

Abstract
The signaling switch of β2-adrenergic and μ1-opioid receptors from stimulatory G-protein (Gαs) to inhibitory G-protein (Gαi) (and vice versa) influences adenylyl cyclase (AC) and extracellular-regulated kinase (ERK)1 ⁄ 2 activation. Post-translational modifications, including dephosphorylation of Gαs, enhance opioid receptor coupling to Gαs. In the present study, we substituted the Ser ⁄ Thr residues of Gαs at the α3 ⁄ β5 and α4 ⁄ β6 loops aiming to study the role of Gαs lacking Ser ⁄ Thr phosphorylation with respect to AC sensitization and mitogen-activated protein kinase activation. Isoproterenol increased the cAMP concentration (EC50 = 22.8 ± 3.4 μM) in Gαs-transfected S49 cyc– cells but not in nontransfected cells. However, there was no significant difference between the Gαs-wild-type (wt) and mutants. Morphine (10 μM) inhibited AC activity more efficiently in cyc– compared to Gαs-wt introduced cells (P < 0.05); however, we did not find a notable difference between Gαs-wt and mutants. Interestingly, Gαs-wt transfected cells showed more sensitization with respect to AC after chronic morphine compared to nontransfected cells (101 ± 12% versus 34 ± 6%; P < 0.001); μ1-opioid receptor interacted with Gαs, and both co-immunoprecipitated after chronic morphine exposure. Furthermore, mutation of T270A and S272A (P < 0.01), as well as T270A, S272A and S261A (P < 0.05), in α3 ⁄ β5, resulted in a higher level of AC supersensitization. ERK1⁄ 2 phosphorylation was rapidly induced by isoproterenol (by 9.5 ± 2.4-fold) and morphine (22 ± 2.2-fold) in Gαs-transfected cells; mutations of α3 ⁄ β5 and α4 ⁄ β6 did not affect the pattern or extent of mitogen-activated protein kinase activation. The findings of the present study show that Gαs interacts with the μ1-opioid receptor, and the Ser ⁄ Thr mutation to Ala at the α3 ⁄ β5 loop of Gαs enhances morphine-induced AC sensitization. In addition, Gαs was required for the rapid phosphorylation of ERK1⁄ 2 by isoproterenol but not morphine.
Keywords: adrenergic, Gαs, MAPK, opioid, signaling switch
Introduction
G-protein coupled receptors (GPCRs) are transmembrane proteins that convey signals via interaction with several G-proteins [1]. Coupling with a particular G-protein is important in the specificity of signaling to downstream effectors and determines the fate and type of the ligand-induced cellular event [2]. Several domains of both receptor and G-protein are involved in their selective connection [3–7]; the α3 ⁄ β5 and α4 ⁄ β6 loops of stimulatory G-protein (Gαs) are suggested to be crucial for its interaction with receptor and effector [6,8].
Recently, a switch in G-protein coupling has been reported for several GPCRs [9–12]. β1 and β2 adrenergic receptors (ADRB1 and 2), which generally couple to Gαs and activate adenylyl cyclase (AC), can switch to inhibitory G-protein (Gi) upon phosphorylation with protein kinase A. This shift in G-protein interaction appears to be responsible for the activation of mitogen-activated protein kinase (MAPK) and the alteration from pro- to anti-apoptotic signaling in cardiomyocytes [13–16].
An excitatory response of opioid receptors in chronic treatment is well recognized [17–22], and a variety of mechanisms have been suggested [23–27], including the direct coupling of opioid receptor with Gαs [28–31]. Post-translational modifications in Gαs including depalmitoylation [32] or dephosphorylation [33] enhance its interaction with μ-opioid receptor (OPRM). Gαs phosphorylation is diminished after chronic morphine exposure and in vitro dephosphorylation of Gαs improves its interaction with OPRM [33].
In the present study, we substituted the Ser ⁄ Thr residues at the α3 ⁄ β5 and α4 ⁄ β6 regions of Gαs with Ala to prepare constructs that cannot be phosphorylated. Subsquently, we aimed to investigate the role of Gαs and its mutants in opioid and adrenergic receptor signaling to AC and MAPK.
Results
Expression of Gαs mutants, OPRM1 and ADRB2, in S49 cyc– cells
To set up the cell system, a western blot analysis was performed to detect the expression of receptors and Gαs in S49 cyc– cells. As shown in Fig. 1B, C, OPRM1 and ADRB2 proteins are endogenously expressed in S49 cyc– cells, although they do not express a detectable amount of Gαs (Fig. 1A); PC12 cell lysates were used as a positive control. The Gαs sequence with helices and sheets is shown in Fig. 1E and residues that have been substituted with Ala are also underlined. The mutated Gαs constructs were confirmed by restriction enzyme mapping and sequencing, and S49 cyc– cells were then transfected with wild-type (wt) and mutants. As shown in Fig. 1D, S49 cyc– cells expressed Gαs-Flag after transfection for up to 72 h.
Fig. 1.
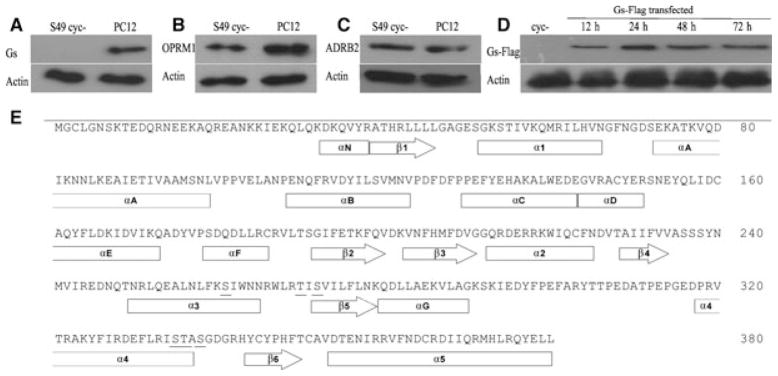
Expression of Gαs, ADRB2, OPRM1 and sequence alignment of Gαs. S49 cyc– cell lysates were prepared and (A) Gαs, (B) OPRM1 and (C) ADRB2 protein expression was analyzed by western blotting; PC12 cell lysates were used as a positive control. (D) Expression of Flag tagged-Gαs in cyc– cells was studied at different times after transient transfection. (E) Protein sequence of the short isoform of Gαs with the point mutations underlined.
