

Abstract
Background
Integrating transcriptomic experiments within drug development is increasingly advocated for the early detection of toxicity. This is partly to reduce costs related to drug failures in the late, and expensive phases of clinical trials. Such an approach has proven useful both in the study of toxicology and carcinogenicity. However, general lack of translation of in vitro findings to in vivo systems remains one of the bottle necks in drug development. This paper proposes a method for identifying disconnected genes between in vitro and in vivo toxicogenomic rat experiments. The analytical framework is based on the joint modeling of dose-dependent in vitro and in vivo data using a fractional polynomial framework and biclustering algorithm.
Results
Most disconnected genes identified belonged to known pathways, such as drug metabolism and oxidative stress due to reactive metabolites, bilirubin increase, glutathion depletion and phospholipidosis. We also identified compounds that were likely to induce disconnect in gene expression between in vitro and in vivo toxicogenomic rat experiments. These compounds include: sulindac and diclofenac (both linked to liver damage), naphtyl isothiocyanate (linked to hepatoxocity), indomethacin and naproxen (linked to gastrointestinal problem and damage of intestines).
Conclusion
The results confirmed that there are important discrepancies between in vitro and in vivo toxicogenomic experiments. However, the contribution of this paper is to provide a tool to identify genes that are disconnected between the two systems. Pathway analysis of disconnected genes may improve our understanding of uncertainties in the mechanism of actions of drug candidates in humans, especially concerning the early detection of toxicity.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-015-1726-7) contains supplementary material, which is available to authorized users.
Keywords: in vitro, in vivo, Toxicogenomics, Gene expression, Dose-response relationship, Liver toxicity
Background
Introduction
Pharmaceutical companies are facing an urgent need to increase their lead compound and clinical candidate portfolios, to satisfy market demands for continued innovation and revenue growth [1]. A relatively small number of drugs are being approved, while research costs are increasing, patents are expiring, and both governments and health insurance companies are pushing for cheaper medications [2]. Moreover, 20–40 % of novel drug candidates fail because of safety issues [3, 4], increasing the costs of bringing new drugs to the market [5]. Drug development costs could be reduced substantially if undesirable toxicity of a drug candidate could be predicted at earlier stages of the drug development process [6]. Integrating transcriptomics within drug development pipelines is being increasingly considered to help the early discovery of potential safety issues during preclinical phase and toxicology studies [7–10]. Such an approach has proven useful both in toxicology [11, 12] and carcinogenicity studies [13, 14].
Toxicogenomics studies mostly focus on network building for rat in vivo experiments [15] or the connection between rat in vivo and human in vitro transcriptomics experiments, particularly in relation to drug induced liver injury (e.g., [16–18]). Zhang et al. [19] developed a consensus early response toxicity signatures of in vitro and in vivo toxicity in human and rat using time-dependent gene expressions. For the hepatotoxicant hydrazine, Timbrell et al. [20] reported that the effects on various parameters do not always show a quantitative or qualitative correlation between in vivo and in vitro gene signatures. Enayetallah et al. [4] profiled nine compounds for in vitro and in vivo cardiotoxicity, and reported that while there were common biological pathways for in vivo and in vitro rat experiments for drugs like dexamethasone, most of the biological pathways identified in vivo for the drug amiodarone were not detected in vitro. Early prediction of safety issues for hit or lead compounds would benefit not only from consensus signatures, but also from disconnect signatures between in vivo and in vitro toxicogenomics experiments. These disconnect signatures can indicate which biological pathways are less likely to translate from a simplified in vitro model to a complex and holistic in vivo system.
Toxicity signatures developed from in vitro models most probably reflect protein modulations or pathway changes resulting from direct effects of compounds upon cells instead of the more complex interactions found in in vivo systems. In vitro signatures could also show excessive toxicity not to be detected in vivo due to compensatory mechanisms found in in vivo systems. Thus the framework is proposed to detect genes that are disconnected between in vitro and in vivo dose-dependent toxicogenomics experiments using fractional polynomial models. Biclustering is applied to find subsets of disconnected genes that are common to several compounds. Finally, the identified groups of disconnected genes are interpreted by their most probable biological pathways.
Data set
The ’Toxicogenomics Project - Genomics Assisted Toxicity Evaluation system’ (TG-GATEs, TGP, [21]) is a collaborative initiative between Japanese governmental bodies and fifteen pharmaceutical companies. It offers a rich source of transcriptomics data related to toxicology, providing human in vitro experiments together with in vitro and in vivo rat experiments [22–24]. We focus on a subset of the TG-GATEs data set consisting of 128 therapeutic drugs from a wide range of chemotypes. Gene expression were quantified using Affymetrix chip Rat 230_2 arrays. Six weeks old male Sprague-Dawley rats were used for both experiments and a single dose study design was used. Each rat was administered a placebo (the vehicle) or one of three active doses of a compound. For in vivo experiment, the rats were sacrificed after a fixed time period and liver tissue was subsequently profiled for gene expression. For the in vitro experiments, a modified two-step collagenase perfusion method was used to isolate liver cells from 6-week-old rats. These primary cultured hepatocytes were then exposed (in duplo) to a compound and gene expression changes were investigated at multiple time points. The analysis in this manuscript focuses on gene expression data at single time point, after exposure to a therapeutic drug for 24 hours, as gene expression signals are likely to be stronger at this time point in a single-dose study design [18]. The final data set for the rat in vitro experiments contains 5,914 genes and 1024 arrays (2 arrays per dose per compound), while the data set for the in vivo experiments contains 5,914 genes and 1536 arrays (3 arrays per dose per compound). The gene expression data were pre-filtered using I/NI calls to minimise false positives [25, 26]. The actual response variable represents the fold change of log2 mRNA intensities between the doses and the control dose. Hereafter, referred to as ’gene expression’ for simplicity. An example of a dose-response profile of gene A2m for compound sulindac is shown in Fig. 1.
