

Abstract
Since their inception in the 1980's, oligonucleotide-based (ON-based) therapeutics have been recognized as powerful tools that can treat a broad spectrum of diseases. The discoveries of novel regulatory methods of gene expression with diverse mechanisms of action are still driving the development of novel ON-based therapeutics. Difficulties in the delivery of this class of therapeutics hinder their in vivo applications, which forces drug delivery systems to be a prerequisite for clinical translation. This review discusses the strategy of using lipid nanoparticles as carriers to deliver therapeutic ONs to target cells in vitro and in vivo. A discourse on how chemical and physical properties of the lipid materials could be utilized during formulation and the resulting effects on delivery efficiency constitutes the major part of this review.
Graphical Abstract

1 Introduction
Nucleic acid-based macromolecules have paved a new avenue for the development of therapies to treat a wide spectrum of inherited and acquired diseases. The broad potential of this class of therapeutics arises from the precision and effectiveness of the intervention of cellular protein expression levels. This intervention results from the introduction of exogenous genes for expression products or regulatory non-coding RNAs to suppress the target gene expression. The development of nucleic acid therapeutics has also shifted the conventional drug development paradigm by skipping the tedious and time-consuming drug screening process for particular therapeutic targets. Drugs are instead designed completely based on the genetic sequence of the target gene, which the Human Genome Project has helped to reveal. However, these therapeutics face a principal barrier to clinical translation: delivery.
The composition and physicochemical properties of nucleic-acid based therapeutics leave them susceptibile to nuclease-mediated degradation. In addition, high molecular weight and negative charges make nucleic acids impermeable to the cellular membrane. Therefore, an efficient delivery system is a requirement for therapeutic efficacy. To address pharmaceutical issues such as stability, cellular uptake, and targeted delivery, a series of delivery carriers have been developed. Particularly for cell-targeted delivery, nanosized carriers are the most investigated systems. These carriers can enter certain cell populations with specificity by recognizing the biomarkers on the cell surface.
Despite their identical building blocks, the biological activities and mechanisms of action of nucleic acid-based therapeutics are diverse. Based on the length and structure of these biological polymers, nucleic acid therapeutic agents are divided into several categories: (1) double stranded plasmid DNA with linear or circular forms that are generally used for a “gain of function” purpose by introducing exogenous genes; (2) single-stranded messenger RNA that are mainly used for a more direct source of gene products; (3) double-stranded RNAs such as small interfering RNAs (siRNAs) or micro RNAs (miRNAs) that are exploited in gene regulation by down-regulating the expression of target genes; and (4) oligonucleotides, defined as short, single-stranded DNA or RNA that have a wider range of biomedical applications than their predecessors.
It is important to note that the last decade has been labeled as the age of siRNA, as scientists have been excited about the elegance of siRNA-mediated gene silencing that was discovered by Fire and Mello in 1998 [1]. Moreover, the 2006 Nobel Prize in Physiology and Medicine given to these two scientists boosted enthusiasm for the development and clinical translation of siRNA based therapeutics. Due to the appealing nature of siRNA, much of the attention has been driven toward the development of siRNA-based therapies. However, ON-based therapies could not be replaced by siRNA therapy owing to its much broader biological applications.
Generally, six major classes of ON-based therapeutics with diverse functionalities are currently under investigation: (1) DNA-based ONs that are designed to hybridize with target mRNA transcripts and block translation [2-4]; (2) triple-helix-forming ONs that are designed to be inserted into double-stranded DNA and inhibit transcription elongation [5]; (3) ribozymes or RNA ONs with a broad spectrum of biological and catalytic activities[6-8]; (4) Anti-miR molecules, which are ONs designed to specifically block the activity of endogenous miRNA to regulate gene expression [9-11]; (5) CpG-containing ONs that are intended to boost the immune response as adjuvants [12-14]; and (6) splice-switching ONs that are able to control pre-mRNA splicing patterns [15-17].
Despite differences in their mechanisms of action, these six types of molecules share similar physicochemical characteristics and molecular weights. Therefore, once the delivery system is established on one of these classes of ONs, any class of these ON-based therapeutics are readily adapted. An increased efficiency of delivering ONs into the cells after systemic administration can be achieved by incorporating the therapeutics into liposomes. Ever since Loke et al. reported one of the earliest liposome-mediated ON delivery studies [18], a number of groups have reported using liposomal ON formulations to interfere withbiological cell activities. The intent of this review is to act as an overview of encapsulation strategies for ONs in lipid nanoparticles and to analyze the critical issues in delivering ON-based therapeutics using lipid-based nanoparticles, which are the most investigated delivery system in the field.
2 Barriers to efficient ON delivery
The pursuit of efficient delivery of ONs has not ceased since it began in the 1980s. Chemical modifications and carrier-assisted delivery have been the two primary strategies that are used to achieve successful delivery as biological studies have unraveled the critical intracellular and extracellular barriers to ON delivery. These findings provide a theoretical basis that supports the rational design and engineering of carriers so that those barriers can be circumvented or overcome by the functional modalities in the carrier.
There are some extensive reviews that discuss these delivery barriers in detail [19-23]. In brief, nanoparticle-mediated ONs, after intravenous injection, will have to maintain colloidal stability before entering target cells. The high concentration of serum proteins tend to adsorb onto the surface of the nanoparticles causing premature release of encapsulated therapeutics. Moreover, the adsorption of complements in the blood will enhance the clearance of the nanoparticles by the reticuloendothelial system (RES) residing in the liver and spleen. This leads to a shorter blood circulation half-life and low bioavailability of the administered therapeutics. The nanoparticles that escape RES capture must extravasate the endothelium of the blood vessels to access the parenchymal cells of the target tissue or the cancer cells in the tumors. Particularly in tumors, the crowded extracellular matrix and accumulated interstitial fluid pressure due to the lack of functional lymphatics hinder the passive diffusion of the nano-scale particles into the deeper areas of the tissue.
Once the nanoparticle accesses the periphery of the target cell, the remaining intracellular obstacles to successful delivery are just as challenging as the extracellular barriers (Figure 1). Nano-sized carriers are still too large for passive diffusion into the cells. The nanoparticles therefore have to enter the cells via endocytosis, pinocytosis or phagocytosis pathwayswhich often leave the nanoparticles in endosomal compartments. Entrapment of the nanoparticles in endosomes is another critical intracellular barrier that dramatically reduces the bioavailability of the therapeutics. The payload in the nanoparticles must also be released from the carrier to provide therapeutic effect once the nanoparticles reach the cytosol of the target cells. If the ON functions by working on the DNA transcriptional level, it must also cross the nuclear membrane to access genomic DNA via active transport or passive diffusion during cell mitosis.
Figure 1.

Schematic illustration of trafficking of lipid nanoparticles based antisense-ON delivery to the cells.
3 Lipid nanoparticles mediated ON delivery
As ON molecules are susceptible to degradation by nucleases, encapsulation in lipid nanoparticles keeps these agents intact and biologically active after administration. Lipid nanoparticles represent one of the best optimized and characterized drug delivery systems that have shown extended blood circulation profiles. The encapsulation strategy also endows the ON with the physicochemical properties of the carrier, and therefore converts the pharmacokinetic behavior of the ON to that of the lipid nanoparticles. The blood circulation time is especially important for tumor-targeted ON delivery, as the increased circulation half-life allows the therapeutics to accumulate in the tumor tissue through a tumor's leaky vasculature. The addition of targeting ligands on the carrier can achieve delivery selectivity and enhance cellular uptake. Numerous methods have been reported to formulate ONs into lipid nanoparticles that can be generalized based on the physicochemical properties of the lipids used in the formulation (Table 1).
Table 1.
Representative formulations and characterizations of Lipid NP for delivery of ON in vitro or in vivo.
| Cationic lipoplexes | Preparation Procedure |
Lipid Composition |
Trapping Efficiency |
Particle size |
In vivo PK or in vitro Serum Stability |
In vitro or In vivo Delivery Efficiency |
Reference |
| Bulk mixing | DOTMA/DOPE (Lipofectin™) | N.D. | N.D. | N.D. | ED50<30nm for inhibition of intracellular adhesion molecule (ICAM-1) expression in HUVEC cells | [38] | |
| Lipid film hydration | DDAB: PC: Chol (5:16:8) | >90% | Heterogeneous size distribution (200nm-10μm) | Two-compartment PK model t1/2α=24.5 min t1/2β=11.3 6 h | Inhibition of Raf-1 (52%) expression in SQ-20B cells dosed at 10μM ON; Inhibition of Raf-1 in liver (51%), Kidney (42%) and variable levels in SQ-20B xenograft (37%-57%) after 5 daily iv injections (6mg/kg) | [27] | |
| Bulk mixing | DE: DOPE: Chol (2:1:1) | N.D. | <200nm | Aggregation in the presence of 10% serum | Reducing NF-κB/DNA binding activity by 58% in RAW264.7 macrophages after LPS stimulation dosed at 400nM | [25] | |
| Bulk mixing of lipidoid NP and ON in 35% ethanol (pH5.2) followed by buffer exchange and dialysis | 98N12-5: Chol: C16 Ceramide-PEG (42:48:10) |
>95% | 50nm | Stable in serum | Decreased miR-122 level in the liver after three consecutive injections of anti-miR122 dosed at 5mg/kg | [24] | |
| Neutral Liposome | Lyophilization-Rehydration | DOPC | ≥95% | 100nm | Two-compartment PK model t1/2α=8.1 min t1/2β=3.9 h | Inhibition of Bcl-2 expression by 44% in lymphoma cells. No in vivo knockdown study has been performed | [39, 40] |
| Minimal Volume Entrapment | EPC: Chol: DSPE-PEG (2:1:0.1) | 20% | 200nm | Stable in the presence of 10% serum at 37°C | N.D. | [41] | |
| Reverse Phase evaporation | DSPC: Chol: DOTAP: DSPE-PGE | 80%-90% | 70-120nm | T1/2=4h | Inhibition of c-myb expression by 70% in neuroblastom a cells dosed at 100μg/ml [42] Reduction of c-myc expression in melanoma xenograft when dosed at 2.5mg/kg [43] |
[41-44] | |
| Reverse phase evaporation | DOPE: OA: Chol (10:5:2) | 10% | 170nm | N.D | ED50=40nM against Friend retrovirus infection in Dunni Cells | [45] | |
| Reverse phase evaporation with detergent dialysis | DOPE: OA: Chol (2:1:2) | 10-12% | N.D. | N.D | Enhanced uptake of ON delivered by streptavidin-biotin-coupled immunoliposomes by two fold compared with ordinary liposomes | [46] | |
| Lipid Nanoparticles | Step-wise bulk mixing | DOTAP/Chol/DSPE-PGE/Protamine | >90% | 64nm | Stable in serum | Down-regulation of surviving expression in H1299 cells by 87% when dosed at 1uM | [47] |
| Step-wise bulk mixing | EPC/DC-Chol/DSPE-PEG/Protamine | 90% | 90nm | Stable in serum | Inhibition of Bcl-2 expression in transferrin receptor expressing leukemia cell lines MV4-11 (41%), K562 (62%) and Raji (50%) | [29] |
DDAB: Dimethyldioctadecylammonium bromide; DE: (2,3-didodecyloxypropyl) (2-hydroxyethyl) dimethylammonium bromide; OA: oleic acid; EPC: egg-PC
3.1 Cationic liposomes
Cationic liposomes are frequently used for nucleic acid delivery studies due to the multiple cationic groups presented on the surface of the liposomes [24-29]. These cations naturally interact with polyanionic nucleic acids and form lipoplexes. Generally, cationic lipids are classified into three major categories based on the head group structure [30]: 1) monovalent lipids such as N (1-(2,3-dioleyloxy) propyl)-N,N,N-trimethylammonium chloride (DOTMA) [31] and 1,2-dioleyl-3-trimethylammonium-propane (DOTAP) [32]; 2) multivalent lipids such as dioctadecylamidoglycylspermine (DOGS) [33] and 3) cationic lipid derivatives such as 3β-(N-(N',N'-dimethylaminoethane)-carbamoyl) cholesterol (DC-Chol) [34]. The hydrophobic chains of the lipids also provide the liposomes with different features. It has been demonstrated that the myristoyl (C14) chain is optimal for transfection compared to C16 and C18 chains [35, 36]. Longer chains increase the phase transition temperature and reduce the fluidity of the lipid membrane, which is unfavorable for lipid membrane fusion and ion-pair formation. For the same reason, unsaturated alkyl chains with considerably higher lipid fluidity often lead to a higher transfection efficiency compared to saturated alkyl chain lipids [37].