Mutation in Ser and Thr at the α3 ⁄ β5 and α4 ⁄ β6 loops of Gαs did not change cAMP production by isoproterenol
S49 cyc– cells were transiently transfected with Gαs constructs and subjected to different treatments. Isoproterenol did not alter cAMP level in nontransfected S49 cyc– cells; however, in Gαs-transfected cells, isoproterenol elevated cAMP levels by 7.6 ± 0.8-fold compared to control (Fig. 2B; P < 0.001, n = 7). Dose–response analysis with Gαs-wt showed that acute administration of isoproterenol for 45 min produced a significant increase in cAMP levels (r2 = 0.96, EC50 = 22.8 ± 3.4 μM), whereas treatment for 24 h resulted in a small elevation of cAMP content [Fig. 2A; P < 0.001; two-way analysis of variance (ANOVA) compared to acute treatment, n = 5]. Furthermore, there was no significant difference among Gαs-wt and mutants either after acute or in chronic isoproterenol exposure (Fig. 2B, C; n = 7). The basal level of cAMP in transfected cells was comparable to nontransfected and neither Gαs-wt, nor its mutants demonstrated constitutive activity (data not shown).
Fig. 2.

cAMP accumulation in response to Isoproterenol. S49 cyc– cells transfected with Gαs-wt were exposed to different doses of isoproterenol for 45 min (acute) and 24 h (chronic), and then the cAMP concentration was assayed with a competitive ELISA method (A; n = 5). The effects of (B) acute and (C) chronic isoproterenol (10 μM) on the cAMP concentration were studied in cyc– cells transfected with indicated constructs. The data are presented as the ratio to control. *Compared to cyc–; mean ± SD; n = 7.
Double (T270A, S272A) as well as triple (T270A, S272A, S261A) mutation of Gαs at α3 ⁄ β5 loop enhanced the stimulatory response to chronic morphine
As shown in Fig. 3A, morphine, dose-dependently, reduced forskolin (Fsk)-augmented cAMP concentration in acute treatment (r2 = 0.83, EC50 = 5.2 ± 1.7 μM, maximum inhibition = 77.2 ± 8%). In addition, acute treatment with morphine (10 μM) diminished AC activity more efficiently in cyc– (59 ± 5%) compared to Gαs-wt transfected cells (43 ± 8%; P < 0.05) although we did not identify a notable difference between Gαs-wt and mutant groups (Fig. 3B; n = 7).
Fig. 3.

cAMP accumulation in response to morphine. S49 cyc– cells transfected with Gαs-wt were exposed to different doses of morphine for 45 min (acute) and 24 h (chronic), and then the cAMP concentration was assayed with a competitive ELISA method (A; n = 5). The effects of (B) acute and (C) chronic morphine (10 μM) on cAMP concentration were studied in cyc– cells transfected with indicated constructs. The data are normalized to Fsk. *Compared to cyc–. #Compared to Gαs-wt; mean ± SD; n = 7.
Morphine treatment of Gαs-transfected cells for 24 h resulted in a rebound increase in Fsk-induced cAMP accumulation (r2 = 0.9, EC50 = 20.02 ± 3.9 μM). There was only a 34 ± 6% rise in the AC response to Fsk in morphine (10 μM) treated cyc– cells, whereas the Gαs-wt group demonstrated 101 ± 12% of the AC superactivation (P < 0.001). Comparing the mutant groups, double mutation of T270A and S272A (P < 0.01), as well as triple mutation of T270A, S272A and S261A (P < 0.05), in the α3 ⁄ β5 regions of Gαs promoted a higher level of Fsk-induced cAMP concentration after chronic morphine exposure, whereas there was no significant difference between Gαs-wt and α4 ⁄ β6 mutants (Fig. 3C; n = 7).
OPRM1 co-immunoprecipitated with Gαs after chronic morphine treatment
To study the interaction of opioid receptor with Gαs, cells were treated with morphine (10 μM, for 24 h) and then a pull-down experiment was conducted with Flag and OPRM antibodies. We observed that OPRM1 co-immunoprecipitates with Gαs-Flag after chronic morphine treatment where the agonist was present in all solubilizing and washing buffers (OPRM ratio to Gαs-Flag: 0.4 ± 0.12). Similarly, when OPRM1 was pulled down, a Gαs interaction was observed by western blotting (Gαs-Flag ratio to OPRM: 0.34 ± 0.1); however, the receptor : G-protein ratio was not affected by the α3 ⁄ β5 or α4 ⁄ β6 mutants in either the OPRM or Gαs-Flag pull-down experiments (Fig. 4; n = 2).
Fig. 4.

Co-immunoprecipitation of Gαs with OPRM1. S49 cyc–, Gαs-wt and mutant transfected cells were treated with morphine (10 μM) for 24 h and pull-down experiments with (A) Flag or (B) OPRM1 antibodies were conducted, and western blotting was used for the detection of protein bands. The ratio of receptor to G-protein (and vice versa) are presented in (C) (mean ± SD; n = 2).
Ser and Thr mutations in α3 ⁄ β5 and α4 ⁄ β6 did not alter extracellular-regulated kinase (ERK)1 ⁄ 2 phosphorylation after isoproterenol or morphine treatment
S49 cyc– and Gαs-transfected cells were serum starved for 24 h and subsequently treated with isoproterenol or morphine; subsequently, a time course study for MAPK activation was accomplished by western blotting. Isoproterenol (10 μM) rapidly induced ERK1⁄ 2 phosphorylation by up to 9.5 ± 2.4-fold of basal levels in Gαs-transfected cells (P < 0.001). However, there was no significant change either in the pattern or extent of MAPK activity between the Gαs-wt and mutant groups (Fig. 5; n = 2). Unexpectedly, isoproterenol elevated the phosphorylated ERK (pERK)1 ⁄ 2 band intensity (by 4.7 ± 0.7-fold of basal levels) in cyc– cells at 1 h after treatment (Fig. 5; P < 0.001).
Fig. 5.

Time course of isoproterenol-induced MAPK activation. S49 cyc–, Gαs-wt and mutant transfected cells were serum starved for 24 h and then treated with isoproterenol (10 μM), cell lysates were prepared at the indicated times, and phosphorylation of ERK1 ⁄ 2 was studied by western blotting; blots were stripped and reprobed for ERK1 ⁄ 2 (A) and β-actin (not shown). (B) The pERK1 ⁄ 2 band intensities were normalized to corresponding ERK1 ⁄ 2 and β-actin, and are presented as the mean ± SD of the ratio to time 0 (t0) (n = 2).
Morphine (10 μM) induced a fast phosphorylation of ERK1⁄ 2 by up to 22 ± 2.2-fold of basal levels in Gs-transfected and nontransfected S49 cyc– cells (P < 0.001). Moreover, we did not find a notable difference among cyc–, Gαs-wt or mutant groups with respect to ERK1⁄ 2 activation (Fig. 6; n = 2). Furthermore, the pERK1⁄ 2 band intensity was diminished very quickly in Gαs-transfected cells exposed to isoproterenol (t1 ⁄ 2 = 9.9 min, r2 = 0.9), in contrast to opioid-treated groups, which showed a sustained and two phase decay [t1 ⁄ 2 (fast) = 6.1 min, t1 ⁄ 2 (slow) = 271.9 min, r2 = 0.92; P < 0.001].
Fig. 6.