Fig. 1.
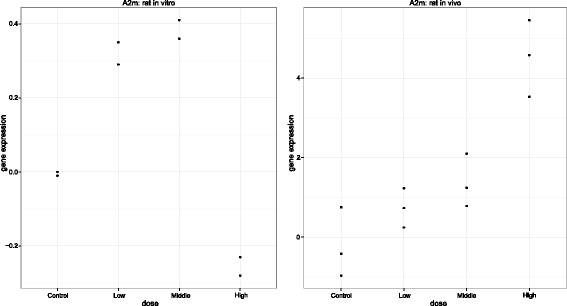
Data set example: Using gene A2m and compound sulindac. Observed gene expression profile for gene A2m under the activity of sulindac. Left panel: in vitro data. Right panel: in vivo data
Methods
A flexible fractional polynomial modeling framework is proposed to: (1) identify genes with significant dose-response relationships in an in vitro or in vivo experiments and (2) identify genes that are disconnected between the two systems. The in vitro and in vivo gene expression matrices were analysed jointly by compound and the resulting disconnected genes from the separate analyses were integrated using the Bimax biclustering algorithm [27] in order to identify subsets of disconnected genes that are common to several compounds.
The fractional polynomial framework
The fractional polynomial modeling framework aims to capture non-linear relationship between a predictor and a response variable. It assumes that most non-linear profiles can be captured by a combination of two polynomial powers [28]. It is particularly appealing for modeling dose-response relationships since it does not impose monotonicity apparent in most dose-response modeling methods [29, 30]. For a single gene, let Yij denote the gene expression from an in vitro experiment, where i=1,2,…,m represents dose levels and j=1,2,…,ni denotes the number of replicates per dose. The fractional polynomial framework assumes that relationships between gene expression and the compound dose can be captured by a polynomial function;
| (1) |
where εij∼N(0,σ2) and the polynomial powers p1,p2∈P, where P={−3,−2.5,…,1.5,2}. This range of values provides enough flexibility to capture different forms of dose-response profile [28]. The functions fij(p1) and gij(p1,p2) are defined as
and
| (2) |
Note that for p1≠0, p2≠0 and p1≠p2, the fractional polynomial model is given by . An example of fitting different combinations of powers for one particular gene is shown in Fig. 2.
Fig. 2.
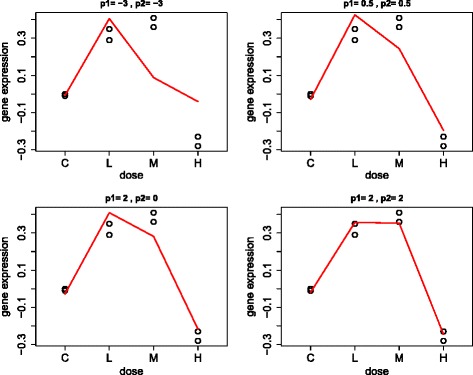
Fractional polynomial framework example: Using gene A2m and compound sulindac. Illustration of changes in predicted profiles by fitting fractional polynomial models with different powers on the same gene expression data. The model in the bottom right panel with p 1,p 2=2 is the best predictive model with the minimum Akaike’s Information Criterion (AIC)
Akaike’s information criterion (AIC, [31]) is used to select the optimal combination of p1 and p2 that best reflects the observed dose-response relationship. Optimal solutions are denoted by . In order to identify genes with a significant dose-response relationship in vitro, a likelihood ratio test (LRT, [32]) is used to compare model (1), that best fits the data and model (3), the null model that assumes no dose effect:
| (3) |
This additional testing is necessary to adjust for the relativity of the minimum AIC criterion.
To identify disconnected genes when comparing in vitro and in vivo data, the optimal fractional polynomial function selected per gene (with , as fixed above) from in vitro data set is projected to in vivo data set under the assumptions that both in vitro and in vivo dose-response relationships are similar. For a single gene, let Xijk denote gene expression in vitro and in vivo, where i=1,2,…,m represents dose levels, j=1,2,…,ni denotes number of replicates per dose and k=1 or k=2 depending on whether the data is from in vitro or in vivo experiment. The in vitro - in vivo projected fractional polynomial model is specified as
| (4) |
where εijk∼N(0,σ2). A LRT is used to quantify the dissimilarity in in vivo - in vitro dose-response relationships. It compares model (4), which assumes that dose-response relationships from in vitro and in vivo experiments are the same, with model (5), which assumes different dose-response relationships.
| (5) |
The comparison translates into testing if γ0=γ1=γ2=0 in model (5). An example of a projected fractional polynomial model is shown in Fig. 3. A significant result obtained from LRT comparison of model (4) and model (5) can be interpreted as a disconnect in gene expression between in vitro and in vivo rat experiments. The significance level was specified as 10 % after correction for multiplicity [33]. Resulting disconnected genes were subjected to fold change filtering by excluding genes with maximal dose-specific fold change between in vitro and in vivo data set less than 1. The fold change filtering further reduces false positives due to small variance genes [34, 35].
