

Abstract
Metalloproteases (MPs) are a large and diverse class of enzymes implicated in numerous physiological and pathological processes, including tissue remodeling, peptide hormone processing, and cancer. MPs are tightly regulated by multiple posttranslational mechanisms in vivo, hindering their functional analysis by conventional genomic and proteomic methods. Here we describe a general strategy for creating activity-based proteomic probes for MPs by coupling a zinc-chelating hydroxamate to a benzophenone photocrosslinker, which promote selective binding and modification of MP active sites, respectively. These probes labeled active MPs but not their zymogen or inhibitor-bound counterparts and were used to identify members of this enzyme class up-regulated in invasive cancer cells and to evaluate the selectivity of MP inhibitors in whole proteomes. Interestingly, the matrix metalloproteinase inhibitor GM6001 (ilomastat), which is currently in clinical development, was found to also target the neprilysin, aminopeptidase, and dipeptidylpeptidase clans of MPs. These results demonstrate that MPs can display overlapping inhibitor sensitivities despite lacking sequence homology and stress the need to evaluate MP inhibitors broadly across this enzyme class to develop agents with suitable target selectivities in vivo. Activity-based profiling offers a powerful means for conducting such screens, as this approach can be carried out directly in whole proteomes, thereby facilitating the discovery of disease-associated MPs concurrently with inhibitors that selectively target these proteins.
Amajor goal of proteomics is to develop global methods for the analysis of protein function in samples of high biological complexity (1–3). In typical proteomic experiments, the expression levels of proteins in cells/tissues/fluids are compared by techniques, such as two-dimensional electrophoresis (4) or isotope-coded affinity tagging (5), in which variations in protein abundance are used to infer changes in protein activity. However, many proteins and, in particular, enzymes are regulated by a complex array of posttranslational mechanisms (6), meaning that alterations in their abundance may not correlate with changes in activity. To address this problem, a chemical strategy referred to as activity-based protein profiling (ABPP) has been introduced that utilizes active site-directed probes to record variations in the activity of enzymes in whole proteomes (7, 8).
ABPP probes typically possess three general elements: (i) a binding group that promotes interactions with the active sites of specific classes of enzymes, (ii) a reactive group that covalently labels these active sites, and (iii) a reporter group (e.g., fluorophore or biotin) for the visualization and affinity purification of probe-labeled enzymes. To date, ABPP probes have been developed for many biomedically relevant enzyme classes, including serine hydrolases (9–12), cysteine proteases (13–15), and oxidoreductases (16, 17). Notably, these probes have been shown to selectively label active enzymes but not their inactive precursor (e.g., zymogen) (10) or inhibitor-bound forms (10, 18, 19). Additionally, by tagging specific groups of enzymes based on functional properties rather than expression level alone, ABPP probes provide exceptional access to low abundance proteins in complex proteomes (12). Recently, advanced versions of ABPP have been introduced for profiling enzyme activities in living cells (20–22) and animals (20, 21). Nonetheless, despite these advantages of ABPP over conventional proteomic methods, several important enzyme classes remain unaddressed by this approach.
Of the enzyme families beyond the current scope of ABPP, the metalloproteases (MPs) stand out as an exceptionally important target class. First, MPs are an extremely large and diverse group of enzymes that play key roles in many physiological and pathological processes (23, 24), including tissue remodeling (25), peptide hormone signaling (26), and cancer (23, 25). Additionally, MPs are subject to numerous forms of posttranslational regulation in vivo (23), including production as inactive zymogens and inhibition by endogenous proteins [e.g., TIMP, tissue inhibitors of matrix metalloproteinases (MMPs)]. These posttranslational events hinder the functional analysis of MPs by conventional, abundance-based genomic and proteomic methods. Although zymography has been used for many years to evaluate MP activities (27), these gel-based assays are performed after the denaturation/renaturation of proteomes and therefore do not account for key protein–protein or protein–small molecule interactions that may regulate MP function in solution. Finally, several MP inhibitors have entered clinical trials but have failed because of toxicities of unknown molecular mechanism (28), highlighting the need for global methods to evaluate the selectivity of compounds that target this complex family of proteases.
In considering strategies for the activity-based profiling of MPs, one might initially look to the design of probes for other protease classes, such as serine (9–12) and cysteine proteases (13–15). However, in these cases, ABPP probes were designed to target conserved nucleophiles in protease active sites, an approach that cannot be directly applied to MPs, which use a zinc-activated water molecule (rather than a protein-bound nucleophile) for catalysis (29). As such, an alternative approach is required to generate chemical probes that label the active sites of MPs with sufficient potency and specificity to enable functional profiling of these enzymes in whole proteomes. Here, we describe a general strategy for the design of ABPP probes for MPs that incorporate a zinc-chelating hydroxamate and a benzophenone photocrosslinking group, which promote selective binding and modification of MP active sites, respectively. We apply these probes to profile the activity and inhibitor sensitivity of MPs in cell and tissue proteomes, resulting in the identification of MPs that are highly up-regulated in invasive cancer cells and the discovery of targets of MP inhibitors currently in clinical development.