Time course of morphine-induced MAPK activation. S49 cyc–, Gαs-wt and mutant transfected cells were serum starved for 24 h and then treated with morphine (10 μM), cell lysates were prepared at the indicated times, and phosphorylation of ERK1 ⁄ 2 was studied by western blotting; blots were stripped and reprobed for ERK1 ⁄ 2 (A) and β-actin (not shown). (B) The pERK1 ⁄ 2 band intensities were then normalized to corresponding ERK1 ⁄ 2 and β-actin, and are presented as the mean ± SD of the ratio to time 0 (t0) (n = 2).
Discussion
There is not a single structure in receptor or G-proteins that guarantees the fidelity of their interaction, and specific coupling is intermediated through several contact regions between the receptor and G-protein [1]. Although many residues in Gαs are known to directly or indirectly affect protein interactions [7,34–36], the α3 ⁄ β5 and α4 ⁄ β6 loops and the α5 helix appear to be crucial for a successful and selective coupling with receptor and effector [1,6]. In this regard, post-translational modification including phosphorylation of Gαs has been implied in coupling. Hence, in the present study, we investigated the role of Ser ⁄ Thr residues at the α3 ⁄ β5 and α4 ⁄ β6 loops with respect to Gαs coupling to ADRB2 and OPRM1.
The results obtained indicate that all the mutated constructs in the α3 ⁄ β5 and α4 ⁄ β6 loops are functional in stimulating AC. In two consecutive studies, Marsh et al. [7] and Grishina & Berlot [8] have reported that S349A, T350A and S352A mutants in the α4 ⁄ β6 loop of Gαs-long did not exhibit defects related to effector activation or receptor-mediated AC stimulation. These mutations correspond to S335, T336 and S338 in the short isoform of Gαs; hence, they are consistent with the results obtained in the present study regarding isoproterenol-stimulated cAMP accumulation in cyc– cells transfected with α4 ⁄ β6 mutants. In addition, they demonstrated that the substitution of corresponding α3 ⁄ β5 residues of Gαs with those of Gαi2 hinders receptor-mediated AC activation, and suggest that α3 ⁄ β5 is the contact site of G-protein with the second and third intracellular loops of ADRB2 [8]. We mutated T270A, S272A and S261A in the α3 helix and α3 ⁄ β5 loop and found that the mutations in this region did not affect isoproterenol-mediated cAMP accumulation (Fig. 2). Such controversy regarding the T270 mutation may result from Grishina et al. [8] substituting the corresponding Thr to Asp, which is a nonconservative change, to a negatively-charged amino acid; furthermore, they also mutated several other residues in α3 ⁄ β5 loop and α3 helix (N271K ⁄K274D⁄R280K⁄ T284D ⁄ I285T) [8]. Although the α3 ⁄ β5 loop is important for receptor interaction, it appears that the ability to fulfill such a role is derived from structural conformation but not amino acid sequences [5] or the state of phosphorylation at this site. In a very recent study, Rasmussen et al. [37] investigated the crystal structure of ADRB2-Gαs complex and showed that the α5-helix of Gαs is critical in receptor interaction and that the β1-α1 and β6-α5 loops are reorganized in the nucleotide-free structure of ADRB2–Gαs.
Opioid receptors preferentially interact with Gαi and Gαo, to inhibit the AC pathway [38]. A stimulatory response to chronic opioid [20,24,26,39–41] and other Gαi coupled receptors [10,12,42] is well documented. Pharmacological evidence in which the excitatory effects of opioids were resistant to pertussis toxin but sensitive to cholera toxin gave rise to the idea that Gαs might play a role in altered opioid signaling [43–47]. We observed that acute morphine produced a greater reduction of Fsk-stimulated cAMP content in cyc– cells compared to Gαs-wt transfected cells (59 ± 5% versus 43 ± 8%; Fig. 3). This may be the result of a shift of balance toward inhibitory regulators in the absence of Gαs in cyc– cells or the possibility that OPRM1 may slightly couple to Gαs even after acute opioid exposure [28]. There was an approximately two-fold increase in Fsk-mediated cAMP accumulation in Gαs-transfected cells after chronic morphine (10 μM) treatment, whereas nontransfected cells showed an AC superactivation of only 34 ± 6%, suggesting a role for Gαs in excitatory opioid signaling. Accordingly, Watts et al. [48] have reported that heterologous sensitization of Fsk-induced cAMP accumulation by dopamine D2 receptor does not occur in Gαs-insensitive mutants of AC type V; however, Thomas et al. [42] have reported a 76% and 87% increase in the cAMP synthesis rate as a result of long-term somatostatin in S49 cyc– and S49 wt cells, respectively [42]. This inconsistency with respect to cAMP accumulation might be explained by dissimilarities between opioid and somatostatin receptors, as well as the level of receptor and effector expression. Feldman et al. [49] have reported that a peptide encoding the C-terminal of Gαs could selectively inhibit its activity; this might represent a useful tool for clarifying the role of Gαs in the opioid signaling switch in future investigations.
Dephosphorylation of Gαs with protein phosphatases 1 and 2A was shown to augment its interaction with OPRM [33]. However, mutation of Ser and Thr residues to Ala in two putative contact sites of Gαs with receptor and effector (α3 ⁄ β5 and α4 ⁄ β6) did not alter the inhibition of AC by morphine (Fig. 3). Interestingly, mutations adjacent to α3 ⁄ β5 loop (T270A, S272A and S261A) increased Fsk-stimulated cAMP production after chronic morphine treatment, suggesting a regulatory role for this domain in the activation of Gαs by OPRM. Because these substitutions did not affect the acute inhibition of AC, it appears that opioid receptor modifications as a result of chronic treatment such as phosphorylation with GPCR kinases [26] and Src [50] are critical for a switch in signaling. Moreover, changes in the α4 ⁄ β6 residues had no effect on the cAMP production induced by OPRM, suggesting a minimal regulatory role for this domain. In addition, dephosphorylation of other residues such as contact sites with escort proteins are worthy of evaluation. A series of investigations have demonstrated that naloxone, which disrupts OPRM-Gαs coupling [51,52], connects with a scaffold protein (filamin A), which is known to interact with OPRM [53,54]. It appears that OPRM coupling to Gαs is governed through a network of mechanisms in which receptor conformation, Gαs depalmitoylation, Gαs dephosphorylation and scaffold proteins have important roles.
Our pull-down experiments indicate that OPRM interacts with Gαs after chronic morphine treatment, consistent with the findings of studies by Chakrabarti et al. [28,33]; these experiments were conducted in the presence of morphine (10 μM) in all solubilizing and washing buffers. In addition, we did not find a notable difference in the Gαs-OPRM interaction among the mutants compared to wild type Gαs (Fig. 4). This discrepancy regarding pull-down and cAMP results might be a result of differences between the two procedures. It may also indicate that a Ser ⁄ Thr mutation at α3 ⁄ β5 does not change the direct coupling of Gs to OPRM, whereas it alters the interaction of Gs with scaffolding proteins such as filamin A, as previously shown to play a crucial role in the OPRM signaling switch [53,54]. Another possible explanation might be the interaction between Gs derived Gβγ and OPRM, as also revealed to be important with respect to the excitatory signaling of opioid receptors [30].