Fig. 3.
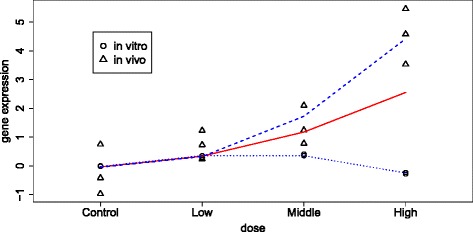
Projected fractional polynomial framework example: Using gene A2m and compound sulindac. Illustration of a projected fractional polynomial model from one system to the other. Red solid line shows the projected fractional polynomial model. The blue lines shows the results of fitting fractional polynomial models with different parameters for in vitro (dotted line) and in vivo data (dashed line), respectively
Biclustering of genes and compounds
A biclustering framework was introduced in order to find subsets of genes and conditions with a similar pattern [36]. Biclustering methods [37, 38] are designed to cluster in two dimensions simultaneously to produce sub-matrices of the original data that behave consistently in both dimensions. The resulting sub-matrices are called biclusters. Based on the identified disconnected genes from the fractional polynomial models, a disconnect matrix D(G×C) of binary values was created with gcth such that:
| (6) |
where G is the number of genes that are significant for at least one compound (i.e., G≤5914) and C=128 is the number of compounds. The Bimax algorithm [27] for binary data is applied to the disconnect matrix (G) to find subsets of the disconnected genes that are common to several compounds.
Results
The data were analysed in two ways depending on the direction of the projected fractional polynomial models. The first set of models (in vitro disconnects) defined the fractional polynomial powers based on the in vitro data set and projected its dose-response profiles to the in vivo data set. The second set of models (in vivo disconnects) defined the fractional polynomial powers based on the in vivo data set and projected its dose-response profiles to the in vitro data set. The resulting number of in vitro and in vivo disconnects for Sulindac and Indomethacin are illustrated in Fig. 4. The analyses were performed in statistical software R version 3.0.1 [39]. The R scripts are available upon request from the authors.
Fig. 4.
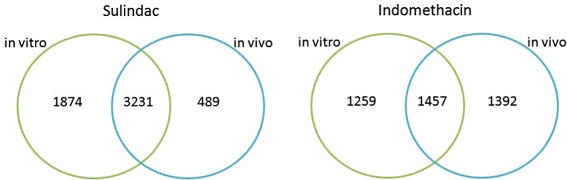
Sulindac and indomethacin compounds. Number of genes with significant dose-response relationships in in vitro data only, in in vivo data only and in both data simultaneously
In vitro disconnects
The number of the identified disconnected genes per compound ranged from 0 to 1,276. Three genes (Aldh1a1, Cyp1a1 and Fam25a) were consistently identified in 56 compounds whilst 446 genes were detected in more than 10 compounds. The 446 genes were analysed further for common biological pathways using GO [40], KEGG [41] and Janssen pharmaceutica in-house gene databases. As expected, many of the genes are involved in drug metabolism (e.g. acetaminophen metabolism, Benzo[a]pyrene metabolism, CAR/RXR activation, PXR/RXR activation), as well as endogenous compound metabolism (e.g. butanoate metabolism, alanine, cysteine and methionine metabolism, nitrogen metabolism, fatty acid metabolism, cholesterol biosynthesis). Additionally, some of the genes are also involved in toxicity related pathways such as oxidative stress due to reactive metabolites, bilirubin increase, glutathion depletion and phospholipidosis as well as complex pathways such as immune response, classical complement and coagulation. Only pathways containing more than five genes and with coverage of more than 10 % (i.e., more than 10 % of their genes were disconnected genes) were considered.
We further identified 188 unique genes that were consistently defined as disconnected genes in seven compounds based on the first 10 biclusters from the Bimax algorithm (left panel on Fig. 5). Sulindac and diclofenac are both anti-inflammatory drugs, acetic acid derivatives that are likely to damage liver [42]. Naphthyl isothiocyanate was shown to cause direct hepatotoxicity [43]. Among the 188 genes, the top genes (with respect to fold change) are associated with liver toxicity. Genes A2m and Lcn2 were validated for being affected in case of hepatotoxicity [44]. Other toxicity associated genes found were Cyp1a1, Pcsk9, Car3, Gstm3 or Ccnd1. Table 1 shows the results of pathway analysis for the first bicluster (compounds: sulindac, naphthyl isothiocyanate, diclofenac and colchicine). For complete results of biclustering in vitro, see Additional files 1 and 2.
Fig. 5.
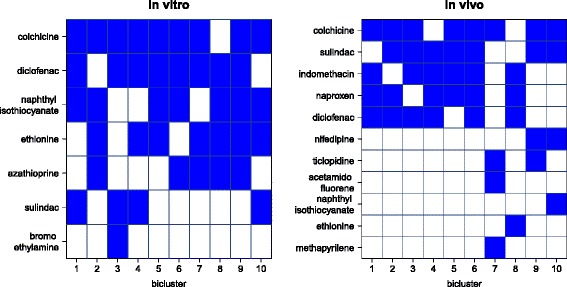
Biclustering results. Appearance of compounds across 10 biclusters. The blue colour indicates membership of a bicluster. Left panel: An example of in vitro disconnects. Right panel: An example of in vivo disconnects
Table 1.