Although numerous cationic lipids with distinct structures and high delivery efficiency have been synthesized in laboratories across the world, the lack of systematic investigation makes it difficult to generalize the structure-activity correlation of the cationic lipids. Akinc et al. have taken an empirical and combinatorial chemistry approach to develop a library of around 1,200 lipid-like compounds for the delivery of nucleic acid therapeutics. All the compounds were synthesized based on one-step conjugation addition of alkyl-acrylates or alkyl-acrylamides to primary or secondary amines [24]. The compound library was used to deliver siRNA to HeLa cells as a screening process. It is deduced from the result that the top performance compounds share some structural similarities such as (1) amide linkages, (2) more than two alkyl tails, (3) an 8-12 carbon chain tail and (4) one tail that is not substituted by amine reactants and contains one secondary amine[24]. It is also noteworthy that some top performance compounds are not structurally similar to any of the conventional, effective lipids. Although their research goal mainly targets the delivery of siRNA therapeutics, they also tested the delivery efficiency of 2'-O-Methyl ON targeting miRNA122. After three consecutive injections of 5mg/kg ON, the protein expression levels regulated by the miRNA122 were significantly elevated in the liver, suggesting successful delivery of the ON. The approach to establish a carrier compound library and screen the effective compounds for nucleic acid delivery expanded the dimensions of available delivery solutions while also providing the parameters for rational design of novel carrier compounds.
A similar one-step synthesis approach was adapted by Love et al. who built an epoxide-derived lipidoid library using a rapid ring-opening reaction between amine substrates and epoxide [48]. The resulting compounds are amine-containing alcohols with nonpolar hydrocarbon tails. One of the top compounds, namely C12-200, demonstrated a potency two magnitudes higher than the top compound (LNP01) screened by Akinc et al. regarding the hepatocyte-targeted siRNA delivery efficiency. The C12-200 formulated nanoparticles were able to knockdown Factor VII expression by 50% in the liver at a dose of 0.01mg/kg via tail vein injection. The knockdown duration lasted up to 20 days when dosed at 0.1mg/kg. This high siRNA delivery efficiency was attributed to the special cellular entry pathway that the nanoparticle harnessed. The C12-200 nanoparticle entered the cells via micropinocytosis rather than the classic endocytic pathway. This effectively avoided the lysosomal degradation of its cargo, overcoming a major barrier to gene delivery. Later, Dong et al. took a similar approach and created a compound library by reacting amino acids or lysine-based dipeptides with aldehydes, acrylates or epoxides [49]. Out of this library, the lead compound cKK-E12 could silence the Factor VII in mouse livers with an ED50 of approximately 0.002mg/kg, a five-fold increase compared to C12-200. Moreover, this compound showed a 500-fold increase in specificity for gene silencing in hepatocytes compared to endothelial cells or immune cells. The development of these three libraries has demonstrated improved efficacy and specificity for the siRNA delivery system. Although these two publications have not shown any application in ON delivery, the strategy of one-step synthesis and screening can be used to develop novel materials with high specificity and potency for the delivery of ON.
Formulation
Two decades ago Capaccioli et al. demonstrated that cationic liposomes can facilitate cellular uptake and enhance the stability of ONs in cell culture [50]. Electrostatic interaction between the cationic lipid headgroup and the backbone of nucleic acids is the main driving force for the encapsulation of ONs in cationic liposomes. The conventional methods for preparation of these lipoplexes is direct mixing between cationic liposomes and anionic ON solutions, or rehydration of a thin-layer lipid membrane with ON solutions. The dispersion of cationic lipid/ON complexes in the aqueous solution often results in heterogeneous complexes, sometimes still referred to as cationic liposomes, but more accurately called lipoplexes. Lipoplexes can encapsulate nucleic acid cargos up to 90% of the input dose.
The physicochemical properties of lipoplexes not only depends on the composition and structure of the lipids, but also varies as a result of the ON sequence. Meidan et al. observed that the maximal level of DOTAP liposome neutralization by the homo-polyA ON is much lower than that for homo-polyT or homo-polyC. Therefore, DOTAP/polyA lipoplexes are much more positively charged [51].
As was mentioned, lipoplexes are formed by mixing preformed liposomes with ON solution or by rehydration of a lipid film with ON solution. The size and polydispersity of lipoplexes formed with either method depends largely on the operator's mixing speed. This affects reproducibility and hinders the industrialization of this delivery system. To address the issue, microfluidics technology can be employed in lipoplex formulation. A microfluidics system provides a more homogenous solvent environment and allows rapid mixing of the lipid and nucleic acids so that the hydrophilic portions of the complexes formed can more easily stabilize the system [52]. Yu et al. have reported the characterization of ON-lipoplexes formed using a 3-inlet microfluidic chip-based device [53]. In their study, an ON solution and preformed cationic liposome composed of DOTAP/Egg Phosphatidylcholine (EPC)/Cholesterol/1-O-(2P-(w-methoxypolyethyleneglycol) succinoyl)-2-N-myristoylsphingosine (Cer-PEG2000) (45:18:35:2) were injected into the chip in different channels with the same rate (50-1100 μl/min). The lipoplexes formed with this method showed relatively lower polydispersity compared to those formed by bulk mixing. It is unfortunate, however, that the author did not compare the gene silencing activity of the lipoplexes prepared with different formulations. In addition, testing lipoplex formation in this manner might not be the most novel way to test a microfluidics system, since microfluidic devices are already expected to address the uncontrolled aggregation due to a heterogeneous solvent environment.
3.1.1 Structure of Lipoplexes
Neither the mechanism of action nor the resulting lipoplex structures formed by mixing nucleic acids and cationic liposomes have been completely elucidated. Investigators have used electron microscopy to observe the diverse structures of lipoplexes, including string-like structures[54], oligolamellar structures [55] and tube-like structures [56]. Joachim et al. combined in situ optical microscopy and x-ray diffraction to study lipoplexes formed with cationic liposomes (DOTAP/DOPC, 1:1 mol ratio) and linear or plasmid DNA (Figure 2A). Regardless of the DNA structure, the complexes consisted of a higher order multilamellar structure with DNA sandwiched between cationic bilayers. This multilayer lipid/DNA/lipid structure is formed as a result of polyanionic DNA screening the charge between two cationic lipid membranes [57].
Figure 2.
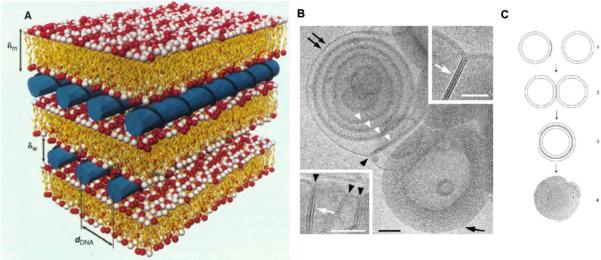
Schematic pictures of the local arrangement in the interior of the lipid/DNA complex (A); Cryo-TEM images of fusion of DOTAP/Cholesterol (1:1) liposomes induced by the addition of ONs. Black arrowheads indicates membrane junctions and white arrowheads in dicates a paired membrane. Scale bar: 50nm (B); Schematic model of lipoplex formation (C). Figure 2A was adapted from ref (45) and Figure 2B and 2C was adapted from ref (46) with permission.
Despite the difference in structure and molecular weight, the lipoplexes formed between ONs and cationic liposomes are quite similar to that of lipoplexes composed of plasmid DNA. In addition to the experiments performed by Joachim et al., Weisman et al. studied the lipoplexes formed by cationic liposomes (DOTAP/Cholesterol) and 18-mer anti-bcl2 ONs using cryo-electron microscopy and small-angle x-ray scattering. The electron microscopic images showed that the addition of ONs into the cationic liposomes induced both liposome aggregation and the formation of a multilamellar structure (Figure 2B and 2C). The aggregation of liposomes was due to the removal of the electrostatic repulsion by ON molecules whose negative charges act as bridges between the membranes. The only structural difference between lipoplexes formed by large molecular weight DNA or ONs is the lack of intervkesicle fusion in the lipid/ON lipoplexes [58].
Remaut et al. published their finding regarding nuclease protection in PEGylated cationic liposomes versus non-PEGylated ones. By monitoring the degradation rate of phosphodiester ONs loaded in DOTAP/DOPE lipoplexes in the presence of DNAse, it was demonstrated that non-PEGylated cationic liposomes provided stronger protection compared with PEGylated liposomes when complexed with ONs. This is due to thePEG chain on the liposomes prohibiting the fusion of the lipid membrane and therefore not trapping the ONs between the lipid bilayers. The surface adsorption of ONs to PEGylated liposomes allows direct exposure to the nuclease which leads to a fast degradation rate [59].
3.1.2 Intracellular Release Mechanism
Evidence has widely indicated that cationic liposomes deliver nucleic acids into cells predominately through an endocytosis pathway [60, 61] rather than fusion with the plasma membrane as suggested by some reports [62, 63]. Zelphati et al. demonstrated that cationic charges on liposomes are required for efficient ON delivery. In their study, the majority of the cationic liposome/ON lipoplexes entered into cells via an endocytic pathway. The inclusion of neutral lipids such as DOPE or cholesterol in the cationic liposomes reduces the optimal ratio (+/−) for high delivery efficiency, although the formulation did not affect the delivery efficiency. [64]. Later, Zelphati et al. measured fluorescence resonance energy transfer between fluorescein-labeled ONs and rhodamine-labeled cationic liposomes to study the release mechanism of cationic liposome-loaded ONs [65]. It was demonstrated that the release of ONs is mainly by displacement and is independent of ionic strength or pH. The displacement could be triggered by highly negatively-charged, linear, and water soluble molecules such as heparin and dextran sulfate, but not DNA or albumin. It was also shown that anionic phospholipids efficiently induced the release of ONs from cationic liposomes, occurring within the time scale of an endocytic event. Therefore, it is hypothesized that the release of ONs in the endosomes is realized in several steps (Figure 3): (1) the interaction between the cationic lipoplexes and the endosome membrane allowing the flip-flop of anionic lipids that are predominantly located on the cytoplasmic side of the membrane. (2) The lateral diffusion of anionic lipidsinto the cationic lipoplexes, forming ion-pairs with cationic lipids in the lipoplexes. (3) The displacement and release of the ON into the cytoplasm [65].
Figure 3.

Schematic illustration of the uptake pathway of the cationic lipid/ON lipoplex and the mechanism of ON endosomal release. Adapted from ref (53) with permission.
3.1.3 Stealth cationic liposomes
The critical issue that hinders the in vivo application of cationic lipoplexes is the fast serum clearance and major distribution in the lung, liver and spleen after intravenous administration [26, 27]. As mentioned above, anionic serum proteins tend to adsorb onto cationic liposomes, destabilizing the lipoplexes and resulting in early release of the therapeutics. The adsorption of complements on the lipoplexes also leads to clearance by the scavenger cells that reside in the spleen and liver. As stated by Allen TM: “If you want to be invisible, look like water” [66]. This strategy of coating cationic liposomes with neutral hydrophilic polymers has been proven to be the most successful resolution to this issue and has been adapted by various liposomal and non-liposomal drug delivery systems to date.