Methods
Synthesis of a Rhodamine-Tagged Hydroxamate Benzophenone Probe (HxBP-Rh). Details on the synthesis and characterization of the HxBP-Rh and trifunctional HxBP probes are provided as Supporting Methods and Schemes 1 and 2, which are published as supporting information on the PNAS web site.
Analysis of the Inhibition of MMPs by HxBP-Rh. The substrate, DABCYL-Gaba-ProAsnGlyLeuGlu-EDANS, and purified MMPs (MMP-2, MMP-7, and MMP-9) were purchased from Calbiochem. The final concentrations in the assay buffer, buffer 1 (100 mM Tricine, pH 7.5/100 mM NaCl/10 mM CaCl2/50 μM ZnCl2/0.005% Brij 35) were 0.5 ng of MMP, 12.5 μM substrate, and 0–5,000 nM HxBP-Rh. Fluorescence measurements (excitation, 340 nm; emission, 465 nm) were performed by using a GENios fluorescence plate reader from Tecan (Maennedorf, Switzerland). Reactions were initiated by adding the substrate last to the mixture and measuring the fluorescence increase every 2 min for 1 h. IC50 values for HxBP-Rh were determined from dose–response curves of three independent trials by using prism software (GraphPad, San Diego).
Labeling and Detection of MPs by Using HxBP-Rh. Standard conditions for HxBP-labeling reactions were as follows. Purified MMP-2 was diluted in buffer 1 (30 ng of enzyme) and mixed with 100 nM HxBP-Rh in the presence or absence of 5 μM GM6001 or TIMP-1 (80 ng). These mixtures were preincubated on ice for 15 min before irradiation at 365 nm for 1 h (on ice) followed by quenching with 1 vol of standard 2× SDS/PAGE loading buffer (reducing). Kidney and cancer cell proteomes, prepared as described in refs. 10 and 12, were adjusted to 1 mg/ml in 50 mM Tris·HCl (pH 8.0) before labeling as described above. Where indicated, a portion of each cancer cell line proteome sample was treated with peptide N-glycosidase F (PNGaseF) (New England Biolabs) to provide deglycosylated proteomes by following the method described in ref. 12. Labeled samples were separated by SDS/PAGE and visualized in-gel with a Hitachi FMBio IIe flatbed scanner (MiraiBio, Alameda, CA). Integrated band intensities were calculated from two to three independent labeling reactions and averaged to provide the level of each enzyme activity in each sample.
Isolation and Molecular Characterization of HxBP-Rh-Labeled Proteins. Isolation of HxBP-labeled proteins was achieved by using a trifunctional HxBP probe (biotin- and rhodamine-coupled) and an avidin-based affinity purification procedure by following methods described in ref. 30. Avidin-enriched probe-labeled proteins were separated by SDS/PAGE, and protein bands were excised and digested with trypsin (Promega). The resulting peptide mixture was then analyzed by microcapillary liquid chromatography-electrospray tandem MS [1100 HPLC (Agilent, Palo Alto, CA) combined with a Finnigan LCQ Deca mass spectrometer (Thermo Finnigan, Woburn, MA)]. The MS data were used to search public databases to identify the HxBP-labeled proteins.
Measurement of IC50 Values for MP Inhibitors by Using HxBP-Rh. IC50 values for neprilysin, dipeptidylpeptidase III (DPPIII), and leucine aminopeptidase (LAP) were determined from dose–response curves of three trials at concentrations of GM6001 ranging from 0.001 to 20 μM and 50 nM HxBP-Rh probe by using prism software. Neprilysin, DPPIII, and LAP assays were conducted by using MUM-2B-membrane, MCF7-soluble, and mouse kidney-soluble proteomes, respectively.
Results
Design and Synthesis of ABPP Probes for MPs. To date, the development of ABPP probes for proteases has relied on electrophilic reactive groups to label the conserved active site nucleophiles of specific classes of these enzymes, such as the serine (9–12) and cysteine proteases (13–15), and subunits of the proteasome (31). In contrast, MPs do not use a protein-bound nucleophile but rather a zinc-activated water molecule for catalysis (29). As such, the design of electrophilic affinity-labeling reagents for MPs is not straightforward. Although select examples exist of electrophile-based covalent inhibitors of MPs (32), the potential conversion of these agents into ABPP probes is compromised by either weak potency (33) or restricted target selectivity (34).