Examples of the clustered disconnected genes and their probable pathways. The pathways were identified using KEGG [41]. The genes are members of first bicluster of the analysis starting with in vitro data. The compounds in first bicluster are sulindac, naphthyl isothiocyanate, diclofenac and colchicine
| Pathway | Genes |
|---|---|
| Complement and coagulation cascades | A2m C1s C5 C8a C4bpb Cfh F5 |
| Chemical carcinogenesis | Cyp1a1 Gstm3 Gsta5 |
| Metabolism of xenobiotics | Akr7a3 Cyp1a1 Gstm3 Gsta5 |
| Pathways in cancer | Ccnd1 Fn1 Lamc2 |
In vivo disconnects
There were 175 genes that showed disconnect in gene expression from in vivo to in vitro rat experiments for more than 10 compounds. Similar pathways as in the previous section (i.e projection from in vitro to in vivo) were also discovered, although more of the pathways were related to exogenous compound metabolism. Oxidative stress endpoints related pathways were more common in vivo. Complex pathways such as complement and coagulation identified in the in vitro data set were not discovered in the analysis of the in vivo data set, which may be due to differences between the prescribed dose and actual exposure in liver tissue in vivo.
The Bimax algorithm identified 163 unique genes common to 11 distinct compounds based on the first 10 biclusters (right panel on Fig. 5). Five compounds were identified both in in vitro and in vivo analyses of disconnects: sulindac, colchicine, diclofenac, ethionine and naphthyl isothiocyanate. The most interesting of the additional compounds are indomethacin and naproxen. They are both members of a group of non-steroidal anti-inflammatory drugs (NSAIDs), the former an acetic acid derivative and the latter a propionic acid derivative. Both drugs are nonselective cyclooxygenase (COX) isozyme inhibitors, i.e. with undesired targeting of COX-1 that leads to gastrointestinal adverse effects [45, 46]. Specifically, both drugs are indicated as high risk drugs for general upper gastrointestinal complications [47]. All of the compounds are connected to toxicity events. Most of the toxicity related genes (A2m, Lcn2, Car3, Pcsk9, Acsl1, Lamc2, Selenbp1 and Serpina10) from the previous in vitro analysis were also identified from the analysis of the in vivo data set. Other toxicity related genes were Cyp2e1 [48], Upp1, Gss, Ddc, Gstm7 and Srebf1. For complete results of biclustering in vivo, see Additional files 3 and 4.
Simulation study
The empirical validation of the fractional polynomial method in the context of in vitro and in vivo disconnects was done through two simulation studies. In the first simulation study, data were generated according to seven possible scenarios. First setting corresponded to the null model of no disconnect between in vitro and in vivo experiments. The other six settings corresponded to three groups of genes: genes with opposite dose-response profiles for in vitro and in vivo, genes with dose effect only for in vivo and dose effect only for in vitro. The settings followed either linear model or second order fractional polynomial model. For each setting, 10,000 data sets were generated.
According to the simulation results, the proposed projected fractional polynomial method under the null model resulted in 90 % specificity using the same number of dose and the same number of observations per dose as in TGP data set. When number of observations per dose was increased to four, specificity increased up to 98 %. Under the alternative hypothesis of a disconnected dose-response profiles between in vitro and in vivo experiments, the method resulted in 100 % sensitivity for the disconnected linear profiles. For nonlinear profiles, sensitivity of 80–95 % was achieved, for the maximum fold change between the in vitro and in vivo settings greater than 1.2. Sensitivity increased up to 98–100 % when the fold change was greater than 1.6.
The second simulation study mimicked the structure of the TGP experiment. In total, 6,000 genes were generated to create one data set. Half of them contained no dose effects for both in vitro and in vivo. The other half exhibited clear dose-response effect in vitro and opposite dose-response effect in vivo, creating a disconnect between two data sets. Specifically, the model used for in vitro was second order polynomial model with fold change of one (that was the minimal fold change of interest in our analysis). Standard deviation and the number of observations per dose correspond to the TGP data set. LRTs for dose-response and interaction were applied for each gene. The resulting p-values were adjusted for multiplicity using Benjamini-Hochberg procedure to control false discovery rate (BH-FDR) at 10 %. The sensitivity and specificity was computed as amount of correctly identified genes from both categories (null model and true disconnect). The whole procedure was repeated for 1,000 simulated data sets, computing sensitivity and specificity for each of them.
The ROC curves of all the simulated data sets are shown in left panel of Fig. 6, together with the averaged ROC curve. The spread of ROC curves is very low, suggesting stability of the method across the simulated data sets. When FDR was controlled at 10 %, average specificity and sensitivity were 93 % and 95 %, respectively. The box plots of false positives and false negatives for the simulated data set are shown in Fig. 6 (right panel). The FDR is well controlled at the desired level of 10 % and false negatives rate is very low.
Fig. 6.