Polyethylene glycol (PEG) is the most widely used hydrophilic polymer in the drug delivery field owing to its low immunogenicity and cytotoxicity. Surface coating with this kind of neutral polymer screens the charges of the delivery systems. The carriers therefore demonstrate better colloidal stability as a result of low charge and surface steric hindrance that prevents the adsorption of serum proteins. The most conventional way to modify the surface of cationic liposomes is through incubation of liposomes with micelles composed of lipid-PEG conjugates. During incubation, the lipid chain of the amphiphilic molecules will insert into the lipid bilayer with the PEG chain sticking out. This method of PEGylation is called “post insertion”. PEGylated liposomes have an extended blood circulation half-life, which was reported by a landmark publication by Klibanov et al in 1990 [67]. Extended circulation time is particularly important for the tumor-targeted delivery of ON therapeutics, as it increases the chance of the liposome to extravasate from the tumor's leaky endothelium and accumulate in the tumor [68].
The development of liposome-based delivery systems for nucleic-acid based therapeutics often face charge-related dilemmas, such as low encapsulation efficiency (<10%) of cargo in neutral or anionic liposomal formulations and unfavorable pharmacokinetic and toxicity issues in cationic liposomal formulations [69]. The dilemma is resolved from two aspects: (1) by developing formulation methods that increase the encapsulation efficiency of the nucleic acid; or (2) by developing conditionally charged cationic liposomes to minimize the charge related issues.
3.2 Neutral liposomes
As an alternative approach to avoid cationic liposome related issues such as poor PK profiles and cytotoxicity, neutral liposomes are employed as carriers to load ONs [41, 70, 71]. Commonly used neutral lipids are phosphatidylcholine (PC), phosphatidylethanolamine (PE), and cholesterol. Neutral lipids have been demonstrated as effective helper lipids when incorporated in cationic liposomes to achieve higher transfection efficiency. Neutral liposomes have also been investigated as independent carriers for the delivery of nucleic acids. In contrast to cationic lipids, neutral lipids lack the positive charges that promote attractive interaction with nucleic acids to efficiently encapsulate ON into the liposomes. It is due to this same reason, however, that neutral liposomes show less interaction with serum proteins and hence possess higher serum stability. Early studies once implicated that the fusion between liposome and plasma membrane allows the contents to enter the cells [72]. Later research indicated that the fusion does not happen without exogenous perturbations such as polyethylene glycol treatment [73] or some viral fusion protein [74] that promotes this process. Nowadays, it is generally believed that the endocytic pathway is the dominant way for the liposomes to enter the cells. Straubinger et al. have demonstrated that the acidic endosomal environment is a critical factor that causes the leakage of liposomal contents into the cytoplasm [75]. In their observation, more charged and larger molecules seem to escape to the cytoplasm at a slower rate, which could account for the entrapment of nucleic acids in the endosomal compartment. To address this issue, DOPE is often included in the neutral liposomal formulation to enhance the transfection efficiency. It is believed that the acidic endosomal compartment could induce DOPE to transform to an inverted hexagonal phase which more readily fuses with the anionic lipid layer and destabilizes the endosome membranes [76]. This disruption allows the entrapped nucleic acid molecules to efficiently escape to the cytoplasm.
3.2.1 Formulations
The lack of driving forces such as electrostatic or hydrophobic interactions results in a low encapsulation of ONs in neutral liposomes with a conventional thin-film lipid rehydration protocol. One of the strategies to increase encapsulation efficiency of ONs is chemical modification which makes the therapeutics more lipophilic. Lopez-Berestein and colleagues have demonstrated the effective encapsulation of P-ethoxy ONs in neutral liposomes composed of DOPC [39, 40]. The P-ethoxy ONs can be easily incorporated into liposomes with the film rehydration method with an efficiency up to 95% [39]. The mechanism of high encapsulation is unknown. The alternative strategy is to harness the strength of formulation technologies to increase the encapsulation efficiency.
3.2.1.1 Minimal volume entrapment
Minimal volume entrapment (MVE) liposomes are prepared by thin-layer lipid hydration and sonication in highly concentrated ON solutions [77]. In order to enhance the encapsulation efficiency, the hydrated ON/liposome and free ONs could be further mixed with free liposomes and sonicated. This preparation method has resulted in a relatively high encapsulation efficiency in cardiolipin/phosphatidylcholine/cholesterol (0.5:10:7) liposomes with 50%-60% encapsulation of the initial input [77]. The inclusion of negatively charged lipid cardiolipin in the formulation adds a repelling force between the lamellar layers and thus increases the entrapment volumes of the liposomes. Although high encapsulation efficiency was achieved with the MVE technology, the neutral liposomes only rendered limited benefits regarding the intracellular accumulation of the ONs compared with free ONs and are cell line dependent. An eighteen fold increase of intracellular accumulation of ONs was observed in lymphoma MOLT-3 cells compared with free ON treated cells, whereas only a fourfold increase was observed in SKVLB cells [78].
3.2.1.2 Reverse-Phase Evaporation
Liposomes prepared by reverse-phase evaporation are prepared by direct hydration of the lipid from an organic solvent [45, 79]. The lipids are dissolved in a partially water-miscible solvent such as ether or mixtures of chloroform and methanol. Aqueous solutions with ONs are mixed with the organic solvent/lipids to form an emulsion, offering a large interface for lipid and ONs to interact. The liposomes are then formed by removal of the organic solvent under liquid nitrogen or a rotary evaporator. This method can generate relatively smaller unilamellar liposomes with high entrapment volumes [80]. Neutral liposomes containing dipalmitoylphosphocholine/cholesterol/dipalmitoyl-DL-α-phosphatidyl-L-serine (4:3:4) could encapsulate as much as 20% of the input dose of ONs [81].
3.2.1.3 Detergent Dialysis
Detergent dialysis liposomes are prepared by co-solubilizing the lipids and ONs in an aqueous solvent in the presence of detergent, followed by removal of the detergent by dialysis [82]. The thin-layer lipid film is rehydrated by the buffer system containing ONs and the appropriate detergent to generate a micellar structure. Non-ionic detergents with a high critical micellar concentration such as octylglucoside are employed so that they do not interact with nucleotides or lipids and can be easily removed through dialysis [83]. However, this formulation method usually results in a low entrapment efficiency of ONs with only about 10% encapsulation in dioleylphosphatidylethanolamine/cholesterol/oleic acid liposomes [46].
3.3 Ionizable Lipids
Although the use of neutral liposomes as ON carriers often leads to extended serum clearance time, the delivery efficiency is often inferior to that of cationic liposomes due to the lack of endosomal disruption capacity. To address this dilemma, an ideal liposome would be one that (1) carries a positive charge during the nucleic acid loading, (2) loses its charge after administration and before entry into the cells and (3) regains its positive charge after entering the endosomal compartment for the formation of ion-pairs between the endosomal membrane and the carrier lipid. The “smart” lipid will have to be protonated or deprotonated based on the environmental conditions. pH is one of the parameters that can distinguish these conditions. pH-sensitive ionizable lipids have therefore been developed to accommodate the need to deliver siRNAs [24, 84-86] and ONs [69, 82, 87]. Generally, two important factors are considered in the rational design of ionizable lipids: (1) the pKa value of the head group which determines the pH condition at which the lipid gets protonated or deprotonated; and (2) the lipid capacity to induce the hexagonal HII phase structure when the ionized lipid interacts with membrane lipids [88].
Semple et al. have adapted the medicinal chemistry approach which uses a structure-activity relationship as the guideline to direct rational lipid design. The design was based on the putative mechanism of action that ionizable cationic lipids disrupt the endosome through ion-pair formation with anionic lipids in the endosomal membrane. A lipid head group pKa of <7.0 achieves better encapsulation at acidic pH and neutral surface charge at physiological pH. A series of derivatives were developed based on the ionizable cationic lipid 1,2-dilinoleyloxy-3-dimethylaminopropane (DLinDMA) which is highly effective at delivering siRNA in rodents and non-human primates [86, 89]. The derivatives were synthesized by changing the hydrocarbon chains, the linker, and the headgroup of DLinDMA. By adjusting the number of cis double bonds in the chain, it was determined that a linoleyl lipid containing two double bonds per hydrocarbon chain was the most potent. By introducing chemical structures that were expected to exhibit different levels of chemical or enzymatic stability and different hydrophobicities, it was determined that alkoxy-containing lipids showed higher activity than ester-containing lipids. This is probably due to the fast hydrolysis rate in vivo. The distance between the head group and the linker can affect pKa of the molecules as well as the charge presentation. For example, Insertion of a methylene group into DLin-K-DMA makes a four times more potent lipid (DLin-KC2-DMA) [88]. By adjusting the size, acid-dissociation constant and number of ionizable groups, it was determined that the dimethylamino groups in the DLinDMA show the highest activity compared to piperazino, morpholino, trimethylamino and bis-dimethylamino groups. The rational approach, as opposed to a library-based screening process, has systematically explored how changing functional elements in the lipid structure affect delivery efficiency.
In follow-up studies, Jayaraman et al. maintained the unsaturated dilinoleyl chain and modified the head groups to investigate the structure-activity correlation [90]. A pKa-activity relationship emerged based on the Factor VII silencing study performed in vivo. The compound (DLin-MC3-DMA) with the highest delivery potency (ED50=0.03mg/kg) contained a head group of pKa=6.44. This pKa allowed the lipid nanoparticles to display minimal charges during blood circulation (pH 7.4) and reach maximum charges in the acidic endosome (pH 5.5). Unfortunately, this trend has its limitations and may not apply to other cationic lipid systems. In an effort to increase the biocompatibility of these lipid nanoparticles, Maier et al. incorporated the ester bond as a biodegradable feature in the lipid structure [91]. The lipid nanoparticle comprised of such lipids demonstrated rapid elimination rates in plasma and tissue while maintaining its delivery potency.
3.3.1 Formulation
Ionizable lipids show the properties of cationic lipids and neutral lipids at different pH conditions. This feature has been well harnessed to encapsulate nucleic acids. In 2001, Semple et al. reported a formulation that prepares “stabilized antisense-lipid particles” (SALP) [69]. In this formulation, the ionizable lipid dioleoyldimethylammonium chloride (DODAC), the neutral lipid distearoylphosphatidylcholine (DSPC), cholesterol and PEG-CerC14 were solubilized in an ethanol solution while the ONs were dissolved in 100% ethanol. The lipid mix stock (DSPC/Cholesterol/DODAP/PEG-CerC14, 25:45:20:10) was then slowly added to the ONs in pH 4.0 acetate buffer at 65°C. The mixture was dialyzed against acetate buffer to remove the ethanol followed by dialysis against HEPEs buffer to neutralize the pH and remove surface-adsorbed ON. This formulation could encapsulate up to 70% of the input dose. The lipid nanoparticles were typically 110±30 nm in diameter. Since this work is mostly done commercially, the formulation process is conducted in a way that can be scaled up to an industrial level.
3.4 Lipid Composite Nanoparticle
As nanotechnology for biomedical applications progresses, a series of novel materials with distinguished biological and physicochemical properties have been developed. While there may never be one omnipotent carrier material that can overcome all extracellular and intracellular barriers in vivo while realizing delivery selectivity, several materials of diverse functionalities and physicochemical properties may be incorporated into a single nano-formulation to overcome these numerous barriers. This approach allows the use of materials that have low transfection capacity, to tackle other challenges during the transfection process in vivo. Formulations that adhere to this multicomponent strategy are thus referred to as composite nanoparticles. Lipid composite nanoparticles are some of the most investigated formulations due to the versatility of the lipids used. Virtually all of the physicochemical properties of each material are considered when preparing a reproducible and well-controlled fabrication protocol.
