structures such as telomeric displacement loops (T-loops). Emerging evidence suggests that DNA helicases that unwind non-conventional DNA structures have important roles in the replication or repair of telomeres.
Replication forks in rapidly dividing cancer cells are likely to encounter DNA lesions that perturb fork progression. Evidence suggests that certain DNA helicases help cells to cope with replicative lesions by remodelling the fork, restoring the integrity of broken replication forks, or having a role in the signalling mechanism for the intra-S-phase checkpoint.
Elevated expression of DNA helicases in rapidly proliferating cells and tumours suggests that they have a role in resistance to DNA-damaging agents and may represent good biomarkers for response to chemotherapies.
High-throughput screening of chemical libraries may prove to be beneficial for the discovery of small molecules that modulate helicase function in vivo. Such compounds may be useful in synthetic lethal approaches used in anticancer strategies that target tumours with existing DNA repair deficiencies.
Acknowledgments
This research was supported by the Intramural Research Program of the US National Institutes of Health (NIH), National Institute on Aging (NIA). I thank J. Burril of the NIA Visual Media Section for artwork in drafts of figures 2,3 and 5. I express gratitude to M. Seidman, Y. Liu, V. Bohr and W. Wang (NIA-NIH) for critically reading the manuscript, and S. Matson (University of North Carolina at Chapel Hill) for introducing me to the helicase field and for providing strong mentorship. I apologize to those researchers whose published work on DNA helicases was not cited owing to length restrictions.
Glossary
- Poikiloderma
A skin condition that consists of areas of increased and decreased pigmentation, prominent blood vessels and thinning of the skin
- Homologous recombination (HR)
A type of genetic recombination in which nucleotide sequences are exchanged between two similar or identical molecules of DNA. HR is most widely used by cells to accurately repair damage that involves both strands, such as double-strand breaks or interstrand DNA crosslinks
- Base-excision repair (BER)
A DNA repair pathway that operates on small DNA lesions such as oxidized or reduced bases, fragmented or non-bulky adducts, or those produced by methylating agents. The resulting single-strand break can be processed by either short-patch BER (through which a single nucleotide is replaced) or long-patch BER (through which 2–10 new nucleotides are synthesized)
- Transcription Factor IIH (TFIIH)
A general transcription factor complex composed of multiple protein subunits that enables formation of the RNA polymerase II pre-initiation complex. TFIIH is also implicated in nucleotide-excision repair (NER)
- Nucleotide-excision repair (NER)
This pathway recognizes bulky distortions in the DNA that occur after ultraviolet radiation or chemotherapy. Recognition of these distortions leads to the removal of a short single-stranded DNA segment that includes the lesion, creating a single-strand gap in the DNA, which is subsequently filled by a DNA polymerase
- G-quadruplex (G4)
A four-stranded nucleic acid structure stabilized by non-Watson–Crick Hoogsteen base-pairing within stacks of four planar-orientated guanosine nucleotides. G-quadruplex structures can form within or between G-rich strands of telomeric DNA or other G-rich sequences
- Intra-S-phase checkpoint
Single-stranded DNA created at the stalled replication fork generates a signal mediated by phosphorylation of target proteins (for example, mammalian ATR) that prevents the cell cycle from progressing to the G2 phase until replication is complete
- DNA charge transport
The process of transporting electrons along the axis of a double helical DNA molecule through the overlapping π-orbitals of stacked DNA base pairs
- Slippage
When a helicase loses its firm grasp on single-stranded DNA, causing it to slide backwards owing to re-annealing of complementary strands at the DNA fork
- Translesion synthesis
A DNA damage tolerance process that allows replication past DNA lesions. If the normal replicative polymerase cannot insert a base owing to damage in the template strand, it is often replaced by a lower-fidelity translesion polymerase
- Non-homologous end-joining (NHEJ)
Unlike homologous recombination-mediated repair, NHEJ rejoins broken ends of DNA following double-strand breaks without using a homologous DNA template and can therefore be accompanied by loss of nucleotides and errors
- Mismatch repair (MMR)
A process that acts during DNA replication to correct base pairing errors made by the DNA polymerases
- RAD51–double-stranded DNA filaments
During an early stage of homologous recombination, the major eukaryotic recombinase RAD51 forms nucleoprotein filaments on double-stranded DNA to initiate the homology search and exchange of DNA strands. Filaments can also occur on single-stranded DNA
- Synthetic lethality
Cell death resulting from the combined inactivation or inhibition of two genes or gene products that are non-lethal when inactivated individually. Synthetic lethality can occur between genes and small molecules, which may lead to anticancer therapies
- Sister-chromatid exchange (SCE)
A crossing-over event between sister chromatids, leading to the exchange of homologous stretches of DNA sequence
- G4 ligand
A typically planar aromatic small molecule that preferentially binds G-quadruplex (G4) DNA with high affinity. There are also proteins that preferentially bind G4 DNA
- Shelterin complex
A six-protein complex that localizes to the terminal TTAGGG repeats of mammalian chromosome ends, enabling cells to distinguish their natural chromosome ends from DNA breaks. The shelterin complex represses DNA repair reactions and regulates telomerase-based telomere maintenance
- T-loops
The 3′ single-stranded telomere DNA overhang circles around and hybridizes to the complementary strand of the adjacent double-stranded DNA to create a displacement loop (D-loop) by displacing one of the strands. T-loops are stabilized by telomere-binding proteins
- Alternative lengthening of telomeres (ALT)
A recombination-based mechanism that allows telomere length maintenance in the absence of telomerase activity
Footnotes
Competing interests statement
The author declares no competing financial interests.
