

Abstract
Leukemias harboring mixed lineage leukemia (MLL1) gene abnormalities are associated with poor clinical outcomes and new therapeutic approaches are desperately needed. Rearrangement of the MLL1 gene generates chimeric proteins that fuse the NH3-terminus of MLL1 to the COOH-terminus of its translocation partners. These MLL1-fusion oncoproteins drive the expression of homeobox genes such as HOXA cluster genes and MEIS1, which are known to induce leukemic transformation of hematopoietic progenitors. Genome-wide histone methylation studies have revealed that the abnormal expression of MLL1-fusion target genes is associated with high levels of H3K79 methylation at these gene loci. The only known enzyme that catalyzes methylation of H3K79 is disruptor of telomeric-silencing 1-like (DOT1L). Loss-of-function mouse models as well as small molecular inhibitors of DOT1L demonstrate that leukemias driven by MLL1-translocations are dependent on DOT1L enzymatic activity for proliferation and for the maintenance of HOXA gene expression. Furthermore, DOT1L also appears to be important for HOXA gene expression in other settings including leukemias with select genetic abnormalities. These discoveries have established a foundation for disease-specific therapies that target chromatin modifications in highly malignant leukemias harboring specific genetic abnormalities. This review focuses on the molecular mechanisms underlying MLL1-translocation-driven leukemogenesis, and the latest progress on DOT1L-targeted epigenetic therapies for MLL1-rearranged and other leukemias.
Characteristics of mixed lineage leukemias
It has been recognized for more than thirty years that 11q23 chromosomal translocations are found in both acute lymphoid leukemias (ALL) and acute myeloid leukemias (AML) [1–4]. In many cases a mix of cell surface markers of lymphoid and myeloid lineages can be found on leukemia cells or a phenotypic lineage switch (for example, initially diagnosed ALL could relapse as AML) is observed in patients with leukemias harboring 11q23 translocations [5, 6]. The presence of 11q23 translocations led to the cloning of a critical gene that resides on 11q23 known as the mixed lineage leukemia gene (MLL1; also known as MLL, ALL-1, HRX, and TRX1) [7–10]. Rearrangement of the MLL1 gene is found in approximately 5% of ALL, and around 5–10% of AML cases in adults [11–15]. MLL1-rearrangements are frequently found in leukemias of childhood, particularly in infant (defined as < 1-year old) leukemias whether they are diagnosed as AML or ALL [16]. MLL1-translocations are detected in more than 70% of infant ALL cases, and about 35–50% of infant AML patients [17–24]. In addition, MLL1 gene translocation is found in therapy-related leukemias that develop in patients that have been previously treated with topoisomerase II inhibitors [25–29].
Although a recent genome-scale sequencing project revealed that MLL1-rearranged leukemias harbor relatively low frequencies of somatic mutations compared to other types of cancer [30], patients with 11q23 rearrangements generally have poor prognosis and are treated according to high-risk protocols. The 5-year survival rate of infant ALL patients with MLL1-rearrangements (15–50%) is significantly lower than the infant ALL patients without MLL1 gene abnormalities (event free survival 70–80%) [19, 21, 31, 32]. The survival rate of infant leukemias with MLL1 translocations remains low (approximately 50% with worse out come for the youngest patients) even with intensive chemotherapies and allogeneic hematopoietic stem cell transplantation [33–35]. The World Health Organization (WHO) classifies myeloid leukemias with 11q23 abnormalities as one group, with an estimated 4-year event free survival of 24–55% [16, 18, 36]. The unmet clinical problem presented by MLL1-rearranged leukemias emphasizes the need for novel therapeutic strategies targeting these difficult-to-treat hematopoietic malignancies.
Molecular basis of MLL1-rearranged leukemias
Structure and function of wild-type MLL1
Wild-type MLL1 is a mammalian homologue of drosophila trithorax and yeast Set1, the first H3K4 methyltransferase identified in Saccharomyces cerevisiae in the early 2000s [37–39]. Structurally, the MLL1 protein (3,972 amino acids) contains three AT-hook DNA binding domains at its N-terminal region [40], two speckled nuclear localization motifs (SNL1 and SNL2), a CxxC zine-finger motif that binds un-methylated CpG-containing DNA [41], four plant homology domains (PHDs) [42], a bromodomain, a transcription activation (TA) domain that recruits CREB-binding protein (CBP), and a SET (Su(var)3–9, enhancer of zeste, trithorax) domain located at the extreme C-terminus that contains the H3K4 methyltransferase activity (Fig. 1A) [13, 43, 44]. The full length MLL1 protein is cleaved into MLL1-N (320kDa) and MLL1-C (180kDa) fragments by Taspase 1, and reassembled to form a stable dimer through the F/Y-rich N terminal (FYRN) and F/Y-rich C terminal (FYRC) domains [45, 46].
Figure 1.
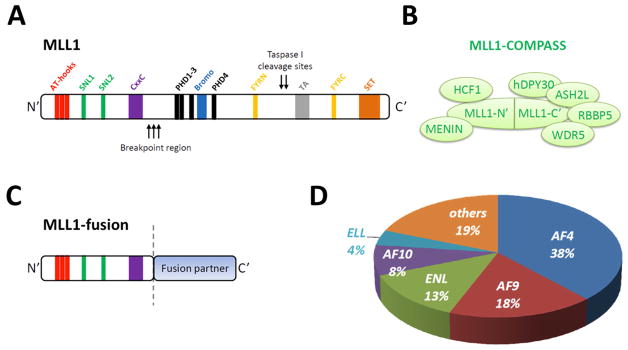
Schematic summary of the MLL1 and MLL1-fusion proteins. (A) The functional domains in wild-type MLL1 protein include three AT-hook motifs that mediate binding to the AT-rich DNA sequences (red), two speckled nuclear localization motifs (SNL; green), a DNMT homology domain (CxxC; purple), four zinc-finger plant homology domains (PHD; black) and a bromodomain (Bromo; cyan) for chromatin binding, two F/Y-rich domains (FYRN and FYRC; yellow) that mediate dimerization of the MLL1-N and MLL1-C fragments generated by taspase 1 cleavage, a transcription activation domain (TA; gray) that recruits CREB-binding protein, and a Su(var)3–9, enhancer of zeste, trithorax domain (SET; orange) that possesses the H3K4 methyltransferase activity. The most frequently observed breakpoints in MLL1 locate to the region between the CxxC domain and PHD domains. (B) The subunits of the MLL1-containing complex of proteins associated with Set1 (MLL1-COMPASS) include multiple endocrine neoplasia I (MENIN), host cell factor C1 (HCF1), WD-repeat containing protein 5 (WDR5), Rb-binding protein 5 (RBBP5), absent, small, or homeotic 2-like (ASH2L), and Dpy-30-Like Protein (hDPY30) [53, 54, 57]. (C) Structure of MLL1-fusion proteins caused by MLL1 gene translocations. The recurrent chimeric MLL1-fusion proteins are composed of the N-terminal sequence of MLL1 up to the breakpoint (indicated by the dotted line), followed by the in-frame translation of the C-terminal sequence of one of over 60 different MLL1 fusion partners. (D) Frequency of the most common MLL1 translocation partner genes including AF4, AF9, ENL, AF10 and ELL in patients with MLL1-rearranged acute leukemia (n = 1590) [28].
The binding of MLL1 to chromatin is associated with transcriptionally active genes [47]. Studies revealed that MLL1 (now officially known as lysine-specific methyltransferase 2A or KMT2A) is a histone H3K4 methyltransferase that is part of a protein complex, which is involved in transcriptional regulation [48]. The MLL1 complex consists of multiple members, some of which are shared among all related MLL complexes (MLL1–4) and some of which are unique to a subset. The components that are shared include WDR5, RBBP5, hDPY30 and ASH2L, all of which interact with the C terminal region of MLL1. The unique members included MENIN and HCF1, both bind to the N terminal region of MLL1 (Fig. 1B) [49–57]. Analysis of H3K4 methylation in MLL1−/− mouse embryonic fibroblasts, which indirectly defines the MLL1 genomic targets, points to approximately 1.8% of genes (299 out of 16,327 promoter loci assessed) that are dependent on MLL1 for recruiting RNA polymerase II-mediated transcriptional initiation [58].