Isoproterenol failed to rapidly induce ERK1⁄ 2 phosphorylation in S49 cyc– cells; however, after Gαs transfection, we observed a quick rise in the pERK1⁄ 2 band intensity (Fig. 5). It appears that Gαs-dependent cellular events such as protein kinase A-mediated activation of Rap1 and Raf [55,56], βγ dissociation [57] and Src family kinases [58] are essential for rapid MAPK phosphorylation. The delayed phosphorylation of ERK1⁄ 2 even in S49 cyc– cells at 1 h might be a result of ADRB2 coupling with Gi [59] or Gαs-independent recruitment of β-arrestin followed by receptor internalization and MAPK activation [60,61]. In this regard, Sun et al. [62] have also reported Gαs-dependent and -independent activation of MAPK by ADRB2 after low-dose and high-dose isoproterenol treatment, respectively. The substitution of Ser and Thr with Ala in α3 ⁄ β5 or α4 ⁄ β6 did not change the AC response to ADRB2 (Fig. 2); it should be noted that these residues are not involved in the association of Gαs with βγ subunits [5], which might explain why mutations failed to alter MAPK activation by isoproterenol.
Morphine induced a rapid phosphorylation of ERK1⁄ 2 in both cyc– and Gαs-transfected cells (Fig. 6). These results are consistent with previous reports of opioid receptor-induced MAPK activation [24,63]. Opioid receptors transmit signals to MAPK through a variety of pathways involving βγ dissociation [64], β-arrestin [65,66], receptor internalization [67,68], tyrosine kinase transactivation [69,70] and Gz [71]. We observed that neither Gαs-wt, nor Gαs-mutants changed the pattern or extent of ERK1⁄ 2 phosphorylation; therefore, the stimulatory opioid response might be downstream to MAPK activation [24,72], although blockade of ERK phosphorylation has not abolished AC supersensitization by opioids [73].
In summary, Gαs appeared to be involved in excitatory opioid signaling, and Ser ⁄ Thr mutation to Ala at α3 ⁄ β5 loop of Gαs enhanced chronic morphine-mediated AC superactivation. In addition, Gαs was required for rapid phosphorylation of ERK1⁄ 2 by isoproterenol but not morphine, and mutations of α3 ⁄ β5 and α4 ⁄ β6 did not affect the pattern or extent of MAPK activation by either isoproterenol or morphine.
Materials and methods
Reagents
Isoproterenol-HCl, forkolin and 3-isobutyl-1-methylxanthine (IBMX) were purchased from Sigma (St Louis, MO, USA); morphine sulfate was obtained from Temad (Tehran, Iran).
Site-directed mutagenesis
Gαs-pcDNA 3.1 was obtained from Missouri S&T cDNA Resource Center (Rolla, MO, USA) and a sequence containing a start codon, as well as kozak and nucleotides coding DYKDDDDK, was introduced into the N-terminal of Gαs at the NheI and HindIII sites.
Site-directed mutagenesis was performed using Quick-change (Stratagene, La Jolla, CA, USA) to substitute the Ser ⁄ Thr residues with Ala in the α3 ⁄ β5 and α4 ⁄ β6 loops of Gαs. These mutants cannot be phosphorylated and render in dephosphorylate state. We prepared α3 ⁄ β5 double (T270A, S272A), α3 ⁄ β5 triple (T270A, S272A, S261A), α4 ⁄ β6 double (S335A, T336A) and α4 ⁄ β6 triple (S335A, T336A, S338A) mutations. Different primers with appropriate mismatch were used to introduce the desired mutation using a long-PCR method; the template plasmid was then digested with DpnI, and amplified DNA was transformed into Escherichia coli XL-blue cells. Restriction enzyme mapping and DNA sequencing (Seqlab, Gottingen, Germany) were used to confirm mutations.
Cell culture and transfection
S49 cyc– cells (obtained from cell culture facility of the University of California, San Francisco, CA, USA) were cultured in DMEM (Gibco, Gaithersburg, MD, USA) supplemented with 10% horse serum (ΔHS) and penicillin ⁄ streptomycin (100 IU·mL−1; 100 μg·mL−1) at 37 °C and 5% CO2. Cells were transiently transfected with N-terminal Flag tagged Gαs-pcDNA3.1 by electroporation using Gene-Pulser Xcell apparatus (Bio-Rad, Hercules, CA, USA). Cells were transfected in their exponential phase of growth; therefore, they were subdivided 24 h before transfection and cultured in antibiotic-free medium. Cells were then washed tree-times with NaCl ⁄ Pi and 1 × 106 cells were electroporated (square wave, 160 V, 25 ms) in 100 μL of OPTIMEM (Gibco) containing 3 μg of DNA. Immediately after electroporation, cells were transferred into OPTIMEM supplemented with 10% ΔHS.
Western blotting
Cells were lysed with a lysis buffer containing 10 mM Tris (pH 7.5), 2% SDS, 0.25% Na-deoxycolate, 100 mM dithiothreitol, 1% glycerol, 1 μM phenylmethanesulfonyl fluoride and 10 μM leupeptin. Protein samples were boiled for 7 min, resolved by SDS ⁄PAGE using a 4% stacking and 12% separating gels and then electroblotted (2.5 mA·cm−2, 70 min) onto poly(vinylidene difluoride) membranes (Roche, Mannheim, Germany) using a semidry apparatus (Peqlab, Erlangen, Germany). After blocking with casein 1%, blots were incubated overnight with different primary antibodies against pERK1 ⁄ 2 (dilution 1 : 1000; Cell Signaling Technology, Beverly, MA, USA), Flag (dilution 1 : 1000; Cell Signaling Technology), Gαs (dilution 1 : 2000; Santa Cruz Biotechnology, Santa Cruz, CA, USA), OPRM1 (dilution 1 : 1000; Santa Cruz Biotechnology) or ADRB2 (dilution 1 : 500; Novus Biologicals, Littleton, CO, USA). The primary antibody was detected using horseradish peroxidase-conjugated secondary antibodies (dilution 1 : 10 000; Bio-Rad) and protein bands were revealed using a chemiluminescence kit (Roche) on X-ray Films (Fujifilm, Tokyo, Japan). Membranes were stripped and re-probed with ERK1⁄ 2 (dilution 1 : 2000; Cell Signaling Technology) and β-actin (dilution 1 : 2000; Santa Cruz Biotechnology) antibodies, when appropriate. The protein bands were analyzed by densitometry scanning with IMAGEJ software (National Institutes of Health, Bethesda, MD, USA) and expressed as the mean ± SD.
cAMP assay
Cells were seeded in 96-well plates (Nunc, Roskilde, Denmark), 48 h after transfection, and then cells were treated with morphine for 45 min (acute) or 24 h (chronic) [19,20] under normal conditions (37 °C, DMEM, 10% serum). Before cAMP measurement, cells were incubated with Fsk (5 μM) plus IBMX (a nonselective phosphodiesterase inhibitor; 0.1 mM) for 10 min. For isoproterenol, cells were incubated with the drug for 45 min (acute) or 24 h (chronic) and IBMX was added for the last 10 min. At the end of drug treatments, cells were centrifuged in a plate centrifuge (300 g for 5 min) and were subsequently lysed with a buffer containing 0.1 M HCl, 0.5% Triton X-100 and 0.1 mM IBMX. A competitive ELISA assay (Cayman Chemical, Ann Arbor, MI, USA) was conducted to determine the cAMP concentration; The cAMP measurements were further normalized to total protein content of samples determined by the Bradford assay [74] and Gαs expression. The results are presented as the mean ± SD of tree independent experiments.