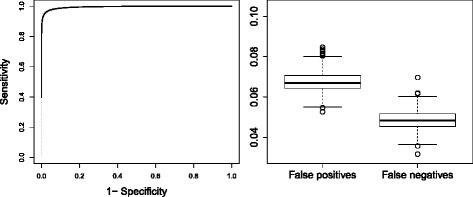
Validation of fractional polynomials: Results of simulation study. Left panel: ROC curves for sensitivity and specificity of all simulated data set (grey dotted lines) and averaged ROC curve (black solid line). Right panel: Box plot of false positives and false negatives of all 1,000 simulated data sets
The simulation studies indicated that the method may perform better in other settings than the reported results for the TGP experiment due to its limited number of replicates per dose and the weak signals. The full description of the simulations settings and results can be found in the Additional file 5.
Discussion
The analytic framework identified three broad groups of genes from a joint analyses of in vitro and in vivo rats toxicogenomic experiments. The first group showed a significant dose-response relationship in both the in vitro and in vivo toxicogenomic experiments. These types of genes are shown in the top panels of Fig. 7. Whilst some of the genes in this group showed contradictory dose-responses profiles between the in vitro and in vivo data, others showed similar dose-response profiles, but with different magnitude of gene expression values. The second group contains genes that showed a significant dose-response relationship in in vitro experiments, but not in in vivo experiments. Example of such genes are presented in the top panels of Fig. 8. This set of genes may represent the difference in biological complexity between in vivo and in vitro systems. The third group are those genes that showed significant dose-response relationship in in vivo experiments, but not in in vitro experiments. For complete results, see Additional files 6 and 7.
Fig. 7.
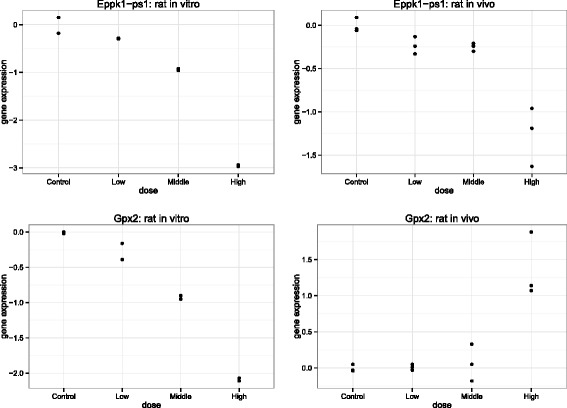
Group 1 example: compound sulindac. Two genes from Group 1. Top panels: gene Eppk1-ps1 with the same direction of dose-response relationships, but different magnitude of fold change. Bottom panels: gene Gpx2 with different direction of dose-response relationships across platforms. Left panels: in vitro. Right panels: in vivo
Fig. 8.
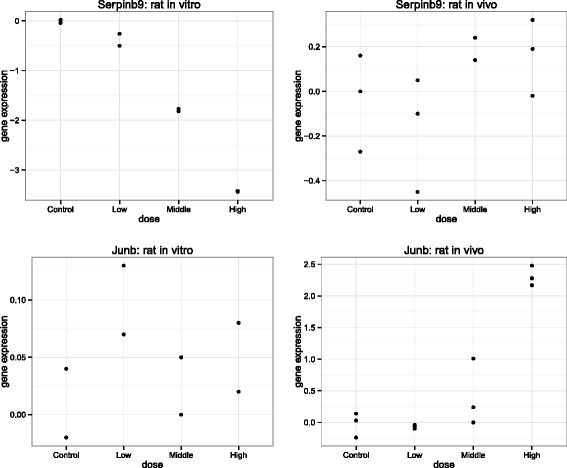
Group 2 and 3 examples: compound sulindac. Top panels: gene Serpinb9 from Group 2, with significant dose-response relationship only in in vitro data. Bottom panels: gene Junb from Group 3, with significant dose-response relationship only in in vivo data. Left panels: in vitro. Right panels: in vivo
This set of genes may occur due to the mechanism of action (MoA) in vitro of a drug candidate not being representative of in vivo. Most of the compounds in our specific case study that triggered the expression of the identified disconnected genes are members of a group of anti-inflammatory drugs and all of them are related to hepatotoxicity, nephrotoxicity or gastro-intestinal toxicity.
If additional data about experiments are available both for in vitro and in vivo, such data can be included in the proposed methodology. The adjustment can be done by adding the new variables in the fractional polynomial model as covariates. Note that in this type of gene expression studies, the rats are specially bred to ensure baseline comparability across all rats.
Conclusion
The findings demonstrated that substantial differences may be identified between in vitro and in vivo gene expression. While this result is not surprising, the importance of the analysis is in the identification of different groups of the disconnected genes. Genes were identified that showed significant dose-response relationships with compounds in vitro that were absent in vivo, and vice-versa. Moreover, there was a group of genes with a different direction of dose-response relationship across the two systems. These finding confirms possibility of important discrepancies between in vitro experiments and in vivo experiments. Pathway analysis of the identifying disconnected genes between in vivo and in vitro rat system may improve our understanding of uncertainties in mechanism of action of a drug candidate in human, especially for early toxicity detection.
Availability of supporting data
The data sets supporting the results of this article are available in the TG-GATEs Toxicogenomics Project repository (http://toxico.nibio.go.jp/english/index.html).