MLL1 rearrangement drives expression of an oncogenic program including homeobox genes
A hallmark of MLL1-rearranged leukemia is high expression of the homeobox-A (HOXA) cluster genes [59–66]. The HOX gene family is a highly conserved group of homeodomain-containing transcription factors that specify cell identity during organismal development, including body patterning and hematopoiesis [67]. In normal hematopoietic stem and progenitor cells, the expression of developmentally important HOX cluster genes is regulated by MLL1 [65]. Mice that lack MLL1 die during embryonic development and have altered Hox gene expression [68]. These MLL1-null embryos show defects in yolk sac hematopoiesis with reduced proliferation and/or survival of hematopoietic progenitors [69, 70] and defective hematopoietic stem cell activity in the aorta-gonad-mesonephros region [71]. Mechanistically, MLL1 and its complex partner MENIN bind to genomic DNA at HOX loci and modulate HOX gene expression [44, 72–76]. Unlike the embryonic lethality caused by complete deletion of MLL1, mice that harbor homozygous MLL1 with a SET domain deletion (MLL1ΔSET) survive into adulthood. These mice exhibit skeletal abnormalities with altered Hox gene expression [77], however, they show normal hematopoiesis into adulthood [78]. Intriguingly, the profile of H3K4 methylation at Hoxa loci remains normal in hematopoietic stem/progenitor cells isolated from MLL1ΔSET mice, suggesting that the SET-domain activity of MLL1 is either compensated for by other SET-domain containing histone H3K4 methyltransferases in the hematopoietic system or MLL1 is not the dominant H3K4 methyltransferase that controls Hox gene expression.
Numerous examples of dysregulated HOX gene expression are found to drive tumorigenesis [79]. Because MLL1 is a critical regulator of HOX genes in hematopoietic tissue, it is believed that the aberrant expression of HOXA genes in MLL1-rearranged leukemias is attributable to the targeting of chimeric MLL1 fusion proteins to these loci [61]. In fact, the genomic loci directly bound by a MLL1-fusion protein (MLL-AF9) was defined in a mouse AML model using chromatin immunoprecipitation and high-throughput sequencing [80]. These direct targets of MLL1-fusion protein in AML include several homeobox-containing genes such as HOXA cluster genes (HOXA7, HOXA9 and HOXA10) and MEIS1. Importantly, ectopic expression of HOXA and MEIS1 genes transforms hematopoietic progenitors and induces leukemia in mouse models [81, 82]. These results reveal that at least one fundamental role of MLL1 translocation in hematopoietic malignancy is the direct regulation of homeobox gene expression. Of note, the MLL1-fusion proteins appear to regulate only a subset of genes recognized by wild-type MLL1, suggesting a differential preference of MLL1-fusion versus wild-type MLL1 in chromatin recognition and localization [83]. Interestingly, the MLL1 fusion-induced homeobox gene expression may require co-expression of the wild-type MLL1 allele in a SET-domain-independent manner [78], suggesting an essential cooperation between a fusion oncogene and its wild-type counterpart in MLL1-rearranged leukemogenesis, but not as a result of H3K4 methylation [84].
MLL1 abnormalities link transcriptional initiation to elongation
The MLL1 gene contains 36 coding exons spread throughout a 90 kb genomic region on human chromosome 11 (Chr11: 118,436,490–118,526,832). More than 70 different recurrent MLL1 translocation partner genes have been identified [28]. The most frequently observed breakpoints are in introns 9, 10 and 11 of the MLL1 gene. All of the chromosomal rearrangements generate a fusion protein containing the N-terminus of MLL1 fused, in-frame, to the C-terminus of the fusion partner (Fig. 1C). Amongst these MLL1 abnormalities, the most frequent translocation product found in patients is MLL-AF4 [encoded by t(4;11)(q21;q23)], which is mostly associated with CD19-positive B-ALL [8, 24, 85]. The second most common translocation product is MLL-AF9 [encoded by t(9:11)(p22;q23)], which is found in approximately 2–5% of all AML patients and a high percentage of de novo AML in children [35]. Other recurrent translocation products including MLL-ENL [encoded by t(11;19)(q23;p13.3)], MLL-AF10 [encoded by t(10;11)(p12;q23)], and MLL-ELL [encoded by t(11;19)(q23;p13.1)] each contribute to between 4–13% of the MLL1-rearranged leukemia cases. These five most common MLL1 translocations account for nearly 80% of all MLL1-rearranged leukemias in patients (Fig. 1D)[28]. Many of these MLL1-fusion proteins (including MLL-AF9, MLL-ENL and MLL-AF4) can induce leukemias in genetically manipulated knock-in mouse models [86–91].
It is also the case that several of the most common MLL1 fusion partners, including AF4 (also known as AFF1 and MLLT2), AF9 (also known as MLLT3), ENL (also known as MLLT1) and ELL (elongation factor RNA polymerase II), are found in a complex called the super elongation complex (SEC) that also includes the RNA polymerase II kinase CDK9 [17]. The function of SEC is to regulate the release of paused RNA polymerase II for active elongation [92–100]. The direct fusion of MLL1 with the SEC component genes may increase the recruitment of the transcriptional elongation activity to the MLL1-regulated genes, thereby bypassing the normal initiation-to-elongation checkpoints [97, 101, 102]. Indeed, normal SEC function is required for the expression of the leukemic program (including HOXA genes) driven by MLL1 translocations [103, 104].
Epigenetic mechanisms underlying MLL1-rearranged leukemia
Eukaryotic genomes are packaged in chromatin that is regulated in a highly orchestrated fashion. Such intricate chromatin organization ensures that the integrity of the genome is maintained during cell division and that appropriate gene expression programs can be initiated and maintained during organismal development. One such mechanism of regulation is via covalent modification, such as methylation and acetylation, of specific amino acid residues in histone subunits found within the nucelosomes. These posttranslational histone modifications are mediated by several classes of epigenetic modifying enzymes, namely lysine methyltransferases (KMTs), lysine demethylases (KDMs), histone acetyltransferases (HATs) and histone deacetylase (HDACs). Recent genome-wide epigenetic landscape studies have shown that many of these histone modifications are selectively associated with either transcriptionally active (H3K4me3, H3K9ac, H3K79me2) or silent (H3K9me2/3 and H3K27me3) gene loci, and in many cases modulate gene expression through regulating the local chromatin structure [105–107].
In addition to the ability of MLL1-fusion proteins to recruit transcriptional machinery such as the SEC, genome-wide epigenetic landscape studies have revealed that MLL1-rearrangement may promote gene expression through elevating local H3K79me2 levels [89, 108]. Further studies found that the genes directly bound by the MLL1 fusion proteins are selectively associated with aberrant levels of H3K79me2 in leukemias harboring MLL1 translocation, whereas several other modifications such as H3K4me3, H3K27me3, and H3K36me3 remain unaffected [80]. Because H3K79 methylation is broadly associated with actively transcribed genes [80, 109–113], it has been hypothesized that the excessive H3K79me2 level observed at the MLL1-fusion target genes, including HOXA cluster genes and MEIS1, may contribute to the continued expression of these developmental genes in cell types that would not normally express them at high level.
The only known enzyme in mammals that catalyzes methylation of H3K79 is DOT1L (disruptor of telomeric silencing 1 like; also known as lysine N-methyltransferase 4 or KMT4) [114, 115], which was initially identified through a screen for genes that disrupt telomeric silencing in Saccharomyces cerevisiae [116, 117]. Genetic ablation of the orthologues of DOT1L in mouse, fly and yeast models leads to a complete loss of H3K79 methylation, which suggests that DOT1L is the single dominant enzyme for H3K79 methylation in eukaryotes [80, 114, 115, 118, 119]. In addition to controlling telomeric silencing, DOT1L is involved in the regulation of tissue development, DNA damage response, cell cycle checkpoint and transcription [120]. Mouse embryos lacking DOT1L fail to progress through normal development and die between E9.5 and E10.5 [119], suggesting that H3K79 methylation might be required for the establishment or maintenance of specific cell fate decisions that underlie tissue specification and body patterning. DOT1L also plays an important role in normal hematopoiesis [121]. DOT1L-deficient hematopoietic stem cells minimally reconstitute recipient bone marrow in competitive transplantation experiments [122]. Mice harboring Vav-Cre mediated deletion of Dot1L in hematopoietic cells can survive to adulthood, however, these mice are significantly anemic and thrombocytopenic [80]. Other reports using tamoxifen-inducible Cre recombinase (Cre-ER) also suggest an essential rule of Dot1L in adult hematopoiesis [122, 123].
DOT1L was found to interact with several recurrent MLL1 translocation partners such as AF9, ENL, and AF10, suggesting that MLL1-fusion proteins may directly recruit DOT1L to MLL1-fusion target loci through the C-terminal portion of the chimeric proteins [96, 97, 124–130]. Later studies using loss of function mouse models demonstrated that leukemias driven by diverse MLL1 fusion proteins (including MLL-AF9, MLL-AF10 and MLL-AF6 leukemia) are selectively dependent on DOT1L for leukemia initiation and maintenance, whereas many other types of transformed hematopoietic cells (such as HOXA9/MEIS1, E2A-HLF or AML-ETO leukemia) are insensitive to complete loss of DOT1L and H3K79 methylation [80, 122, 131–133]. A genome-wide study showed that inactivation of DOT1L leads to down-regulation of MLL-AF9 direct targets and an MLL1 translocation-associated gene expression signature [80]. Importantly, the AF9-binding site in DOT1L was mapped to 2 regions of human DOT1L (628–653 and 863–900 residues), and the interaction of DOT1L with the C-terminus of AF9 (present in MLL-AF9 fusion protein) is required for transformation by MLL-AF9 [127, 134]. These data suggest that MLL1-translocations create chimeric proteins that link MLL1 to other functionally distinct protein complexes including the super elongation complex (SEC) and the DOT1L-H3K79 methyltransferase complex and thus contribute to the ectopic expression of a homeobox gene-centric leukemic program (Fig. 2) [17, 104, 135].