FURTHER INFORMATION
Author’s homepage: http://www.irp.nia.nih.gov/branches/lmg/rbrosh.htm
Fanconi Anemia Mutation Database: http://www.rockefeller.edu/fanconi/mutate
The International Registry of Werner Syndrome: http://www.wernersyndrome.org
The Bloom’s Syndrome Registry: http://www.med.cornell.edu/bsr
ALL LINKS ARE ACTIVE IN THE ONLINE PDF
References
- 1.Singleton MR, Dillingham MS, Wigley DB. Structure and mechanism of helicases and nucleic acid translocases. Annu Rev Biochem. 2007;76:23–50. doi: 10.1146/annurev.biochem.76.052305.115300. [DOI] [PubMed] [Google Scholar]
- 2.Umate P, Tuteja N, Tuteja R. Genome-wide comprehensive analysis of human helicases. Commun Integr Biol. 2011;4:118–137. doi: 10.4161/cib.4.1.13844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abdel-Monem M, Hoffmann-Berling H. Enzymic unwinding of DNA. 1 Purification and characterization of a DNA-dependent ATPase from Escherichia coli. Eur J Biochem. 1976;65:431–440. doi: 10.1111/j.1432-1033.1976.tb10358.x. [DOI] [PubMed] [Google Scholar]
- 4.Suhasini AN, Brosh RM., Jr Disease-causing missense mutations in human DNA helicase disorders. Mutat Res. 2013;752:138–152. doi: 10.1016/j.mrrev.2012.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Futami K, Ogasawara S, Goto H, Yano H, Furuichi Y. RecQL1 DNA repair helicase: a potential tumor marker and therapeutic target against hepatocellular carcinoma. Int J Mol Med. 2010;25:537–545. doi: 10.3892/ijmm_00000375. [DOI] [PubMed] [Google Scholar]
- 6.Kawabe T, et al. Differential regulation of human RecQ family helicases in cell transformation and cell cycle. Oncogene. 2000;19:4764–4772. doi: 10.1038/sj.onc.1203841. [DOI] [PubMed] [Google Scholar]
- 7.Su Y, et al. Human RecQL4 helicase plays critical roles in prostate carcinogenesis. Cancer Res. 2010;70:9207–9217. doi: 10.1158/0008-5472.CAN-10-1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Turley H, Wu L, Canamero M, Gatter KC, Hickson ID. The distribution and expression of the Bloom’s syndrome gene product in normal and neoplastic human cells. Br J Cancer. 2001;85:261–265. doi: 10.1054/bjoc.2001.1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chu WK, Hickson ID. RecQ helicases: multifunctional genome caretakers. Nature Rev Cancer. 2009;9:644–654. doi: 10.1038/nrc2682. [DOI] [PubMed] [Google Scholar]
- 10.Monnat RJ., Jr Human RECQ helicases: roles in DNA metabolism, mutagenesis and cancer biology. Semin Cancer Biol. 2010;20:329–339. doi: 10.1016/j.semcancer.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ellis NA, et al. The Bloom’s syndrome gene product is homologous to RecQ helicases. Cell. 1995;83:655–666. doi: 10.1016/0092-8674(95)90105-1. [DOI] [PubMed] [Google Scholar]
- 12.Yu CE, et al. Positional cloning of the Werner’s syndrome gene. Science. 1996;272:258–262. doi: 10.1126/science.272.5259.258. References 11 and 12 represent seminal discoveries in the helicase field: the discoveries of the predicted DNA helicase genes that are genetically linked to the cancer predisposition disorders Bloom syndrome and Werner syndrome, the latter of which is characterized by premature ageing. [DOI] [PubMed] [Google Scholar]
- 13.Kitao S, et al. Mutations in RECQL4 cause a subset of cases of Rothmund-Thomson syndrome. Nature Genet. 1999;22:82–84. doi: 10.1038/8788. [DOI] [PubMed] [Google Scholar]
- 14.Siitonen HA, et al. Molecular defect of RAPADILINO syndrome expands the phenotype spectrum of RECQL diseases. Hum Mol Genet. 2003;12:2837–2844. doi: 10.1093/hmg/ddg306. [DOI] [PubMed] [Google Scholar]
- 15.Van Maldergem L, et al. Revisiting the craniosynostosis-radial ray hypoplasia association: Baller-Gerold syndrome caused by mutations in the RECQL4 gene. J Med Genet. 2006;43:148–152. doi: 10.1136/jmg.2005.031781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lauper JM, Krause A, Vaughan TL, Monnat RJ., Jr Spectrum and risk of neoplasia in Werner syndrome: a systematic review. PLoS ONE. 2013;8:e59709. doi: 10.1371/journal.pone.0059709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Siitonen HA, et al. The mutation spectrum in RECQL4 diseases. Eur J Hum Genet. 2009;17:151–158. doi: 10.1038/ejhg.2008.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li D, et al. Single nucleotide polymorphisms of RecQ1, RAD54L and ATM genes are associated with reduced survival of pancreatic cancer. J Clin Oncol. 2006;24:1720–1728. doi: 10.1200/JCO.2005.04.4206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li D, et al. Significant effect of homologous recombination DNA repair gene polymorphisms on pancreatic cancer survival. Cancer Res. 2006;66:3323–3330. doi: 10.1158/0008-5472.CAN-05-3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sharma S, et al. RECQL, a member of the RecQ family of DNA helicases, suppresses chromosomal instability. Mol Cell Biol. 2007;27:1784–1794. doi: 10.1128/MCB.01620-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sharma S, Brosh RM., Jr Human RECQ1 Is a DNA damage responsive protein required for genotoxic stress resistance and suppression of sister chromatid exchanges. PLoS ONE. 2007;2:e1297. doi: 10.1371/journal.pone.0001297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parvathaneni S, et al. Human RECQ1 interacts with Ku70/80 and modulates end-joining of double-strand breaks. PLoS ONE. 2013;8:e62481. doi: 10.1371/journal.pone.0062481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sharma S, Phatak P, Stortchevoi A, Jasin M, Larocque JR. RECQ1 plays a distinct role in cellular response to oxidative DNA damage. DNA Repair. 2012;11:537–549. doi: 10.1016/j.dnarep.2012.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Popuri V, Croteau DL, Brosh RM, Jr, Bohr VA. RECQ1 is required for cellular resistance to replication stress and catalyzes strand exchange on stalled replication fork structures. Cell Cycle. 2012;11:4252–4265. doi: 10.4161/cc.22581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arai A, et al. RECQL1 and WRN proteins are potential therapeutic targets in head and neck squamous cell carcinoma. Cancer Res. 2011;71:4598–4607. doi: 10.1158/0008-5472.CAN-11-0320. [DOI] [PubMed] [Google Scholar]
- 26.Futami K, et al. Induction of mitotic cell death in cancer cells by small interference RNA suppressing the expression of RecQL1 helicase. Cancer Sci. 2008;99:71–80. doi: 10.1111/j.1349-7006.2007.00647.x. [DOI] [PubMed] [Google Scholar]
- 27.Futami K, et al. Anticancer activity of RecQL1 helicase siRNA in mouse xenograft models. Cancer Sci. 2008;99:1227–1236. doi: 10.1111/j.1349-7006.2008.00794.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mendoza-Maldonado R, et al. The human RECQ1 helicase is highly expressed in glioblastoma and plays an important role in tumor cell proliferation. Mol Cancer. 2011;10:83. doi: 10.1186/1476-4598-10-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ramamoorthy M, et al. RECQL5 cooperates with topoisomerase IIα in DNA decatenation and cell cycle progression. Nucleic Acids Res. 2012;40:1621–1635. doi: 10.1093/nar/gkr844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hu Y, et al. RECQL5/Recql5 helicase regulates homologous recombination and suppresses tumor formation via disruption of Rad51 presynaptic filaments. Genes Dev. 2007;21:3073–3084. doi: 10.1101/gad.1609107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Islam MN, et al. RecQL5 promotes genome stabilization through two parallel mechanisms—interacting with RNA polymerase II and acting as a helicase. Mol Cell Biol. 2010;30:2460–2472. doi: 10.1128/MCB.01583-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tadokoro T, et al. Human RECQL5 participates in the removal of endogenous DNA damage. Mol Biol Cell. 2012;23:4273–4285. doi: 10.1091/mbc.E12-02-0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu Y, Brosh RM., Jr DNA helicase and helicase-nuclease enzymes with a conserved iron-sulfur cluster. Nucleic Acids Res. 2012;40:4247–4260. doi: 10.1093/nar/gks039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Digiovanna JJ, Kraemer KH. Shining a light on xeroderma pigmentosum. J Invest Dermatol. 2012;132:785–796. doi: 10.1038/jid.2011.426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Levitus M, et al. The DNA helicase BRIP1 is defective in Fanconi anemia complementation group. J Nature Genet. 2005;37:934–935. doi: 10.1038/ng1625. [DOI] [PubMed] [Google Scholar]