Immunoprecipitation
To analyze protein interaction, 3 × 105 cells were plated in six-well plates and treated with morphine (10 μM) for 24 h. Cells were lysed with 0.5 mL of a solubilizing buffer containing 20 mM Hepes (pH 7.4), 150 mM NaCl, 1% NP-40, 0.1% SDS, 0.5% Na-deoxycolate, 1% glycerol, 5 μM phenylmethanesulfonyl fluoride and 50 μM leupeptin. Samples were incubated on ice for 1 h, passed several times through a 29-gauge syringe and centrifuged for 5 min at 14 000 g at 4 °C; supernatant was transferred to a new tube and 25 μL of pre-washed protein-A agarose beads (Roche) was added. Samples were incubated overnight at 4 °C with either anti-Flag (dilution 1 : 200; Cell Signaling) or anti-OPRM1 (dilution 1 : 200; Santa Cruz) antibodies. The next day, beads were washed with NaCl ⁄ Pi (3 × 5 min) and centrifuged; The pellet was boiled for 7 min in loading buffer (10 mM Tris, pH 7.5, 2% SDS, 50 mM dithiothreitol, 1% glycerol, 1 μM phenylmethanesulfonyl fluoride, 10 μM leupeptin, 0.5 mg·mL−1 bromophenol blue) and subjected to western blotting analysis. Morphine (10 μM) was present in all solubilizing and washing buffers. A cross examination with the other antibody was performed to confirm the interaction.
Statistical analysis
Linear or nonlinear regressions with least square root were applied for the determination of EC50 and t1 ⁄ 2 values, as well as protein and cAMP concentrations. Two-way and one-way ANOVA followed by Tukey’s test were used for the analysis of cAMP levels in dose–response experiments and potential differences among mutants, respectively. Repeated-measure ANOVA was applied for phosphorylated ERK1⁄ 2 time course experiments. Statistical analysis and curve fitting were carried out using PRISM 5 (GraphPad Software Inc., San Diego, CA, USA). P < 0.05 was considered statistically significant.
Acknowledgments
This work was financially supported by grant no. 87-01-33-6840 to M. H. Ghahremani and no. 123 ⁄ 458 to M. Ghazi-Khansari from the deputy of research of Tehran University of Medical Sciences. The authors would like to express their gratitude to Professors H. Bourne, C. Berlot and H. Ammer for their helpful comments regarding S49 cyc– cells and Gαs transfection, as well as thank Mr H. Akbari and Mr A. Kazemi for providing technical assistance.
Abbreviations
- AC
adenylyl cyclase
- ADRB2
β2-adrenergic receptor
- ERK
extracellular-regulated kinase
- Fsk
forskolin
- GPCR
G-protein coupled receptor
- Gαi
inhibitory G-protein
- Gαs
stimulatory G-protein
- IBMX
3-isobutyl-1-methylxanthine
- MAPK
mitogen-activated protein kinase
- OPRM
μ-opioid receptor
- pERK
phosphorylated ERK
- wt
wild-type
References
- 1.Cabrera-Vera TM, Vanhauwe J, Thomas TO, Medkova M, Preininger A, Mazzoni MR, Hamm HE. Insights into G protein structure, function, and regulation. Endocr Rev. 2003;24:765–781. doi: 10.1210/er.2000-0026. [DOI] [PubMed] [Google Scholar]
- 2.Muramatsu T, Suwa M. Statistical analysis and prediction of functional residues effective for GPCR-G-protein coupling selectivity. Protein Eng Des Sel. 2006;19:277–283. doi: 10.1093/protein/gzl010. [DOI] [PubMed] [Google Scholar]
- 3.Slessareva JE, Ma H, Depree KM, Flood LA, Bae H, Cabrera-Vera TM, Hamm HE, Graber SG. Closely related G-protein-coupled receptors use multiple and distinct domains on G-protein alpha-subunits for selective coupling. J Biol Chem. 2003;278:50530–50536. doi: 10.1074/jbc.M304417200. [DOI] [PubMed] [Google Scholar]
- 4.Heydorn A, Ward RJ, Jorgensen R, Rosenkilde MM, Frimurer TM, Milligan G, Kostenis E. Identification of a novel site within G protein alpha subunits important for specificity of receptor-G protein interaction. Mol Pharmacol. 2004;66:250–259. doi: 10.1124/mol.66.2.250. [DOI] [PubMed] [Google Scholar]
- 5.Sunahara RK, Tesmer JJ, Gilman AG, Sprang SR. Crystal structure of the adenylyl cyclase activator Gsalpha. Science. 1997;278:1943–1947. doi: 10.1126/science.278.5345.1943. [DOI] [PubMed] [Google Scholar]
- 6.Grishina G, Berlot CH. Identification of common and distinct residues involved in the interaction of alphai2 and alphas with adenylyl cyclase. J Biol Chem. 1997;272:20619–20626. doi: 10.1074/jbc.272.33.20619. [DOI] [PubMed] [Google Scholar]
- 7.Marsh SR, Grishina G, Wilson PT, Berlot CH. Receptor-mediated activation of Gsalpha: evidence for intramolecular signal transduction. Mol Pharmacol. 1998;53:981–990. [PubMed] [Google Scholar]
- 8.Grishina G, Berlot CH. A surface-exposed region of G(salpha) in which substitutions decrease receptor-mediated activation and increase receptor affinity. Mol Pharmacol. 2000;57:1081–1092. [PubMed] [Google Scholar]
- 9.Kilts JD, Gerhardt MA, Richardson MD, Sreeram G, Mackensen GB, Grocott HP, White WD, Davis RD, Newman MF, Reves JG, et al. Beta(2)-adrenergic and several other G protein-coupled receptors in human atrial membranes activate both G(s) and G(i) Circ Res. 2000;87:705–709. doi: 10.1161/01.res.87.8.705. [DOI] [PubMed] [Google Scholar]
- 10.Malmberg A, Strange PG. Site-directed mutations in the third intracellular loop of the serotonin 5-HT(1A) receptor alter G protein coupling from G(i) to G(s) in a ligand-dependent manner. J Neurochem. 2000;75:1283–1293. doi: 10.1046/j.1471-4159.2000.751283.x. [DOI] [PubMed] [Google Scholar]
- 11.Abel A, Wittau N, Wieland T, Schultz G, Kalkbrenner F. Cell cycle-dependent coupling of the vasopressin V1a receptor to different G proteins. J Biol Chem. 2000;275:32543–32551. doi: 10.1074/jbc.M002171200. [DOI] [PubMed] [Google Scholar]
