

Abstract
The hairpin ribozyme is a minimalist paradigm for studying RNA folding and function. In this enzyme, two domains dock by induced fit to form a catalytic core that mediates a specific backbone cleavage reaction. Here, we have fully dissected its reversible reaction pathway, which comprises two structural transitions (docking/undocking) and a chemistry step (cleavage/ligation), by applying a combination of single-molecule fluorescence resonance energy transfer (FRET) assays, ensemble cleavage assays, and kinetic simulations. This has allowed us to quantify the effects that modifications of essential functional groups remote from the site of catalysis have on the individual rate constants. We find that all ribozyme variants show similar fractionations into effectively noninterchanging molecule subpopulations of distinct undocking rate constants. This leads to heterogeneous cleavage activity as commonly observed for RNA enzymes. A modification at the domain junction additionally leads to heterogeneous docking. Surprisingly, most modifications not only affect docking/undocking but also significantly impact the internal chemistry rate constants over a substantial distance from the site of catalysis. We propose that a network of coupled molecular motions connects distant parts of the RNA with its reaction site, which suggests a previously undescribed analogy between RNA and protein enzymes. Our findings also have broad implications for applications such as the action of drugs and ligands distal to the active site or the engineering of allostery into RNA.
RNA enzymes (ribozymes) have been recognized as ideal model systems for studying the relationship of structure and function in RNA, because their catalytic activity directly reports the extent of native structure formation (1–3). This provides the basis for powerful modification-interference experiments in which the activity of site-specifically modified ribozymes is compared to the unmodified WT to map functionally important residues of the catalytic core. Such chemical modifications, however, may impact ribozyme function through reaction chemistry, structure formation, or both. Distinguishing these mechanisms has long been an experimental challenge. A typical example is the hairpin ribozyme, derived from the self-cleaving 359-nt negative strand of the tobacco ringspot virus satellite RNA, a member of a family of plant pathogens (4–6). A wealth of modification-interference experiments has helped to define the residues important for function of the minimal two-way junction form of this catalytic RNA, designed as the sequence with highest enzymatic activity in external substrate cleavage (4, 5, 7, 8). Many functional groups of the 24 non-Watson–Crick base-paired nucleotides in the two internal loops of domains A and B were shown to be essential for catalytic activity (Fig. 1A). However, recent crystallographic and biochemical experiments have suggested that only two nucleobases in the ribozyme, G8 and A38, are directly involved in reaction chemistry (Fig. 1 A) (9–12). An open question is therefore how all of the other functional groups exert their influence on activity.
Fig. 1.

Dissecting the impact of single functional group modifications on hairpin ribozyme catalysis. (A) Secondary and tertiary structure of the docked WT ribozyme used in this study (9). (Left) Watson–Crick and noncanonical base pairs are indicated as solid and dashed lines, respectively. Orange, substrate; cyan arrow, cleavage site; color-coded nucleotides and junction connection were modified in this study (purple, dC12; red, dA38; green, C39S3; blue, RzAS3). (Right) 3D ribbon-and-stick representation with same color coding (www.pymol.org). Yellow arrows, modified functional groups distant from the (cyan) cleavage site; pink dashed tubes, modified hydrogen bonds. (B) Schematic of the reaction pathway. Gray boxes, observed effects of the color-coded functional group modifications on individual rate constants relative to those of the WT; black box, effects on the overall cleavage rate constant.
Modification-interference analyses in protein enzymes have recently shown that essential functional groups remote from the catalytic site often have an impact on catalysis, presumably exerted through networks of coupled molecular motions; this has led to a more holistic view of protein structural dynamics and function (13). To test such a model for RNA, techniques that measure the rate constants of all steps on the kinetic reaction pathway are required. In previous work, we have established bulk and single-molecule fluorescence resonance energy transfer (FRET) assays as tools for obtaining such information on the hairpin ribozyme (2, 14–16). In particular, we have dissected the WT reaction pathway into five distinct steps (Fig. 1 A): (i) The substrate binds to the ribozyme, forming the secondary structure of domain A in an initially extended (undocked) and inactive ribozyme–substrate complex. (ii) Domains A and B dock by induced fit, forming the tertiary structure of a compact, catalytically active conformer (9). (iii) The ribozyme cleaves the substrate. (iv) The ribozyme–product complex undocks. (v) The undocked product complex releases the 5′ and 3′ products. Both substrate binding and product dissociation are thermodynamically highly favorable, making these steps essentially irreversible under standard conditions, in contrast to the readily reversible docking and cleavage steps (Fig. 1B). In addition, our single-molecule FRET studies have revealed four subpopulations of molecules with distinct undocking kinetics and a memory effect, explaining the previously observed heterogeneous overall cleavage kinetics (15).