- 36.Levran O, et al. The BRCA1-interacting helicase BRIP1 is deficient in Fanconi anemia. Nature Genet. 2005;37:931–933. doi: 10.1038/ng1624. [DOI] [PubMed] [Google Scholar]
- 37.Litman R, et al. BACH1 is critical for homologous recombination and appears to be the Fanconi anemia gene product FANC. J Cancer Cell. 2005;8:255–265. doi: 10.1016/j.ccr.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 38.Capo-chichi JM, et al. Identification and biochemical characterization of a novel mutation in DDX11 causing Warsaw breakage syndrome. Hum Mut. 2013;34:103–107. doi: 10.1002/humu.22226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van der LP, et al. Warsaw breakage syndrome, a cohesinopathy associated with mutations in the XPD helicase family member DDX11/ChlR1. Am J Hum Genet. 2010;86:262–266. doi: 10.1016/j.ajhg.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ballew BJ, et al. Germline mutations of regulator of telomere elongation helicase 1, RTEL1, in dyskeratosis congenita. Hum Genet. 2013;132:473–480. doi: 10.1007/s00439-013-1265-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Le Guen T, et al. Human RTEL1 deficiency causes Hoyeraal-Hreidarsson syndrome with short telomeres and genome instability. Hum Mol Genet. 2013 Apr 15; doi: 10.1093/hmg/ddt178. [DOI] [PubMed] [Google Scholar]
- 42.Walne AJ, Vulliamy T, Kirwan M, Plagnol V, Dokal I. Constitutional mutations in RTEL1 cause severe dyskeratosis congenita. Am J Hum Genet. 2013;92:448–453. doi: 10.1016/j.ajhg.2013.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Flejter WL, McDaniel LD, Askari M, Friedberg EC, Schultz RA. Characterization of a complex chromosomal rearrangement maps the locus for in vitro complementation of xeroderma pigmentosum group D to human chromosome band 19q13. Genes Chromosomes Cancer. 1992;5:335–342. doi: 10.1002/gcc.2870050409. [DOI] [PubMed] [Google Scholar]
- 44.Flejter WL, McDaniel LD, Johns D, Friedberg EC, Schultz RA. Correction of xeroderma pigmentosum complementation group D mutant cell phenotypes by chromosome and gene transfer: involvement of the human ERCC2 DNA repair gene. Proc Natl Acad Sci USA. 1992;89:261–265. doi: 10.1073/pnas.89.1.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kee Y, D’Andrea AD. Molecular pathogenesis and clinical management of Fanconi anemia. J Clin Invest. 2012;122:3799–3806. doi: 10.1172/JCI58321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cantor SB, Guillemette S. Hereditary breast cancer and the BRCA1-associated FANCJ/BACH1/BRIP1. Future Oncol. 2011;7:253–261. doi: 10.2217/fon.10.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Uringa EJ, et al. RTEL1 contributes to DNA replication and repair and telomere maintenance. Mol Biol Cell. 2012;23:2782–2792. doi: 10.1091/mbc.E12-03-0179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vannier JB, Pavicic-Kaltenbrunner V, Petalcorin MI, Ding H, Boulton SJ. RTEL1 dismantles T loops and counteracts telomeric G4-DNA to maintain telomere integrity. Cell. 2012;149:795–806. doi: 10.1016/j.cell.2012.03.030. This work shows that the RTEL1 helicase has a crucial role in telomere maintenance by disassembling T-loops and counteracting telomeric G-quadruplexes, which potentially impede replication or DNA repair and pose a source of telomere instability. [DOI] [PubMed] [Google Scholar]
- 49.Barber LJ, et al. RTEL1 maintains genomic stability by suppressing homologous recombination. Cell. 2008;135:261–271. doi: 10.1016/j.cell.2008.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Egan KM, et al. Cancer susceptibility variants and the risk of adult glioma in a US case-control study. J Neurooncol. 2011;104:535–542. doi: 10.1007/s11060-010-0506-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shete S, et al. Genome-wide association study identifies five susceptibility loci for glioma. Nature Genet. 2009;41:899–904. doi: 10.1038/ng.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wrensch M, et al. Variants in the CDKN2B and RTEL1 regions are associated with high-grade glioma susceptibility. Nature Genet. 2009;41:905–908. doi: 10.1038/ng.408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Weeda G, et al. Molecular cloning and biological characterization of the human excision repair gene ERCC-3. Mol Cell Biol. 1990;10:2570–2581. doi: 10.1128/mcb.10.6.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Weeda G, et al. A presumed DNA helicase encoded by ERCC-3 is involved in the human repair disorders xeroderma pigmentosum and Cockayne’s syndrome. Cell. 1990;62:777–791. doi: 10.1016/0092-8674(90)90122-u. [DOI] [PubMed] [Google Scholar]
- 55.Gagou ME, et al. Suppression of apoptosis by PIF1 helicase in human tumor cells. Cancer Res. 2011;71:4998–5008. doi: 10.1158/0008-5472.CAN-10-4404. [DOI] [PubMed] [Google Scholar]
- 56.Chisholm KM, et al. A genomewide screen for suppressors of Alu-mediated rearrangements reveals a role for PIF1. PLoS ONE. 2012;7:e30748. doi: 10.1371/journal.pone.0030748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Alzu A, et al. Senataxin associates with replication forks to protect fork integrity across RNA-Polymerase-II-transcribed genes. Cell. 2012;151:835–846. doi: 10.1016/j.cell.2012.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yuce-Petronczki O, West SC. Senataxin, defective in the neurogenerative disorder AOA-2, lies at the interface of transcription and the DNA damage response. Mol Cell Biol. 2012;33:406–417. doi: 10.1128/MCB.01195-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kang YH, Lee CH, Seo YS. Dna2 on the road to Okazaki fragment processing and genome stability in eukaryotes. Crit Rev Biochem Mol Biol. 2010;45:71–96. doi: 10.3109/10409230903578593. [DOI] [PubMed] [Google Scholar]
- 60.Duxin JP, et al. Human Dna2 is a nuclear and mitochondrial DNA maintenance protein. Mol Cell Biol. 2009;29:4274–4282. doi: 10.1128/MCB.01834-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nimonkar AV, et al. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev. 2011;25:350–362. doi: 10.1101/gad.2003811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zheng L, et al. Human DNA2 is a mitochondrial nuclease/helicase for efficient processing of DNA replication and repair intermediates. Mol Cell. 2008;32:325–336. doi: 10.1016/j.molcel.2008.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Copeland WC. Defects in mitochondrial DNA replication and human disease. Crit Rev Biochem Mol Biol. 2012;47:64–74. doi: 10.3109/10409238.2011.632763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Brosh RM, Jr, Waheed J, Sommers JA. Biochemical characterization of the DNA substrate specificity of Werner syndrome helicase. J Biol Chem. 2002;277:23236–23245. doi: 10.1074/jbc.M111446200. [DOI] [PubMed] [Google Scholar]