As an alternative strategy for the design of ABPP probes for MPs, we considered the possibility of converting tight-binding reversible MP inhibitors into active site-directed affinity labels by incorporating a photocrosslinking group into these agents. Indeed, a similar approach has proven effective for creating covalent probes for specific enzymes, such as presenilin (35) and creatine kinase (36). To test this strategy for MPs, we synthesized a photoactivatable probe containing a hydroxamate group, which has been shown to chelate the conserved zinc atom in MP-active sites in a bidendate manner (Fig. 1A) and serve as the basis for high-affinity inhibitors of numerous members of this enzyme class (37). As molecular templates for this MP-directed probe, we selected GM6001 and marimastat, two broad-spectrum hydroxamate inhibitors of MMPs, an important subclass of MPs implicated in tissue remodeling and cancer (23, 25). Both of these inhibitors possess large hydrophobic groups in the P2 position (Table 1), suggesting that a photoreactive benzophenone could be incorporated at this site without significant effects on binding to MMPs. Accordingly, a candidate activity-based probe for MPs, HxBP-Rh (Table 1), was synthesized as shown in Fig. 1B.
Fig. 1.

Design and synthesis of an MP-directed activity-based probe, HxBP-Rh. (A) General interactions between a broad-spectrum reversible hydroxamate inhibitor (GM6001) and MMP active sites based on a combination of structure-activity (45) and crystallographic data (51). (B) Synthesis of HxBP-Rh for activity-based profiling of MPs. Hydroxamate, benzophenone, and rhodamine groups are shown in magenta, blue, and red, respectively. Details regarding the synthesis and characterization of HxBP-Rh are provided in the supporting information. HBTU, 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate; DIEA, diisopropylethylamine.
Table 1. Comparison of the IC50 values (in nM) of HxBP-Rh and known broad-spectrum MMP inhibitors marimastat and GM6001.
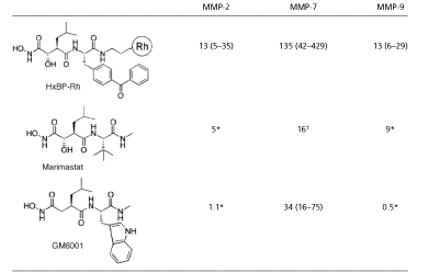 |
HxBP-Rh Selectively Labels Active, but Not Inactive (Zymogen or Inhibitor-Bound), MMPs. Under reversible binding conditions (i.e., in the absence of photocrosslinking), HxBP-Rh was found to inhibit several MMPs with potencies that were only slightly lower than those reported for the parent compounds GM6001 and marimastat (Table 1), indicating that the incorporation of the benzophenone and rhodamine groups into HxBP-Rh did not significantly impair binding to MMPs. HxBP-Rh was next tested for its ability to covalently label MMPs in an activity-based manner. Incubation of this agent (100 nM) with both active and inactive (zymogen and inhibitor-bound) variants of purified MMP-2 for 15 min, followed by photocrosslinking for 60 min, led to the selective labeling of active MMP-2 (Fig. 2A). In contrast, HxBP-Rh did not modify pro-MMP-2 or MMP-2 treated with a small molecule (GM6001) or protein (TIMP-1) inhibitor. Examination of the concentration dependence of the MMP-2–HxBP-Rh reaction revealed that maximal labeling was achieved with as little as a 100-nM probe (Fig. 2B). Collectively, these data demonstrate that HxBP-Rh acts as a high-affinity activity-based probe for MMP-2.
Fig. 2.

Activity-based labeling of purified MMP-2 by HxBP-Rh. (A) HxBP-Rh (100 nM) was incubated with purified samples of pro-MMP-2 (30 ng) or MMP-2 (30 ng) with or without inhibitors [GM6001 (5 μM) or TIMP-1 (80 ng)] for 15 min before photocrosslinking by exposure to UV light (hν). Samples were then analyzed by SDS/PAGE and in-gel fluorescence scanning. HxBP-Rh labeled active MMP-2 but not pro-MMP-2 or inhibitor-bound MMP-2. (B) Concentration dependence of MMP-2 labeling by HxBP-Rh. Labeling of MMP-2 was saturated at ≈100 nM HxBP-Rh. Labeling was measured by in-gel fluorescence scanning (integrated band intensities are given in arbitrary units). Each data point corresponds to the average of two independent trials.