Here, we combine single-molecule FRET with functional assays and kinetic simulations to investigate how a set of representative essential functional groups remote from the cleavage site specifically impact the docking, undocking, and chemistry rate constants. Surprisingly, most of these functional group modifications significantly affect not only the docking and undocking rate constants but also the rate constants of catalytic chemistry over a substantial distance from the cleavage site. This leads us to propose that extensive networks of coupled molecular motions connect distant parts of the RNA with its active site, as is the case for some protein enzymes. This has important implications for an expanded role of the overall fold of RNA in its function and for applications such as the design of allosteric ribozymes and of drugs and ligands binding distal to RNA active sites.
Materials and Methods
RNA Preparation. Synthetic RNA oligonucleotides (sequences given in Fig. 1 A) were purchased with 2′-protection groups from the Howard Hughes Medical Institute Biopolymer/Keck Foundation Biotechnology Resource Laboratory (Yale University, New Haven, CT) and were deprotected as suggested by the manufacturer (http://info.med.yale.edu/wmkeck) (17). Deprotected RNA was purified by denaturing 20% polyacrylamide gel electrophoresis and C8-reverse-phase HPLC chromatography, as described (17). Cy5 was attached to the RNA during synthesis, whereas Cy3 was incorporated postsynthetically as described (14, 15, 17).RNA concentrations were calculated from their absorption at 260 nm.
Single-Molecule FRET. The RzA and RzB strands (Fig. 1 A) were annealed at each 500 nM in a buffer containing 50 mM Tris·HCl, pH 7.5; 100 mM NaCl; 1 mM EDTA; and 1% 2-mercaptoethanol. The annealing solution was heated to 80°C for 45 sec before cooling at room temperature over ≈5 min. The annealed biotinylated and FRET-labeled ribozyme was diluted to ≈50–100 pM and bound to a streptavidin-coated quartz slide surface via the biotin–streptavidin interaction to generate a surface density of ≈0.1 molecules per μm2. Then either 100 nM noncleavable substrate analog [with a 2′-O-methyl modification at the cleavage site to suppress catalysis without altering the docking behavior (14)] or each 5 μM nonligatable 5′ product analog with a 3′ phosphate and 3′ product were added to the ribozyme. The docking and undocking kinetics did not change when the authentic 5′ product with 2′,3′-cyclic phosphate was used instead of the analog (data not shown). The donor (ID) and acceptor (IA) fluorescence signals of optically resolved single molecules (characterized by single-step photobleaching) were detected on a total internal reflection fluorescence microscope as described (15). The FRET ratio [defined as IA/(IA + ID)] was followed in real time for each individual molecule. Measurements were performed under standard buffer conditions (50 mM Tris·HCl, pH 7.5/12 mM MgCl2) at 25°C, with an oxygen scavenging system consisting of 10% (wt/vol) glucose, 2% (vol/vol) 2-mercaptoethanol, 750 μg/ml glucose oxidase, and 90 μg/ml catalase to reduce photobleaching. The dwell times of each docked and undocked event were calculated, histograms constructed, and the rate constants for docking and undocking determined as detailed in Fig. 4 and Supporting Text, which are published as supporting information on the PNAS web site.
Cleavage and Ligation Assays. All cleavage reactions were conducted under single turnover (presteady state) conditions in standard buffer (50 mM Tris·HCl, pH 7.5/12 mM Mg2+)at25°C, essentially as described (18). Details can be found in Supporting Text. Error bars in Fig. 3 are calculated from at least two independent cleavage assays. Time traces of product formation were fit to the double-exponential first-order rate equation y(t) = y0 + Af (1 - e(-kcleav,obs,ft)) + As (1 - e(-kcleav,obs,st)), using Marquardt–Levenberg nonlinear least-squares regression, where Af + As is the final extent of cleavage and the two kcleav,obs are the first-order rate constants of the fast and slow phases (Table 2).
Fig. 3.

Experimental and simulated cleavage time courses of WT and variant ribozymes. Analytical kinetic simulations (black lines) are least-squares fits to the experimental data (open circles). The derived chemistry rate constants for cleavage and ligation vary significantly with the modification. The subpopulation contributions are also included (I, light gray dashed line; II, gray dash-dotted line; III, dark gray short-dashed line; IV, black dashed line), except for the many minor subpopulations of the RzAS3-containing variants.