- 12.Lawler OA, Miggin SM, Kinsella BT. Protein kinase A-mediated phosphorylation of serine 357 of the mouse prostacyclin receptor regulates its coupling to G(s)-, to G(i)-, and to G(q)-coupled effector signaling. J Biol Chem. 2001;276:33596–33607. doi: 10.1074/jbc.M104434200. [DOI] [PubMed] [Google Scholar]
- 13.Martin NP, Whalen EJ, Zamah MA, Pierce KL, Lefkowitz RJ. PKA-mediated phosphorylation of the beta1-adrenergic receptor promotes Gs ⁄ Gi switching. Cell Signal. 2004;16:1397–1403. doi: 10.1016/j.cellsig.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 14.Hasseldine AR, Harper EA, Black JW. Cardiac-specific overexpression of human beta2 adrenoceptors in mice exposes coupling to both Gs and Gi proteins. Br J Pharmacol. 2003;138:1358–1366. doi: 10.1038/sj.bjp.0705191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zamah AM, Delahunty M, Luttrell LM, Lefkowitz RJ. Protein kinase A-mediated phosphorylation of the beta 2-adrenergic receptor regulates its coupling to Gs and Gi. Demonstration in a reconstituted system. J Biol Chem. 2002;277:31249–31256. doi: 10.1074/jbc.M202753200. [DOI] [PubMed] [Google Scholar]
- 16.Zhu WZ, Zheng M, Koch WJ, Lefkowitz RJ, Kobilka BK, Xiao RP. Dual modulation of cell survival and cell death by beta(2)-adrenergic signaling in adult mouse cardiac myocytes. Proc Natl Acad Sci USA. 2001;98:1607–1612. doi: 10.1073/pnas.98.4.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ammer H, Schulz R. Chronic activation of inhibitory delta-opioid receptors cross-regulates the stimulatory adenylate cyclase-coupled prostaglandin E1 receptor system in neuroblastoma x glioma (NG108-15) hybrid cells. J Neurochem. 1995;64:2449–2457. doi: 10.1046/j.1471-4159.1995.64062449.x. [DOI] [PubMed] [Google Scholar]
- 18.Ammer H, Schulz RD. Chronic morphine treatment increases stimulatory beta-2 adrenoceptor signaling in A431 cells stably expressing the mu opioid receptor. J Pharmacol Exp Ther. 1997;280:512–520. [PubMed] [Google Scholar]
- 19.Copeland RL, Jr, Pradhan SN, Dillon-Carter O, Chuang DM. Rebound increase of basal cAMP level in NG108-15 cells during chronic morphine treatment: effects of naloxone and chloramphenicol. Life Sci. 1989;44:1107–1116. doi: 10.1016/0024-3205(89)90338-x. [DOI] [PubMed] [Google Scholar]
- 20.Mouledous L, Neasta J, Uttenweiler-Joseph S, Stella A, Matondo M, Corbani M, Monsarrat B, Meunier JC. Long-term morphine treatment enhances proteasome-dependent degradation of G beta in human neuroblastoma SH-SY5Y cells: correlation with onset of adenylate cyclase sensitization. Mol Pharmacol. 2005;68:467–476. doi: 10.1124/mol.105.013391. [DOI] [PubMed] [Google Scholar]
- 21.Chao J, Nestler EJ. Molecular neurobiology of drug addiction. Annu Rev Med. 2004;55:113–132. doi: 10.1146/annurev.med.55.091902.103730. [DOI] [PubMed] [Google Scholar]
- 22.Nestler EJ. Molecular neurobiology of addiction. Am J Addict. 2001;10:201–217. doi: 10.1080/105504901750532094. [DOI] [PubMed] [Google Scholar]
- 23.Williams JT, Christie MJ, Manzoni O. Cellular and synaptic adaptations mediating opioid dependence. Physiol Rev. 2001;81:299–343. doi: 10.1152/physrev.2001.81.1.299. [DOI] [PubMed] [Google Scholar]
- 24.Yue X, Varga EV, Stropova D, Vanderah TW, Yamamura HI, Roeske WR. Chronic morphine-mediated adenylyl cyclase superactivation is attenuated by the Raf-1 inhibitor, GW5074. Eur J Pharmacol. 2006;540:57–59. doi: 10.1016/j.ejphar.2006.04.033. [DOI] [PubMed] [Google Scholar]
- 25.Wang L, Milne B, Jhamandas K. Involvement of excitatory amino acid pathways in the expression of precipitated opioid withdrawal in the rostral ventrolateral medulla: an in vivo voltammetric study. Brain Res. 1995;697:130–142. doi: 10.1016/0006-8993(95)00803-x. [DOI] [PubMed] [Google Scholar]
- 26.Bailey CP, Connor M. Opioids: cellular mechanisms of tolerance and physical dependence. Curr Opin Pharmacol. 2005;5:60–68. doi: 10.1016/j.coph.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 27.Liu JG, Anand KJ. Protein kinases modulate the cellular adaptations associated with opioid tolerance and dependence. Brain Res Brain Res Rev. 2001;38:1–19. doi: 10.1016/s0165-0173(01)00057-1. [DOI] [PubMed] [Google Scholar]
- 28.Chakrabarti S, Regec A, Gintzler AR. Biochemical demonstration of mu-opioid receptor association with Gsalpha: enhancement following morphine exposure. Brain Res Mol Brain Res. 2005;135:217–224. doi: 10.1016/j.molbrainres.2004.12.016. [DOI] [PubMed] [Google Scholar]
- 29.Cruciani RA, Dvorkin B, Morris SA, Crain SM, Makman MH. Direct coupling of opioid receptors to both stimulatory and inhibitory guanine nucleotide-binding proteins in F-11 neuroblastoma-sensory neuron hybrid cells. Proc Natl Acad Sci USA. 1993;90:3019–3023. doi: 10.1073/pnas.90.7.3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang HY, Burns LH. Gbetagamma that interacts with adenylyl cyclase in opioid tolerance originates from a Gs protein. J Neurobiol. 2006;66:1302–1310. doi: 10.1002/neu.20286. [DOI] [PubMed] [Google Scholar]
- 31.Chakrabarti S, Chang A, Gintzler AR. Subcellular localization of mu-opioid receptor G(s) signaling. J Pharmacol Exp Ther. 2010;333:193–200. doi: 10.1124/jpet.109.165142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ammer H, Schulz R. Enhanced stimulatory adenylyl cyclase signaling during opioid dependence is associated with a reduction in palmitoylated Gs alpha. Mol Pharmacol. 1997;52:993–999. doi: 10.1124/mol.52.6.993. [DOI] [PubMed] [Google Scholar]
- 33.Chakrabarti S, Gintzler AR. Phosphorylation of Galphas influences its association with the micro-opioid receptor and is modulated by long-term morphine exposure. Mol Pharmacol. 2007;72:753–760. doi: 10.1124/mol.107.036145. [DOI] [PubMed] [Google Scholar]