HxBP-Rh Labels Several MMPs in an Activity-Based Manner in Whole Proteomes. We next examined whether HxBP-Rh could detect active MMPs in complex proteomes. Aliquots of 100 ng of purified pro-MMP-2, MMP-2, or MMP-2 plus GM6001/TIMP-1 were added to a mouse kidney proteome (15 μl of 1 μg/μl protein), followed by HxBP-Rh (100 nM), and the mixture was incubated for 15 min before photocrosslinking. Direct analysis of the reactions by in-gel fluorescence scanning revealed that HxBP-Rh labeled active MMP-2 but not pro-MMP-2 or inhibitor-bound MMP-2 (Fig. 3A). Next, the sensitivity of HxBP-Rh was examined by incubating this reagent with a serial dilution of active MMP-2 added to the mouse kidney proteome. These data revealed that HxBP-Rh could detect as few as 3 ng of active MMP-2 in the kidney proteome (Fig. 3B). This quantity of active MMP-2, which corresponded to ≈3 nM enzyme and 0.02% of the tissue proteome, approximates the sensitivity that other ABPP probes display for their respective enzyme classes (12, 21). Finally, HxBP-Rh also labeled in an activity-based manner other MMPs introduced into kidney homogenates, including MMP-7 and MMP-9 (Fig. 3C), indicating that this agent serves as an effective functional proteomic probe for several members of this enzyme family.
Fig. 3.

Activity-based labeling of MMPs in whole proteomes by HxBP-Rh. (A) Purified MMP-2 (100 ng) or pro-MMP-2 (100 ng) was added to a mouse kidney proteome (15 μl, 1 μg of protein per microliter) and treated with HxBP-Rh (100 nM) for 15 min before photocrosslinking and analysis by SDS/PAGE and in-gel fluorescence scanning. Only MMP-2 was labeled by HxBP-Rh, and this labeling was blocked by GM6001 (5 μM) and TIMP-1 (200 ng). No protein labeling was observed in the absence of exposure to UV light. Δ, heat-denatured proteome (containing 100 ng of MMP-2). Also highlighted in this profile is an endogenous GM6001-sensitive enzyme activity labeled by HxBP-Rh, which was identified by using a trifunctional HxBP probe as LAP (see Fig. 5B). (B) HxBP-Rh labeling of a serial dilution of purified MMP-2 added to a mouse kidney proteome. HxBP-Rh could detect as few as 3 ng of active MMP-2 (corresponding to 3 nM enzyme in a background of 15 μlof1 μg/μl proteome). HxBP-Rh did not label pro-MMP-2 (150 ng). (C) HxBP-Rh labeling of MMP-7 and MMP-9 in proteomes. MMP-7 and MMP-9 (30 ng) were added to the mouse kidney proteome (15 μl, 1 μg/μl), and the samples were treated with HxBP-Rh (100 nM) and analyzed as described above. HxBP-Rh labeled active, but not GM6001-inhibited, MMP-7 and MMP-9.
ABPP Identifies a Membrane-Associated MP That Is Highly Up-Regulated in Invasive Melanoma Cells and Potently Inhibited by GM6001. Several MPs have been proposed to support the development and progression of human cancer (23, 28); however, the specific members of this large enzyme class that contribute to tumorigenesis and metastasis remain mostly unknown (23). A persistent challenge in deciphering the function of MPs in cancer (as well as in other pathophysiological processes) has been the dearth of available techniques to measure changes in the activity of these enzymes in samples of high biological complexity. Having determined that HxBP-Rh can distinguish active from inactive MPs in whole proteomes (Fig. 3), we next applied this probe to functionally profile MP activities across a panel of human melanoma cell lines that differ in their invasive properties (12, 38). These studies identified an HxBP-Rh-labeled, membrane-associated glycoprotein that was dramatically up-regulated in invasive melanoma lines compared with their noninvasive counterparts (Fig. 4). By using a trifunctional HxBP probe (containing both biotin and rhodamine groups), this glycoprotein was affinity-purified and identified as neprilysin, an integral membrane MP that metabolizes several peptide hormones in vivo, including enkephalins and bombesin-like peptides (26). Interestingly, neprilysin shares no sequence homology with MMPs, thus demonstrating that HxBP-Rh can profile enzymes from different subgroups of the MP superfamily. Additionally, the labeling of neprilysin by HxBP-Rh was inhibited by the MMP-directed agent GM6001, which blocked this reaction with an IC50 value of 17 nM (Fig. 5A). These data indicate that GM6001 potently inhibits MPs outside of its intended target family (MMPs). Consistent with this premise, two additional GM6001-sensitive targets were identified in cell and tissue proteomes as LAP (Fig. 5B, IC50 = 111 nM) and DPPIII (Fig. 5C, IC50 = 76 nM), both of which represent MPs unrelated in sequence to MMPs, neprilysin, or each other. Collectively, these data demonstrate that HxBP-Rh can identify MP activities differentially expressed in human cancer cells and discover unanticipated targets of MP inhibitors directly in whole proteomes.