Table 2. Bulk docking and overall cleavage rate constants of modified hairpin ribozymes compared to WT.
| Variant | kdock,obs (s-1) | kcleav,obs,f (s-1) [Af] | kcleav,obs,s (s-1) [As] | klig/kcleav |
|---|---|---|---|---|
| WT | 0.014 ± 0.002 | 0.0017 ± 0.0002 [0.83] | 0.0002 ± 0.0001 [0.16] | 2.5 ± 0.1 |
| dC12 | 0.033 ± 0.012 | 0.0020 ± 0.0002 [0.78] | 0.0003 ± 0.0001 [0.21] | 0.32 ± 0.03 |
| dA38 | n.d. | 0.0029 ± 0.0002 [0.68] | 0.0004 ± 0.0001 [0.27] | 0.15 ± 0.01 |
| C39S3 | 0.056 ± 0.017 | 0.0048 ± 0.0002 [0.71] | 0.0005 ± 0.0002 [0.26] | 1.25 ± 0.04 |
| RzAS3 | 0.102 ± 0.034 | 0.0026 ± 0.0002 [0.70] | 0.0005 ± 0.0002 [0.24] | 3.4 ± 0.3 |
| RzAS3/C39S3 | 0.114 ± 0.088 | 0.0034 ± 0.00004 [0.80] | 0.0007 ± 0.0003 [0.18] | 2.65 ± 0.03 |
Docking rate constants were observed by steady-state FRET in bulk solution under standard conditions (50 mM Tris·HCl, pH 7.5/12 mM Mg2+/25°C/100 nM strand RzA, 400 nM strand RzB). Cleavage rate constants were derived from single-turnover reactions as described in Materials and Methods. The internal equilibrium for the chemical step (klig/kcleav) was determined by ligating radioactive 2′,3′-cyclic phosphorylated 5′ product to completion, as described in Materials and Methods and Supporting Text and Fig. 5.
Ligation assays were performed as detailed in Supporting Text and Fig. 5, which is published as supporting information on the PNAS web site, yielding the equilibrium fraction ligated, flig. The internal equilibrium of the chemical step was determined as 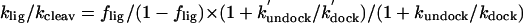 (Table 2). Forward cleavage assays yielded very similar final product/substrate distributions, showing that the internal overall equilibrium is indeed reached in both the cleavage and ligation experiments.
(Table 2). Forward cleavage assays yielded very similar final product/substrate distributions, showing that the internal overall equilibrium is indeed reached in both the cleavage and ligation experiments.
Steady-State FRET Kinetic Assays. Steady-state FRET measurements were performed as described in Supporting Text. The resulting time traces were fit to a single exponential increase function of the form y(t) = y0 + A(1 - e(-kdock,obst)), where A and kdock,obs are the extent and rate constant of the FRET increase, respectively (Table 2).
Kinetic Simulations of Steady-State FRET Assays. The (un)docking rate constants of each hairpin ribozyme variant, kdock,i and kundock,i, and their subpopulation fractions, fi, measured by single-molecule FRET, were used to generate a kinetic simulation that was compared to an experimental steady-state FRET time course (Fig. 6, which is published as supporting information on the PNAS web site, and Supporting Text).
Kinetic Simulations of Cleavage Reactions. Assuming that substrate binding and product release are irreversible (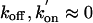 ), and that kcleav and klig are shared among all noninterchanging subpopulations of a given ribozyme variant, the cleavage reaction pathway of the hairpin ribozyme (Fig. 1B) can be approximated by four sequential unimolecular reactions, three of which are reversible, one (product dissociation) irreversible, as indicated in Fig. 1B. The analytical solution for the resulting set of coupled rate equations was derived by matrix algebra (using master equations) as described in detail in Supporting Text (19). The ratio r = klig/kcleav was experimentally determined as the internal chemistry equilibrium (see above) and kept constant throughout the simulation. The cleavage and ligation rate constants were then obtained by a single-variable fit of the simulated cleavage time courses to the experimental ones using the experimentally measured docking/undocking rate constants, the subpopulation fractions, and the internal equilibrium constant r. It should be noted that the high reaction extent in both cleavage and ligation reactions suggests that most, if not all, of the noninterchanging subpopulations are active, although they may not necessarily share the same kcleav and klig. Our analysis therefore yields values for kcleav and klig that are not necessarily specific for any of the molecular subpopulations but are averaged over all active subpopulations of a given ribozyme variant (see also Supporting Text).
), and that kcleav and klig are shared among all noninterchanging subpopulations of a given ribozyme variant, the cleavage reaction pathway of the hairpin ribozyme (Fig. 1B) can be approximated by four sequential unimolecular reactions, three of which are reversible, one (product dissociation) irreversible, as indicated in Fig. 1B. The analytical solution for the resulting set of coupled rate equations was derived by matrix algebra (using master equations) as described in detail in Supporting Text (19). The ratio r = klig/kcleav was experimentally determined as the internal chemistry equilibrium (see above) and kept constant throughout the simulation. The cleavage and ligation rate constants were then obtained by a single-variable fit of the simulated cleavage time courses to the experimental ones using the experimentally measured docking/undocking rate constants, the subpopulation fractions, and the internal equilibrium constant r. It should be noted that the high reaction extent in both cleavage and ligation reactions suggests that most, if not all, of the noninterchanging subpopulations are active, although they may not necessarily share the same kcleav and klig. Our analysis therefore yields values for kcleav and klig that are not necessarily specific for any of the molecular subpopulations but are averaged over all active subpopulations of a given ribozyme variant (see also Supporting Text).