- 34.Codina J, Birnbaumer L. Requirement for intramolecular domain interaction in activation of G protein alpha subunit by aluminum fluoride and GDP but not by GTP gamma S. J Biol Chem. 1994;269:29339–29342. [PubMed] [Google Scholar]
- 35.Conklin BR, Herzmark P, Ishida S, Voyno-Yasenetskaya TA, Sun Y, Farfel Z, Bourne HR. Carboxyl-terminal mutations of Gq alpha and Gs alpha that alter the fidelity of receptor activation. Mol Pharmacol. 1996;50:885–890. [PubMed] [Google Scholar]
- 36.Hildebrandt JD, Day R, Farnsworth CL, Feig LA. A mutation in the putative Mg(2+)-binding site of Gs alpha prevents its activation by receptors. Mol Cell Biol. 1991;11:4830–4838. doi: 10.1128/mcb.11.10.4830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rasmussen SG, Devree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, et al. Crystal structure of the beta(2) adrenergic receptor-Gs protein complex. Nature. 2011;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Laugwitz KL, Offermanns S, Spicher K, Schultz G. mu and delta opioid receptors differentially couple to G protein subtypes in membranes of human neuroblastoma SH-SY5Y cells. Neuron. 1993;10:233–242. doi: 10.1016/0896-6273(93)90314-h. [DOI] [PubMed] [Google Scholar]
- 39.Avidor-Reiss T, Nevo I, Saya D, Bayewitch M, Vogel Z. Opiate-induced adenylyl cyclase superactivation is isozyme-specific. J Biol Chem. 1997;272:5040–5047. doi: 10.1074/jbc.272.8.5040. [DOI] [PubMed] [Google Scholar]
- 40.Ferrer-Alcon M, Garcia-Fuster MJ, La Harpe R, Garcia-Sevilla JA. Long-term regulation of signalling components of adenylyl cyclase and mitogen-activated protein kinase in the pre-frontal cortex of human opiate addicts. J Neurochem. 2004;90:220–230. doi: 10.1111/j.1471-4159.2004.02473.x. [DOI] [PubMed] [Google Scholar]
- 41.Liu JG, Gong ZH, Qin BY. Effects of opioid receptor agonists on cAMP second messenger system. Zhongguo Yao Li Xue Bao. 1999;20:452–456. [PubMed] [Google Scholar]
- 42.Thomas JM, Meier-Davis SR, Hoffman BB. Prolonged activation of inhibitory somatostatin receptors increases adenylate cyclase activity in wild-type and Gs alpha-deficient (cyc-) S49 mouse lymphoma cells. Cell Signal. 1992;4:571–581. doi: 10.1016/0898-6568(92)90026-5. [DOI] [PubMed] [Google Scholar]
- 43.Crain SM, Shen KF. Modulation of opioid analgesia, tolerance and dependence by Gs-coupled, GM1 ganglioside-regulated opioid receptor functions. Trends Pharmacol Sci. 1998;19:358–365. doi: 10.1016/s0165-6147(98)01241-3. [DOI] [PubMed] [Google Scholar]
- 44.Gintzler AR, Xu H. Different G proteins mediate the opioid inhibition or enhancement of evoked [5-methionine]enkephalin release. Proc Natl Acad Sci USA. 1991;88:4741–4745. doi: 10.1073/pnas.88.11.4741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shen KF, Crain SM. Cholera toxin-A subunit blocks opioid excitatory effects on sensory neuron action potentials indicating mediation by Gs-linked opioid receptors. Brain Res. 1990;525:225–231. doi: 10.1016/0006-8993(90)90868-c. [DOI] [PubMed] [Google Scholar]
- 46.Szucs M, Boda K, Gintzler AR. Dual effects of DAMGO [D-Ala2,N-Me-Phe4,Gly5-ol]-enkephalin and CTAP (D-Phe-Cys-Tyr-D-Trp-Arg-Thr-Pen-Thr-NH2) on adenylyl cyclase activity: implications for mu-opioid receptor Gs coupling. J Pharmacol Exp Ther. 2004;310:256–262. doi: 10.1124/jpet.104.066837. [DOI] [PubMed] [Google Scholar]
- 47.Wang L, Gintzler AR. Altered mu-opiate receptor-G protein signal transduction following chronic morphine exposure. J Neurochem. 1997;68:248–254. doi: 10.1046/j.1471-4159.1997.68010248.x. [DOI] [PubMed] [Google Scholar]
- 48.Watts VJ, Taussig R, Neve RL, Neve KA. Dopamine D2 receptor-induced heterologous sensitization of adenylyl cyclase requires Galphas: characterization of Galphas-insensitive mutants of adenylyl cyclase V. Mol Pharmacol. 2001;60:1168–1172. doi: 10.1124/mol.60.6.1168. [DOI] [PubMed] [Google Scholar]
- 49.Feldman DS, Zamah AM, Pierce KL, Miller WE, Kelly F, Rapacciuolo A, Rockman HA, Koch WJ, Luttrell LM. Selective inhibition of heterotrimeric Gs signaling. Targeting the receptor-G protein interface using a peptide minigene encoding the Galpha(s) carboxyl terminus. J Biol Chem. 2002;277:28631–28640. doi: 10.1074/jbc.M204753200. [DOI] [PubMed] [Google Scholar]
- 50.Zhang L, Zhao H, Qiu Y, Loh HH, Law PY. Src phosphorylation of micro-receptor is responsible for the receptor switching from an inhibitory to a stimulatory signal. J Biol Chem. 2009;284:1990–2000. doi: 10.1074/jbc.M807971200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tsai RY, Tai YH, Tzeng JI, Cherng CH, Yeh CC, Wong CS. Ultra-low dose naloxone restores the antinociceptive effect of morphine in pertussis toxin-treated rats by reversing the coupling of mu-opioid receptors from Gs-protein to coupling to Gi-protein. Neuroscience. 2009;164:435–443. doi: 10.1016/j.neuroscience.2009.08.015. [DOI] [PubMed] [Google Scholar]
- 52.Wang HY, Friedman E, Olmstead MC, Burns LH. Ultra-low-dose naloxone suppresses opioid tolerance, dependence and associated changes in mu opioid receptor-G protein coupling and Gbetagamma signaling. Neuroscience. 2005;135:247–261. doi: 10.1016/j.neuroscience.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 53.Wang HY, Burns LH. Naloxone’s pentapeptide binding site on filamin A blocks Mu opioid receptor-Gs coupling and CREB activation of acute morphine. PLoS ONE. 2009;4:e4282. doi: 10.1371/journal.pone.0004282. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 54.Wang HY, Frankfurt M, Burns LH. High-affinity naloxone binding to filamin a prevents mu opioid receptor-Gs coupling underlying opioid tolerance and dependence. PLoS ONE. 2008;3:e1554. doi: 10.1371/journal.pone.0001554. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 55.Wan Y, Huang XY. Analysis of the Gs ⁄ mitogen-activated protein kinase pathway in mutant S49 cells. J Biol Chem. 1998;273:14533–14537. doi: 10.1074/jbc.273.23.14533. [DOI] [PubMed] [Google Scholar]