Figure 2.

Proposed mechanism of MLL1-fusion super complex in MLL1-rearranged leukemias. (A) MLL1-fusion protein (here using MLL-AF9 as an example) and its interacting protein MENIN are involved in chromatin localization of the complex. Several of the most common MLL1-fusion partners including AF4, AF9, ENL and ELL are components of the super elongation complex (SEC). These MLL1-fusion partners and the positive transcription elongation factor b (P-TEFb; including cyclin-dependent kinases CDK9 and cyclin-T) assemble SEC, which plays an essential role in the release of paused RNA polymerase II for transcriptional elongation. The SEC subunits AF9 and ENL are known to interact with DOT1L. The core components of the DOT1L complex, including AF10 and AF17, are recurrent fusion partners of MLL1 found in MLL1-rearranged leukemia patients. AF10 serves as a cofactor of DOT1L to enhance the H3K79 methyltransferase activity, which promotes the accumulation of H3K79me2. The ability of MLL1-fusion proteins to link MLL1 to other functionally distinct protein complexes including SEC and DOT1L is thought to contribute to the aberrant expression of oncogenic programs in MLL1-rearranged leukemias. (B) Profiles of MLL-AF9 (black), H3K79me1 (pink) and H3K79me2 (red) at HOXA cluster (a MLL1-fusion bound target) and ACTB (a normal active gene) loci in mouse MLL-AF9 leukemia. The core occupied regions of MLL-AF9 fusion protein span through the Hoxa5–10 genomic region, highlighted by a dotted green rectangle. The localization of the MLL1-fusion super complex to chromatin recruits excessive DOT1L activity to create a hyper-methylated H3K79 status (H3K79me2high and H3K79me1low), which is distinct from the H3K79 methylation patterns observed for non-MLL1-fusion target genes [129]. Of note, transition from a lower to higher degree of H3K79 methylation at a given gene locus is known to correlate with increased gene expression.
DOT1L complex members are critical for MLL1-rearranged leukemia
It is known that the degree of H3K79 methylation (i.e. mono-, di- and tri-methylation) dynamically responds to changes in transcriptional activity [110]. Transition from a lower to higher degree of H3K79 methylation at a given gene locus is correlated with increased mRNA abundance. In addition to the recruitment of DOT1L by MLL1-fusion proteins, the hyper-methylated H3K79 (H3K79me2high and H3K79me1low) regions overlap with the MLL1-fusion bound loci, suggesting an optimized catalytic efficiency of DOT1L in the MLL1-fusion super complex (Fig. 2) [129]. Studies using mass spectroscopy found that two MLL1 fusion partners, AF10 (also known as MLLT10) and AF17 (also known as MLLT6), are core members of the DOT1L-H3K79 methyltransferase complex [124, 126, 129]. While AF17 is likely not required for embryogenesis, hematopoiesis, and animal survival [136], inactivation of AF10 is embryonic lethal and exhibits strong defects in hematopoiesis and MLL1-rearranged leukemogenesis [129]. DOT1L interacts with the octapeptide motif-leucine-zipper (OM-LZ) region of AF10, which is required for MLL-AF10-mediated leukemogenesis [125]. Recent studies reveal that genetic ablation of the OM-LZ domain in AF10 inhibits the ability of DOT1L to convert H3K79me1 to H3K79me2, and impairs cell survival and HOXA gene expression in MLL1-rearranged leukemia cells [129]. In addition to the DOT1L core complex members, the ring finger protein 20 (RNF20; also known as BRE1A) is important for DOT1L function. RNF20 is the major H2B-specific histone E3 ubiquitin ligase in mammalian cells that targets lysine 120 for monoubiquitination [137–140]. H2B ubiquitinylation was found to facilitate DOT1L activity [141], and RNF20 is required to maintain gene expression and local levels of H3K79 methylation at HOXA9 and MEIS1 in MLL1-rearranged leukemia [142]. These findings suggest that the modulation of DOT1L complex members can control progressive H3K79 methylation and HOX gene expression in MLL1-rearranged leukemias.
Regulation of homeobox genes by DOT1L in normal hematopoietic progenitors and non-MLL1-rearranged leukemia
It is known that the transition of hematopoietic stem cells toward granulocyte-macrophage progenitors during normal hematopoiesis is associated with a drastic decrease in the expression of HOXA cluster genes [80, 143]. The essential role of DOT1L and H3K79 methylation in the expression of HOXA genes in MLL1-rearranged leukemia prompts analyses of DOT1L’s function in the regulation of homeobox genes in normal hematopoietic cells. Recent studies showed that the reduction of HOXA genes during hematopoietic differentiation is accompanied by a diminution in higher order H3K79 methylation (i.e. H3K79me2/3) at HOXA gene locus, whereas the lower order H3K79me1 is minimally affected [129]. Genetic studies further revealed that both Hoxa9 and Meis1 genes are remarkably downregulated in normal hematopoietic stem/progenitor cells isolated from mice with genetic ablation of Dot1l, thus demonstrating a role for DOT1L in the normal regulation of the homeobox genes [129]. Such discovery sheds light on the possibility that in addition to MLL1-rearranged leukemias, DOT1L may also play an important role in other types of hematopoietic malignancies with aberrant HOX gene expression. Indeed, our studies identified that leukemia driven by NUP98-NSD1, a hematopoietic malignancy observed in 16% of cytogenetically normal pediatric AML patients with poor prognosis, is dependent on DOT1L for the maintenance of high-level HOXA gene expression as well as the proliferation of NUP98-NSD1 transformed cells [129]. Additionally, mutations in isocitrate dehydrogenase 1 (IDH1) and 2 (IDH2) genes, which are found in more than 75% of lower grade gliomas and secondary glioblastoma multiforme and about 20% of AML cases, are associated with elevated expression of HOXA cluster genes [144]. A recent report demonstrates that primary AML cells with IDH1 or IDH2 mutations are sensitive to DOT1L inhibitor treatment [145], which is likely due to an inhibition of HOXA gene expression [146, 147]. These recent discoveries expand the potential application of DOT1L inhibitory therapies to diverse cancer types associated with elevated level of HOX genes.
Structural and chemical basis of DOT1L-mediated H3K79 methylation
DOT1L is a non-SET domain containing histone methyltransferase, which is distinct from almost all other histone methyltransferases most of which contain a conserved SET (Su(var)3–9, enhancer of zeste, trithorax) domain [116, 148, 149]. Unlike most other histone methyltransferases that modify the lysine residues in the flexible histone tails, methylation of H3K79 by DOT1L occurs in the globular domain of nucleosomal histone H3 [114]. Additionally, DOT1L’s activity is specific to nucleosomes instead of free histone H3 or the short peptides, suggesting a context dependent interaction between DOT1L and its chromatin substrates. DOT1L catalyzes mono-, di- and tri-methylation of the ε-amino group of H3K79 using S-adenosylmethionine (SAM) as a cofactor. This reaction produces methylated lysine and S-adenosyl-L-homocysteine (SAH). The first 416 amino acid residues in human DOT1L are highly conserved during evolution and contain the catalytic active site. The co-crystal structure of the human DOT1L catalytic domain in complex with SAM has been solved, which identified five peptide segments within the N terminal region of DOT1L (amino acid residues 161–169, 186–191, 239–245, 133–139 and 221–224) that bind the cofactor SAM (Fig. 3A–C) [150]. The first three segments are consensus sequence motifs also found on other SAM-dependent methyltransferases, whereas the last two segments are unique to DOT1L [151]. Additionally, a flexible and positively charged region between amino acid residues 390 and 407 is critical for nucleosome/DNA binding and methyltransferase activity [150]. Structurally, DOT1L does not clearly fit within the protein lysine methyltransferase family, but is more similar to the protein arginine methyltransferases including the METTL and NOP2/Sun domain family proteins [152].
Figure 3.

DOT1L protein and DOT1L inhibitors. (A) A schematic representation of DOT1L protein. (B) Topological diagram of the DOT1L catalytic domain (amino acid residues 1–416) showing secondary structure elements including α helices (cylinders) and β sheets (arrow ribbons) [150]. The N-terminal domain (amino acid residues 1–126; green) consists of five α helices and two β strand hairpins. The open α/β domain (amino acid residues 141–332; red) contains a seven-strand central β sheet and five α helices. An S-adenosylmethionine (SAM; white hexagon) binding pocket (highlighted in gray) is formed by part of the open α/β domain and a flexible loop region (amino acid residues 127–140; blue). (C) A schematic diagram showing the SAM–DOT1L interaction in the SAM binding pocket (gray). Amino acid residues in close contact with SAM (white area) are highlighted (loop region shown in blue; open α/β domain shown in red). (D) Structures and chemical/pharmacological properties (Ki or IC50) of DOT1L inhibitors including S-adenosyl-L-homocysteine (SAH; a non-selective inhibitor to most SAM-dependent methyltransferases), EPZ004777, EPZ-5676, SYC-522, SYC-534 and SGC0946.