Ethical approval
There was no ethical approval needed for this manuscript, because it was based on publicly available data sets. The ethical approval for the original TGP study was granted by the Ethics Review Committee for Animal Experimentation of the National Institute of Health Sciences, Japan, and by the respective contract research organizations [49].
Acknowledgements
Martin Otava and Ziv Shkedy gratefully acknowledge the support from the IAP Research Network P7/06 of the Belgian State (Belgian Science Policy). Martin Otava gratefully acknowledge the financial support of the Research Project BOF11DOC09 of Hasselt University. Authors are grateful to Vladimir Nerandzic for helpful advice about biological background and to Suzanne Boyd (WRIHW, Durham University, UK) and Prof James Mason (DCTU, Durham University, UK) for helpful language corrections and editing. The computational resources and services used for the simulation studies were provided by the VSC (Flemish Supercomputer Center), funded by the Hercules Foundation and the Flemish Government - department EWI.
Abbreviations
- TG-GATEs
Toxicogenomics Project - Genomics assisted toxicity evaluation system
- TGP
The Toxicogenomics Project
- I/NI
Informative/non-informative
- AIC
Akaike’s information criterion
- LRT
Likelihood ratio test
- GO
The gene ontology consortium
- KEGG
Kyoto encyclopedia of genes and genomes
Additional files
Genes of top 10 biclusters in vitro . List of disconnected genes that appeared in the top 10 biclusters in vitro.
Top 10 biclusters in vitro . List of top 10 biclusters in vitro with all the disconnected genes and compounds that are bicluster members.
Genes of top 10 biclusters in vivo . List of disconnected genes that appeared in the top 10 biclusters in vivo.
Top 10 biclusters in vivo . List of top 10 biclusters in vivo with all the disconnected genes and compounds that are bicluster members.
Supplementary appendix. Additional figures on gene expression profiles. Simulation study based validation of the methodology.
Integrated results - sulindac. List of disconnected genes for sulindac organized in three groups (see Discussion).
Integrated results - all. List of disconnected genes for all compounds organized in three groups (see Discussion).
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MO performed the analyses. AK and ZS developed the methodological framework. WT and GRV contributed to interpretation of results, focusing on biological pathways and context. All the authors contributed significantly in writing the manuscript. All authors read and approved the final manuscript.
Contributor Information
Martin Otava, Email: martin.otava@uhasselt.be.
Ziv Shkedy, Email: ziv.shkedy@uhasselt.be.
Willem Talloen, Email: WTALLOEN@its.jnj.com.
Geert R Verheyen, Email: Geert.Verheyen@thomasmore.be.
Adetayo Kasim, Email: a.s.kasim@durham.ac.uk.
References
- 1.Davidov E, Holland J, Marple E, Naylor S. Advancing drug discovery through systems biology. Drug Discov Today. 2003;8(4):175–83. doi: 10.1016/S1359-6446(03)02600-X. [DOI] [PubMed] [Google Scholar]
- 2.Scannell JW, Blanckley A, Boldon H, Warrington B. Diagnosing the decline in pharmaceutical R&D efficiency. Nat Rev Drug Discov. 2012;11(3):191–200. doi: 10.1038/nrd3681. [DOI] [PubMed] [Google Scholar]
- 3.Arrowsmith J. Trial watch: phase III and submission failures: 2007-2010. Nat Rev Drug Discov. 2011;10(2):87. doi: 10.1038/nrd3375. [DOI] [PubMed] [Google Scholar]
- 4.Enayetallah AE, Puppala D, Ziemek D, Fischer JE, Kantesaria S, Pletcher MT. Assessing the translatability of in vivo cardiotoxicity mechanisms to in vitro models using causal reasoning. BMC Pharmacol Toxicol. 2013;14(46):1–12. doi: 10.1186/2050-6511-14-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Paul SM, Mytelka DS, Dunwiddie CT, Persinger CC, Munos BH, Lindborg SR, Schacht AL. How to improve R&D productivity: the pharmaceutical industry’s grand challenge. Nat Rev Drug Discov. 2010;9(3):203–14. doi: 10.1038/nrd3078. [DOI] [PubMed] [Google Scholar]
- 6.Food and Drug Administration. Innovation or stagnation? Challenge and opportunity on the critical path to new medicinal products: U.S. Department of Health and Human Services; 2004. http://www.fda.gov/ScienceResearch/SpecialTopics/CriticalPathInitiative/CriticalPathOpportunitiesReports/ucm077262.htm.