Fig. 4.

HxBP-Rh identifies neprilysin as an MP activity dramatically upregulated in invasive human melanoma cell lines. (A) HxBP-Rh labeling profiles of membrane proteomes from a panel of human melanoma cell lines. An HxBP-Rh-labeled glycoprotein highly up-regulated in invasive melanoma lines (MUM-2B and C-8161) compared with noninvasive melanoma lines (UACC-62, M14-Mel, and MUM-2C) was identified as neprilysin by using a trifunctional HxBP probe (see Methods for more details). Deglycoslyation was accomplished by treating a portion of each HxBP-Rh-labeled proteome with PNGaseF before analysis. (B) Quantitation of neprilysin activity in melanoma membrane proteomes by in-gel fluorescence scanning (n = 3 per group).
Fig. 5.

HxBP-Rh identifies several MPs outside the MMP family that are inhibited by GM6001, including neprilysin (A), LAP (B), and DPPIII (C). (Left) Shown is representative labeling of MPs in whole proteomes by HxBP-Rh (100 nM) and inhibition by GM6001 (5 μM). Note that PNGaseF lanes are not shown for LAP and DPPIII because treatment with this glycosidase did not alter the migration of these MPs by SDS/PAGE. Neprilysin was identified in the secreted proteome of invasive human melanoma cell lines (see Fig. 4), whereas LAP and DPPIII were identified in soluble proteomes from mouse kidney (see Fig. 3A) and the human breast cancer cell line MCF7, respectively (for a full profile of HxBP-Rh labeling of the MCF7 soluble proteome, see Fig. 6, which is published as supporting information on the PNAS web site). (Right) Shown is the concentration-dependence of inhibition of HxBP-Rh labeling by GM6001 (each data point corresponds to the average of three independent trials and is presented as a percentage of control reactions conducted without GM6001). From these curves, IC50 values of 17 nM (12–23 nM, 95% confidence limits), 111 nM (83–149 nM, 95% confidence limits), and 76 nM (53–109 nM, 95% confidence limits), were calculated for the inhibition of neprilysin, LAP, and DPPIII, respectively, by GM6001.
Discussion
In these studies, we have described a general strategy for the design and application of ABPP probes for the proteomic analysis of MPs. Key to the success of this approach was the incorporation of hydroxamate and benzophenone groups into the chemical probes, which promote selective binding and UV-induced labeling of MP active sites, respectively. A prototype agent HxBP-Rh was found to label several active MPs in cell/tissue proteomes but not their zymogen or inhibitor-bound forms. Both protein (e.g., TIMP-1) and small-molecule inhibitors of MPs were found to block probe labeling, indicating that HxBP-Rh can distinguish active members of this enzyme class from those that are bound by either endogenously produced or exogenously administered inhibitors. Notably, HxBP-Rh was able to detect active MMP-2 at concentrations as low as 3 nM in tissue proteomes, which is a sensitivity limit on par with ABPP probes that target other enzyme families (12, 21) and compatible with profiling low-abundance proteins in biological samples of high complexity. In this regard, HxBP-Rh detected MMPs with much greater sensitivity (>100-fold) than a previously described method for analyzing these enzymes based on binding to an immobilized reversible inhibitor resin and detection by using standard protein-staining techniques (39). Collectively, these data demonstrate that HxBP-Rh acts as a bona fide activity-based probe for several members of the MP superfamily and offers exceptional sensitivity for profiling these enzymes at low levels directly in complex proteomes.
By using HxBP-Rh, we identified neprilysin as an MP activity dramatically up-regulated in invasive human melanoma cell lines. Neprilysin (or neutral endopeptidase) is a metallopeptidase that degrades several mitogenic peptides (26) and has historically been considered a negative regulator of tumorigenesis (40, 41). Interestingly, however, this enzyme is also highly expressed in several advanced tumor types (42, 43), including melanoma (44), suggesting that it may, in some cases, contribute to the progression of cancer. Our finding that neprilysin activity is selectively up-regulated in invasive melanoma cells is consistent with this latter possibility and suggests that further functional studies are warranted to test the role that this enzyme may play in tumorigenesis. Toward this end, inhibitors of neprilysin would be of value, and it is therefore significant that this enzyme was found by ABPP to represent a high-affinity target of GM6001, a compound originally designed as a broad-spectrum inhibitor of MMPs (45). To our knowledge, neither neprilysin nor, for that matter, LAP or DPPIII has been previously identified as a target for GM6001, and the sensitivity that these enzymes show to other MMP inhibitors has only rarely been investigated (46). Our findings suggest that these MPs may be relevant sites for “off-target” activity of MMP-directed inhibitors in vivo. Given that several MMP inhibitors have failed in clinical trials, at least in part because of dose-limiting toxicity (musculoskeletal pain and inflammation) (28), it is intriguing to speculate that some of these side effects may be due to the inhibition of MPs outside the MMP family. This possibility is especially relevant to consider for GM6001, which is currently in preclinical and clinical development (listed as ilomastat) for the treatment of tobacco-related lung damage and mustard gas exposure, respectively. More generally, these data highlight that enzymes from the MP family may share considerable overlap in inhibitor sensitivity despite lacking any discernible sequence homology and therefore stress the need to evaluate MP inhibitors broadly across this enzyme class to develop agents with suitable target selectivities in vivo. The ABPP methods described herein offer a powerful means for conducting such screens, because these functional assays can be carried out directly in whole proteomes without the need to recombinantly express or purify MPs before analysis.