Development of small molecular DOT1L inhibitors
It has been shown that SAH, the product of the DOT1L catalyzed reaction, is a competitive inhibitor of DOT1L. However, SAH inhibits binding of the cofactor SAM to most of the SAM-dependent methyltransferases [151]. In 2011, the first-in-class aminonucleoside-based DOT1L specific inhibitor EPZ004777 that selectively suppresses leukemia cells with MLL1 translocations was identified (Fig. 3D) [153]. Treatment of leukemia cells with EPZ004777 inhibits methylation of H3K79 while methylation of most other histone amino acid residues is not markedly affected. Although the suppression of H3K79me2 is observed within three days of treatment, the maximal antiproliferative efficacy of EPZ004777 on MLL1-rearranged leukemia cells (IC50 0.17–6.47 uM depending on the cell line) is observed between 9–14 days of treatment [153]. This suggests that EPZ004777 does not result in acute apoptosis of the leukemic blasts, but instead induces myeloid differentiation of the MLL1-rearranged leukemic cells similar to the genetic inactivation of DOT1L in mouse models [80]. Crystallographic studies reveal that the binding of EPZ004777 is associated with a conformational change of DOT1L and stabilizes a unique conformation of the DOT1L active site [154].
Despite the potency and selectivity of EPZ004777 toward DOT1L, as well as good in vitro efficacy of EPZ004777 against the proliferation of many types of leukemias with MLL1 abnormalities, the poor pharmacokinetic properties of EPZ004777 precluded it from clinical development [131, 132, 153, 155]. Based on the structures of SAH and EPZ004777, several research laboratories utilized mechanism-guided designs to further optimize the binding affinity, selectivity and in vivo stability of aminonucleoside compounds for DOT1L inhibition (Fig. 3D) [156–161]. One of the second-generation DOT1L inhibitors, EPZ-5676, is a more potent and selective inhibitor of DOT1L compared to EPZ004777. Specifically, it demonstrates a lower Ki (80 pM as compared to 300 pM for EPZ004777) and a higher selectivity (37,000-fold more selective for DOT1L amongst a panel of histone methyltransferases as compared to 1,000-fold for EPZ004777) to DOT1L [156]. EPZ-5676 also shows an increased potency against MLL1-rearranged leukemia cells (IC50 = 3.5nM against MV4–11 cells as compared to 150nM for EPZ004777) and an extended DOT1L-binding residence time as compared to EPZ004777. In animals, EPZ-5676 has a low oral bioavailability; however, a high intraperitoneal and intravenous bioavailability is observed in rodent models. In vivo pharmacokinetics studies showed a terminal elimination half-life (t1/2 ) of 1.1, 3.7 and 13.6 hours following intravenous administration in mouse, rat and dog, respectively [162]. The in vivo clearance of EPZ-5676 shows low renal clearance, instead implicating a hepatic oxidative metabolism as the predominant elimination route in preclinical species. Importantly, EPZ-5676 causes a significant regression of tumor size in rodent subcutaneous xenograft models of MLL1-rearranged leukemia, while no significant weight loss of the animals is observed during the 21 days of treatment [156]. Based on the superior potency, selectivity, and pharmacokinetic behavior of EPZ-5676 over the first-generation DOT1L inhibitor, as well as good physiological tolerance in animals, EPZ-5676 has been selected for further drug development and is currently under Phase I clinical investigation for acute leukemias bearing MLL1-rearrangements in adult (CT.gov: NCT01684150; A First-in-Human Phase 1 and Expanded Cohort Study of EPZ-5676 in Advanced Hematologic Malignancies, Including Acute Leukemia With Rearrangement of the MLL1 Gene) [163] and pediatric (CT.gov: NCT02141828; Dose Escalation Study of EPZ-5676 in Pediatric Patients With Leukemias Bearing a Rearrangement of the MLL1 Gene) patients.
Conclusion and perspectives
DOT1L is the sole methyltransferase capable of catalyzing mono-, di-, and trimethylation of H3K79. Previous genetic and pharmacologic studies provide a compelling rationale for DOT1L-targeted therapy in MLL1-rearranged leukemia, a hematopoietic malignancy that carries a poor prognosis. Several potent and selective inhibitors of DOT1L have been developed, and one is currently undergoing Phase I clinical trials for patients with MLL1 gene rearrangements. Despite the emerging progress of DOT1L inhibition in MLL1-rearranged leukemias, the mechanisms by which DOT1L and H3K79 methylation drive the expression of the MLL1-fusion leukemogenic program remain largely unexplored. Approaches to identify the pathways associated with DOT1L have led to the discovery of other important epigenetic regulators that impact the function of DOT1L in leukemia. In addition to identifying the cofactors of DOT1L (e.g. AF10), our recent attempts using a genome-wide RNAi screen strategy revealed a role for DOT1L-dependent H3K79 methylation in inhibition of SIRT1-mediated histone deacetylation and epigenetic silencing in leukemias driven by MLL1 translocations [164]. This finding leads us to believe that future studies focusing on combinatorial targeting DOT1L and other epigenetic pathways should lead to greater therapeutic responses against difficult-to-treat MLL1-rearranged leukemias. We also foresee that genome-scale high throughput screens using RNAi and CRISPR technologies will facilitate future research to uncover more cross-interactions between epigenetic programs in leukemias and other types of cancer.
Highlights.
The histone methyltransferase DOT1L is required for proliferation of MLL-rearranged leukemias.
DOT1L and H3K79 methylation maintains high level HOXA and MEIS 1 gene expression
DOT1L inhibition induces differentiation of MLL-rearranged and other high HOXA leukemias
DOT1L inhibitors are in clinical trials.
Acknowledgments
This work was supported by the Leukemia and Lymphoma Society, Gabrielle’s Angel Research Foundation and NIH Grants CA66996, CA140575, and CA176745 to S.A.A..
Footnotes
Conflict of interest disclosure
S.A.A. is a consultant for Epizyme Inc.. C.W.C reports no competing financial interest/relationships with the topic of this review.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Morse HG, et al. 4;11 translocation in acute lymphoblastic leukemia: a specific syndrome. Cancer Genet Cytogenet. 1982;7(2):165–72. doi: 10.1016/0165-4608(82)90012-7. [DOI] [PubMed] [Google Scholar]
- 2.Prigogina EL, et al. Chromosomes in acute leukemia. Hum Genet. 1979;53(1):5–16. doi: 10.1007/BF00289443. [DOI] [PubMed] [Google Scholar]
- 3.Van den Berghe H, et al. A new chromosome anomaly in acute lymphoblastic leukemia (ALL) Hum Genet. 1979;46(2):173–80. doi: 10.1007/BF00291919. [DOI] [PubMed] [Google Scholar]
- 4.Mirro J, et al. Clinical and laboratory characteristics of acute leukemia with the 4;11 translocation. Blood. 1986;67(3):689–97. [PubMed] [Google Scholar]
- 5.Mirro J, et al. Acute mixed lineage leukemia: clinicopathologic correlations and prognostic significance. Blood. 1985;66(5):1115–23. [PubMed] [Google Scholar]
- 6.Stass S, Mirro J. Unexpected heterogeneity in acute leukemia: mixed lineages and lineage switch. Hum Pathol. 1985;16(9):864–6. doi: 10.1016/s0046-8177(85)80125-8. [DOI] [PubMed] [Google Scholar]
- 7.Djabali M, et al. A trithorax-like gene is interrupted by chromosome 11q23 translocations in acute leukaemias. Nat Genet. 1992;2(2):113–8. doi: 10.1038/ng1092-113. [DOI] [PubMed] [Google Scholar]
- 8.Gu Y, et al. The t(4;11) chromosome translocation of human acute leukemias fuses the ALL-1 gene, related to Drosophila trithorax, to the AF-4 gene. Cell. 1992;71(4):701–8. doi: 10.1016/0092-8674(92)90603-a. [DOI] [PubMed] [Google Scholar]
- 9.Tkachuk DC, Kohler S, Cleary ML. Involvement of a homolog of Drosophila trithorax by 11q23 chromosomal translocations in acute leukemias. Cell. 1992;71(4):691–700. doi: 10.1016/0092-8674(92)90602-9. [DOI] [PubMed] [Google Scholar]
- 10.Ziemin-van der Poel S, et al. Identification of a gene, MLL, that spans the breakpoint in 11q23 translocations associated with human leukemias. Proc Natl Acad Sci U S A. 1991;88(23):10735–9. doi: 10.1073/pnas.88.23.10735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Armstrong SA, Look AT. Molecular genetics of acute lymphoblastic leukemia. J Clin Oncol. 2005;23(26):6306–15. doi: 10.1200/JCO.2005.05.047. [DOI] [PubMed] [Google Scholar]