- 65.Suhasini AN, Brosh RM., Jr Mechanistic and biological aspects of helicase action on damaged DNA. Cell Cycle. 2010;9:2317–2329. doi: 10.4161/cc.9.12.11902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sontz PA, Mui TP, Fuss JO, Tainer JA, Barton JK. DNA charge transport as a first step in coordinating the detection of lesions by repair proteins. Proc Natl Acad Sci USA. 2012;109:1856–1861. doi: 10.1073/pnas.1120063109. This study elegantly describes how DNA damage can be efficiently recognized by collaboration between DNA repair proteins that are able to carry out DNA charge transport. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Suhasini AN, et al. FANCJ helicase uniquely senses oxidative base damage in either strand of duplex DNA and is stimulated by replication protein A to unwind the damaged DNA substrate in a strand-specific manner. J Biol Chem. 2009;284:18458–18470. doi: 10.1074/jbc.M109.012229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ghosh A, Rossi ML, Aulds J, Croteau D, Bohr VA. Telomeric D-loops containing 8-oxo-2′-deoxyguanosine are preferred substrates for Werner and Bloom syndrome helicases and are bound by POT1. J Biol Chem. 2009;284:31074–31084. doi: 10.1074/jbc.M109.027532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Indiani C, O’Donnell MA. Proposal: source of single strand DNA that elicits the SOS response. Front Biosci. 2013;18:312–323. doi: 10.2741/4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Honda M, Park J, Pugh RA, Ha T, Spies M. Single-molecule analysis reveals differential effect of ssDNA-binding proteins on DNA translocation by XPD helicase. Mol Cell. 2009;35:694–703. doi: 10.1016/j.molcel.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Reardon JT, Sancar A. Nucleotide excision repair. Prog Nucleic Acid Res Mol Biol. 2005;79:183–235. doi: 10.1016/S0079-6603(04)79004-2. [DOI] [PubMed] [Google Scholar]
- 72.Egly JM, Coin F. A history of TFIIH: Two decades of molecular biology on a pivotal transcription/repair factor. DNA Repair. 2011;10:714–721. doi: 10.1016/j.dnarep.2011.04.021. This review by two leading experts provides an interesting discussion of the molecular functions of TFIIH and their relation to diseases that are characterized by defects in DNA repair and transcription. [DOI] [PubMed] [Google Scholar]
- 73.Sugasawa K, Akagi J, Nishi R, Iwai S, Hanaoka F. Two-step recognition of DNA damage for mammalian nucleotide excision repair: directional binding of the XPC complex and DNA strand scanning. Mol Cell. 2009;36:642–653. doi: 10.1016/j.molcel.2009.09.035. This paper describes in a refined system the interlinked mechanism for the initial detection and verification steps of mammalian NER. These steps involve strand-specific DNA binding by XPC and 5′ to 3′ translocation by XPD to search for and authenticate the lesion. [DOI] [PubMed] [Google Scholar]
- 74.Mathieu N, Kaczmarek N, Ruthemann P, Luch A, Naegeli H. DNA quality control by a lesion sensor pocket of the xeroderma pigmentosum group D helicase subunit of TFIIH. Curr Biol. 2013;23:204–212. doi: 10.1016/j.cub.2012.12.032. [DOI] [PubMed] [Google Scholar]
- 75.Fuss JO, Tainer JA. XPB and XPD helicases in TFIIH orchestrate DNA duplex opening and damage verification to coordinate repair with transcription and cell cycle via CAK kinase. DNA Repair. 2011;10:697–713. doi: 10.1016/j.dnarep.2011.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Clarkson SG, Wood RD. Polymorphisms in the human XPD (ERCC2) gene, DNA repair capacity and cancer susceptibility: an appraisal. DNA Repair. 2005;4:1068–1074. doi: 10.1016/j.dnarep.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 77.Deans AJ, West SC. DNA interstrand crosslink repair and cancer. Nature Rev Cancer. 2011;11:467–480. doi: 10.1038/nrc3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Garaycoechea JI, et al. Genotoxic consequences of endogenous aldehydes on mouse haematopoietic stem cell function. Nature. 2012;489:571–575. doi: 10.1038/nature11368. [DOI] [PubMed] [Google Scholar]
- 79.Rosado IV, Langevin F, Crossan GP, Takata M, Patel KJ. Formaldehyde catabolism is essential in cells deficient for the Fanconi anemia DNA-repair pathway. Nature Struct Mol Biol. 2011;18:1432–1434. doi: 10.1038/nsmb.2173. [DOI] [PubMed] [Google Scholar]
- 80.Adamo A, et al. Preventing nonhomologous end joining suppresses DNA repair defects of Fanconi anemia. Mol Cell. 2010;39:25–35. doi: 10.1016/j.molcel.2010.06.026. [DOI] [PubMed] [Google Scholar]
- 81.Pace P, et al. Ku70 corrupts DNA repair in the absence of the Fanconi anemia pathway. Science. 2010;329:219–223. doi: 10.1126/science.1192277. References 80 and 81 describe how the FA DNA repair pathway suppresses toxic NHEJ in the repair of DNA ICLs. [DOI] [PubMed] [Google Scholar]
- 82.Meetei AR, et al. A human ortholog of archaeal DNA repair protein Hef is defective in Fanconi anemia complementation group M. Nature Genet. 2005;37:958–963. doi: 10.1038/ng1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Singh TR, et al. MHF1-MHF2, a histone-fold-containing protein complex, participates in the Fanconi anemia pathway via FANCM. Mol Cell. 2010;37:879–886. doi: 10.1016/j.molcel.2010.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Peng M, et al. The FANCJ/MutLα interaction is required for correction of the cross-link response in FA-J cells. EMBO J. 2007;26:3238–3249. doi: 10.1038/sj.emboj.7601754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Meetei AR, et al. A multiprotein nuclear complex connects Fanconi anemia and Bloom syndrome. Mol Cell Biol. 2003;23:3417–3426. doi: 10.1128/MCB.23.10.3417-3426.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Deans AJ, West SC. FANCM connects the genome instability disorders Bloom’s Syndrome and Fanconi Anemia. Mol Cell. 2009;36:943–953. doi: 10.1016/j.molcel.2009.12.006. References 85 and 86 together demonstrate that FA and Bloom syndrome are linked by proteins that function in a common nuclear complex through protein interaction domains in FANCM that facilitate its role in ICL repair and the maintenance of genomic stability. [DOI] [PubMed] [Google Scholar]
- 87.Larocque JR, et al. Interhomolog recombination and loss of heterozygosity in wild-type and Bloom syndrome helicase (BLM)-deficient mammalian cells. Proc Natl Acad Sci USA. 2011;108:11971–11976. doi: 10.1073/pnas.1104421108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Luo G, et al. Cancer predisposition caused by elevated mitotic recombination in Bloom mice. Nature Genet. 2000;26:424–429. doi: 10.1038/82548. [DOI] [PubMed] [Google Scholar]
- 89.Suhasini AN, et al. Interaction between the helicases genetically linked to Fanconi anemia Group J and Bloom’s syndrome. EMBO J. 2011;30:692–705. doi: 10.1038/emboj.2010.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Suhasini AN, et al. Fanconi anemia Group J helicase and MRE11 nuclease interact to facilitate the DNA damage response. Mol Cell Biol. 2013;33:2212–2227. doi: 10.1128/MCB.01256-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cheng WH, et al. Collaboration of Werner syndrome protein and BRCA1 in cellular responses to DNA interstrand cross-links. Nucleic Acids Res. 2006;34:2751–2760. doi: 10.1093/nar/gkl362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cheng WH, et al. WRN Is required for ATM activation and the S-phase checkpoint in response to interstrand crosslink-induced DNA double strand breaks. Mol Biol Cell. 2008;19:3923–3933. doi: 10.1091/mbc.E07-07-0698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pichierri P, Ammazzalorso F, Bignami M, Franchitto A. The Werner syndrome protein: linking the replication checkpoint response to genome stability. Aging. 2011;3:311–318. doi: 10.18632/aging.100293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Moldovan GL, et al. DNA polymerase POLN participates in cross-link repair and homologous recombination. Mol Cell Biol. 2010;30:1088–1096. doi: 10.1128/MCB.01124-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ward JD, et al. Overlapping mechanisms promote postsynaptic RAD-51 filament disassembly during meiotic double-strand break repair. Mol Cell. 2010;37:259–272. doi: 10.1016/j.molcel.2009.12.026. [DOI] [PubMed] [Google Scholar]
- 96.Tafel AA, Wu L, McHugh PJ. Human HEL308 localizes to damaged replication forks and unwinds lagging strand structures. J Biol Chem. 2011;286:15832–15840. doi: 10.1074/jbc.M111.228189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chan KL, Palmai-Pallag T, Ying S, Hickson ID. Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nature Cell Biol. 2009;11:753–760. doi: 10.1038/ncb1882. [DOI] [PubMed] [Google Scholar]