- 56.Schmitt JM, Stork PJS. β2-adrenergic receptor activates extracellular signal-regulated kinases (ERKs) via the small G protein Rap1 and the serine ⁄ threonine kinase B-Raf. J Biol Chem. 2000;275:25342–25350. doi: 10.1074/jbc.M003213200. [DOI] [PubMed] [Google Scholar]
- 57.Crespo P, Cachero TG, Xu N, Gutkind JS. Dual effect of beta-adrenergic receptors on mitogen-activated protein kinase. Evidence for a beta gamma-dependent activation and a G alpha s-cAMP-mediated inhibition. J Biol Chem. 1995;270:25259–25265. doi: 10.1074/jbc.270.42.25259. [DOI] [PubMed] [Google Scholar]
- 58.Klinger M, Kudlacek O, Seidel MG, Freissmuth M, Sexl V. MAP kinase stimulation by cAMP does not require RAP1 but SRC family kinases. J Biol Chem. 2002;277:32490–32497. doi: 10.1074/jbc.M200556200. [DOI] [PubMed] [Google Scholar]
- 59.Daaka Y, Luttrell LM, Lefkowitz RJ. Switching of the coupling of the beta2-adrenergic receptor to different G proteins by protein kinase A. Nature. 1997;390:88–91. doi: 10.1038/36362. [DOI] [PubMed] [Google Scholar]
- 60.Azzi M, Charest PG, Angers S, Rousseau G, Kohout T, Bouvier M, Pineyro G. Beta-arrestin-mediated activation of MAPK by inverse agonists reveals distinct active conformations for G protein-coupled receptors. Proc Natl Acad Sci USA. 2003;100:11406–11411. doi: 10.1073/pnas.1936664100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shenoy SK, Drake MT, Nelson CD, Houtz DA, Xiao K, Madabushi S, Reiter E, Premont RT, Lichtarge O, Lefkowitz RJ. beta-arrestin-dependent, G protein-independent ERK1⁄ 2 activation by the beta2 adrenergic receptor. J Biol Chem. 2006;281:1261–1273. doi: 10.1074/jbc.M506576200. [DOI] [PubMed] [Google Scholar]
- 62.Sun Y, Huang J, Xiang Y, Bastepe M, Juppner H, Kobilka BK, Zhang JJ, Huang XY. Dosage-dependent switch from G protein-coupled to G protein-independent signaling by a GPCR. EMBO J. 2007;26:53–64. doi: 10.1038/sj.emboj.7601502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Asensio VJ, Miralles A, Garcia-Sevilla JA. Stimulation of mitogen-activated protein kinase kinases (MEK1⁄ 2) by mu-, delta- and kappa-opioid receptor agonists in the rat brain: regulation by chronic morphine and opioid withdrawal. Eur J Pharmacol. 2006;539:49–56. doi: 10.1016/j.ejphar.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 64.Belcheva MM, Vogel Z, Ignatova E, Avidor-Reiss T, Zippel R, Levy R, Young EC, Barg J, Coscia CJ. Opioid modulation of extracellular signal-regulated protein kinase activity is ras-dependent and involves Gbetagamma subunits. J Neurochem. 1998;70:635–645. doi: 10.1046/j.1471-4159.1998.70020635.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Macey TA, Lowe JD, Chavkin C. Mu opioid receptor activation of ERK1⁄ 2 is GRK3 and arrestin dependent in striatal neurons. J Biol Chem. 2006;281:34515–34524. doi: 10.1074/jbc.M604278200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zheng H, Loh HH, Law P-Y. β-Arrestin-dependent μ-opioid receptor-activated extracellular signal-regulated kinases (ERKs) translocate to nucleus in contrast to G protein-dependent ERK activation. Mol Pharmacol. 2008;73:178–190. doi: 10.1124/mol.107.039842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Audet N, Paquin-Gobeil M, Landry-Paquet O, Schiller PW, Pineyro G. Internalization and Src activity regulate the time course of ERK activation by delta opioid receptor ligands. J Biol Chem. 2005;280:7808–7816. doi: 10.1074/jbc.M411695200. [DOI] [PubMed] [Google Scholar]
- 68.Ignatova EG, Belcheva MM, Bohn LM, Neuman MC, Coscia CJ. Requirement of receptor internalization for opioid stimulation of mitogen-activated protein kinase: biochemical and immunofluorescence confocal microscopic evidence. J Neurosci. 1999;19:56–63. doi: 10.1523/JNEUROSCI.19-01-00056.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Post GR, Brown JH. G protein-coupled receptors and signaling pathways regulating growth responses. FASEB J. 1996;10:741–749. doi: 10.1096/fasebj.10.7.8635691. [DOI] [PubMed] [Google Scholar]
- 70.Belcheva MM, Szucs M, Wang D, Sadee W, Coscia CJ. mu-Opioid receptor-mediated ERK activation involves calmodulin-dependent epidermal growth factor receptor transactivation. J Biol Chem. 2001;276:33847–33853. doi: 10.1074/jbc.M101535200. [DOI] [PubMed] [Google Scholar]
- 71.Tso PH, Yung LY, Wong YH. Regulation of adenylyl cyclase, ERK1⁄ 2, and CREB by Gz following acute and chronic activation of the delta-opioid receptor. J Neurochem. 2000;74:1685–1693. doi: 10.1046/j.1471-4159.2000.0741685.x. [DOI] [PubMed] [Google Scholar]
- 72.Polakiewicz RD, Schieferl SM, Dorner LF, Kansra V, Comb MJ. A mitogen-activated protein kinase pathway is required for mu-opioid receptor desensitization. J Biol Chem. 1998;273:12402–12406. doi: 10.1074/jbc.273.20.12402. [DOI] [PubMed] [Google Scholar]
- 73.Tso PH, Wong YH. Role of extracellular signal-regulated kinases in opioid-induced adenylyl cyclase superactivation in human embryonic kidney 293 cells. Neurosci Lett. 2001;316:13–16. doi: 10.1016/s0304-3940(01)02340-0. [DOI] [PubMed] [Google Scholar]
- 74.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]