- 12.Huret JL, Dessen P, Bernheim A. An atlas of chromosomes in hematological malignancies. Example: 11q23 and MLL partners. Leukemia. 2001;15(6):987–9. doi: 10.1038/sj.leu.2402135. [DOI] [PubMed] [Google Scholar]
- 13.Krivtsov AV, Armstrong SA. MLL translocations, histone modifications and leukaemia stem-cell development. Nat Rev Cancer. 2007;7(11):823–33. doi: 10.1038/nrc2253. [DOI] [PubMed] [Google Scholar]
- 14.Meyer C, et al. New insights to the MLL recombinome of acute leukemias. Leukemia. 2009;23(8):1490–9. doi: 10.1038/leu.2009.33. [DOI] [PubMed] [Google Scholar]
- 15.Meyer C, et al. The MLL recombinome of acute leukemias. Leukemia. 2006;20(5):777–84. doi: 10.1038/sj.leu.2404150. [DOI] [PubMed] [Google Scholar]
- 16.Raimondi SC, et al. Chromosomal abnormalities in 478 children with acute myeloid leukemia: clinical characteristics and treatment outcome in a cooperative pediatric oncology group study-POG 8821. Blood. 1999;94(11):3707–16. [PubMed] [Google Scholar]
- 17.Mohan M, et al. Licensed to elongate: a molecular mechanism for MLL-based leukaemogenesis. Nat Rev Cancer. 2010;10(10):721–8. doi: 10.1038/nrc2915. [DOI] [PubMed] [Google Scholar]
- 18.Chessells JM, et al. Clinical features, cytogenetics and outcome in acute lymphoblastic and myeloid leukaemia of infancy: report from the MRC Childhood Leukaemia working party. Leukemia. 2002;16(5):776–84. doi: 10.1038/sj.leu.2402468. [DOI] [PubMed] [Google Scholar]
- 19.Hilden JM, et al. Analysis of prognostic factors of acute lymphoblastic leukemia in infants: report on CCG 1953 from the Children’s Oncology Group. Blood. 2006;108(2):441–51. doi: 10.1182/blood-2005-07-3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pieters R, et al. A treatment protocol for infants younger than 1 year with acute lymphoblastic leukaemia (Interfant-99): an observational study and a multicentre randomised trial. Lancet. 2007;370(9583):240–50. doi: 10.1016/S0140-6736(07)61126-X. [DOI] [PubMed] [Google Scholar]
- 21.Tomizawa D, et al. Outcome of risk-based therapy for infant acute lymphoblastic leukemia with or without an MLL gene rearrangement, with emphasis on late effects: a final report of two consecutive studies, MLL96 and MLL98, of the Japan Infant Leukemia Study Group. Leukemia. 2007;21(11):2258–63. doi: 10.1038/sj.leu.2404903. [DOI] [PubMed] [Google Scholar]
- 22.van der Linden MH, et al. Outcome of congenital acute lymphoblastic leukemia treated on the Interfant-99 protocol. Blood. 2009;114(18):3764–8. doi: 10.1182/blood-2009-02-204214. [DOI] [PubMed] [Google Scholar]
- 23.Biondi A, et al. Biological and therapeutic aspects of infant leukemia. Blood. 2000;96(1):24–33. [PubMed] [Google Scholar]
- 24.Huret JL, et al. The “Atlas of genetics and cytogenetics in oncology and haematology” on the internet and a review on infant leukemias. Cancer Genet Cytogenet. 2000;120(2):155–9. doi: 10.1016/s0165-4608(99)00250-2. [DOI] [PubMed] [Google Scholar]
- 25.Felix CA, et al. ALL-1 gene rearrangements in DNA topoisomerase II inhibitor-related leukemia in children. Blood. 1995;85(11):3250–6. [PubMed] [Google Scholar]
- 26.Felix CA. Secondary leukemias induced by topoisomerase-targeted drugs. Biochim Biophys Acta. 1998;1400(1–3):233–55. doi: 10.1016/s0167-4781(98)00139-0. [DOI] [PubMed] [Google Scholar]
- 27.Pui CH, Relling MV. Topoisomerase II inhibitor-related acute myeloid leukaemia. Br J Haematol. 2000;109(1):13–23. doi: 10.1046/j.1365-2141.2000.01843.x. [DOI] [PubMed] [Google Scholar]
- 28.Meyer C, et al. The MLL recombinome of acute leukemias in 2013. Leukemia. 2013;27(11):2165–76. doi: 10.1038/leu.2013.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schoch C, et al. AML with 11q23/MLL abnormalities as defined by the WHO classification: incidence, partner chromosomes, FAB subtype, age distribution, and prognostic impact in an unselected series of 1897 cytogenetically analyzed AML cases. Blood. 2003;102(7):2395–402. doi: 10.1182/blood-2003-02-0434. [DOI] [PubMed] [Google Scholar]
- 30.Andersson AK, et al. The landscape of somatic mutations in infant MLL-rearranged acute lymphoblastic leukemias. Nat Genet. 2015;47(4):330–337. doi: 10.1038/ng.3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen CS, et al. Molecular rearrangements on chromosome 11q23 predominate in infant acute lymphoblastic leukemia and are associated with specific biologic variables and poor outcome. Blood. 1993;81(9):2386–93. [PubMed] [Google Scholar]
- 32.Pui CH, et al. Improved outcome for children with acute lymphoblastic leukemia: results of Total Therapy Study XIIIB at St Jude Children’s Research Hospital. Blood. 2004;104(9):2690–6. doi: 10.1182/blood-2004-04-1616. [DOI] [PubMed] [Google Scholar]
- 33.Kosaka Y, et al. Infant acute lymphoblastic leukemia with MLL gene rearrangements: outcome following intensive chemotherapy and hematopoietic stem cell transplantation. Blood. 2004;104(12):3527–34. doi: 10.1182/blood-2004-04-1390. [DOI] [PubMed] [Google Scholar]
- 34.Mann G, et al. Improved outcome with hematopoietic stem cell transplantation in a poor prognostic subgroup of infants with mixed-lineage-leukemia (MLL)-rearranged acute lymphoblastic leukemia: results from the Interfant-99 Study. Blood. 2010;116(15):2644–50. doi: 10.1182/blood-2010-03-273532. [DOI] [PubMed] [Google Scholar]
- 35.Muntean AG, Hess JL. The pathogenesis of mixed-lineage leukemia. Annu Rev Pathol. 2012;7:283–301. doi: 10.1146/annurev-pathol-011811-132434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stark B, et al. Classical and molecular cytogenetic abnormalities and outcome of childhood acute myeloid leukaemia: report from a referral centre in Israel. Br J Haematol. 2004;126(3):320–37. doi: 10.1111/j.1365-2141.2004.05038.x. [DOI] [PubMed] [Google Scholar]
- 37.Krogan NJ, et al. COMPASS, a histone H3 (Lysine 4) methyltransferase required for telomeric silencing of gene expression. J Biol Chem. 2002;277(13):10753–5. doi: 10.1074/jbc.C200023200. [DOI] [PubMed] [Google Scholar]
- 38.Miller T, et al. COMPASS: a complex of proteins associated with a trithorax-related SET domain protein. Proc Natl Acad Sci U S A. 2001;98(23):12902–7. doi: 10.1073/pnas.231473398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roguev A, et al. The Saccharomyces cerevisiae Set1 complex includes an Ash2 homologue and methylates histone 3 lysine 4. EMBO J. 2001;20(24):7137–48. doi: 10.1093/emboj/20.24.7137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zeleznik-Le NJ, Harden AM, Rowley JD. 11q23 translocations split the “AT-hook” cruciform DNA-binding region and the transcriptional repression domain from the activation domain of the mixed-lineage leukemia (MLL) gene. Proc Natl Acad Sci U S A. 1994;91(22):10610–4. doi: 10.1073/pnas.91.22.10610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Birke M, et al. The MT domain of the proto-oncoprotein MLL binds to CpG-containing DNA and discriminates against methylation. Nucleic Acids Res. 2002;30(4):958–65. doi: 10.1093/nar/30.4.958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fair K, et al. Protein interactions of the MLL PHD fingers modulate MLL target gene regulation in human cells. Mol Cell Biol. 2001;21(10):3589–97. doi: 10.1128/MCB.21.10.3589-3597.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Briggs SD, et al. Histone H3 lysine 4 methylation is mediated by Set1 and required for cell growth and rDNA silencing in Saccharomyces cerevisiae. Genes Dev. 2001;15(24):3286–95. doi: 10.1101/gad.940201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Milne TA, et al. MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol Cell. 2002;10(5):1107–17. doi: 10.1016/s1097-2765(02)00741-4. [DOI] [PubMed] [Google Scholar]
- 45.Hsieh JJ, et al. Proteolytic cleavage of MLL generates a complex of N- and C-terminal fragments that confers protein stability and subnuclear localization. Mol Cell Biol. 2003;23(1):186–94. doi: 10.1128/MCB.23.1.186-194.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yokoyama A, et al. Leukemia proto-oncoprotein MLL is proteolytically processed into 2 fragments with opposite transcriptional properties. Blood. 2002;100(10):3710–8. doi: 10.1182/blood-2002-04-1015. [DOI] [PubMed] [Google Scholar]