Results
Choice of Hairpin Ribozyme Modifications. To make the acquisition of statistically reliable single-molecule data feasible, we prescreened nine specific functional group modifications, particularly ones that were previously found to exhibit accelerated cleavage (4, 5, 7, 8). Among these, four modifications, dC12, dA38, C39S3, and RzAS3, exemplified the following particular effects on docking and undocking and were chosen for detailed analysis by single-molecule FRET [two of these are standard 2′-deoxy modifications (dC12, dA38), whereas the other two (C39S3, RzAS3) are propyl-spacer modifications (Fig. 7, which is published as supporting information on the PNAS web site)]:
dC12, or 2′-deoxy-C12, specifically affects the U42-binding pocket of the docked structure. U42 is bulged out from domain B to be captured between loops A and B, establishing a hydrogen-bonding network that connects the 2′ hydroxyl of C12 (U12 in the crystal structure) with A22 and A23 (Fig. 1 A) (9). Our single-molecule FRET data reveal that the dC12 modification only slightly decelerates docking (by 43%) but significantly accelerates undocking (5.8-fold, Table 1 and Fig. 1B).
dA38, or 2′-deoxy-A38, specifically affects the vicinity of the g+1-binding pocket of the docked structure, in which g+1 of domain A forms a base pair with C25 of domain B. The 2′ hydroxyl of A38 forms a hydrogen bond to the nonbridging pro-RP-oxygen of the downstream phosphate so that the base is held in place to stack on g+1, forming the roof of the g+1-binding pocket (Fig. 1 A) (9). The dA38 modification only modestly accelerates docking (2.3-fold) but drastically accelerates undocking (65-fold; Table 1 and Fig. 1B).
C39S3 replaces the nucleotide in position 39 (Fig. 1 A), which is involved in the substantial conformational rearrangement of loop B upon docking (9), with a propyl linker (S3-spacer) that lacks the base but has a three-carbon spacing similar to that of the WT ribose. The C39S3 modification results in a moderate rate increase (3.8-fold) in docking and a more significant one (8.5-fold) in undocking (Table 1 and Fig. 1B).
RzAS3 affects the domain junction by inserting a propyl linker (S3-spacer) between A14 and A15, at the flexible hinge connecting domains A and B (Fig. 1 A). This modification leads to significantly (35-fold) faster docking than the WT, whereas undocking is accelerated only by 1.5-fold (Table 1 and Fig. 1B).
Table 1. Single-molecule docking and undocking rate constants of modified hairpin ribozyme–substrate and –product analog complexes compared to WT.
| Variant (number of trajectories) | Subpopulation | Fraction | kdock (s-1) | kundock (s-1) | k′undock (s-1) | k′dock (s-1) |
|---|---|---|---|---|---|---|
| WT (454) | I | 0.62 | 0.0086 ± 0.0010 | 0.004 ± 0.001 | 0.011 ± 0.001 | 0.020 ± 0.001 |
| II | 0.08 | 0.079 ± 0.020 | 0.076 ± 0.002 | |||
| III | 0.13 | 0.59 ± 0.10 | 0.4 ± 0.2 | |||
| IV | 0.17 | 4.5 ± 1.0 | 5 ± 3 | |||
| dC12 (201) | I | 0.62 | 0.0049 ± 0.0006 | 0.0223 ± 0.0004 | 0.142 ± 0.001 | 0.016 ± 0.001 |
| II | 0.11 | 0.83 ± 0.07 | 5 ± 5 | |||
| III (u.a.) | 0.27 | n.d. | n.d. | |||
| dA38 (252) | I | 0.64 | 0.020 ± 0.002 | 0.26 ± 0.01 | 0.63 ± 0.10 | 0.034 ± 0.003 |
| II | 0.10 | 1.7 ± 0.1 | 5 ± 3 | |||
| III (u.a.) | 0.26 | n.d. | n.d. | |||
| C39S3 (370) | I | 0.64 | 0.033 ± 0.002 | 0.034 ± 0.001 | 0.095 ± 0.003 | 0.071 ± 0.006 |
| II | 0.09 | 0.3 ± 0.1 | 0.23 ± 0.06 | |||
| III | 0.09 | 3 ± 1 | 3.4 ± 0.6 | |||
| IV (u.a.) | 0.18 | n.d. | n.d. | |||
| RzAS3 (835) | I | 0.48 | 0.30 ± 0.04 | 0.0061 ± 0.0001 | 0.020 ± 0.003 | 0.23 ± 0.01 |
| II | 0.06 | 0.05 ± 0.02 | 0.19 ± 0.01 | |||
| III | 0.06 | 0.68 ± 0.40 | 0.91 ± 0.001 | |||
| IV | 0.04 | 4.5 ± 0.3 | 6 ± 2 | |||
| V (u.a.) | 0.07 | n.d. | n.d. | |||
| I′ | 0.09 | 0.038 ± 0.03 | 0.0061 ± 0.0004 | 0.02 ± 0.001 | 0.029 ± 0.005 | |
| II′ | 0.07 | 0.05 ± 0.02 | 0.19 ± 0.01 | |||
| III′ | 0.05 | 0.68 ± 0.40 | 0.91 ± 0.04 | |||