Li et al. formulated ONs into a core-membrane structured lipid nanoparticle [47] based on the lipid-polycation-DNA (LPD) formulation that was developed by Gao et al. [92]. The defined structure of the nanoparticle was achieved by a multiple-step, self-assembly procedure. ONs were complexed by the cationic polypeptide protamine to form a negatively charged polyplex. The charge of this polyplex allowed for a cationic lipid surface coating. The lipid nanoparticles were then further stabilized by post-insertion of DSPE-PEG. The core structure supported the lipid coating of the cationic liposome, allowing up to 20% (mol %) of the DSPE-PEG to be inserted in the membrane. Although the PEGylated LPD showed compromised cellular uptake compared with non-PEGylated cationic LPD, the presence of a targeting ligand could increase the delivery efficiency and selectivity of the system [47]. A similar formulation was also reported by Junghans et al. to deliver a c-myc antisense ON to U937 cells [93].
Ko et al. reported a similar core-membrane formulation for brain-targeted ON delivery [94]. The core of the nanoparticle is the polyplex formed by polyethylenimine and ONs. The PEI/ON core was adjusted to bear a positive charge so that anionic liposomes (1-Palmitoyl-2-Oleoyl-sn-Glycero-3-Phospho-rac-91-glycerol (POPG)/POPC/cholesterol/DSPE-PEG2000 3.7:3:3:0.3 mol ratio) would spontaneously coat the polyplex and form a similar LPD structure. To realize brain-targeted delivery, the distal end of PEG was conjugated with a monoclonal antibody against the transferrin receptor on the blood-brain barrier. This targeting ligand was meant to carry the nanoparticle through the blood-brain barrier via transcytosis [95]. The formulation has shown mildly extended blood circulation, with 10% of the injected dose remaining in the blood 60 minutes after intravenous injection and 0.3% of the dose accumulated in the brain per gram of brain tissue. As an alternative strategy to induce lipid coating, which is normally induced by electrostatic interaction between the polyplex and liposomes, Yang et al. reported the use of hexadecenal-PEI as a condensing agent to complex with ONs [96]. The resulting polyplexes have hexadecenal chains at the surface of the complex which induce and stabilize the liposome coating. It was demonstrated that the hexadecenal-PEI conjugate with pH-sensitive aldehyde groups increased the stability of the nanoparticle and thus resulted in higher protection of ONs in the presence of serum [96].
McMahon et al. fabricated an artificial high density lipoprotein nanoparticle using a 5nm gold nanoparticle template [97]. The gold nanoparticle was mixed with Apolipoprotein A-I which subsequently stabilized the lipid mix on the surface of the gold nanoparticle, forming a lipid layer. The cholesterylated ONs were then loaded on the surface of the nanoparticle by insertion of the hydrophobic part into the lipid membrane of the artificial lipoprotein nanoparticle. The final particle with ONs loaded is around 30nm. The nanoparticle was used to deliver anti-miR210 ONs to the PC-3 cells and caused an 80% reduction in cellular miR-210 levels [97]. Since the nanoparticle has shown serum stability and its capacity to protect ONs despite the payload being attached to the surface of the carrier, it holds potential to deliver ONs to the liver because Apolipoprotein A-I is a natural ligand for hepatocytes.
4 Therapeutic Applications of ONs
4.1 Cancer
Antisense ONs are the most extensively studied therapeutics in ON-based anti-cancer drugs. Over 90 clinical trials have been conducted to evaluate the anti-cancer efficacy of ON-based therapeutics [98]. However, the majority of the trials are in phase I/II, which are only for toxicity evaluation and dose escalation studies. Only a few of the drugs were tested in phase III clinical trials, which used chemically modified ONs administered without carriers. Unfortunately, there are still no ON-based drugs that have been approved by the FDA for the treatment of cancer. The development of liposome assisted ON therapeutics is still in its infancy regarding clinical translation. So far, there are only three kinds of liposomal ONs tested in clinical phase I trials. Since 2001, the National Cancer Institute has initiated a phase I trial in patients with advanced oral squamous cell carcinoma using a DC-Chol liposome to deliver EGFR antisense ON. NeoPharm Inc. has finished three phase I clinical trials to test the efficacy and maximum tolerated dose for a cationic lipoplex formulation loaded with anti-raf1 ON [99]. Bio-Path has exploited neutral liposomes to deliver antisense ON against growth factor receptor bound protein-2, which have been evaluated in phase I for the treatment of leukemia.
Compared to the number of clinical trials that test liposomal ON based therapeutics, more preclinical trials are conducted to evaluate the anti-cancer efficacy in tumor bearing animal models. ProNAi Therapeutics has developed a liposome-based ON formulation that demonstrated anti-tumor efficacy in vivo [100]. The therapeutic ON is a 24-mer, chemically unmodified DNA nucleotide sequence, which is designed to be complementary to a non-coding, non-transcribed region of the BCL-2 gene. The hybridization of DNA led to the inhibition of transcription of BCL-2 and growth inhibition in cancer cells. In order to achieve systemic delivery, the ON, namely PNT100, was encapsulated in a series of so-called amphoteric liposomal formulations with similar technology to that reported by Semple et al. [69]. These formulations included pH-sensitive cationic, anionic, fusogenic and bilayer-stabilizing lipids. A formulation composed of 1-palmitoyl-2-oleoyl-sn-glycero-3 phosphocholine (POPC)/ 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE)/ cholesteryl hemisuccinate (CHEMS)/ cholesteryl-4-[[2-(4-morpholinyl)ethyl]amino]-4-oxobutanoate (MOCHOL) (6:24:23:47 mol ratio) was selected based on the antitumor activity tested in a PC-3 tumor-bearing animal model when used in combination with docetaxel. Since the formulation was developed for clinical evaluation, sophisticated optimization studies were conducted to test the dosing schedules, dose-escalation, and the resulting pharmacokinetics, pharmacodynamics, and immune response [100].
Griveau et al. reported the use of neutral solid lipid nanoparticles in an oil/water microemulsion system formed by polyethylene glycol hydroxystearate (surfactant) and lecithin (surfactant)-stabilized triglycerides (oil) [101]. The microemulsion was prepared above the phase inversion temperature of the oil by vigorous mixing, and the nanoparticles were formed when the temperature was dropped below the phase inversion temperature. The surface of the nanoparticles was post-inserted with DSPE-PEG chains that were functionalized with an arginine and lysine papillomavirus-derived peptide. This allows the adsorption of ONs on the surface of nanoparticles, but has two inherent defects. Firstly, surface-loaded ONs are directly exposed to serum nucleases that could cause payload degradation. Secondly, charge-based loading of ONs could be easily displaced by negatively charged proteins in the serum, causing early release of the payload. The author attempted to resolve the first issue by using nuclease-resistant locked nucleic acid ONs with methylene bridges connecting the 2’ oxygen and 4’ carbon of the ribose, but the second issue remained unsolved. This nanoparticle has demonstrated delivery of anti-miR210, anti-miR221 and anti-miR21 to glioblastoma UM87 cells in vitro in a proof of concept study. Structurally similar lipid nanoparticles have been developed by Siddiqui et al. to co-deliver doxorubicin and antisense ONs against glucosylceramide synthase (GCS), an enzyme related to drug resistance [102]. The nanoparticle was tested in an NCI/ADR-RES ovarian cancer cell line. The delivery of ONs against GCS reversed the drug resistance status and sensitized the cells to the doxorubicin treatment.
4.2 Immunotherapy
ONs containing unmethylated CpG dinucleotide motifs are recognized by the innate immune system through the pattern recognition receptor Toll Like Receptor 9 (TLR9) located inside the endosome [103]. Activation of TLR9 will elicit strong cellular and humoral immune responses against a variety of antigens [104]. Although CpG ON is licensed as a clinical adjuvant, it is a well-established adjuvant that can be used as a stand-alone agent or vaccine adjuvant for the treatment of cancer [105] or infectious disease [106-108]. In contrast to the delivery of ONs which often require intravenous administration to access target sites, delivery of CpG-containing ONs is insensitive to the route of administration and pharmacokinetic profiles. Even the presence of low dose lipid nanoparticle-encapsulated CpG could be efficiently taken up by immune cells (mostly macrophages, dendritic cells) and induce an immune response regardless of intravenous or subcutaneous administration [109]. Therefore, the therapeutic efficacy of CpG ON delivery stems mostly from the protection of ONs by lipid nanoparticles as well as the enhanced uptake of CpG ONs by the immune cells.
It is also interesting to note that some lipid formulations could further boost the immune response elicited by the CpG motif in the ON as demonstrated by Bramson et al. They reported that the intravenous administration of naked mitogenic ON (INX-6295) or SALP-formulated INX-6295 could both activate the expansion and boost the cytolytic activity of natural killer (NK) cells in the liver of immune-competent mice. Moreover, a weakly mitogenic ON (INX-6300) could also elicit such effects when formulated in SALP, whereas the INX-6300 or SALP alone had no effect. The result indicated the formulation of SALP, combined with ON, may activate an additional pathway for immunostimulation regardless of the CpG motif. The formulation and ON-based therapy could synergistically work to provide a successful platform for the development of novel therapeutics for immunotherapy [110]. Inex Pharmaceuticals Corporation has since adapted this SALP technology as an adjuvant and have attempted to conjugate antigenic proteins to this adjuvant formulation as a vaccine therapy [111].
4.3 Liver metabolic disorders
The liver plays an important role in carbohydrate, lipid, and protein metabolism. Therefore many metabolic disorders can be corrected by directly delivering therapeutics to hepatocytes. The liver has sinusoidal blood vessels with fenestrated endothelium, allowing delivered nanoparticles to access the hepatocytes as long as they are not captured by Kupfer cells. The liver anatomy therefore provides a great chance to develop liver-targeted nano-formulations for medical intervention of many metabolic disorders.
Hatakeyama and Harashima reported the delivery of anti-miRNA122 to the hepatocytes using a pH-sensitive multifunctional envelope-type nano device (MEND). This lipid based formulation was composed of an ionizable lipid, namely YSK05, which contained one tertiary amine that confers the pH-sensitive property and a long, unsaturated carbon chain to achieve a cone-shaped lipid structure [112]. That cone-shaped structure could facilitate endosomal escape and promote transfection efficiency. Anti-miRNA oligos were encapsulated into the ionizable lipid using a similar approach to that reported by Semple et al. [69]. Briefly, YSK05, cholesterol and 1,2-dimyristoyl-sn-glycerol-methoxypolyethylene-glyco 2000 ether (PEG-DMG) (70:30:3 mol ratio) was solubilized in 90% (V/V) t-BuOH solution. The lipid was mixed with ONs in the presence of citrate buffer (pH 4.0) with vigorous agitation. The t-BuOH was removed by ultrafiltration and the buffer was exchanged with PBS (pH 7.4) [113]. The anti-miRNA encapsulation efficiency in the system was measured as up to 90% of the input dose. Three injected doses of YSK05-MEND loaded with anti-miR122 resulted in 90% downregulation of miR122. It is notable that this downregulation continued until 12 days after the first injection. The therapeutic effect, which is the decline of serum cholesterol levels, lasted until day 18. However, the longevity of the anti-miRNA effect is probably due to the chemical modification of ONs with 2-O-Me and phophorothioate linkages which allowed the anti-miRNA to be stable in the hepatocytes for two weeks [113].