- 47.Milne TA, et al. MLL associates specifically with a subset of transcriptionally active target genes. Proc Natl Acad Sci U S A. 2005;102(41):14765–70. doi: 10.1073/pnas.0503630102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nakamura T, et al. ALL-1 is a histone methyltransferase that assembles a supercomplex of proteins involved in transcriptional regulation. Mol Cell. 2002;10(5):1119–28. doi: 10.1016/s1097-2765(02)00740-2. [DOI] [PubMed] [Google Scholar]
- 49.Couture JF, Collazo E, Trievel RC. Molecular recognition of histone H3 by the WD40 protein WDR5. Nat Struct Mol Biol. 2006;13(8):698–703. doi: 10.1038/nsmb1116. [DOI] [PubMed] [Google Scholar]
- 50.Dou Y, et al. Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat Struct Mol Biol. 2006;13(8):713–9. doi: 10.1038/nsmb1128. [DOI] [PubMed] [Google Scholar]
- 51.Steward MM, et al. Molecular regulation of H3K4 trimethylation by ASH2L, a shared subunit of MLL complexes. Nat Struct Mol Biol. 2006;13(9):852–4. doi: 10.1038/nsmb1131. [DOI] [PubMed] [Google Scholar]
- 52.Crawford BD, Hess JL. MLL core components give the green light to histone methylation. ACS Chem Biol. 2006;1(8):495–8. doi: 10.1021/cb600367v. [DOI] [PubMed] [Google Scholar]
- 53.Zhang P, et al. A phosphorylation switch on RbBP5 regulates histone H3 Lys4 methylation. Genes Dev. 2015;29(2):123–8. doi: 10.1101/gad.254870.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shinsky SA, et al. Biochemical Reconstitution and Phylogenetic Comparison of Human SET1 Family Core Complexes Involved in Histone Methylation. J Biol Chem. 2015;290(10):6361–75. doi: 10.1074/jbc.M114.627646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shinsky SA, et al. A non-active-site SET domain surface crucial for the interaction of MLL1 and the RbBP5/Ash2L heterodimer within MLL family core complexes. J Mol Biol. 2014;426(12):2283–99. doi: 10.1016/j.jmb.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Patel A, et al. Automethylation activities within the mixed lineage leukemia-1 (MLL1) core complex reveal evidence supporting a “two-active site” model for multiple histone H3 lysine 4 methylation. J Biol Chem. 2014;289(2):868–84. doi: 10.1074/jbc.M113.501064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shilatifard A. The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annu Rev Biochem. 2012;81:65–95. doi: 10.1146/annurev-biochem-051710-134100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang P, et al. Global analysis of H3K4 methylation defines MLL family member targets and points to a role for MLL1-mediated H3K4 methylation in the regulation of transcriptional initiation by RNA polymerase II. Mol Cell Biol. 2009;29(22):6074–85. doi: 10.1128/MCB.00924-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Armstrong SA, et al. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat Genet. 2002;30(1):41–7. doi: 10.1038/ng765. [DOI] [PubMed] [Google Scholar]
- 60.Dorrance AM, et al. Mll partial tandem duplication induces aberrant Hox expression in vivo via specific epigenetic alterations. J Clin Invest. 2006;116(10):2707–16. doi: 10.1172/JCI25546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Milne TA, et al. Leukemogenic MLL fusion proteins bind across a broad region of the Hox a9 locus, promoting transcription and multiple histone modifications. Cancer Res. 2005;65(24):11367–74. doi: 10.1158/0008-5472.CAN-05-1041. [DOI] [PubMed] [Google Scholar]
- 62.Ferrando AA, et al. Gene expression signatures in MLL-rearranged T-lineage and B-precursor acute leukemias: dominance of HOX dysregulation. Blood. 2003;102(1):262–8. doi: 10.1182/blood-2002-10-3221. [DOI] [PubMed] [Google Scholar]
- 63.Yeoh EJ, et al. Classification, subtype discovery, and prediction of outcome in pediatric acute lymphoblastic leukemia by gene expression profiling. Cancer Cell. 2002;1(2):133–43. doi: 10.1016/s1535-6108(02)00032-6. [DOI] [PubMed] [Google Scholar]
- 64.Ross ME, et al. Classification of pediatric acute lymphoblastic leukemia by gene expression profiling. Blood. 2003;102(8):2951–9. doi: 10.1182/blood-2003-01-0338. [DOI] [PubMed] [Google Scholar]
- 65.Kawagoe H, et al. Expression of HOX genes, HOX cofactors, and MLL in phenotypically and functionally defined subpopulations of leukemic and normal human hematopoietic cells. Leukemia. 1999;13(5):687–98. doi: 10.1038/sj.leu.2401410. [DOI] [PubMed] [Google Scholar]
- 66.Abramovich C, Humphries RK. Hox regulation of normal and leukemic hematopoietic stem cells. Curr Opin Hematol. 2005;12(3):210–6. doi: 10.1097/01.moh.0000160737.52349.aa. [DOI] [PubMed] [Google Scholar]
- 67.Alharbi RA, et al. The role of HOX genes in normal hematopoiesis and acute leukemia. Leukemia. 2013;27(5):1000–8. doi: 10.1038/leu.2012.356. [DOI] [PubMed] [Google Scholar]
- 68.Yu BD, et al. Altered Hox expression and segmental identity in Mll-mutant mice. Nature. 1995;378(6556):505–8. doi: 10.1038/378505a0. [DOI] [PubMed] [Google Scholar]
- 69.Hess JL, et al. Defects in yolk sac hematopoiesis in Mll-null embryos. Blood. 1997;90(5):1799–806. [PubMed] [Google Scholar]
- 70.Yagi H, et al. Growth disturbance in fetal liver hematopoiesis of Mll-mutant mice. Blood. 1998;92(1):108–17. [PubMed] [Google Scholar]
- 71.Ernst P, et al. Definitive hematopoiesis requires the mixed-lineage leukemia gene. Dev Cell. 2004;6(3):437–43. doi: 10.1016/s1534-5807(04)00061-9. [DOI] [PubMed] [Google Scholar]
- 72.Guenther MG, et al. Global and Hox-specific roles for the MLL1 methyltransferase. Proc Natl Acad Sci U S A. 2005;102(24):8603–8. doi: 10.1073/pnas.0503072102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hughes CM, et al. Menin associates with a trithorax family histone methyltransferase complex and with the hoxc8 locus. Mol Cell. 2004;13(4):587–97. doi: 10.1016/s1097-2765(04)00081-4. [DOI] [PubMed] [Google Scholar]
- 74.Yokoyama A, et al. The menin tumor suppressor protein is an essential oncogenic cofactor for MLL-associated leukemogenesis. Cell. 2005;123(2):207–18. doi: 10.1016/j.cell.2005.09.025. [DOI] [PubMed] [Google Scholar]
- 75.Yokoyama A, et al. Leukemia proto-oncoprotein MLL forms a SET1-like histone methyltransferase complex with menin to regulate Hox gene expression. Mol Cell Biol. 2004;24(13):5639–49. doi: 10.1128/MCB.24.13.5639-5649.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Borkin D, et al. Pharmacologic Inhibition of the Menin-MLL Interaction Blocks Progression of MLL Leukemia In Vivo. Cancer Cell. 2015 doi: 10.1016/j.ccell.2015.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Terranova R, et al. Histone and DNA methylation defects at Hox genes in mice expressing a SET domain-truncated form of Mll. Proc Natl Acad Sci U S A. 2006;103(17):6629–34. doi: 10.1073/pnas.0507425103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mishra BP, et al. The histone methyltransferase activity of MLL1 is dispensable for hematopoiesis and leukemogenesis. Cell Rep. 2014;7(4):1239–47. doi: 10.1016/j.celrep.2014.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shah N, Sukumar S. The Hox genes and their roles in oncogenesis. Nat Rev Cancer. 2010;10(5):361–71. doi: 10.1038/nrc2826. [DOI] [PubMed] [Google Scholar]
- 80.Bernt KM, et al. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell. 2011;20(1):66–78. doi: 10.1016/j.ccr.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kroon E, et al. Hoxa9 transforms primary bone marrow cells through specific collaboration with Meis1a but not Pbx1b. EMBO J. 1998;17(13):3714–25. doi: 10.1093/emboj/17.13.3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Buske C, Humphries RK. Homeobox genes in leukemogenesis. Int J Hematol. 2000;71(4):301–8. [PubMed] [Google Scholar]
- 83.Wang QF, et al. MLL fusion proteins preferentially regulate a subset of wild-type MLL target genes in the leukemic genome. Blood. 2011;117(25):6895–905. doi: 10.1182/blood-2010-12-324699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Thiel AT, et al. MLL-AF9-induced leukemogenesis requires coexpression of the wild-type Mll allele. Cancer Cell. 2010;17(2):148–59. doi: 10.1016/j.ccr.2009.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Domer PH, et al. Acute mixed-lineage leukemia t(4;11)(q21;q23) generates an MLL-AF4 fusion product. Proc Natl Acad Sci U S A. 1993;90(16):7884–8. doi: 10.1073/pnas.90.16.7884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Corral J, et al. An Mll-AF9 fusion gene made by homologous recombination causes acute leukemia in chimeric mice: a method to create fusion oncogenes. Cell. 1996;85(6):853–61. doi: 10.1016/s0092-8674(00)81269-6. [DOI] [PubMed] [Google Scholar]