| IV′ | 0.05 | 4.5 (3) | 6 ± 2 | |||
| V′ (u.a.) | 0.03 | n.d. | n.d. | |||
| RzAS3/C39S3 (560) | I | 0.53 | 1.0 ± 0.1 | 0.022 ± 0.001 | 0.17 ± 0.01 | 0.51 ± 0.03 |
| II | 0.05 | 0.55 ± 0.04 | 0.83 ± 0.08 | |||
| III | 0.02 | 5 ± 1 | 5 ± 5 | |||
| IV (u.a.) | 0.08 | n.d. | n.d. | |||
| I′ | 0.13 | 0.13 ± 0.03 | 0.022 ± 0.001 | 0.17 ± 0.01 | 0.10 ± 0.03 | |
| II′ | 0.08 | 0.55 ± 0.04 | 0.83 ± 0.08 | |||
| III′ | 0.07 | 5 ± 1 | 5 ± 5 | |||
| IV′ (u.a.) | 0.04 | n.d. | n.d. |
Docking and undocking rate constants were determined under standard conditions (50 mM Tris-HCl, pH 7.5/12 mM Mg2+/25°C) by statistical analysis of single-molecule FRET time trajectories, as described in Materials and Methods and Supporting Text. The estimated confidence intervals have the same accuracy as the last digit. In case of the dC12, dA38, C39S3 and RzAS3/C39S3 variants, undocking is significantly faster than that of the WT. It is thus likely that the fastest undocking rate constants become faster than our experimental time resolution of 100 ms. This is consistent with larger unassigned subpopulations in these variants compared to the WT. u.a., unassigned subpopulation; n.d., undetermined rate constant, in kinetic simulations assumed to be equivalent to our lower-limit estimate of 10 s-1.
Single Hairpin Ribozymes Show Heterogeneous Docking and Undocking Kinetics. Fig. 2 shows representative single-molecule FRET time traces for the WT and each modified hairpin ribozyme-noncleavable substrate analog complex (dC12, dA38, C39S3, RzAS3, and the double mutant RzAS3/C39S3), where the FRET ratio stochastically jumps between consistently ≈0.2 (undocked states) and ≈0.8 (docked states). The dwell times in the undocked and docked conformations are calculated for each event, and the resultant dwell time distributions are used to deduce the rate constants for docking and undocking, respectively, of all substrate and product complexes (Fig. 4 and Supporting Text) (15). Our analysis, performed on two or three independent data sets of different time resolutions for maximal confidence level (Supporting Text), shows that all modified ribozymes undock with multiple rate constants (Table 1), consistent with previous observations for the WT (15). Moreover, they all show evidence of a conformational memory effect in which the molecules essentially do not switch between different undocking behaviors over the time course of our experiments (typically 10–20 min) (15). Notably, the subpopulation distributions for dC12, dA38, and C39S3, which carry modifications in different positions resulting in a variety of docking/undocking rate constants, mirror those of the WT (Table 1; note that the partially unassigned subpopulations III of dC12 and dA38 have fractions that equal, within error, the sum of those of subpopulations III and IV of the WT). This is consistent with the notion that the observed kinetic folding heterogeneity is not influenced by any changes introduced by these modifications.
Fig. 2.

Exemplary single-molecule FRET time trajectories of the major subpopulations of the WT and variant hairpin ribozyme-noncleavable substrate analog complexes.
Significantly, the RzAS3 modification, while accelerating docking, also gives rise to heterogeneity in the docking rate constant, in addition to that observed for undocking (Table 1). This is consistent with observations for the four-way junction form of the hairpin ribozyme, which also shows accelerated and heterogeneous docking (20). In our two-way junction RzAS3 variant, we discern two docking and four undocking rate constants, with the dominant (48%) subpopulation displaying the fastest docking and the slowest undocking rate constant. In addition, small subpopulations of molecules representing each possible combination of docking and undocking rate constants are found (Table 1), suggesting that the molecular origins for the two heterogeneities are not necessarily the same.