- 98.Naim V, Rosselli F. The FANC pathway and BLM collaborate during mitosis to prevent micro-nucleation and chromosome abnormalities. Nature Cell Biol. 2009;11:761–768. doi: 10.1038/ncb1883. [DOI] [PubMed] [Google Scholar]
- 99.Bernstein KA, Gangloff S, Rothstein R. The RecQ DNA helicases in DNA repair. Annu Rev Genet. 2010;44:393–417. doi: 10.1146/annurev-genet-102209-163602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Schwendener S, et al. Physical interaction of RECQ5 helicase with RAD51 facilitates its anti-recombinase activity. J Biol Chem. 2010;285:15739–15745. doi: 10.1074/jbc.M110.110478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nimonkar AV, Ozsoy AZ, Genschel J, Modrich P, Kowalczykowski SC. Human exonuclease 1 and BLM helicase interact to resect DNA and initiate DNA repair. Proc Natl Acad Sci USA. 2008;105:16906–16911. doi: 10.1073/pnas.0809380105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bugreev DV, Yu X, Egelman EH, Mazin AV. Novel pro- and anti-recombination activities of the Bloom’s syndrome helicase. Genes Dev. 2007;21:3085–3094. doi: 10.1101/gad.1609007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chapman JR, Taylor MR, Boulton SJ. Playing the end game: DNA double-strand break repair pathway choice. Mol Cell. 2012;47:497–510. doi: 10.1016/j.molcel.2012.07.029. [DOI] [PubMed] [Google Scholar]
- 104.Cooper MP, et al. Ku complex interacts with and stimulates the Werner protein. Genes Dev. 2000;14:907–912. [PMC free article] [PubMed] [Google Scholar]
- 105.Kusumoto R, et al. Werner protein cooperates with the XRCC4-DNA ligase IV complex in end-processing. Biochemistry. 2008;47:7548–7556. doi: 10.1021/bi702325t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sallmyr A, Tomkinson AE, Rassool FV. Up-regulation of WRN and DNA ligase IIIα in chronic myeloid leukemia: consequences for the repair of DNA double-strand breaks. Blood. 2008;112:1413–1423. doi: 10.1182/blood-2007-07-104257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chen L, et al. WRN, the protein deficient in Werner syndrome, plays a critical structural role in optimizing DNA repair. Aging Cell. 2003;2:191–199. doi: 10.1046/j.1474-9728.2003.00052.x. [DOI] [PubMed] [Google Scholar]
- 108.Perry JJ, et al. WRN exonuclease structure and molecular mechanism imply an editing role in DNA end processing. Nature Struct Mol Biol. 2006;13:414–422. doi: 10.1038/nsmb1088. [DOI] [PubMed] [Google Scholar]
- 109.Swanson C, Saintigny Y, Emond MJ, Monnat RJ., Jr The Werner syndrome protein has separable recombination and survival functions. DNA Repair. 2004;3:475–482. doi: 10.1016/j.dnarep.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 110.Kennedy RD, et al. Fanconi anemia pathway-deficient tumor cells are hypersensitive to inhibition of ataxia telangiectasia mutated. J Clin Invest. 2007;117:1440–1449. doi: 10.1172/JCI31245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.von Kobbe C, et al. Central role for the Werner syndrome protein/poly(ADP-ribose) polymerase 1 complex in the poly(ADP-ribosyl)ation pathway after DNA damage. Mol Cell Biol. 2003;23:8601–8613. doi: 10.1128/MCB.23.23.8601-8613.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Woo LL, Futami K, Shimamoto A, Furuichi Y, Frank KM. The Rothmund-Thomson gene product RECQL4 localizes to the nucleolus in response to oxidative stress. Exp Cell Res. 2006;312:3443–3457. doi: 10.1016/j.yexcr.2006.07.023. [DOI] [PubMed] [Google Scholar]
- 113.Lebel M, Lavoie J, Gaudreault I, Bronsard M, Drouin R. Genetic cooperation between the Werner syndrome protein and poly(ADP-ribose) polymerase-1 in preventing chromatid breaks, complex chromosomal rearrangements, and cancer in mice. Am J Pathol. 2003;162:1559–1569. doi: 10.1016/S0002-9440(10)64290-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Rossi ML, Ghosh AK, Bohr VA. Roles of Werner syndrome protein in protection of genome integrity. DNA Repair. 2010;9:331–344. doi: 10.1016/j.dnarep.2009.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hewish M, Lord CJ, Martin SA, Cunningham D, Ashworth A. Mismatch repair deficient colorectal cancer in the era of personalized treatment. Nature Rev Clin Oncol. 2010;7:197–208. doi: 10.1038/nrclinonc.2010.18. [DOI] [PubMed] [Google Scholar]
- 116.Lahue RS, Au KG, Modrich P. DNA mismatch correction in a defined system. Science. 1989;245:160–164. doi: 10.1126/science.2665076. [DOI] [PubMed] [Google Scholar]
- 117.Song L, Yuan F, Zhang Y. Does a helicase activity help mismatch repair in eukaryotes? IUBMB Life. 2010;62:548–553. doi: 10.1002/iub.349. [DOI] [PubMed] [Google Scholar]
- 118.Sharma S, et al. The exonucleolytic and endonucleolytic cleavage activities of human exonuclease 1 are stimulated by an interaction with the carboxyl-terminal region of the Werner syndrome protein. J Biol Chem. 2003;278:23487–23496. doi: 10.1074/jbc.M212798200. [DOI] [PubMed] [Google Scholar]
- 119.Doherty KM, et al. RECQ1 helicase interacts with human mismatch repair factors that regulate genetic recombination. J Biol Chem. 2005;280:28085–28094. doi: 10.1074/jbc.M500265200. [DOI] [PubMed] [Google Scholar]
- 120.Helleday T. Amplifying tumour-specific replication lesions by DNA repair inhibitors — a new era in targeted cancer therapy. Eur J Cancer. 2008;44:921–927. doi: 10.1016/j.ejca.2008.02.044. [DOI] [PubMed] [Google Scholar]
- 121.Hegnauer AM, et al. An N-terminal acidic region of Sgs1 interacts with Rpa70 and recruits Rad53 kinase to stalled forks. EMBO J. 2012;31:3768–3783. doi: 10.1038/emboj.2012.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bjergbaek L, Cobb JA, Tsai-Pflugfelder M, Gasser SM. Mechanistically distinct roles for Sgs1p in checkpoint activation and replication fork maintenance. EMBO J. 2005;24:405–417. doi: 10.1038/sj.emboj.7600511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Frei C, Gasser SM. The yeast Sgs1p helicase acts upstream of Rad53p in the DNA replication checkpoint and colocalizes with Rad53p in S-phase-specific foci. Genes Dev. 2000;14:81–96. [PMC free article] [PubMed] [Google Scholar]
- 124.Doherty KM, et al. Physical and functional mapping of the RPA interaction domain of the Werner and Bloom syndrome helicases. J Biol Chem. 2005;280:29494–29505. doi: 10.1074/jbc.M500653200. [DOI] [PubMed] [Google Scholar]
- 125.Sidorova JM, Li N, Folch A, Monnat RJ., Jr The RecQ helicase WRN is required for normal replication fork progression after DNA damage or replication fork arrest. Cell Cycle. 2008;7:796–807. doi: 10.4161/cc.7.6.5566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Machwe A, Xiao L, Groden J, Orren DK. The Werner and Bloom syndrome proteins catalyze regression of a model replication fork. Biochemistry. 2006;45:13939–13946. doi: 10.1021/bi0615487. [DOI] [PubMed] [Google Scholar]
- 127.Machwe A, Karale R, Xu X, Liu Y, Orren DK. The Werner and Bloom syndrome proteins help resolve replication blockage by converting (regressed) Holliday junctions to functional replication forks. Biochemistry. 2011;50:6774–6788. doi: 10.1021/bi2001054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ralf C, Hickson ID, Wu L. The Bloom’s syndrome helicase can promote the regression of a model replication fork. J Biol Chem. 2006;281:22839–22846. doi: 10.1074/jbc.M604268200. [DOI] [PubMed] [Google Scholar]
- 129.Berti M, et al. Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nature Struct Mol Biol. 2013;20:347–354. doi: 10.1038/nsmb.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Fugger K, et al. FBH1 co-operates with MUS81 in inducing DNA double-strand breaks and cell death following replication stress. Nature Commun. 2013;4:1423. doi: 10.1038/ncomms2395. [DOI] [PubMed] [Google Scholar]