4.4 Pulmonary diseases
Lipid nanoparticles have also been used to deliver ON therapeutics to treat respiratory disease. The anatomical structure of the lung, allows delivery of therapeutics via the intravenous or pulmonary pathway. Ma et al. have attempted to use the LPD formulation to systemically deliver ONs against intercellular adhesion molecule-1 (ICAM-1) as a therapy to suppress lung inflammation. Three kinds of PEG-free lipid coatings were used to coat the protamine/ON polyplex. These included DOTAP alone, DOTAP/cholesterol (1:1 mol ratio) and DOTAP/DOPE (1:1 mol ratio). The biodistribution study indicated that DOTAP/DOPE-coated LPD nanoparticles showed the highest lung accumulation (23%). It was also demonstrated that the delivery of CpG-free ONs in the cationic LPD formulation did not induce pro-inflammatory cytokines such as TNF-α, IL-1 and IFN-γ. This suggested that the cationic liposome-related immunostimulatory activity was mostly related to the CpG motif of the cargo. The systemic delivery of anti-ICAM-1 ONs using LPD nanoparticles efficiently suppressed the ICAM-1 mRNA that is induced by endotoxin, which suggested that the system could be adapted to treat pulmonary disease [114]. However, a failure has also been reported by Griesenbach et al., who tested the capacity of a cationic lipid Genzyme lipid 67 (GL67) to deliver siRNA and ONs to the airway epithelium [115]. GL67 is a “gold-standard” agent for lung gene transfer and was considered an opportunity to treat cystic fibrosis. However, no consistent target gene downregulation by either siRNA or antisense ON was observed after intranasal instillation of the lipoplex. In order to extend the drug exposure time, they used a catheter-mediated perfusion system to administer lipoplexes to the nasal cavity. This approach resulted in the transfection of nasal airway epithelial cells using ONs, but not siRNA. In contrast, when an antisense ON against the epithelial sodium channel, a therapeutic target of cystic fibrosis, was delivered using GL67 liposomes, no transfection or function was detected. Based on the high transfection efficiency in vitro by GL67 liposomes, the failure of in vivo transfection is most likely due to the incompatibility of the liposome formulation for pulmonary delivery. Stenton et al. delivered antisense ONs against the SKY gene encapsulated in aerosolized DOTAP/DOPE lipid nanoparticles. The tyrosine kinase SKY is involved in the signaling pathway of allergic asthma. Treatment of rats with a nebulized liposome formulation inhibited mRNA and protein expression in alveolar macrophages [116]. The therapeutic efficacy of this formulation was also evaluated in two animal models of diseased airway inflammation. It was demonstrated that the treatment suppressed the inflammation and reduced immune cell infiltration in the airway [117].
4.5 Infectious disease
ON-based therapeutics are proven to be promising in treating infectious disease [118, 119]. The use of antisense ONs as an antiviral agent was first demonstrated in 1978 by Zamecnik and Stephenson when they observed that a 13-mer ON could block Rous sarcoma virus replication [120]. Since then, ON-based agents have been widely tested as anti-viral agents in different viral diseases in vitro or in vivo, such as Hepatitis B [121, 122], human papillomavirus [123, 124] , human immunodeficiency virus [71, 125-129], and influenza virus [130-134]. Antisense ONs are usually developed based on some defined viral elements including structural, translational, and assembly elements. Therefore, inhibition of viral translation, replication or assembly is expected to have some critical therapeutic effect for treating viral infection. Due to the safety concerns, most of these studies were performed in cell culture and the pharmacokinetics of ON-based antiviral agents still remains an issue in drug development.
5 Conclusion and Future Directions
Ever since researchers came to realize that lipid nanoparticles could be utilized as effective delivery systems for nucleic-acid based therapeutics such as DNA, RNA and siRNA, an explosion of studies have been conducted to seek the optimal materials and formulations to achieve higher delivery efficiency. Early studies mainly focused on transfection in cell culture, which mechanistically unraveled the critical intracellular barriers to transport of nucleic acids from the cellular membrane to the cytoplasm or nucleus. Novel cationic lipids were synthesized with the intention to overcome these barriers. However, transfection mediated by these cationic lipids was only observed in local administration sites or in the “first-pass” organs such as the lung and liver after systemic administration. This was followed by the development of stealth liposomes and ionizable liposomes with higher serum stability and extended circulation time, which are required for liposomes to circulate long enough to access disease sites. This progress clearly paved the way for the successful clinical translation of the technology.
In retrospect, clinical trials of ON-based therapy rarely exploited liposomal carriers for enhanced delivery, although many liposomal formulations have shown great potential in the pre-clinical trials. Instead, these studies focused on chemical modifications to increase serum stability and cellular permeability of the molecules. An extremely high dose of the therapeutics needed to be administered in order to achieve efficacy, which was often accompanied with adverse effects owing to non-targeted delivery. One of the most important reasons for this phenomenon is probably the difficulty of large scale production of liposomal ON with good manufacturing practice (GMP) standards. These formulations are often synthesized via multiple compositions and certain preparation procedures, which pose obstacles to commercial manufacture and clinical translation.
Despite these hurdles, the recent success of rational lipid design and library-based screening has demonstrated a model approach that could streamline the process of novel material synthesis and testing. It is anticipated that a myriad of sophisticated lipids will emerge in the near future with high delivery efficiency, tissue-targeting specificity, controlled-release properties, and biocompatibility. From a manufacturing perspective, exploitation of a microfluidics system for nanoparticle preparation ensures upscale manufacturing with minimal batch to batch variation. These sophisticated technologies and approaches will significantly boost the progression of ON-based therapies, potentially making them mainstream drugs that could match or even exceed the achievements of small molecule drugs.
Acknowledgment
The work in this lab was supported by NIH grants CA149363, CA151652, CA149387 and DK100664. Andrew M. Blair is acknowledged for his help in preparing the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference
- 1.Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 2.Skutella T, Stohr T, Probst JC, Ramalho-Ortigao FJ, Holsboer F, Jirikowski GF. Antisense oligodeoxynucleotides for in vivo targeting of corticotropin-releasing hormone mRNA: comparison of phosphorothioate and 3'-inverted probe performance. Hormone and metabolic research = Hormon- und Stoffwechselforschung = Hormones et metabolisme. 1994;26:460–464. doi: 10.1055/s-2007-1001733. [DOI] [PubMed] [Google Scholar]
- 3.Souza P, Sedlackova L, Kuliszewski M, Wang J, Liu J, Tseu I, et al. Antisense oligodeoxynucleotides targeting PDGF-B mRNA inhibit cell proliferation during embryonic rat lung development. Development. 1994;120:2163–2173. doi: 10.1242/dev.120.8.2163. [DOI] [PubMed] [Google Scholar]
- 4.Biro S, Fu YM, Yu ZX, Epstein SE. Inhibitory effects of antisense oligodeoxynucleotides targeting c-myc mRNA on smooth muscle cell proliferation and migration. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:654–658. doi: 10.1073/pnas.90.2.654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Young SL, Krawczyk SH, Matteucci MD, Toole JJ. Triple helix formation inhibits transcription elongation in vitro. Proceedings of the National Academy of Sciences of the United States of America. 1991;88:10023–10026. doi: 10.1073/pnas.88.22.10023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Konopka K, Rossi JJ, Swiderski P, Slepushkin VA, Duzgunes N. Delivery of an anti-HIV-1 ribozyme into HIV-infected cells via cationic liposomes. Biochimica et biophysica acta. 1998;1372:55–68. doi: 10.1016/s0005-2736(98)00046-7. [DOI] [PubMed] [Google Scholar]
- 7.Brown SA, Jarvis TC. Optimization of lipid-mediated ribozyme delivery to cells in culture. Methods in molecular biology. 1997;74:441–449. doi: 10.1385/0-89603-389-9:441. [DOI] [PubMed] [Google Scholar]
- 8.Kariko K, Megyeri K, Xiao Q, Barnathan ES. Lipofectin-aided cell delivery of ribozyme targeted to human urokinase receptor mRNA. FEBS letters. 1994;352:41–44. doi: 10.1016/0014-5793(94)00914-7. [DOI] [PubMed] [Google Scholar]
- 9.Dai W, Wang C, Wang F, Wang Y, Shen M, Chen K, et al. Anti-miR-197 inhibits migration in HCC cells by targeting KAI 1/CD82. Biochemical and biophysical research communications. 2014;446:541–548. doi: 10.1016/j.bbrc.2014.03.006. [DOI] [PubMed] [Google Scholar]
- 10.Meng W, Jiang L, Lu L, Hu H, Yu H, Ding D, et al. Anti-miR-155 oligonucleotide enhances chemosensitivity of U251 cell to taxol by inducing apoptosis. Cell biology international. 2012;36:653–659. doi: 10.1042/CBI20100918. [DOI] [PubMed] [Google Scholar]
- 11.Ru P, Steele R, Hsueh EC, Ray RB. Anti-miR-203 Upregulates SOCS3 Expression in Breast Cancer Cells and Enhances Cisplatin Chemosensitivity. Genes & cancer. 2011;2:720–727. doi: 10.1177/1947601911425832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim TG, Kim CH, Won EH, Bae SM, Ahn WS, Park JB, et al. CpG-ODN-stimulated dendritic cells act as a potent adjuvant for E7 protein delivery to induce antigen-specific antitumour immunity in a HPV 16 E7-associated animal tumour model. Immunology. 2004;112:117–125. doi: 10.1111/j.1365-2567.2004.01851.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Prince GA, Mond JJ, Porter DD, Yim KC, Lan SJ, Klinman DM. Immunoprotective activity and safety of a respiratory syncytial virus vaccine: mucosal delivery of fusion glycoprotein with a CpG oligodeoxynucleotide adjuvant. Journal of virology. 2003;77:13156–13160. doi: 10.1128/JVI.77.24.13156-13160.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wilson A, Pitt B, Li S. Complex roles of CpG in liposomal delivery of DNA and oligonucleotides. Bioscience reports. 2002;22:309–322. doi: 10.1023/a:1020146924504. [DOI] [PubMed] [Google Scholar]
- 15.Kole R, Williams T, Cohen L. RNA modulation, repair and remodeling by splice switching oligonucleotides. Acta biochimica Polonica. 2004;51:373–378. [PubMed] [Google Scholar]
- 16.Mercatante DR, Mohler JL, Kole R. Cellular response to an antisense-mediated shift of Bcl-x pre-mRNA splicing and antineoplastic agents. The Journal of biological chemistry. 2002;277:49374–49382. doi: 10.1074/jbc.M209236200. [DOI] [PubMed] [Google Scholar]