- 87.Collins EC, et al. Inter-chromosomal recombination of Mll and Af9 genes mediated by cre-loxP in mouse development. EMBO Rep. 2000;1(2):127–32. doi: 10.1093/embo-reports/kvd021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Forster A, et al. Engineering de novo reciprocal chromosomal translocations associated with Mll to replicate primary events of human cancer. Cancer Cell. 2003;3(5):449–58. doi: 10.1016/s1535-6108(03)00106-5. [DOI] [PubMed] [Google Scholar]
- 89.Krivtsov AV, et al. H3K79 methylation profiles define murine and human MLL-AF4 leukemias. Cancer Cell. 2008;14(5):355–68. doi: 10.1016/j.ccr.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chen W, et al. A murine Mll-AF4 knock-in model results in lymphoid and myeloid deregulation and hematologic malignancy. Blood. 2006;108(2):669–77. doi: 10.1182/blood-2005-08-3498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Metzler M, et al. A conditional model of MLL-AF4 B-cell tumourigenesis using invertor technology. Oncogene. 2006;25(22):3093–103. doi: 10.1038/sj.onc.1209636. [DOI] [PubMed] [Google Scholar]
- 92.Muse GW, et al. RNA polymerase is poised for activation across the genome. Nat Genet. 2007;39(12):1507–11. doi: 10.1038/ng.2007.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Margaritis T, Holstege FC. Poised RNA polymerase II gives pause for thought. Cell. 2008;133(4):581–4. doi: 10.1016/j.cell.2008.04.027. [DOI] [PubMed] [Google Scholar]
- 94.Shilatifard A, et al. An RNA polymerase II elongation factor encoded by the human ELL gene. Science. 1996;271(5257):1873–6. doi: 10.1126/science.271.5257.1873. [DOI] [PubMed] [Google Scholar]
- 95.Shilatifard A, Conaway RC, Conaway JW. The RNA polymerase II elongation complex. Annu Rev Biochem. 2003;72:693–715. doi: 10.1146/annurev.biochem.72.121801.161551. [DOI] [PubMed] [Google Scholar]
- 96.Mueller D, et al. A role for the MLL fusion partner ENL in transcriptional elongation and chromatin modification. Blood. 2007;110(13):4445–54. doi: 10.1182/blood-2007-05-090514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bitoun E, Oliver PL, Davies KE. The mixed-lineage leukemia fusion partner AF4 stimulates RNA polymerase II transcriptional elongation and mediates coordinated chromatin remodeling. Hum Mol Genet. 2007;16(1):92–106. doi: 10.1093/hmg/ddl444. [DOI] [PubMed] [Google Scholar]
- 98.Eissenberg JC, et al. dELL is an essential RNA polymerase II elongation factor with a general role in development. Proc Natl Acad Sci U S A. 2002;99(15):9894–9. doi: 10.1073/pnas.152193699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gerber M, et al. Drosophila ELL is associated with actively elongating RNA polymerase II on transcriptionally active sites in vivo. EMBO J. 2001;20(21):6104–14. doi: 10.1093/emboj/20.21.6104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Peterlin BM, Price DH. Controlling the elongation phase of transcription with P-TEFb. Mol Cell. 2006;23(3):297–305. doi: 10.1016/j.molcel.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 101.Mueller D, et al. Misguided transcriptional elongation causes mixed lineage leukemia. PLoS Biol. 2009;7(11):e1000249. doi: 10.1371/journal.pbio.1000249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Monroe SC, et al. MLL-AF9 and MLL-ENL alter the dynamic association of transcriptional regulators with genes critical for leukemia. Exp Hematol. 2011;39(1):77–86. e1–5. doi: 10.1016/j.exphem.2010.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yokoyama A, et al. A higher-order complex containing AF4 and ENL family proteins with P-TEFb facilitates oncogenic and physiologic MLL-dependent transcription. Cancer Cell. 2010;17(2):198–212. doi: 10.1016/j.ccr.2009.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lin C, et al. AFF4, a component of the ELL/P-TEFb elongation complex and a shared subunit of MLL chimeras, can link transcription elongation to leukemia. Mol Cell. 2010;37(3):429–37. doi: 10.1016/j.molcel.2010.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403(6765):41–5. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 106.Tessarz P, Kouzarides T. Histone core modifications regulating nucleosome structure and dynamics. Nat Rev Mol Cell Biol. 2014;15(11):703–8. doi: 10.1038/nrm3890. [DOI] [PubMed] [Google Scholar]
- 107.Rea S, et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature. 2000;406(6796):593–9. doi: 10.1038/35020506. [DOI] [PubMed] [Google Scholar]
- 108.Guenther MG, et al. Aberrant chromatin at genes encoding stem cell regulators in human mixed-lineage leukemia. Genes Dev. 2008;22(24):3403–8. doi: 10.1101/gad.1741408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Barski A, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129(4):823–37. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 110.Steger DJ, et al. DOT1L/KMT4 recruitment and H3K79 methylation are ubiquitously coupled with gene transcription in mammalian cells. Mol Cell Biol. 2008;28(8):2825–39. doi: 10.1128/MCB.02076-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Im H, et al. Dynamic regulation of histone H3 methylated at lysine 79 within a tissue-specific chromatin domain. J Biol Chem. 2003;278(20):18346–52. doi: 10.1074/jbc.M300890200. [DOI] [PubMed] [Google Scholar]
- 112.Schubeler D, et al. The histone modification pattern of active genes revealed through genome-wide chromatin analysis of a higher eukaryote. Genes Dev. 2004;18(11):1263–71. doi: 10.1101/gad.1198204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Guenther MG, et al. A chromatin landmark and transcription initiation at most promoters in human cells. Cell. 2007;130(1):77–88. doi: 10.1016/j.cell.2007.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lacoste N, et al. Disruptor of telomeric silencing-1 is a chromatin-specific histone H3 methyltransferase. J Biol Chem. 2002;277(34):30421–4. doi: 10.1074/jbc.C200366200. [DOI] [PubMed] [Google Scholar]
- 115.van Leeuwen F, Gafken PR, Gottschling DE. Dot1p modulates silencing in yeast by methylation of the nucleosome core. Cell. 2002;109(6):745–56. doi: 10.1016/s0092-8674(02)00759-6. [DOI] [PubMed] [Google Scholar]
- 116.Feng Q, et al. Methylation of H3-lysine 79 is mediated by a new family of HMTases without a SET domain. Curr Biol. 2002;12(12):1052–8. doi: 10.1016/s0960-9822(02)00901-6. [DOI] [PubMed] [Google Scholar]
- 117.Singer MS, et al. Identification of high-copy disruptors of telomeric silencing in Saccharomyces cerevisiae. Genetics. 1998;150(2):613–32. doi: 10.1093/genetics/150.2.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Shanower GA, et al. Characterization of the grappa gene, the Drosophila histone H3 lysine 79 methyltransferase. Genetics. 2005;169(1):173–84. doi: 10.1534/genetics.104.033191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Jones B, et al. The histone H3K79 methyltransferase Dot1L is essential for mammalian development and heterochromatin structure. PLoS Genet. 2008;4(9):e1000190. doi: 10.1371/journal.pgen.1000190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Nguyen AT, Zhang Y. The diverse functions of Dot1 and H3K79 methylation. Genes Dev. 2011;25(13):1345–58. doi: 10.1101/gad.2057811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Feng Y, et al. Early mammalian erythropoiesis requires the Dot1L methyltransferase. Blood. 2010;116(22):4483–91. doi: 10.1182/blood-2010-03-276501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Jo SY, et al. Requirement for Dot1l in murine postnatal hematopoiesis and leukemogenesis by MLL translocation. Blood. 2011;117(18):4759–68. doi: 10.1182/blood-2010-12-327668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Nguyen AT, et al. Essential role of DOT1L in maintaining normal adult hematopoiesis. Cell Res. 2011;21(9):1370–3. doi: 10.1038/cr.2011.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Mohan M, et al. Linking H3K79 trimethylation to Wnt signaling through a novel Dot1-containing complex (DotCom) Genes Dev. 2010;24(6):574–89. doi: 10.1101/gad.1898410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Okada Y, et al. hDOT1L links histone methylation to leukemogenesis. Cell. 2005;121(2):167–78. doi: 10.1016/j.cell.2005.02.020. [DOI] [PubMed] [Google Scholar]
- 126.Park G, et al. Characterization of the DOT1L network: implications of diverse roles for DOT1L. Protein J. 2010;29(3):213–23. doi: 10.1007/s10930-010-9242-8. [DOI] [PubMed] [Google Scholar]