- 7.Bajorath J. Rational drug discovery revisited: interfacing experimental programs with bio- and chemo-informatics. Drug Discov Today. 2001;6(9):989–95. doi: 10.1016/S1359-6446(01)01961-4. [DOI] [PubMed] [Google Scholar]
- 8.Fanton CP, Rowe MW, Moler EJ, Ison-Dugenny M, De Long SK, Rendahl K, et al. Development of a screening assay for surrogate markers of CHK1 inhibitor-induced cell cycle release. J Biomol Screen. 2006;11(7):792–806. doi: 10.1177/1087057106289808. [DOI] [PubMed] [Google Scholar]
- 9.Baum P, Schmid R, Ittrich C, Rust W, Fundel-Clemens K, Siewert S, et al. Phenocopy-a strategy to qualify chemical compounds during hit-to-lead and/or lead optimization. PloS One. 2010;5(12):14272. doi: 10.1371/journal.pone.0014272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Amaratunga D, Cabrera J, Shkedy Z. Exploration and Analysis of DNA Microarray and Other High-Dimensional Data. New Jersey: Wiley; 2014. [Google Scholar]
- 11.Pognan F. Toxicogenomics applied to predictive and exploratory toxicology for the safety assessment of new chemical entities: a long road with deep potholes. Prog Drug Res. 2007;64(217):219–38. doi: 10.1007/978-3-7643-7567-6_9. [DOI] [PubMed] [Google Scholar]
- 12.Afshari CA, Hamadeh HK, Bushel PR. The evolution of bioinformatics in toxicology: advancing toxicogenomics. Toxicol Sci. 2011;120:225–37. doi: 10.1093/toxsci/kfq373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nie AY, McMillian M, Parker JB, Leone A, Bryant S, Yieh L, et al. Predictive toxicogenomics approaches reveal underlying molecular mechanisms of nongenotoxic carcinogenicity. Mol Carcinog. 2006;45:914–33. doi: 10.1002/mc.20205. [DOI] [PubMed] [Google Scholar]
- 14.Ellinger-Ziegelbauer H, Gmuender H, Bandenburg A, Ahr HJ. Prediction of a carcinogenic potential of rat hepatocarcinogens using toxicogenomics analysis of short-term in vivo studies. Mutat Res. 2008;637(1-2):23–39. doi: 10.1016/j.mrfmmm.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 15.Kiyosawa N, Manabe S, Sanbuissho A, Yamoto T. Gene set-level network analysis using a toxicogenomics database. Genomics. 2010;96:39–49. doi: 10.1016/j.ygeno.2010.03.014. [DOI] [PubMed] [Google Scholar]
- 16.Uehara T, Kiyosawa N, Shimizu T, Omura K, Hirode M, Imazawa T, et al. Species-specific differences in coumarin-induced hepatotoxicity as an example toxicogenomics-based approach to assessing risk of toxicity to humans. Hum Exp Toxicol. 2008;27(1):23–35. doi: 10.1177/0960327107087910. [DOI] [PubMed] [Google Scholar]
- 17.Clevert DA, Heusel M, Mitterecker A, Talloen W, Göhlmann HWH, Wegner J, et al.Exploiting the Japanese Toxicogenomics Project for Predictive Modelling of Drug Toxicity. In: CAMDA 2012, Satellite Meeting of ISMB/ECCB 2012, Long Beach CA, USA, July 13-14: 2012. http://www.bioinf.jku.at/publications/2012.html.
- 18.Otava M, Shkedy Z, Kasim A. Prediction of gene expression in human using rat in vivo gene expression in Japanese Toxicogenomics Project. Syst Biomed. 2014;2:29412. [Google Scholar]
- 19.Zhang JD, Berntenis N, Roth A, Ebeling M. Data mining reveals a network of early-response genes as a consensus signature of drug-induced in vitro and in vivo toxicity. Pharmacogenomics J. 2014;14:208–16. doi: 10.1038/tpj.2013.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Timbrell JA, Delaney J, Waterfield CJ. Correlation between in vivo and in vitro toxic effects of foreign compounds. Comparative Haematol Int. 1996;6:232–6. doi: 10.1007/BF00378116. [DOI] [Google Scholar]
- 21.Uehara T, Ono A, Maruyama T, Kato I, Yamada H, Ohno Y, Urushidani T. The Japanese Toxicogenomics Project: application of toxicogenomics. Mol Nutr Food Res. 2010;54(2):218–77. doi: 10.1002/mnfr.200900169. [DOI] [PubMed] [Google Scholar]
- 22.Ganter B, Tugendreich S, Pearson CI, Ayanoglu E, Baumhueter S, Bostian KA, et al. Development of a large-scale chemogenomics database to improve drug candidate selection and to understand mechanisms of chemical toxicity and action. J Biotechnol. 2005;119(3):219–44. doi: 10.1016/j.jbiotec.2005.03.022. [DOI] [PubMed] [Google Scholar]
- 23.Suter L, Schroeder S, Meyer K, Gautier JC, Amberg A, Wendt M, et al. EU framework 6 project: predictive toxicology (PredTox) - overview and outcome. Toxicol Appl Pharmacol. 2011;252(2):73–84. doi: 10.1016/j.taap.2010.10.008. [DOI] [PubMed] [Google Scholar]
- 24.Briggs K, Cases M, Heard DJ, Pastor M, Pognan F, Sanz F, et al. Inroads to predict in vivo toxicology - an introduction to the eTOX project. Int J Mol Sci. 2012;13:3820–846. doi: 10.3390/ijms13033820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Talloen W, Clevert DA, Hochreiter S, Amaratunga D, Bijnens L, Kass S, Göhlmann HWH. I/NI-calls for the exclusion of non-informative genes: a highly effective filtering tool for microarray data. Bioinformatics. 2007;23(21):2897–902. doi: 10.1093/bioinformatics/btm478. [DOI] [PubMed] [Google Scholar]