- 17.Sazani P, Kole R. Modulation of alternative splicing by antisense oligonucleotides. Progress in molecular and subcellular biology. 2003;31:217–239. doi: 10.1007/978-3-662-09728-1_8. [DOI] [PubMed] [Google Scholar]
- 18.Loke SL, Stein CA, Zhang XH, Mori K, Nakanishi M, Subasinghe C, et al. Characterization of oligonucleotide transport into living cells. Proceedings of the National Academy of Sciences of the United States of America. 1989;86:3474–3478. doi: 10.1073/pnas.86.10.3474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jones CH, Chen CK, Ravikrishnan A, Rane S, Pfeifer BA. Overcoming nonviral gene delivery barriers: perspective and future. Molecular pharmaceutics. 2013;10:4082–4098. doi: 10.1021/mp400467x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hsu CY, Uludag H. Nucleic-acid based gene therapeutics: delivery challenges and modular design of nonviral gene carriers and expression cassettes to overcome intracellular barriers for sustained targeted expression. Journal of drug targeting. 2012;20:301–328. doi: 10.3109/1061186X.2012.655247. [DOI] [PubMed] [Google Scholar]
- 21.Bergen JM, Pun SH. Analysis of the intracellular barriers encountered by nonviral gene carriers in a model of spatially controlled delivery to neurons. The journal of gene medicine. 2008;10:187–197. doi: 10.1002/jgm.1137. [DOI] [PubMed] [Google Scholar]
- 22.Wiethoff CM, Middaugh CR. Barriers to nonviral gene delivery. Journal of pharmaceutical sciences. 2003;92:203–217. doi: 10.1002/jps.10286. [DOI] [PubMed] [Google Scholar]
- 23.Nishikawa M, Huang L. Nonviral vectors in the new millennium: delivery barriers in gene transfer. Human gene therapy. 2001;12:861–870. doi: 10.1089/104303401750195836. [DOI] [PubMed] [Google Scholar]
- 24.Akinc A, Zumbuehl A, Goldberg M, Leshchiner ES, Busini V, Hossain N, et al. A combinatorial library of lipid-like materials for delivery of RNAi therapeutics. Nat Biotechnol. 2008;26:561–569. doi: 10.1038/nbt1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.De Rosa G, De Stefano D, Laguardia V, Arpicco S, Simeon V, Carnuccio R, et al. Novel cationic liposome formulation for the delivery of an oligonucleotide decoy to NF-kappaB into activated macrophages. European journal of pharmaceutics and biopharmaceutics : official journal of Arbeitsgemeinschaft fur Pharmazeutische Verfahrenstechnik eV. 2008;70:7–18. doi: 10.1016/j.ejpb.2008.03.012. [DOI] [PubMed] [Google Scholar]
- 26.Geary RS, Leeds JM, Fitchett J, Burckin T, Truong L, Spainhour C, et al. Pharmacokinetics and metabolism in mice of a phosphorothioate oligonucleotide antisense inhibitor of C-raf-1 kinase expression. Drug metabolism and disposition: the biological fate of chemicals. 1997;25:1272–1281. [PubMed] [Google Scholar]
- 27.Gokhale PC, Soldatenkov V, Wang FH, Rahman A, Dritschilo A, Kasid U. Antisense raf oligodeoxyribonucleotide is protected by liposomal encapsulation and inhibits Raf-1 protein expression in vitro and in vivo: implication for gene therapy of radioresistant cancer. Gene therapy. 1997;4:1289–1299. doi: 10.1038/sj.gt.3300543. [DOI] [PubMed] [Google Scholar]
- 28.Beisner J, Dong M, Taetz S, Piotrowska K, Kleideiter E, Friedel G, et al. Efficient telomerase inhibition in human non-small cell lung cancer cells by liposomal delivery of 2'-O-methyl-RNA. Journal of pharmaceutical sciences. 2009;98:1765–1774. doi: 10.1002/jps.21553. [DOI] [PubMed] [Google Scholar]
- 29.Yang X, Koh CG, Liu S, Pan X, Santhanam R, Yu B, et al. Transferrin receptor-targeted lipid nanoparticles for delivery of an antisense oligodeoxyribonucleotide against Bcl-2. Molecular pharmaceutics. 2009;6:221–230. doi: 10.1021/mp800149s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morille M, Passirani C, Vonarbourg A, Clavreul A, Benoit JP. Progress in developing cationic vectors for non-viral systemic gene therapy against cancer. Biomaterials. 2008;29:3477–3496. doi: 10.1016/j.biomaterials.2008.04.036. [DOI] [PubMed] [Google Scholar]
- 31.Felgner PL, Gadek TR, Holm M, Roman R, Chan HW, Wenz M, et al. Lipofection: a highly efficient, lipid-mediated DNA-transfection procedure. Proceedings of the National Academy of Sciences of the United States of America. 1987;84:7413–7417. doi: 10.1073/pnas.84.21.7413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alexander MY, Akhurst RJ. Liposome-medicated gene transfer and expression via the skin. Human molecular genetics. 1995;4:2279–2285. doi: 10.1093/hmg/4.12.2279. [DOI] [PubMed] [Google Scholar]
- 33.Remy JS, Sirlin C, Vierling P, Behr JP. Gene transfer with a series of lipophilic DNA-binding molecules. Bioconjugate chemistry. 1994;5:647–654. doi: 10.1021/bc00030a021. [DOI] [PubMed] [Google Scholar]
- 34.Gao X, Huang L. A novel cationic liposome reagent for efficient transfection of mammalian cells. Biochemical and biophysical research communications. 1991;179:280–285. doi: 10.1016/0006-291x(91)91366-k. [DOI] [PubMed] [Google Scholar]
- 35.Felgner JH, Kumar R, Sridhar CN, Wheeler CJ, Tsai YJ, Border R, et al. Enhanced gene delivery and mechanism studies with a novel series of cationic lipid formulations. The Journal of biological chemistry. 1994;269:2550–2561. [PubMed] [Google Scholar]
- 36.Lenssen K, Jantscheff P, von Kiedrowski G, Massing U. Combinatorial synthesis of new cationic lipids and high-throughput screening of their transfection properties. Chembiochem : a European journal of chemical biology. 2002;3:852–858. doi: 10.1002/1439-7633(20020902)3:9<852::AID-CBIC852>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 37.Yuba E, Nakajima Y, Tsukamoto K, Iwashita S, Kojima C, Harada A, et al. Effect of unsaturated alkyl chains on transfection activity of poly(amidoamine) dendron-bearing lipids. Journal of controlled release : official journal of the Controlled Release Society. 2012;160:552–560. doi: 10.1016/j.jconrel.2012.04.002. [DOI] [PubMed] [Google Scholar]
- 38.Bennett CF, Chiang MY, Chan H, Shoemaker JE, Mirabelli CK. Cationic lipids enhance cellular uptake and activity of phosphorothioate antisense oligonucleotides. Molecular pharmacology. 1992;41:1023–1033. [PubMed] [Google Scholar]
- 39.Tormo M, Tari AM, McDonnell TJ, Cabanillas F, Garcia-Conde J, Lopez-Berestein G. Apoptotic induction in transformed follicular lymphoma cells by Bcl-2 downregulation. Leukemia & lymphoma. 1998;30:367–379. doi: 10.3109/10428199809057548. [DOI] [PubMed] [Google Scholar]
- 40.Gutierrez-Puente Y, Tari AM, Stephens C, Rosenblum M, Guerra RT, Lopez-Berestein G. Safety, pharmacokinetics, and tissue distribution of liposomal P-ethoxy antisense oligonucleotides targeted to Bcl-2. The Journal of pharmacology and experimental therapeutics. 1999;291:865–869. [PubMed] [Google Scholar]
- 41.Stuart DD, Allen TM. A new liposomal formulation for antisense oligodeoxynucleotides with small size, high incorporation efficiency and good stability. Biochimica et biophysica acta. 2000;1463:219–229. doi: 10.1016/s0005-2736(99)00209-6. [DOI] [PubMed] [Google Scholar]
- 42.Pagnan G, Stuart DD, Pastorino F, Raffaghello L, Montaldo PG, Allen TM, et al. Delivery of c-myb antisense oligodeoxynucleotides to human neuroblastoma cells via disialoganglioside GD(2)-targeted immunoliposomes: antitumor effects. Journal of the National Cancer Institute. 2000;92:253–261. doi: 10.1093/jnci/92.3.253. [DOI] [PubMed] [Google Scholar]
- 43.Pastorino F, Brignole C, Marimpietri D, Pagnan G, Morando A, Ribatti D, et al. Targeted liposomal c-myc antisense oligodeoxynucleotides induce apoptosis and inhibit tumor growth and metastases in human melanoma models. Clinical cancer research : an official journal of the American Association for Cancer Research. 2003;9:4595–4605. [PubMed] [Google Scholar]
- 44.Brignole C, Pastorino F, Marimpietri D, Pagnan G, Pistorio A, Allen TM, et al. Immune cell-mediated antitumor activities of GD2-targeted liposomal c-myb antisense oligonucleotides containing CpG motifs. Journal of the National Cancer Institute. 2004;96:1171–1180. doi: 10.1093/jnci/djh221. [DOI] [PubMed] [Google Scholar]
- 45.Ropert C, Lavignon M, Dubernet C, Couvreur P, Malvy C. Oligonucleotides encapsulated in pH sensitive liposomes are efficient toward Friend retrovirus. Biochemical and biophysical research communications. 1992;183:879–885. doi: 10.1016/0006-291x(92)90565-3. [DOI] [PubMed] [Google Scholar]
- 46.Ma DD, Wei AQ. Enhanced delivery of synthetic oligonucleotides to human leukaemic cells by liposomes and immunoliposomes. Leukemia research. 1996;20:925–930. doi: 10.1016/s0145-2126(96)00062-8. [DOI] [PubMed] [Google Scholar]
- 47.Li SD, Huang L. Targeted delivery of antisense oligodeoxynucleotide and small interference RNA into lung cancer cells. Molecular pharmaceutics. 2006;3:579–588. doi: 10.1021/mp060039w. [DOI] [PubMed] [Google Scholar]
- 48.Love KT, Mahon KP, Levins CG, Whitehead KA, Querbes W, Dorkin JR, et al. Lipid-like materials for low-dose, in vivo gene silencing. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:1864–1869. doi: 10.1073/pnas.0910603106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dong Y, Love KT, Dorkin JR, Sirirungruang S, Zhang Y, Chen D, et al. Lipopeptide nanoparticles for potent and selective siRNA delivery in rodents and nonhuman primates. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:3955–3960. doi: 10.1073/pnas.1322937111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Capaccioli S, Di Pasquale G, Mini E, Mazzei T, Quattrone A. Cationic lipids improve antisense oligonucleotide uptake and prevent degradation in cultured cells and in human serum. Biochemical and biophysical research communications. 1993;197:818–825. doi: 10.1006/bbrc.1993.2552. [DOI] [PubMed] [Google Scholar]
- 51.Meidan VM, Glezer J, Amariglio N, Cohen JS, Barenholz Y. Oligonucleotide lipoplexes: the influence of oligonucleotide composition on complexation. Biochimica et biophysica acta. 2001;1568:177–182. doi: 10.1016/s0304-4165(01)00216-1. [DOI] [PubMed] [Google Scholar]
- 52.Valencia PM, Farokhzad OC, Karnik R, Langer R. Microfluidic technologies for accelerating the clinical translation of nanoparticles. Nature nanotechnology. 2012;7:623–629. doi: 10.1038/nnano.2012.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yu B, Zhu J, Xue W, Wu Y, Huang X, Lee LJ, et al. Microfluidic assembly of lipid-based oligonucleotide nanoparticles. Anticancer research. 2011;31:771–776. [PMC free article] [PubMed] [Google Scholar]
- 54.Gershon H, Ghirlando R, Guttman SB, Minsky A. Mode of formation and structural features of DNA-cationic liposome complexes used for transfection. Biochemistry. 1993;32:7143–7151. doi: 10.1021/bi00079a011. [DOI] [PubMed] [Google Scholar]
- 55.Gustafsson J, Arvidson G, Karlsson G, Almgren M. Complexes between cationic liposomes and DNA visualized by cryo-TEM. Biochimica et biophysica acta. 1995;1235:305–312. doi: 10.1016/0005-2736(95)80018-b. [DOI] [PubMed] [Google Scholar]