- 127.Shen C, et al. Targeting recruitment of disruptor of telomeric silencing 1-like (DOT1L): characterizing the interactions between DOT1L and mixed lineage leukemia (MLL) fusion proteins. J Biol Chem. 2013;288(42):30585–96. doi: 10.1074/jbc.M113.457135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zhang W, et al. Dot1a-AF9 complex mediates histone H3 Lys-79 hypermethylation and repression of ENaCalpha in an aldosterone-sensitive manner. J Biol Chem. 2006;281(26):18059–68. doi: 10.1074/jbc.M601903200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Deshpande AJ, et al. AF10 regulates progressive H3K79 methylation and HOX gene expression in diverse AML subtypes. Cancer Cell. 2014;26(6):896–908. doi: 10.1016/j.ccell.2014.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Biswas D, et al. Function of leukemogenic mixed lineage leukemia 1 (MLL) fusion proteins through distinct partner protein complexes. Proc Natl Acad Sci U S A. 2011;108(38):15751–6. doi: 10.1073/pnas.1111498108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Chen L, et al. Abrogation of MLL-AF10 and CALM-AF10-mediated transformation through genetic inactivation or pharmacological inhibition of the H3K79 methyltransferase Dot1l. Leukemia. 2013;27(4):813–22. doi: 10.1038/leu.2012.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Deshpande AJ, et al. Leukemic transformation by the MLL-AF6 fusion oncogene requires the H3K79 methyltransferase Dot1l. Blood. 2013;121(13):2533–41. doi: 10.1182/blood-2012-11-465120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Nguyen AT, et al. DOT1L, the H3K79 methyltransferase, is required for MLL-AF9-mediated leukemogenesis. Blood. 2011;117(25):6912–22. doi: 10.1182/blood-2011-02-334359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kuntimaddi A, et al. Degree of Recruitment of DOT1L to MLL-AF9 Defines Level of H3K79 Di- and Tri-methylation on Target Genes and Transformation Potential. Cell Rep. 2015;11(5):808–20. doi: 10.1016/j.celrep.2015.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Deshpande AJ, Bradner J, Armstrong SA. Chromatin modifications as therapeutic targets in MLL-rearranged leukemia. Trends Immunol. 2012;33(11):563–70. doi: 10.1016/j.it.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Zhang Z, et al. Widely expressed Af17 is likely not required for embryogenesis, hematopoiesis, and animal survival. Genesis. 2010;48(12):693–706. doi: 10.1002/dvg.20679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hwang WW, et al. A conserved RING finger protein required for histone H2B monoubiquitination and cell size control. Mol Cell. 2003;11(1):261–6. doi: 10.1016/s1097-2765(02)00826-2. [DOI] [PubMed] [Google Scholar]
- 138.Wood A, et al. Bre1, an E3 ubiquitin ligase required for recruitment and substrate selection of Rad6 at a promoter. Mol Cell. 2003;11(1):267–74. doi: 10.1016/s1097-2765(02)00802-x. [DOI] [PubMed] [Google Scholar]
- 139.Kim J, Hake SB, Roeder RG. The human homolog of yeast BRE1 functions as a transcriptional coactivator through direct activator interactions. Mol Cell. 2005;20(5):759–70. doi: 10.1016/j.molcel.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 140.Zhu B, et al. Monoubiquitination of human histone H2B: the factors involved and their roles in HOX gene regulation. Mol Cell. 2005;20(4):601–11. doi: 10.1016/j.molcel.2005.09.025. [DOI] [PubMed] [Google Scholar]
- 141.Chatterjee C, et al. Disulfide-directed histone ubiquitylation reveals plasticity in hDot1L activation. Nat Chem Biol. 2010;6(4):267–9. doi: 10.1038/nchembio.315. [DOI] [PubMed] [Google Scholar]
- 142.Wang E, et al. Histone H2B ubiquitin ligase RNF20 is required for MLL-rearranged leukemia. Proc Natl Acad Sci U S A. 2013;110(10):3901–6. doi: 10.1073/pnas.1301045110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Krivtsov AV, et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature. 2006;442(7104):818–22. doi: 10.1038/nature04980. [DOI] [PubMed] [Google Scholar]
- 144.Xu W, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19(1):17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Sarkaria SM, et al. Primary acute myeloid leukemia cells with IDH1 or IDH2 mutations respond to a DOT1L inhibitor in vitro. Leukemia. 2014;28(12):2403–6. doi: 10.1038/leu.2014.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Chaturvedi A, et al. Mutant IDH1 promotes leukemogenesis in vivo and can be specifically targeted in human AML. Blood. 2013;122(16):2877–87. doi: 10.1182/blood-2013-03-491571. [DOI] [PubMed] [Google Scholar]
- 147.Kats LM, et al. Proto-oncogenic role of mutant IDH2 in leukemia initiation and maintenance. Cell Stem Cell. 2014;14(3):329–41. doi: 10.1016/j.stem.2013.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Cheng X, Collins RE, Zhang X. Structural and sequence motifs of protein (histone) methylation enzymes. Annu Rev Biophys Biomol Struct. 2005;34:267–94. doi: 10.1146/annurev.biophys.34.040204.144452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Dillon SC, et al. The SET-domain protein superfamily: protein lysine methyltransferases. Genome Biol. 2005;6(8):227. doi: 10.1186/gb-2005-6-8-227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Min J, et al. Structure of the catalytic domain of human DOT1L, a non-SET domain nucleosomal histone methyltransferase. Cell. 2003;112(5):711–23. doi: 10.1016/s0092-8674(03)00114-4. [DOI] [PubMed] [Google Scholar]
- 151.Anglin JL, Song Y. A medicinal chemistry perspective for targeting histone H3 lysine-79 methyltransferase DOT1L. J Med Chem. 2013;56(22):8972–83. doi: 10.1021/jm4007752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Richon VM, et al. Chemogenetic analysis of human protein methyltransferases. Chem Biol Drug Des. 2011;78(2):199–210. doi: 10.1111/j.1747-0285.2011.01135.x. [DOI] [PubMed] [Google Scholar]
- 153.Daigle SR, et al. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell. 2011;20(1):53–65. doi: 10.1016/j.ccr.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Basavapathruni A, et al. Conformational adaptation drives potent, selective and durable inhibition of the human protein methyltransferase DOT1L. Chem Biol Drug Des. 2012;80(6):971–80. doi: 10.1111/cbdd.12050. [DOI] [PubMed] [Google Scholar]
- 155.Kuhn MW, et al. MLL partial tandem duplication leukemia cells are sensitive to small molecule DOT1L inhibition. Haematologica. 2015 doi: 10.3324/haematol.2014.115337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Daigle SR, et al. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood. 2013;122(6):1017–25. doi: 10.1182/blood-2013-04-497644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Yao Y, et al. Selective inhibitors of histone methyltransferase DOT1L: design, synthesis, and crystallographic studies. J Am Chem Soc. 2011;133(42):16746–9. doi: 10.1021/ja206312b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Anglin JL, et al. Synthesis and structure-activity relationship investigation of adenosine-containing inhibitors of histone methyltransferase DOT1L. J Med Chem. 2012;55(18):8066–74. doi: 10.1021/jm300917h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Deng L, et al. Synthesis, Activity and Metabolic Stability of Non-Ribose Containing Inhibitors of Histone Methyltransferase DOT1L. Medchemcomm. 2013;4(5):822–826. doi: 10.1039/C3MD00021D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Yu W, et al. Bromo-deaza-SAH: a potent and selective DOT1L inhibitor. Bioorg Med Chem. 2013;21(7):1787–94. doi: 10.1016/j.bmc.2013.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Yu W, et al. Catalytic site remodelling of the DOT1L methyltransferase by selective inhibitors. Nat Commun. 2012;3:1288. doi: 10.1038/ncomms2304. [DOI] [PubMed] [Google Scholar]
- 162.Basavapathruni A, et al. Nonclinical pharmacokinetics and metabolism of EPZ-5676, a novel DOT1L histone methyltransferase inhibitor. Biopharm Drug Dispos. 2014;35(4):237–52. doi: 10.1002/bdd.1889. [DOI] [PubMed] [Google Scholar]
- 163.Stein EM, et al. The DOT1L Inhibitor EPZ-5676: safety and activity in relapsed/refractory patients with MLL-rearranged leukemia. Blood. 2014;124(21):387. [Google Scholar]
- 164.Chen CW, et al. DOT1L inhibits SIRT1-mediated epigenetic silencing to maintain leukemic gene expression in MLL-rearranged leukemia. Nat Med. 2015;21(4):335–43. doi: 10.1038/nm.3832. [DOI] [PMC free article] [PubMed] [Google Scholar]