- 131.Jeong YT, et al. FBH1 promotes DNA double-strand breakage and apoptosis in response to DNA replication stress. J Cell Biol. 2013;200:141–149. doi: 10.1083/jcb.201209002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kumaraswamy E, Shiekhattar R. Activation of BRCA1/BRCA2-associated helicase BACH1 is required for timely progression through S phase. Mol Cell Biol. 2007;27:6733–6741. doi: 10.1128/MCB.00961-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Gong Z, Kim JE, Leung CC, Glover JN, Chen J. BACH1/FANCJ acts with TopBP1 and participates early in DNA replication checkpoint control. Mol Cell. 2010;37:438–446. doi: 10.1016/j.molcel.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Gari K, Decaillet C, Delannoy M, Wu L, Constantinou A. Remodeling of DNA replication structures by the branch point translocase FANCM. Proc Natl Acad Sci USA. 2008;105:16107–16112. doi: 10.1073/pnas.0804777105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Yan Z, et al. A histone-fold complex and FANCM form a conserved DNA-remodeling complex to maintain genome stability. Mol Cell. 2010;37:865–878. doi: 10.1016/j.molcel.2010.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Betous R, et al. SMARCAL1 catalyzes fork regression and Holliday junction migration to maintain genome stability during DNA replication. Genes Dev. 2012;26:151–162. doi: 10.1101/gad.178459.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Yusufzai T, Kadonaga JT. HARP is an ATP-driven annealing helicase. Science. 2008;322:748–750. doi: 10.1126/science.1161233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Euskirchen G, Auerbach RK, Snyder M. SWI/SNF chromatin-remodeling factors: multiscale analyses and diverse functions. J Biol Chem. 2012;287:30897–30905. doi: 10.1074/jbc.R111.309302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Lai AY, Wade PA. Cancer biology and NuRD: a multifaceted chromatin remodelling complex. Nature Rev Cancer. 2011;11:588–596. doi: 10.1038/nrc3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.McGlynn P, Savery NJ, Dillingham MS. The conflict between DNA replication and transcription. Mol Microbiol. 2012;85:12–20. doi: 10.1111/j.1365-2958.2012.08102.x. [DOI] [PubMed] [Google Scholar]
- 141.de Septenville AL, Duigou S, Boubakri H, Michel B. Replication fork reversal after replication-transcription collision. PLoS Genet. 2012;8:e1002622. doi: 10.1371/journal.pgen.1002622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lutzmann M, et al. MCM8- and MCM9-deficient mice reveal gametogenesis defects and genome instability due to impaired homologous recombination. Mol Cell. 2012;47:523–534. doi: 10.1016/j.molcel.2012.05.048. [DOI] [PubMed] [Google Scholar]
- 143.Nishimura K, et al. Mcm8 and Mcm9 form a complex that functions in homologous recombination repair induced by DNA interstrand crosslinks. Mol Cell. 2012;47:511–522. doi: 10.1016/j.molcel.2012.05.047. References 142 and 143 demonstrate that two MCM proteins that are traditionally known to be involved in DNA replication have a role in HR-mediated DNA repair that is important for cancer suppression and fertility. [DOI] [PubMed] [Google Scholar]
- 144.Hartford SA, et al. Minichromosome maintenance helicase paralog MCM9 is dispensible for DNA replication but functions in germ-line stem cells and tumor suppression. Proc Natl Acad Sci USA. 2011;108:17702–17707. doi: 10.1073/pnas.1113524108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Aguilera A, Garcia-Muse T. R loops: from transcription byproducts to threats to genome stability. Mol Cell. 2012;46:115–124. doi: 10.1016/j.molcel.2012.04.009. [DOI] [PubMed] [Google Scholar]
- 146.Sabouri N, McDonald KR, Webb CJ, Cristea IM, Zakian VA. DNA replication through hard-to-replicate sites, including both highly transcribed RNA Pol II and Pol III genes, requires the S. pombe Pfh1 helicase. Genes Dev. 2012;26:581–593. doi: 10.1101/gad.184697.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Steinacher R, Osman F, Dalgaard JZ, Lorenz A, Whitby MC. The DNA helicase Pfh1 promotes fork merging at replication termination sites to ensure genome stability. Genes Dev. 2012;26:594–602. doi: 10.1101/gad.184663.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Bochman ML, Paeschke K, Zakian VA. DNA secondary structures: stability and function of G-quadruplex structures. Nature Rev Genet. 2012;13:770–780. doi: 10.1038/nrg3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Wu Y, Brosh RM., Jr G-quadruplex nucleic acids and human disease. FEBS J. 2010;277:3470–3488. doi: 10.1111/j.1742-4658.2010.07760.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Paeschke K, Capra JA, Zakian VA. DNA replication through G-quadruplex motifs is promoted by the Saccharomyces cerevisiae Pif1 DNA helicase. Cell. 2011;145:678–691. doi: 10.1016/j.cell.2011.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Ribeyre C, et al. The yeast Pif1 helicase prevents genomic instability caused by G-quadruplex-forming CEB1 sequences in vivo. PLoS Genet. 2009;5:e1000475. doi: 10.1371/journal.pgen.1000475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Piazza A, et al. Genetic instability triggered by G-quadruplex interacting Phen-DC compounds in Saccharomyces cerevisiae. Nucleic Acids Res. 2010;38:4337–4348. doi: 10.1093/nar/gkq136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Lopes J, et al. G-quadruplex-induced instability during leading-strand replication. EMBO J. 2011;30:4033–4046. doi: 10.1038/emboj.2011.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Cheung I, Schertzer M, Rose A, Lansdorp PM. Disruption of dog-1 in Caenorhabditis elegans triggers deletions upstream of guanine-rich DNA. Nature Genet. 2002;31:405–409. doi: 10.1038/ng928. [DOI] [PubMed] [Google Scholar]
- 155.Wu Y, Shin-Ya K, Brosh RM., Jr FANCJ helicase defective in Fanconia anemia and breast cancer unwinds G-quadruplex DNA to defend genomic stability. Mol Cell Biol. 2008;28:4116–4128. doi: 10.1128/MCB.02210-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Shin-Ya K, et al. Telomestatin, a novel telomerase inhibitor from Streptomyces anulatus. J Am Chem Soc. 2001;123:1262–1263. doi: 10.1021/ja005780q. [DOI] [PubMed] [Google Scholar]
- 157.London TB, et al. FANCJ is a structure-specific DNA helicase associated with the maintenance of genomic G/C tracts. J Biol Chem. 2008;283:36132–36139. doi: 10.1074/jbc.M808152200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Balasubramanian S, Hurley LH, Neidle S. Targeting G-quadruplexes in gene promoters: a novel anticancer strategy? Nature Rev Drug Discov. 2011;10:261–275. doi: 10.1038/nrd3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Johnson JE, Cao K, Ryvkin P, Wang LS, Johnson FB. Altered gene expression in the Werner and Bloom syndromes is associated with sequences having G-quadruplex forming potential. Nucleic Acids Res. 2010;38:1114–1122. doi: 10.1093/nar/gkp1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Corpet A, Almouzni G. Making copies of chromatin: the challenge of nucleosomal organization and epigenetic information. Trends Cell Biol. 2009;19:29–41. doi: 10.1016/j.tcb.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 161.Sarkies P, Reams C, Simpson LJ, Sale JE. Epigenetic instability due to defective replication of structured DNA. Mol Cell. 2010;40:703–713. doi: 10.1016/j.molcel.2010.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Sarkies P, et al. FANCJ coordinates two pathways that maintain epigenetic stability at G-quadruplex DNA. Nucleic Acids Res. 2012;40:1485–1498. doi: 10.1093/nar/gkr868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Schwab RA, Nieminuszczy J, Shin-Ya K, Niedzwiedz W. FANCJ couples replication past natural fork barriers with maintenance of chromatin structure. J Cell Biol. 2013;201:33–48. doi: 10.1083/jcb.201208009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Kamath-Loeb AS, Lan L, Nakajima S, Yasui A, Loeb LA. Werner syndrome protein interacts functionally with translesion DNA polymerases. Proc Natl Acad Sci USA. 2007;104:10394–10399. doi: 10.1073/pnas.0702513104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Kamath-Loeb AS, Shen JC, Schmitt MW, Loeb LA. The Werner syndrome exonuclease facilitates DNA degradation and high fidelity DNA polymerization by human DNA polymerase δ. J Biol Chem. 2012;287:12480–12490. doi: 10.1074/jbc.M111.332577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Dejardin J, Kingston RE. Purification of proteins associated with specific genomic loci. Cell. 2009;136:175–186. doi: 10.1016/j.cell.2008.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Sfeir A, et al. Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell. 2009;138:90–103. doi: 10.1016/j.cell.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Opresko PL, et al. The Werner Syndrome helicase and exonuclease cooperate to resolve telomeric D loops in a manner regulated by TRF1 and TRF2. Mol Cell. 2004;14:763–774. doi: 10.1016/j.molcel.2004.05.023. [DOI] [PubMed] [Google Scholar]