Combining the RzAS3 and C39S3 modifications has additive effects on the docking and, to a lesser extent, the undocking rate constants (Table 1 and Fig. 1B). For example, whereas docking of the major subpopulations of the RzAS3 and C39S3 variants is accelerated 35- and 3.8-fold, respectively, compared to the WT ribozyme, the major subpopulation of the doubly modified variant is accelerated 116-fold, close to the theoretical prediction (133-fold), assuming that the effects of the two modifications are independent and additive (Table 1). Again, heterogeneity in both docking and undocking kinetics is observed, inherited from the parent RzAS3 variant (Table 1).
Single-Molecule Docking Kinetics Are Consistent with Solution Average. To verify our single-molecule data, we performed bulk steady-state FRET assays. We then performed kinetic simulations based on the single-molecule rate constants and population distributions in Table 1 to compare with these bulk data. With only the final amplitude as an adjustable parameter (which is ill-defined in steady-state FRET), this yields fits that closely reproduce the observed bulk kinetics (Fig. 6). Plotting the contribution of each effectively noninterchanging WT molecule subpopulation to the overall time course demonstrates that the most stably docked subpopulation dominates the bulk steady-state FRET signal as it is the only one that accumulates in the high-FRET state to a detectable degree (Fig. 6). As a consequence, the observed bulk docking rate constant kdock,obs (Table 2) coincides closely with the sum of the docking and undocking rate constants of the most stably docked subpopulation, but not the others (Table 1). These results underscore that a lack of heterogeneity in bulk folding experiments is not necessarily indicative of the absence of structural heterogeneity at the individual molecule level, which may simply be masked in the ensemble average (15, 21, 22).
Overall Cleavage Activity Depends on both Docking and Undocking Rate Constants. We tested the sensitivity of the overall cleavage rate constant to changes in the rates of docking and undocking by matrix-algebra-assisted fully analytical simulation of the fully reversible reaction pathway in Fig. 1B (Supporting Text). To isolate the effects of (un)docking alone, we kept the rate constants for the chemistry steps fixed at kcleav = 0.15 s-1 and klig = 0.37 s-1, the values obtained for the WT hairpin ribozyme (see below). We further assumed that the (un)docking rate constants of the product complex change proportionally to those of the substrate complex, as experimentally observed (Table 1). Our analysis shows that only a parallel acceleration of both docking and undocking rate constants leads to significant acceleration in the overall cleavage rate constant kcleav,obs (Fig. 8, which is published as supporting information on the PNAS web site). Notably, a hairpin ribozyme subpopulation such as IV where only undocking is strongly accelerated (Table 1) is predicted to have slow overall cleavage, because its residence time in the active docked state is short (see also Fig. 3). Likewise, a variant that specifically accelerates docking (and so stabilizes the docked state), as does the naturally occurring four-way junction hairpin ribozyme (20), is predicted to be a slow cleaver (Fig. 8), essentially due to slow undocking and limiting product release.
Modifications Remote from the Catalytic Site Affect both Structural Dynamics and Chemistry. In the following, we combine results from single-molecule docking/undocking experiments and bulk cleavage/ligation experiments with kinetic simulations to address a central question, whether changes in the (un)docking rate constants alone predict the impact of our specific functional group modifications on the overall reaction kinetics of the hairpin ribozyme. We observe biphasic overall cleavage kinetics with a fast and a slow rate constant for all ribozyme variants (Fig. 3), as expected from the heterogeneous (un)docking with associated memory effect (15). Our site-specific modifications alter both the fast and slow rate constants (kcleav,obs,f and kcleav,obs,s, respectively; Table 2). The C39S3 variant is the fastest overall cleaver, with a rate acceleration in kcleav,obs,f of nearly 3-fold over the WT, followed by the RzAS3/C39S3 double variant, dA38, RzAS3, and dC12. To extract the actual chemistry (cleavage, ligation) rate constants (klig, kcleav), we performed a kinetic analysis based on the experimentally determined docking and undocking rate constants (Table 1) and the experimentally derived equilibrium constant of the chemical step (klig/kcleav) (Table 2 and Fig. 5). We analytically fit the resulting simulated reaction time courses to the experimental ones using a single fitting parameter, kcleav, assuming that all molecular subpopulations of a given ribozyme variant share a single set of klig and kcleav values (for details of the analysis and the dependence of our results on the latter assumption, see Supporting Text and Table 3, which is published as supporting information on the PNAS web site).