- 56.Sternberg B, Sorgi FL, Huang L. New structures in complex formation between DNA and cationic liposomes visualized by freeze-fracture electron microscopy. FEBS letters. 1994;356:361–366. doi: 10.1016/0014-5793(94)01315-2. [DOI] [PubMed] [Google Scholar]
- 57.Radler JO, Koltover I, Salditt T, Safinya CR. Structure of DNA-cationic liposome complexes: DNA intercalation in multilamellar membranes in distinct interhelical packing regimes. Science. 1997;275:810–814. doi: 10.1126/science.275.5301.810. [DOI] [PubMed] [Google Scholar]
- 58.Weisman S, Hirsch-Lerner D, Barenholz Y, Talmon Y. Nanostructure of cationic lipid-oligonucleotide complexes. Biophysical journal. 2004;87:609–614. doi: 10.1529/biophysj.103.033480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Remaut K, Lucas B, Braeckmans K, Sanders NN, Demeester J, De Smedt SC. Protection of oligonucleotides against nucleases by pegylated and non-pegylated liposomes as studied by fluorescence correlation spectroscopy. Journal of controlled release : official journal of the Controlled Release Society. 2005;110:212–226. doi: 10.1016/j.jconrel.2005.09.048. [DOI] [PubMed] [Google Scholar]
- 60.Zuhorn IS, Hoekstra D. On the mechanism of cationic amphiphile-mediated transfection. To fuse or not to fuse: is that the question? The Journal of membrane biology. 2002;189:167–179. doi: 10.1007/s00232-002-1015-7. [DOI] [PubMed] [Google Scholar]
- 61.Zuhorn IS, Kalicharan R, Hoekstra D. Lipoplex-mediated transfection of mammalian cells occurs through the cholesterol-dependent clathrin-mediated pathway of endocytosis. The Journal of biological chemistry. 2002;277:18021–18028. doi: 10.1074/jbc.M111257200. [DOI] [PubMed] [Google Scholar]
- 62.Zabner J, Fasbender AJ, Moninger T, Poellinger KA, Welsh MJ. Cellular and molecular barriers to gene transfer by a cationic lipid. The Journal of biological chemistry. 1995;270:18997–19007. doi: 10.1074/jbc.270.32.18997. [DOI] [PubMed] [Google Scholar]
- 63.Zhou X, Huang L. DNA transfection mediated by cationic liposomes containing lipopolylysine: characterization and mechanism of action. Biochimica et biophysica acta. 1994;1189:195–203. doi: 10.1016/0005-2736(94)90066-3. [DOI] [PubMed] [Google Scholar]
- 64.Zelphati O, Szoka FC., Jr. Intracellular distribution and mechanism of delivery of oligonucleotides mediated by cationic lipids. Pharmaceutical research. 1996;13:1367–1372. doi: 10.1023/a:1016026101195. [DOI] [PubMed] [Google Scholar]
- 65.Zelphati O, Szoka FC., Jr. Mechanism of oligonucleotide release from cationic liposomes. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:11493–11498. doi: 10.1073/pnas.93.21.11493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Allen TM. Long-circulating (sterically stabilized) liposomes for targeted drug delivery. Trends in pharmacological sciences. 1994;15:215–220. doi: 10.1016/0165-6147(94)90314-x. [DOI] [PubMed] [Google Scholar]
- 67.Klibanov AL, Maruyama K, Torchilin VP, Huang L. Amphipathic polyethyleneglycols effectively prolong the circulation time of liposomes. FEBS letters. 1990;268:235–237. doi: 10.1016/0014-5793(90)81016-h. [DOI] [PubMed] [Google Scholar]
- 68.Campbell RB, Fukumura D, Brown EB, Mazzola LM, Izumi Y, Jain RK, et al. Cationic charge determines the distribution of liposomes between the vascular and extravascular compartments of tumors. Cancer research. 2002;62:6831–6836. [PubMed] [Google Scholar]
- 69.Semple SC, Klimuk SK, Harasym TO, Dos Santos N, Ansell SM, Wong KF, et al. Efficient encapsulation of antisense oligonucleotides in lipid vesicles using ionizable aminolipids: formation of novel small multilamellar vesicle structures. Biochimica et biophysica acta. 2001;1510:152–166. doi: 10.1016/s0005-2736(00)00343-6. [DOI] [PubMed] [Google Scholar]
- 70.Nakamura N, Timmermann SA, Hart DA, Kaneda Y, Shrive NG, Shino K, et al. A comparison of in vivo gene delivery methods for antisense therapy in ligament healing. Gene therapy. 1998;5:1455–1461. doi: 10.1038/sj.gt.3300765. [DOI] [PubMed] [Google Scholar]
- 71.Zelphati O, Zon G, Leserman L. Inhibition of HIV-1 replication in cultured cells with antisense oligonucleotides encapsulated in immunoliposomes. Antisense research and development. 1993;3:323–338. doi: 10.1089/ard.1993.3.323. [DOI] [PubMed] [Google Scholar]
- 72.Weinstein JN, Yoshikami S, Henkart P, Blumenthal R, Hagins WA. Liposome-cell interaction: transfer and intracellular release of a trapped fluorescent marker. Science. 1977;195:489–492. doi: 10.1126/science.835007. [DOI] [PubMed] [Google Scholar]
- 73.Szoka F, Magnusson KE, Wojcieszyn J, Hou Y, Derzko Z, Jacobson K. Use of lectins and polyethylene glycol for fusion of glycolipid-containing liposomes with eukaryotic cells. Proceedings of the National Academy of Sciences of the United States of America. 1981;78:1685–1689. doi: 10.1073/pnas.78.3.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Huang RT, Wahn K, Klenk HD, Rott R. Fusion between cell membrane and liposomes containing the glycoproteins of influenza virus. Virology. 1980;104:294–302. doi: 10.1016/0042-6822(80)90334-7. [DOI] [PubMed] [Google Scholar]
- 75.Straubinger RM, Hong K, Friend DS, Papahadjopoulos D. Endocytosis of liposomes and intracellular fate of encapsulated molecules: encounter with a low pH compartment after internalization in coated vesicles. Cell. 1983;32:1069–1079. doi: 10.1016/0092-8674(83)90291-x. [DOI] [PubMed] [Google Scholar]
- 76.Koltover I, Salditt T, Radler JO, Safinya CR. An inverted hexagonal phase of cationic liposome-DNA complexes related to DNA release and delivery. Science. 1998;281:78–81. doi: 10.1126/science.281.5373.78. [DOI] [PubMed] [Google Scholar]
- 77.Thierry AR, Dritschilo A. Intracellular availability of unmodified, phosphorothioated and liposomally encapsulated oligodeoxynucleotides for antisense activity. Nucleic acids research. 1992;20:5691–5698. doi: 10.1093/nar/20.21.5691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Thierry AR, Dritschilo A. Liposomal delivery of antisense oligodeoxynucleotides. Application to the inhibition of the multidrug resistance in cancer cells. Annals of the New York Academy of Sciences. 1992;660:300–302. doi: 10.1111/j.1749-6632.1992.tb21093.x. [DOI] [PubMed] [Google Scholar]
- 79.Szoka F, Jr., Papahadjopoulos D. Procedure for preparation of liposomes with large internal aqueous space and high capture by reverse-phase evaporation. Proceedings of the National Academy of Sciences of the United States of America. 1978;75:4194–4198. doi: 10.1073/pnas.75.9.4194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Szoka F, Olson F, Heath T, Vail W, Mayhew E, Papahadjopoulos D. Preparation of unilamellar liposomes of intermediate size (0.1-0.2 mumol) by a combination of reverse phase evaporation and extrusion through polycarbonate membranes. Biochimica et biophysica acta. 1980;601:559–571. doi: 10.1016/0005-2736(80)90558-1. [DOI] [PubMed] [Google Scholar]
- 81.Fresta M, Chillemi R, Spampinato S, Sciuto S, Puglisi G. Liposomal delivery of a 30-mer antisense oligodeoxynucleotide to inhibit proopiomelanocortin expression. Journal of pharmaceutical sciences. 1998;87:616–625. doi: 10.1021/js9702978. [DOI] [PubMed] [Google Scholar]
- 82.Semple SC, Klimuk SK, Harasym TO, Hope MJ. Lipid-based formulations of antisense oligonucleotides for systemic delivery applications. Methods in enzymology. 2000;313:322–341. doi: 10.1016/s0076-6879(00)13020-4. [DOI] [PubMed] [Google Scholar]
- 83.Li W, Szoka FC., Jr. Lipid-based nanoparticles for nucleic acid delivery. Pharmaceutical research. 2007;24:438–449. doi: 10.1007/s11095-006-9180-5. [DOI] [PubMed] [Google Scholar]
- 84.Akinc A, Goldberg M, Qin J, Dorkin JR, Gamba-Vitalo C, Maier M, et al. Development of lipidoid-siRNA formulations for systemic delivery to the liver. Mol Ther. 2009;17:872–879. doi: 10.1038/mt.2009.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhang J, Fan H, Levorse DA, Crocker LS. Ionization behavior of amino lipids for siRNA delivery: determination of ionization constants, SAR, and the impact of lipid pKa on cationic lipid-biomembrane interactions. Langmuir. 2011;27:1907–1914. doi: 10.1021/la104590k. [DOI] [PubMed] [Google Scholar]
- 86.Zimmermann TS, Lee AC, Akinc A, Bramlage B, Bumcrot D, Fedoruk MN, et al. RNAi-mediated gene silencing in non-human primates. Nature. 2006;441:111–114. doi: 10.1038/nature04688. [DOI] [PubMed] [Google Scholar]
- 87.Zhou W, Yuan X, Wilson A, Yang L, Mokotoff M, Pitt B, et al. Efficient intracellular delivery of oligonucleotides formulated in folate receptor-targeted lipid vesicles. Bioconjugate chemistry. 2002;13:1220–1225. doi: 10.1021/bc025569z. [DOI] [PubMed] [Google Scholar]
- 88.Semple SC, Akinc A, Chen J, Sandhu AP, Mui BL, Cho CK, et al. Rational design of cationic lipids for siRNA delivery. Nat Biotechnol. 2010;28:172–176. doi: 10.1038/nbt.1602. [DOI] [PubMed] [Google Scholar]
- 89.Akinc A, Querbes W, De S, Qin J, Frank-Kamenetsky M, Jayaprakash KN, et al. Targeted delivery of RNAi therapeutics with endogenous and exogenous ligand-based mechanisms. Mol Ther. 2010;18:1357–1364. doi: 10.1038/mt.2010.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jayaraman M, Ansell SM, Mui BL, Tam YK, Chen J, Du X, et al. Maximizing the potency of siRNA lipid nanoparticles for hepatic gene silencing in vivo. Angewandte Chemie. 2012;51:8529–8533. doi: 10.1002/anie.201203263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Maier MA, Jayaraman M, Matsuda S, Liu J, Barros S, Querbes W, et al. Biodegradable lipids enabling rapidly eliminated lipid nanoparticles for systemic delivery of RNAi therapeutics. Mol Ther. 2013;21:1570–1578. doi: 10.1038/mt.2013.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gao X, Huang L. Potentiation of cationic liposome-mediated gene delivery by polycations. Biochemistry. 1996;35:1027–1036. doi: 10.1021/bi952436a. [DOI] [PubMed] [Google Scholar]
- 93.Junghans M, Loitsch SM, Steiniger SC, Kreuter J, Zimmer A. Cationic lipid protamine-DNA (LPD) complexes for delivery of antisense c-myc oligonucleotides. European journal of pharmaceutics and biopharmaceutics : official journal of Arbeitsgemeinschaft fur Pharmazeutische Verfahrenstechnik eV. 2005;60:287–294. doi: 10.1016/j.ejpb.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 94.Ko YT, Bhattacharya R, Bickel U. Liposome encapsulated polyethylenimine/ODN polyplexes for brain targeting. Journal of controlled release : official journal of the Controlled Release Society. 2009;133:230–237. doi: 10.1016/j.jconrel.2008.10.013. [DOI] [PubMed] [Google Scholar]
- 95.Lee HJ, Engelhardt B, Lesley J, Bickel U, Pardridge WM. Targeting rat anti-mouse transferrin receptor monoclonal antibodies through blood-brain barrier in mouse. The Journal of pharmacology and experimental therapeutics. 2000;292:1048–1052. [PubMed] [Google Scholar]