The ABPP approach for targeting MPs also has a few shortcomings that should be mentioned. First, it is likely that certain MP activities exist in cell/tissue proteomes at levels below the current sensitivity of in-gel fluorescence scanning and therefore go undetected by the methods described herein. However, future efforts to incorporate MP-directed probes into a recently described liquid chromatography-MS platform for ABPP (47) should address this limitation, given that this gel-free approach exhibits significantly improved sensitivity and resolution. Second, although the HxBP-Rh probe targeted a wide range of MPs, labeling members from at least three distinct subfamilies, it is unlikely that this agent (or, for that matter, any single probe) will show sufficient broad-spectrum affinity to bind and label all members of the MP class. Thus, to maximize the coverage that ABPP displays for MPs, additional probes will be needed. With this objective in mind, it is intriguing to note that nearly all zinc-dependent hydrolases, including proteases and histone deacetylases (48), are potently inhibited by hydroxamates; therefore, this class-selective binding group should serve as a useful scaffold for the design of activity-based probes that target many members of this enzyme superfamily. More specifically, we speculate that libraries of HxBP agents in which the position of the benzophenone group and the identity of the substituents on the α and β carbons are varied may serve as a rich source of novel ABPP probes for MPs. These probes can then be used, either individually or collectively, to profile the function of MPs in whole proteomes, thereby facilitating the identification of disease-associated members of this enzyme family concurrently with the discovery of inhibitors that selectively target these proteins.
Supplementary Material
Acknowledgments
We thank A. E. Speers for supplying the Rh-N3 reagent, S. Niessen for assistance with MS analysis of proteins, and E. J. Sorensen and the Cravatt laboratory for helpful discussions. This work was supported by National Cancer Institute Grant CA87660, the California Breast Cancer Research Program (A.J.), a Merck Fellowship of the Life Sciences Research Foundation (A.S.), Activx Biosciences, and The Skaggs Institute for Chemical Biology.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: ABPP, activity-based protein profiling; LAP, leucine aminopeptidase; MP, metalloprotease; MMP, matrix metalloproteinase; HxBP-Rh, rhodamine-tagged hydroxamate benzophenone probe; TIMP, tissue inhibitors of MMP; PNGaseF, N-glycosidase F; DPPIII, dipeptidylpeptidase III.
References
- 1.Patterson, S. D. & Aebersold, R. (2003) Nat. Genet. 33, 311-323. [DOI] [PubMed] [Google Scholar]
- 2.Adam, G. C., Sorensen, E. J. & Cravatt, B. F. (2002) Mol. Cell. Proteomics 1, 781-790. [DOI] [PubMed] [Google Scholar]
- 3.Michnick, S. W. (2004) Drug Discovery Today 9, 262-267. [DOI] [PubMed] [Google Scholar]
- 4.Patton, W. F., Schulenberg, B. & Steinberg, T. H. (2002) Curr. Opin. Biotechnol. 13, 321-328. [DOI] [PubMed] [Google Scholar]
- 5.Gygi, S. P., Rist, B., Gerber, S. A., Turecek, F., Gelb, M. H. & Aebersold, R. (1999) Nat. Biotechnol. 17, 994-999. [DOI] [PubMed] [Google Scholar]