Fig. 3 shows that this analysis yields well defined chemistry rate constants. We find that the cleavage and/or ligation rate constants of most variants differ significantly from those of the WT, illuminating how complex the effects of single functional group modifications on RNA function are. The experimentally determined 7.8-fold increase in the internal cleavage equilibrium is dissected into a 3.7-fold increase and a 2.1-fold decrease in the cleavage and ligation rate constants, respectively, compared to the WT (Fig. 3, summarized in Fig. 1B). Combined with the accelerated undocking kinetics and thus lower docking equilibrium, this leads to the slightly accelerated overall cleavage kinetics (Table 2). The dA38 variant has a cleavage rate constant that is only slightly perturbed (1.9-fold slower), but ligation is 31-fold slower (Figs. 1B and 3), so that overall cleavage is accelerated (Table 2) despite the fact that docking of this variant into the active structure is very unstable (Fig. 2 and Table 1). The cleavage and ligation rate constants of the C39S3 variant are 4- and 7-fold slower, respectively, than those of the WT (Figs. 1B and 3); however, combined with a more rapid docking/undocking (Fig. 2 and Table 1), this variant becomes the fastest in overall cleavage (Table 2). Finally, the RzAS3 and RzAS3/C39S3 variants have the most strongly altered chemistry rate constants, which are ≈20-fold slower than those of the WT (Figs. 1B and 3). Yet, combined with their particularly rapid docking (Fig. 2 and Table 1), this leads to an overall cleavage kinetics that still exceeds that of the WT (Table 2). Notably, the doubly modified variant RzAS3/C39S3 largely inherits the chemistry rate constants of the slower of its two parents, RzAS3 (Figs. 1B and 3).
In the above cases, a single modification is introduced at a location remote from the reaction site (Figs. 1 A and 7), at a distance of 17 (dC12), 11 (dA38), 15 (C39S3), and 26 Å (RzAS3) from the scissile phosphate (9, 12), yet our analysis shows that the chemistry rate constants are affected with some modifications showing substantial effects (see also Fig. 9, which is published as supporting information on the PNAS web site). We present a complete dissection of the specific effects that single-site modifications have on the rate constants of tertiary structure folding and chemistry of an RNA enzyme. Our results thus show that the overall cleavage kinetics are intricately dependent on all individual steps (docking, undocking, cleavage, and ligation) of the reaction pathway, so that no a priori assumptions can be made, and only an analysis of all rate constants as presented here will unequivocally resolve whether a functionally important residue is involved in folding, catalysis, or both.
Discussion
We have used a combination of single-molecule and ensemble FRET assays, functional assays, and analytical kinetic simulations to dissect effects of specific functional group modifications on both docking and chemistry of the hairpin ribozyme. We find that all modified ribozymes exhibit similar fractionations into noninterchanging subpopulations of molecules with distinct undocking rate constants, whereas ones with significantly accelerated docking additionally display multiple docking rate constants. Most strikingly, modifications of essential residues distant from the site of catalysis alter the rate constants not only of docking and/or undocking but also of catalytic chemistry (Fig. 1).
There are two possible mechanisms by which such distal single-functional group modification may impact catalysis (13): (i) Subtle perturbations of the “static” RNA structure at the site of catalysis, not detected by our FRET experiments, modulate catalytic function. (ii) Perturbations in the dynamics of networks of coupled motions of functionally important residues modulate the structural dynamics of the RNA at the site of catalysis and, as a result, impact its function. The second mechanism was recently proposed to explain the transmittance of interference effects from remote modifications to the catalytic site of protein enzymes (13). We propose that it is also well suited to explain, for example, the increase in catalytic rate constant of our dC12 hairpin ribozyme variant, in which the hydrogen bond from the 2′-OH group of C12 to the 2-keto group of U42 is removed at a distance of 17 Å from the site of catalysis (Fig. 1 A). This may lead to additional local structural flexibility propagating into the catalytic site and, in turn, a higher probability for the docked state sampling conformations close to the chemical transition state. By contrast, the higher flexibility at the domain junction of the RzAS3 variant (Figs. 1 A and 7) likely translates into larger global dynamic excursions that are apparently not particularly conducive to catalysis, leading to a higher catalytic barrier. Finally, the dA38 modification may lead to enhanced dynamics of the A38 base (Fig. 1 A), which would explain that cleavage and ligation are affected differentially because the base bridges and aligns the 5′ and 3′ products for ligation by forming two hydrogen bonds that are absent in the substrate structure (12). Future work is needed to identify the specific coupled networks of molecular motions that mediate these effects.
The results presented here provide evidence that distal functional group modifications may have a large impact on the intrinsic chemistry rate constants in RNA catalysis. This suggests that coupled molecular motions connect remote parts of an RNA fold with its functional core, in analogy to recent observations in protein enzymes (13). Our findings thus invoke a view of an expanded role of the overall fold in RNA function. They also have broad implications for applications that seek out RNA as a drug target (23), use ligand binding distal to the active site of an RNA, or engineer allosteric RNA enzymes (24).