- 96.Yang X, Peng Y, Yu B, Yu J, Zhou C, Mao Y, et al. A covalently stabilized lipid polycation-DNA (sLPD) vector for antisense oligonucleotide delivery. Molecular pharmaceutics. 2011;8:709–715. doi: 10.1021/mp100272k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.McMahon KM, Mutharasan RK, Tripathy S, Veliceasa D, Bobeica M, Shumaker DK, et al. Biomimetic high density lipoprotein nanoparticles for nucleic acid delivery. Nano letters. 2011;11:1208–1214. doi: 10.1021/nl1041947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Castanotto D, Stein CA. Antisense oligonucleotides in cancer. Current opinion in oncology. 2014;26:584–589. doi: 10.1097/CCO.0000000000000127. [DOI] [PubMed] [Google Scholar]
- 99.Gokhale PC, Zhang C, Newsome JT, Pei J, Ahmad I, Rahman A, et al. Pharmacokinetics, toxicity, and efficacy of ends-modified raf antisense oligodeoxyribonucleotide encapsulated in a novel cationic liposome. Clinical cancer research : an official journal of the American Association for Cancer Research. 2002;8:3611–3621. [PubMed] [Google Scholar]
- 100.Rodrigueza WV, Woolliscroft MJ, Ebrahim AS, Forgey R, McGovren PJ, Endert G, et al. Development and antitumor activity of a BCL-2 targeted single-stranded DNA oligonucleotide. Cancer chemotherapy and pharmacology. 2014;74:151–166. doi: 10.1007/s00280-014-2476-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Griveau A, Bejaud J, Anthiya S, Avril S, Autret D, Garcion E. Silencing of miR-21 by locked nucleic acid-lipid nanocapsule complexes sensitize human glioblastoma cells to radiation-induced cell death. International journal of pharmaceutics. 2013;454:765–774. doi: 10.1016/j.ijpharm.2013.05.049. [DOI] [PubMed] [Google Scholar]
- 102.Siddiqui A, Gupta V, Liu YY, Nazzal S. Doxorubicin and MBO-asGCS oligonucleotide loaded lipid nanoparticles overcome multidrug resistance in adriamycin resistant ovarian cancer cells (NCI/ADR-RES). International journal of pharmaceutics. 2012;431:222–229. doi: 10.1016/j.ijpharm.2012.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, et al. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–745. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 104.Elkins KL, Rhinehart-Jones TR, Stibitz S, Conover JS, Klinman DM. Bacterial DNA containing CpG motifs stimulates lymphocyte-dependent protection of mice against lethal infection with intracellular bacteria. Journal of immunology. 1999;162:2291–2298. [PubMed] [Google Scholar]
- 105.Krieg AM. Antitumor applications of stimulating toll-like receptor 9 with CpG oligodeoxynucleotides. Current oncology reports. 2004;6:88–95. doi: 10.1007/s11912-004-0019-0. [DOI] [PubMed] [Google Scholar]
- 106.Verthelyi D, Wang VW, Lifson JD, Klinman DM. CpG oligodeoxynucleotides improve the response to hepatitis B immunization in healthy and SIV-infected rhesus macaques. Aids. 2004;18:1003–1008. doi: 10.1097/00002030-200404300-00007. [DOI] [PubMed] [Google Scholar]
- 107.Jiao X, Wang RY, Qiu Q, Alter HJ, Shih JW. Enhanced hepatitis C virus NS3 specific Th1 immune responses induced by co-delivery of protein antigen and CpG with cationic liposomes. The Journal of general virology. 2004;85:1545–1553. doi: 10.1099/vir.0.79896-0. [DOI] [PubMed] [Google Scholar]
- 108.Harandi AM, Eriksson K, Holmgren J. A protective role of locally administered immunostimulatory CpG oligodeoxynucleotide in a mouse model of genital herpes infection. Journal of virology. 2003;77:953–962. doi: 10.1128/JVI.77.2.953-962.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wilson KD, Raney SG, Sekirov L, Chikh G, deJong SD, Cullis PR, et al. Effects of intravenous and subcutaneous administration on the pharmacokinetics, biodistribution, cellular uptake and immunostimulatory activity of CpG ODN encapsulated in liposomal nanoparticles. International immunopharmacology. 2007;7:1064–1075. doi: 10.1016/j.intimp.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 110.Bramson JL, Bodner CA, Johnson J, Semple S, Hope MJ. Intravenous administration of stabilized antisense lipid particles (SALP) leads to activation and expansion of liver natural killer cells. Antisense & nucleic acid drug development. 2000;10:217–224. doi: 10.1089/oli.1.2000.10.217. [DOI] [PubMed] [Google Scholar]
- 111.Takasaki J, Raney SG, Chikh G, Sekirov L, Brodsky I, Tam Y, et al. Methods for the preparation of protein-oligonucleotide-lipid constructs. Bioconjugate chemistry. 2006;17:451–458. doi: 10.1021/bc050052j. [DOI] [PubMed] [Google Scholar]
- 112.Sato Y, Hatakeyama H, Sakurai Y, Hyodo M, Akita H, Harashima H. A pH-sensitive cationic lipid facilitates the delivery of liposomal siRNA and gene silencing activity in vitro and in vivo. Journal of controlled release : official journal of the Controlled Release Society. 2012;163:267–276. doi: 10.1016/j.jconrel.2012.09.009. [DOI] [PubMed] [Google Scholar]
- 113.Hatakeyama H, Murata M, Sato Y, Takahashi M, Minakawa N, Matsuda A, et al. The systemic administration of an anti-miRNA oligonucleotide encapsulated pH-sensitive liposome results in reduced level of hepatic microRNA-122 in mice. Journal of controlled release : official journal of the Controlled Release Society. 2014;173:43–50. [PubMed] [Google Scholar]
- 114.Ma Z, Zhang J, Alber S, Dileo J, Negishi Y, Stolz D, et al. Lipid-mediated delivery of oligonucleotide to pulmonary endothelium. American journal of respiratory cell and molecular biology. 2002;27:151–159. doi: 10.1165/ajrcmb.27.2.4653. [DOI] [PubMed] [Google Scholar]
- 115.Griesenbach U, Kitson C, Escudero Garcia S, Farley R, Singh C, Somerton L, et al. Inefficient cationic lipid-mediated siRNA and antisense oligonucleotide transfer to airway epithelial cells in vivo. Respiratory research. 2006;7:26. doi: 10.1186/1465-9921-7-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Stenton GR, Kim MK, Nohara O, Chen CF, Hirji N, Wills FL, et al. Aerosolized Syk antisense suppresses Syk expression, mediator release from macrophages, and pulmonary inflammation. Journal of immunology. 2000;164:3790–3797. doi: 10.4049/jimmunol.164.7.3790. [DOI] [PubMed] [Google Scholar]
- 117.Ulanova M, Schreiber AD, Befus AD. The future of antisense oligonucleotides in the treatment of respiratory diseases. BioDrugs : clinical immunotherapeutics, biopharmaceuticals and gene therapy. 2006;20:1–11. doi: 10.2165/00063030-200620010-00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Matsukura M, Koike K, Zon G. Antisense phosphorothioates as antivirals against human immunodeficiency virus (HIV) and hepatitis B virus (HBV). Toxicology letters. 1995;82-83:435–438. doi: 10.1016/0378-4274(95)03574-5. [DOI] [PubMed] [Google Scholar]
- 119.Goodchild J. Antisense antivirals. Antisense research and development. 1991;1:361–364. doi: 10.1089/ard.1991.1.361. [DOI] [PubMed] [Google Scholar]
- 120.Zamecnik PC, Stephenson ML. Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proceedings of the National Academy of Sciences of the United States of America. 1978;75:280–284. doi: 10.1073/pnas.75.1.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Deng YB, Nong LG, Huang W, Pang GG, Wang YF. [Inhibition of hepatitis B virus (HBV) replication using antisense LNA targeting to both S and C genes in HBV]. Zhonghua gan zang bing za zhi = Zhonghua ganzangbing zazhi = Chinese journal of hepatology. 2009;17:900–904. [PubMed] [Google Scholar]
- 122.Soni PN, Brown D, Saffie R, Savage K, Moore D, Gregoriadis G, et al. Biodistribution, stability, and antiviral efficacy of liposome-entrapped phosphorothioate antisense oligodeoxynucleotides in ducks for the treatment of chronic duck hepatitis B virus infection. Hepatology. 1998;28:1402–1410. doi: 10.1002/hep.510280532. [DOI] [PubMed] [Google Scholar]
- 123.Cho CW, Poo H, Cho YS, Cho MC, Lee KA, Lee SJ, et al. HPV E6 antisense induces apoptosis in CaSki cells via suppression of E6 splicing. Experimental & molecular medicine. 2002;34:159–166. doi: 10.1038/emm.2002.23. [DOI] [PubMed] [Google Scholar]
- 124.Clawson GA, Miranda GQ, Sivarajah A, Xin P, Pan W, Thiboutot D, et al. Inhibition of papilloma progression by antisense oligonucleotides targeted to HPV11 E6/E7 RNA. Gene therapy. 2004;11:1331–1341. doi: 10.1038/sj.gt.3302303. [DOI] [PubMed] [Google Scholar]
- 125.Renneisen K, Leserman L, Matthes E, Schroder HC, Muller WE. Inhibition of expression of human immunodeficiency virus-1 in vitro by antibody-targeted liposomes containing antisense RNA to the env region. The Journal of biological chemistry. 1990;265:16337–16342. [PubMed] [Google Scholar]
- 126.Zelphati O, Degols G, Loughrey H, Leserman L, Pompon A, Puech F, et al. Inhibition of HIV-1 replication in cultured cells with phosphorylated dideoxyuridine derivatives encapsulated in immunoliposomes. Antiviral research. 1993;21:181–195. doi: 10.1016/0166-3542(93)90027-g. [DOI] [PubMed] [Google Scholar]
- 127.Anazodo MI, Salomon H, Friesen AD, Wainberg MA, Wright JA. Antiviral activity and protection of cells against human immunodeficiency virus type-1 using an antisense oligodeoxyribonucleotide phosphorothioate complementary to the 5'-LTR region of the viral genome. Gene. 1995;166:227–232. doi: 10.1016/0378-1119(95)00582-x. [DOI] [PubMed] [Google Scholar]
- 128.Anazodo MI, Wainberg MA, Friesen AD, Wright JA. Sequence-specific inhibition of gene expression by a novel antisense oligodeoxynucleotide phosphorothioate directed against a nonregulatory region of the human immunodeficiency virus type 1 genome. Journal of virology. 1995;69:1794–1801. doi: 10.1128/jvi.69.3.1794-1801.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Anazodo MI, Wainberg MA, Friesen AD, Wright JA. Inhibition of human immunodeficiency virus type 1 (HIV) gag gene expression by an antisense oligodeoxynucleotide phosphorothioate. Leukemia 9 Suppl. 1995;1:S86–88. [PubMed] [Google Scholar]
- 130.Abe T, Hatta T, Takai K, Nakashima H, Yokota T, Takaku H. Inhibition of influenza virus replication by phosphorothioate and liposomally endocapsulated oligonucleotides. Nucleosides & nucleotides. 1998;17:471–478. doi: 10.1080/07328319808005191. [DOI] [PubMed] [Google Scholar]
- 131.Giannecchini S, Clausi V, Nosi D, Azzi A. Oligonucleotides derived from the packaging signal at the 5' end of the viral PB2 segment specifically inhibit influenza virus in vitro. Archives of virology. 2009;154:821–832. doi: 10.1007/s00705-009-0380-2. [DOI] [PubMed] [Google Scholar]
- 132.Giannecchini S, Wise HM, Digard P, Clausi V, Del Poggetto E, Vesco L, et al. Packaging signals in the 5'-ends of influenza virus PA, PB1, and PB2 genes as potential targets to develop nucleic-acid based antiviral molecules. Antiviral research. 2011;92:64–72. doi: 10.1016/j.antiviral.2011.06.013. [DOI] [PubMed] [Google Scholar]
- 133.Hatta T, Abe T, Takai K, Takaku H. [Antisense nucleic acid therapy of influenza virus] Nihon rinsho Japanese journal of clinical medicine. 1997;55:2765–2771. [PubMed] [Google Scholar]
- 134.Wong JP, Christopher ME, Salazar AM, Dale RM, Sun LQ, Wang M. Nucleic acid-based antiviral drugs against seasonal and avian influenza viruses. Vaccine. 2007;25:3175–3178. doi: 10.1016/j.vaccine.2007.01.051. [DOI] [PubMed] [Google Scholar]