- 6.Kobe, B. & Kemp, B. E. (1999) Nature 402, 373-376. [DOI] [PubMed] [Google Scholar]
- 7.Jessani, N. & Cravatt, B. F. (2004) Curr. Opin. Chem. Biol. 8, 54-59. [DOI] [PubMed] [Google Scholar]
- 8.Speers, A. E. & Cravatt, B. F. (2004) ChemBioChem 5, 41-47. [DOI] [PubMed] [Google Scholar]
- 9.Liu, Y., Patricelli, M. P. & Cravatt, B. F. (1999) Proc. Natl. Acad. Sci. USA 96, 14694-14699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kidd, D., Liu, Y. & Cravatt, B. F. (2001) Biochemistry 40, 6107-6115. [DOI] [PubMed] [Google Scholar]
- 11.Patricelli, M. P., Giang, D. K., Stamp, L. M. & Burbaum, J. J. (2001) Proteomics 1, 1067-1071. [DOI] [PubMed] [Google Scholar]
- 12.Jessani, N., Liu, Y., Humphrey, M. & Cravatt, B. F. (2002) Proc. Natl. Acad. Sci. USA 99, 10335-10340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Greenbaum, D., Medzihradszky, K. F., Burlingame, A. & Bogyo, M. (2000) Chem. Biol. 7, 569-581. [DOI] [PubMed] [Google Scholar]
- 14.Borodovsky, A., Ovaa, H., Kolli, N., Gan-Erdene, T., Wilkinson, K. D., Ploegh, H. L. & Kessler, B. M. (2002) Chem. Biol. 9, 1149-1159. [DOI] [PubMed] [Google Scholar]
- 15.Faleiro, L., Kobayashi, R., Fearnhead, H. & Lazebnik, Y. (1997) EMBO J. 16, 2271-2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adam, G. C., Cravatt, B. F. & Sorensen, E. J. (2001) Chem. Biol. 8, 81-95. [DOI] [PubMed] [Google Scholar]
- 17.Adam, G. C., Cravatt, B. F. & Sorensen, E. J. (2002) Nat. Biotechnol. 20, 805-809. [DOI] [PubMed] [Google Scholar]
- 18.Leung, D., Hardouin, C., Boger, D. L. & Cravatt, B. F. (2003) Nat. Biotechnol. 21, 687-691. [DOI] [PubMed] [Google Scholar]
- 19.Greenbaum, D., Baruch, A., Hayrapetian, L., Darula, Z., Burlingame, A., Medzihradszky, K. F. & Bogyo, M. (2002) Mol. Cell. Proteomics 1, 60-68. [DOI] [PubMed] [Google Scholar]
- 20.Speers, A. E., Adam, G. C. & Cravatt, B. F. (2003) J. Am. Chem. Soc. 125, 4686-4687. [DOI] [PubMed] [Google Scholar]
- 21.Speers, A. E. & Cravatt, B. F. (2004) Chem. Biol. 11, 535-546. [DOI] [PubMed] [Google Scholar]
- 22.Ovaa, H., van Swieten, P. F., Kessler, B. M., Leeuwenburgh, M. A., Fiebiger, E., van den Nieuwendijk, A. M., Galardy, P. J., van der Marel, G. A., Ploegh, H. L. & Overkleeft, H. S. (2003) Angew. Chem. Int. Ed. Engl. 42, 3626-3629. [DOI] [PubMed] [Google Scholar]
- 23.Overall, C. M. & Lopez-Otin, C. (2002) Nat. Rev. Cancer 2, 657-672. [DOI] [PubMed] [Google Scholar]
- 24.Peunte, X. S., Sanchez, L. M., Overall, C. M. & Lopez-Otin, C. (2003) Nat. Rev. Genet. 4, 544-558. [DOI] [PubMed] [Google Scholar]
- 25.Chang, C. & Werb, Z. (2001) Trends Cell Biol. 11, S37-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Turner, A. J., Isaac, R. E. & Coates, D. (2001) BioEssays 23, 261-269. [DOI] [PubMed] [Google Scholar]
- 27.Troeberg, L. & Nagase, H. (2003) Methods Mol. Biol. 225, 77-87. [DOI] [PubMed] [Google Scholar]
- 28.Coussens, L. M., Fingleton, B. & Matrisian, L. M. (2002) Science 295, 2387-2392. [DOI] [PubMed] [Google Scholar]
- 29.Coleman, J. E. (1998) Curr. Opin. Chem. Biol. 2, 222-234. [DOI] [PubMed] [Google Scholar]
- 30.Adam, G. C., Sorensen, E. J. & Cravatt, B. F. (2002) Mol. Cell. Proteomics 1, 828-835. [DOI] [PubMed] [Google Scholar]
- 31.Bogyo, M. & Wang, E. W. (2002) Curr. Top. Microbiol. Immunol. 268, 184-208. [DOI] [PubMed] [Google Scholar]