- 169.Chang S, et al. Essential role of limiting telomeres in the pathogenesis of Werner syndrome. Nature Genet. 2004;36:877–882. doi: 10.1038/ng1389. [DOI] [PubMed] [Google Scholar]
- 170.Du X, et al. Telomere shortening exposes functions for the mouse Werner and Bloom syndrome genes. Mol Cell Biol. 2004;24:8437–8446. doi: 10.1128/MCB.24.19.8437-8446.2004. References 169 and 170 provide evidence using mouse models that WRN has an important role in suppressing chromosomal instability, cellular senescence and premature aging through its role in preserving the integrity of critically short telomeres. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Laud PR, et al. Elevated telomere-telomere recombination in WRN-deficient, telomere dysfunctional cells promotes escape from senescence and engagement of the ALT pathway. Genes Dev. 2005;19:2560–2570. doi: 10.1101/gad.1321305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Crabbe L, Verdun RE, Haggblom CI, Karlseder J. Defective telomere lagging strand synthesis in cells lacking WRN helicase activity. Science. 2004;306:1951–1953. doi: 10.1126/science.1103619. This paper shows that in human cells the WRN helicase is required for the efficient replication of lagging-strand telomeric DNA. The study supports a model in which WRN resolves abnormal DNA structures that perturb replication fork progression. [DOI] [PubMed] [Google Scholar]
- 173.Saharia A, et al. Flap endonuclease 1 contributes to telomere stability. Curr Biol. 2008;18:496–500. doi: 10.1016/j.cub.2008.02.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Sharma S, et al. WRN helicase and FEN-1 form a complex upon replication arrest and together process branch-migrating DNA structures associated with the replication fork. Mol Biol Cell. 2004;15:734–750. doi: 10.1091/mbc.E03-08-0567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Fan Q, Zhang F, Barrett B, Ren K, Andreassen PR. A role for monoubiquitinated FANCD2 at telomeres in ALT cells. Nucleic Acids Res. 2009;37:1740–1754. doi: 10.1093/nar/gkn995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Rhee DB, et al. FANCC suppresses short telomere-initiated telomere sister chromatid exchange. Hum Mol Genet. 2010;19:879–887. doi: 10.1093/hmg/ddp556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Agrelo R, et al. Epigenetic inactivation of the premature aging Werner syndrome gene in human cancer. Proc Natl Acad Sci USA. 2006;103:8822–8827. doi: 10.1073/pnas.0600645103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Zoppoli G, et al. Putative DNA/RNA helicase Schlafen-11 (SLFN11) sensitizes cancer cells to DNA-damaging agents. Proc Natl Acad Sci USA. 2012;109:15030–15035. doi: 10.1073/pnas.1205943109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Curtin NJ. DNA repair dysregulation from cancer driver to therapeutic target. Nature Rev Cancer. 2012;12:801–817. doi: 10.1038/nrc3399. [DOI] [PubMed] [Google Scholar]
- 180.Bouwman P, Jonkers J. The effects of deregulated DNA damage signalling on cancer chemotherapy response and resistance. Nature Rev Cancer. 2012;12:587–598. doi: 10.1038/nrc3342. [DOI] [PubMed] [Google Scholar]
- 181.Bryant HE, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 182.Farmer H, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. References 181 and 182 demonstrate that inhibitors of the single-strand break DNA repair protein PARP1 are synthetic lethal in HR-deficient cells with mutations in breast cancer susceptibility proteins BRCA1 or BRCA2. This discovery paved the way for researchers to study synthetic lethality to improve cancer therapy. [DOI] [PubMed] [Google Scholar]
- 183.Aggarwal M, Brosh RM., Jr Hitting the bull’s eye: novel directed cancer therapy through helicase-targeted synthetic lethality. J Cell Biochem. 2009;106:758–763. doi: 10.1002/jcb.22048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Moser R, et al. MYC-driven tumorigenesis is inhibited by WRN syndrome gene deficiency. Mol Cancer Res. 2012;10:535–545. doi: 10.1158/1541-7786.MCR-11-0508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Aggarwal M, Sommers JA, Shoemaker RH, Brosh RM., Jr Inhibition of helicase activity by a small molecule impairs Werner syndrome helicase (WRN) function in the cellular response to DNA damage or replication stress. Proc Natl Acad Sci USA. 2011;108:1525–1530. doi: 10.1073/pnas.1006423108. This paper provides the first evidence that a human DNA helicase (WRN) could be inhibited by a small molecule in a cell-based system to modulate its role in helicase-dependent pathways that are important for DNA repair and the replication stress response. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Murai J, et al. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012;72:5588–5599. doi: 10.1158/0008-5472.CAN-12-2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Nguyen GH, et al. A small molecule Inhibitor of the BLM helicase modulates chromosome stability in human cells. Chem Biol. 2013;20:55–62. doi: 10.1016/j.chembiol.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Shay JW, Bacchetti S. A survey of telomerase activity in human cancer. Eur J Cancer. 1997;33:787–791. doi: 10.1016/S0959-8049(97)00062-2. [DOI] [PubMed] [Google Scholar]
- 189.Buseman CM, Wright WE, Shay JW. Is telomerase a viable target in cancer? Mutat Res. 2012;730:90–97. doi: 10.1016/j.mrfmmm.2011.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Li Q, Xiang JF, Zhang H, Tang YL. Searching drug-like anti-cancer compound(s) based on G-quadruplex ligands. Curr Pharm Des. 2012;18:1973–1983. doi: 10.2174/138161212799958369. [DOI] [PubMed] [Google Scholar]
- 191.Gomez D, et al. Telomestatin-induced telomere uncapping is modulated by POT1 through G-overhang extension in HT1080 human tumor cells. J Biol Chem. 2006;281:38721–38729. doi: 10.1074/jbc.M605828200. [DOI] [PubMed] [Google Scholar]
- 192.Rodriguez R, et al. A novel small molecule that alters shelterin integrity and triggers a DNA-damage response at telomeres. J Am Chem Soc. 2008;130:15758–15759. doi: 10.1021/ja805615w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Rodriguez R, et al. Small-molecule-induced DNA damage identifies alternative DNA structures in human genes. Nature Chem Biol. 2012;8:301–310. doi: 10.1038/nchembio.780. This study shows that a G4 ligand (pyridostatin) promotes growth arrest in human cancer cells by inducing replication- and transcription-dependent DNA damage in genomic regions prone to forming G-quadruplexes that are recognized by the human G4-resolving helicase PIF1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Muller S, et al. Pyridostatin analogues promote telomere dysfunction and long-term growth inhibition in human cancer cells. Org Biomol Chem. 2012;10:6537–6546. doi: 10.1039/c2ob25830g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Gocha AR, Harris J, Groden J. Alternative mechanisms of telomere lengthening: permissive mutations, DNA repair proteins and tumorigenic progression. Mutat Res. 2013;743–744:142–150. doi: 10.1016/j.mrfmmm.2012.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Mackintosh SG, Raney KD. DNA unwinding and protein displacement by superfamily 1 and superfamily 2 helicases. Nucleic Acids Res. 2006;34:4154–4159. doi: 10.1093/nar/gkl501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Rad B, Kowalczykowski SC. Efficient coupling of ATP hydrolysis to translocation by RecQ helicase. Proc Natl Acad Sci USA. 2012;109:1443–1448. doi: 10.1073/pnas.1119952109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Sarlos K, Gyimesi M, Kovacs M. RecQ helicase translocates along single-stranded DNA with a moderate processivity and tight mechanochemical coupling. Proc Natl Acad Sci USA. 2012;109:9804–9809. doi: 10.1073/pnas.1114468109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Patel SS, Picha KM. Structure and function of hexameric helicases. Annu Rev Biochem. 2000;69:651–697. doi: 10.1146/annurev.biochem.69.1.651. [DOI] [PubMed] [Google Scholar]