- 26.Kasim A, Lin D, Van Sanden S, Clevert DA, Bijnens L, Göhlmann HWH, et al. Informative or noninformative calls for gene expression: a latent variable approach. Stat Appl Genet Mol Biol. 2010;9:4. doi: 10.2202/1544-6115.1460. [DOI] [PubMed] [Google Scholar]
- 27.Prelic A, Bleuler S, Zimmermann P, Wille A, Buhlmann P, Gruissem W, et al. A systematic comparison and evaluation of biclustering methods for gene expression data. Bioinformatics. 2006;22(9):1122–9. doi: 10.1093/bioinformatics/btl060. [DOI] [PubMed] [Google Scholar]
- 28.Royston P, Altman DG. Regression using fractional polynomials of continuous covariates: Parsimonious parametric modelling. J R Stat Soci. Series C (Appl Stat) 1994;43(3):429–67. [Google Scholar]
- 29.Ramsay J. Monotone regression splines in action. Stat Sci. 1988;3:425–41. doi: 10.1214/ss/1177012761. [DOI] [Google Scholar]
- 30.Lin D, Shkedy Z, Yekutieli D, Amaratunga D, Bijnens L, editors. Modeling Dose-response Microarray Data in Early Drug Development Experiments Using R Classification of Monotone Gene Profiles Using Information Theory Selection Methods. Berlin: Springer; 2012. [Google Scholar]
- 31.Akaike H. A new look at the statistical model identification. IEEE Trans Automatic Control. 1974;AC-19:716–23. doi: 10.1109/TAC.1974.1100705. [DOI] [Google Scholar]
- 32.Neyman J, Pearson E. On the problem of the most efficient tests of statistical hypotheses. Philos Trans R Soc Lond. 1933;231:289–337. doi: 10.1098/rsta.1933.0009. [DOI] [Google Scholar]
- 33.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B (Methodological) 1995;57:289–300. [Google Scholar]
- 34.Talloen W, Göhlmann HWH. Gene Expression Studies Using Affymetrix Microarrays. Boca Raton: Chapman & Hall; 2009. [Google Scholar]
- 35.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001;98(9):5116–121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cheng Y, Church GM. Biclustering of expression data. Proc Conf Intell Syst Mol Biol. 2000;55:93–104. [PubMed] [Google Scholar]
- 37.Madeira SC, Oliveira AL. Biclustering algorithms for biological data analysis: a survey. IEEE Trans Comput Biol Bioinformatics. 2004;1(1):24–45. doi: 10.1109/TCBB.2004.2. [DOI] [PubMed] [Google Scholar]
- 38.Eren K, Deveci M, Küçüktunç O, Çatalyürek UV. A comparative analysis of biclustering algorithms for gene expression data. Brief Bioinform. 2013;14(3):279–92. doi: 10.1093/bib/bbs032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.R Core Team . R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2013. [Google Scholar]
- 40.The Gene Ontology Consortium Gene ontology: tool for the unification of biology. Nat Genet. 2000;25(1):25–9. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rodríguez LAG, Williams R, Derby LE, Dean AD, Jick H. Acute liver injury associated with nonsteroidal anti-inflammatory drugs and the role of risk factors. Arch Intern Med. 1994;154(3):311–6. doi: 10.1001/archinte.1994.00420030117012. [DOI] [PubMed] [Google Scholar]
- 43.Williams GM. The direct toxicity of alpha-naphthylisothiocyanate in cell culture. Chemico-Biological Interactions. 1974;8(6):363–9. doi: 10.1016/0009-2797(74)90042-8. [DOI] [PubMed] [Google Scholar]
- 44.Wang EJ, Snyder RD, Fielden MR, Smith RJ, Gu YZ. Validation of putative genomic biomarkers of nephrotoxicity in rats. Toxicology. 2008;246(2-3):91–100. doi: 10.1016/j.tox.2007.12.031. [DOI] [PubMed] [Google Scholar]
- 45.Rao PNP, Knaus EE. Evolution of nonsteroidal anti-inflammatory drugs (NSAIDs): Cyclooxygenase (COX) inhibition and beyond. J Pharm Pharm Sci. 2008;11(2):81–110. doi: 10.18433/j3t886. [DOI] [PubMed] [Google Scholar]
- 46.Brune K, Patrignani P. New insights into the use of currently available non-steroidal anti-inflammatory drugs. J Pain Res. 2015;8:105–18. doi: 10.2147/JPR.S75160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Castellsague J, Riera-Guardia N, Calingaert B, Varas-Lorenzo C, Fourrier-Reglat A, Nicotra F, et al. Individual NSAIDs and upper gastrointestinal complications: A systematic review and meta-analysis of observational studies (the SOS project) Drug Saf. 2012;35(12):1127–1146. doi: 10.1007/BF03261999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Heijne WHM, Jonker D, Stierum RH, van Ommen B, Groten JP. Toxicogenomic analysis of gene expression changes in rat liver after a 28-day oral benzene exposure. Toxicogenomics Genet Toxicol Hazard Determination. 2005;575(1-2):85–101. doi: 10.1016/j.mrfmmm.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 49.Igarashi Y, Nakatsu N, Yamashita T, Ono A, Ohno Y, Urushidani T, et al. Open TG-GATEs: a large-scale toxicogenomics database. Nucleic Acids Res. 2015;43(Database issue):921–7. doi: 10.1093/nar/gku955. [DOI] [PMC free article] [PubMed] [Google Scholar]