Supplementary Material
Acknowledgments
We thank Tsu-Chien Weng for initial help with matlab (MathWorks, Natick, MA). This work was supported in part by grants from the National Institute of General Medical Sciences (GM62357), the American Chemical Society, and Dow Corning (to N.G.W.); the Office of Naval Research and the National Science Foundation (X.Z.); a National Institutes of Health training grant in Molecular, Cellular, and Chemical Biology Program (to G.B.); and two predoctoral fellowships from the National Science Foundation (to M.M.R. and M.J.R.).
Abbreviation: FRET, fluorescence resonance energy transfer.
References
- 1.Treiber, D. K. & Williamson, J. R. (2001) Curr. Opin. Struct. Biol. 11, 309-314. [DOI] [PubMed] [Google Scholar]
- 2.Walter, N. G., Harris, D. A., Pereira, M. J. & Rueda, D. (2001) Biopolymers 61, 224-242. [DOI] [PubMed] [Google Scholar]
- 3.Thirumalai, D., Lee, N., Woodson, S. A. & Klimov, D. (2001) Annu. Rev. Phys. Chem. 52, 751-762. [DOI] [PubMed] [Google Scholar]
- 4.Walter, N. G. & Burke, J. M. (1998) Curr. Opin. Chem. Biol. 2, 24-30. [DOI] [PubMed] [Google Scholar]
- 5.Fedor, M. J. (2000) J. Mol. Biol. 297, 269-291. [DOI] [PubMed] [Google Scholar]
- 6.Ferre-D'Amare, A. R. (2004) Biopolymers 73, 71-78. [DOI] [PubMed] [Google Scholar]
- 7.Shippy, R., Lockner, R., Farnsworth, M. & Hampel, A. (1999) Mol. Biotechnol. 12, 117-129. [DOI] [PubMed] [Google Scholar]
- 8.Ryder, S. P. & Strobel, S. A. (1999) J. Mol. Biol. 291, 295-311. [DOI] [PubMed] [Google Scholar]
- 9.Rupert, P. B. & Ferre-D'Amare, A. R. (2001) Nature 410, 780-786. [DOI] [PubMed] [Google Scholar]
- 10.Pinard, R., Hampel, K. J., Heckman, J. E., Lambert, D., Chan, P. A., Major, F. & Burke, J. M. (2001) EMBO J. 20, 6434-6442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lebruska, L. L., Kuzmine, I. I. & Fedor, M. J. (2002) Chem. Biol. 9, 465-473. [DOI] [PubMed] [Google Scholar]
- 12.Rupert, P. B., Massey, A. P., Sigurdsson, S. T. & Ferre-D'Amare, A. R. (2002) Science 298, 1421-1424. [DOI] [PubMed] [Google Scholar]
- 13.Benkovic, S. J. & Hammes-Schiffer, S. (2003) Science 301, 1196-1202. [DOI] [PubMed] [Google Scholar]
- 14.Walter, N. G., Hampel, K. J., Brown, K. M. & Burke, J. M. (1998) EMBO J. 17, 2378-2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhuang, X., Kim, H., Pereira, M. J., Babcock, H. P., Walter, N. G. & Chu, S. (2002) Science 296, 1473-1476. [DOI] [PubMed] [Google Scholar]
- 16.Bokinsky, G., Rueda, D., Misra, V. K., Rhodes, M. M., Gordus, A., Babcock, H. P., Walter, N. G. & Zhuang, X. (2003) Proc. Natl. Acad. Sci. USA 100, 9302-9307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Walter, N. G. (2001) Methods 25, 19-30. [DOI] [PubMed] [Google Scholar]
- 18.Esteban, J. A., Banerjee, A. R. & Burke, J. M. (1997) J. Biol. Chem. 272, 13629-13639. [DOI] [PubMed] [Google Scholar]
- 19.Gutfreund, H. (1995) Kinetics for the Life Sciences: Receptors, Transmitters and Catalysts (Cambridge Univ. Press, Cambridge, U.K.).
- 20.Tan, E., Wilson, T. J., Nahas, M. K., Clegg, R. M., Lilley, D. M. & Ha, T. (2003) Proc. Natl. Acad. Sci. USA 100, 9308-9313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu, H. P., Xun, L. & Xie, X. S. (1998) Science 282, 1877-1882. [DOI] [PubMed] [Google Scholar]
- 22.Zhuang, X. & Rief, M. (2003) Curr. Opin. Struct. Biol. 13, 88-97. [DOI] [PubMed] [Google Scholar]
- 23.Moore, P. B. & Steitz, T. A. (2003) Annu. Rev. Biochem. 72, 813-850. [DOI] [PubMed] [Google Scholar]
- 24.Silverman, S. K. (2003) RNA 9, 377-383. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.