- 200.Spies M, Amitani I, Baskin RJ, Kowalczykowski SC. RecBCD enzyme switches lead motor subunits in response to chi recognition. Cell. 2007;131:694–705. doi: 10.1016/j.cell.2007.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Wong I, Lohman TM. Allosteric effects of nucleotide cofactors on Escherichia coli Rep helicase-DNA binding. Science. 1992;256:350–355. doi: 10.1126/science.256.5055.350. [DOI] [PubMed] [Google Scholar]
- 202.Wu Y, et al. Fanconi anemia Group J mutation abolishes its DNA repair function by uncoupling DNA translocation from helicase activity or disruption of protein-DNA complexes. Blood. 2010;116:3780–3791. doi: 10.1182/blood-2009-11-256016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Zittel MC, Keck JL. Coupling DNA-binding and ATP hydrolysis in Escherichia coli RecQ: role of a highly conserved aromatic-rich sequence. Nucleic Acids Res. 2005;33:6982–6991. doi: 10.1093/nar/gki999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Wu Y, et al. The Q Motif of FANCJ DNA helicase regulates its dimerization, DNA binding, and DNA repair function. J Biol Chem. 2012;287:21699–21716. doi: 10.1074/jbc.M112.351338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Heyer WD, Ehmsen KT, Liu J. Regulation of homologous recombination in eukaryotes. Annu Rev Genet. 2010;44:113–139. doi: 10.1146/annurev-genet-051710-150955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Myong S, Rasnik I, Joo C, Lohman TM, Ha T. Repetitive shuttling of a motor protein on DNA. Nature. 2005;437:1321–1325. doi: 10.1038/nature04049. [DOI] [PubMed] [Google Scholar]
- 207.Myong S, Bruno MM, Pyle AM, Ha T. Spring-loaded mechanism of DNA unwinding by hepatitis C virus NS3 helicase. Science. 2007;317:513–516. doi: 10.1126/science.1144130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Yodh JG, Stevens BC, Kanagaraj R, Janscak P, Ha T. BLM helicase measures DNA unwound before switching strands and hRPA promotes unwinding reinitiation. EMBO J. 2009;28:405–416. doi: 10.1038/emboj.2008.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Sun B, et al. ATP-induced helicase slippage reveals highly coordinated subunits. Nature. 2011;478:132–135. doi: 10.1038/nature10409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Pike AC, et al. Structure of the human RECQ1 helicase reveals a putative strand-separation pin. Proc Natl Acad Sci USA. 2009;106:1039–1044. doi: 10.1073/pnas.0806908106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Kuper J, Wolski SC, Michels G, Kisker C. Functional and structural studies of the nucleotide excision repair helicase XPD suggest a polarity for DNA translocation. EMBO J. 2011;31:494–502. doi: 10.1038/emboj.2011.374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Brosh RM, Jr, Bohr V. A Human premature aging, DNA repair and RecQ helicases. Nucleic Acids Res. 2007;35:7527–7544. doi: 10.1093/nar/gkm1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Sharma S, et al. Biochemical analysis of the DNA unwinding and strand annealing activities catalyzed by human RECQ1. J Biol Chem. 2005;280:28072–28084. doi: 10.1074/jbc.M500264200. [DOI] [PubMed] [Google Scholar]
- 214.Mazina OM, Rossi MJ, Deakyne JS, Huang F, Mazin AV. Polarity and bypass of DNA heterology during branch migration of Holliday junctions by human RAD54, BLM, and RECQ1 proteins. J Biol Chem. 2012;287:11820–11832. doi: 10.1074/jbc.M112.341347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Wu L, Hickson ID. The Bloom’s syndrome helicase suppresses crossing over during homologous recombination. Nature. 2003;426:870–874. doi: 10.1038/nature02253. This paper provides the first molecular evidence for how the BLM helicase prevents SCE during the repair of replication-associated DNA damage, and has implications for understanding mechanisms that suppress tumorigenesis. [DOI] [PubMed] [Google Scholar]
- 216.Long DT, Raschle M, Joukov V, Walter JC. Mechanism of RAD51-dependent DNA interstrand cross-link repair. Science. 2011;333:84–87. doi: 10.1126/science.1204258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Brosh RM, Jr, et al. Werner syndrome protein interacts with human flap endonuclease 1 and stimulates its cleavage activity. EMBO J. 2001;20:5791–5801. doi: 10.1093/emboj/20.20.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Sharma S, et al. Stimulation of flap endonuclease-1 by the Bloom’s syndrome protein. J Biol Chem. 2004;279:9847–9856. doi: 10.1074/jbc.M309898200. [DOI] [PubMed] [Google Scholar]
- 219.Wang W, Bambara RA. Human Bloom protein stimulates flap endonuclease 1 activity by resolving DNA secondary structure. J Biol Chem. 2005;280:5391–5399. doi: 10.1074/jbc.M412359200. [DOI] [PubMed] [Google Scholar]
- 220.Chan NL, et al. The Werner syndrome protein promotes CAG/CTG repeat stability by resolving large (CAG)(n)/(CTG)(n) hairpins. J Biol Chem. 2012;287:30151–30156. doi: 10.1074/jbc.M112.389791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Henry RA, Balakrishnan L, Ying-Lin ST, Campbell JL, Bambara RA. Components of the secondary pathway stimulate the primary pathway of eukaryotic Okazaki fragment processing. J Biol Chem. 2010;285:28496–28505. doi: 10.1074/jbc.M110.131870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.McGlynn P. Helicases that underpin replication of protein-bound DNA in Escherichia coli. Biochem Soc Trans. 2011;39:606–610. doi: 10.1042/BST0390606. [DOI] [PubMed] [Google Scholar]
- 223.Yin J, et al. BLAP75, an essential component of Bloom’s syndrome protein complexes that maintain genome integrity. EMBO J. 2005;24:1465–1476. doi: 10.1038/sj.emboj.7600622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Mao FJ, Sidorova JM, Lauper JM, Emond MJ, Monnat RJ. The human WRN and BLM RecQ helicases differentially regulate cell proliferation and survival after chemotherapeutic DNA damage. Cancer Res. 2010;70:6548–6555. doi: 10.1158/0008-5472.CAN-10-0475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Popuri V, et al. Recruitment and retention dynamics of RECQL5 at DNA double strand break sites. DNA Repair. 2012;11:624–635. doi: 10.1016/j.dnarep.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Peng M, Litman R, Jin Z, Fong G, Cantor SB. BACH1 is a DNA repair protein supporting BRCA1 damage response. Oncogene. 2006;25:2245–2253. doi: 10.1038/sj.onc.1209257. [DOI] [PubMed] [Google Scholar]
- 227.Farina A, et al. Studies with the human cohesin establishment factor, ChlR1. Association of ChlR1 with Ctf18-RFC and Fen1. J Biol Chem. 2008;283:20925–20936. doi: 10.1074/jbc.M802696200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Parish JL, et al. The DNA helicase ChlR1 is required for sister chromatid cohesion in mammalian cells. J Cell Sci. 2006;119:4857–4865. doi: 10.1242/jcs.03262. [DOI] [PubMed] [Google Scholar]
- 229.Bhattacharya C, Wang X, Becker D. The DEAD/DEAH box helicase, DDX11, is essential for the survival of advanced melanomas. Mol Cancer. 2012;11:82. doi: 10.1186/1476-4598-11-82. [DOI] [PMC free article] [PubMed] [Google Scholar]