- 32.Kim, D. H. & Mobashery, S. (2001) Curr. Med. Chem. 8, 959-965. [DOI] [PubMed] [Google Scholar]
- 33.Rasnick, D. & Powers, J. C. (1978) Biochemistry 17, 4363-4369. [DOI] [PubMed] [Google Scholar]
- 34.Brown, S., Bernardo, M. M., Li, Z.-H., Kotra, L. P., Tanaka, Y., Fridman, R. & Mobashery, S. (2000) J. Am. Chem. Soc. 122, 3410-3411. [Google Scholar]
- 35.Li, Y. M., Xu, M., Lai, M. T., Huang, Q., Castro, J. L., DiMuzio-Mower, J., Harrison, T., Lellis, C., Nadin, A., Neduvelil, J. G., et al. (2000) Nature 405, 689-694. [DOI] [PubMed] [Google Scholar]
- 36.Hagenstein, M. C., Mussgnug, J. H., Lotte, K., Plessow, R., Brockhinke, A., Kruse, O. & Sewald, N. (2003) Angew. Chem. Int. Ed. Engl. 42, 5635-5638. [DOI] [PubMed] [Google Scholar]
- 37.Whittaker, M., Floyd, C. D., Brown, P. & Gearing, A. J. H. (1999) Chem. Rev. 99, 2735-2776. [DOI] [PubMed] [Google Scholar]
- 38.Seftor, E. A., Meltzer, P. S., Kirschmann, D. A., Pe'er, J., Maniotis, A. J., Trent, J. M., Folberg, R. & Hendrix, M. J. (2002) Clin. Exp. Metastasis 19, 233-246. [DOI] [PubMed] [Google Scholar]
- 39.Freije, J. R. & Bischoff, R. (2003) J. Chromatogr. 1009, 3541-3544. [DOI] [PubMed] [Google Scholar]
- 40.Papandreou, C. N., Usmani, B., Geng, Y., Bogenrieder, T., Freeman, R., Wilk, S., FInstad, C. L., Reuter, V. E., Powell, C. T., Scheinberg, D., et al. (1998) Nat. Med. 4, 50-57. [DOI] [PubMed] [Google Scholar]
- 41.Shipp, M. A., Tarr, G. E., Chen, C.-Y., Switzer, S. N., Hersh, L. B., Stein, H., Sunday, M. E. & Reinherz, E. L. (1991) Proc. Natl. Acad. Sci. USA 88, 10662-10666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yao, T., Takata, M., Tustsumi, S., Nishiyama, K., Taguchi, K., Nagai, E. & Tsuneyoshi, M. (2002) Pathology 34, 556-506. [DOI] [PubMed] [Google Scholar]
- 43.Mikami, Y., Hata, S., Kiyokama, T. & Manabe, T. (2002) Mod. Pathol. 15, 923-930. [DOI] [PubMed] [Google Scholar]
- 44.Kanitakis, J., Narvaez, D. & Claudy, A. (2002) Melanoma Res. 12, 241-244. [DOI] [PubMed] [Google Scholar]
- 45.Levy, D. E., Lapierre, F., Liang, W., Ye, W., Lange, C. W., Li, X., Grobelny, D., Casabonne, M., Tyrrell, D., Holme, K., et al. (1998) J. Med. Chem. 41, 199-223. [DOI] [PubMed] [Google Scholar]
- 46.Marcotte, P. A., Elmore, I. N., Guan, Z., Magoc, T. J., Albert, D. H., Morgand, D. W., Curtin, M. L., Garland, R. B., Guo, Y., Heyman, H. R., et al. (1999) J. Enzyme Inhib. 14, 425-435. [DOI] [PubMed] [Google Scholar]
- 47.Adam, G. C., Burbaum, J. J., Kozarich, J. W., Patricelli, M. P. & Cravatt, B. F. (2004) J. Am. Chem. Soc. 126, 1363-1368. [DOI] [PubMed] [Google Scholar]
- 48.Marks, P. A., Richon, V. M., Breslow, R. & Rifkind, R. A. (2001) Curr. Opin. Oncol. 13, 477-483. [DOI] [PubMed] [Google Scholar]
- 49.Nelson, A. R., Fingleton, B., Rothenberg, M. L. & Matrisian, L. M. (2000) J. Clin. Oncol. 18, 1135-1149. [DOI] [PubMed] [Google Scholar]
- 50.Dhanaraj, V., Ye, Q.-Z., Johnson, L. L., Hupe, D. J., Ortwine, D. F., Dunbar, J. B., Jr., Rubin, J. R., Pavlovsky, A., Humblet, C. & Blundell, T. L. (1996) Structure 4, 375-386. [DOI] [PubMed] [Google Scholar]
- 51.Yamamoto, M., Tsujishita, H., Hori, N., Ohishi, Y., Inoue, S., Ikeda, S. & Okada, Y. (1998) J. Med. Chem. 41, 1209-1217. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.