

Summary
The signaling module that specifies nuclear factor-κB (NF-κB) activation is a three-component system: NF-κB, inhibitor of NFκB (IκB), and IκB kinase complex (IKK). IKK receives upstream signals from the surface or inside the cell and converts itself into a catalytically active form leading to the destruction of IκB in the inhibited IκB: NF-κB complex, leaving active NF-κB free to regulate target genes. Hidden within this simple module are family members that all can undergo various modifications resulting in expansion of functional spectrum. Three-dimensional structures representing all three components are now available. These structures have allowed us to interpret cellular observations in molecular terms and at the same time helped us to bring forward new concepts focused towards understanding the specificity in the NF-κB activation pathway.
Keywords: transcription factors, signaling proteins, kinases/phosphatases, inflammation, signal transdcution
Introduction
The dimeric transcription factor family known as the NF-κB (nuclear factor-κB) family has been intensely investigated by several research groups over the past 25 years because of its involvement of several biological programs. The first NF-κB dimer was identified as a heterodimeric protein of two subunits with molecular mass 50 kDa and 65 kDa, now known as the p50 and RelA (p65) subunits of NF-κB. NF-κB is not a single dimer but defines 15 possible homodimers and heterodimers arise from five genes products NF-κB1/p105/p50, NF-κB2/p100/p52, RelA/p65, RelB, and cRel (1, 2). While most of the dimer combinations are abundant in diverse cell types, others are rare. A few have not been detected directly, but it remains likely that the rare ones (such as c-Rel:RelB heterodimer) also exist in some cells under specific regulatory conditions.
The activity of these NF-κB dimers is directly controlled by a set of proteins known as IκB (inhibitor of NF-κB) through the formation of stable IκB:NF-κB complexes (3). The non-covalent association of IκB with NF-κB shifts the steady-state subcellular localization of NF-κB dimers to the cytoplasm (reviewed by Hinz et al., this volume). A complicated and fascinating kinase complex known as the IKK (IκB kinase) complex is responsible for phosphorylating the complex-associated IκB, leading to its targeted ubiquitination by a specific SCF-type E3 ubiquitin-protein ligase and degradation by the 26S proteasome (4, 5, reviewed in Kanarek & Ben-neriah, this volume). Free NF-κB then rapidly accumulates in the nucleus where it binds to a class of related DNA sequences, known as κB DNA/sites, present within the promoters and enhancers of hundreds genes and elevates or represses their expression (6). While the general theme for NF-κB activation and its transcriptional regulation has not changed since it was first laid out by Baltimore and colleagues (Boldin & Baltimore, this volume), participation of multiple family members of each of the three critical families (IKK, IκB and NF−κB) with distinct features resulted in a large number of activities as such that it now appears that each of the nearly 500 genes has found a highly specific regulatory regimen (Fig. 1).
Fig. 1. Schematic representation of the NF-κB activation pathway.

NF-κB dimers are retained inactive in the cytoplasm in the resting cell through binding with inhibitor protein IκB. Different stimuli activate IKK which phosphorylates IκB. P-IκB is degraded by the 26S proteasome. Free NF-κB regulates the expression of target genes.
In this review, we describe the regulation of the NF-κB family of transcription factors in terms of combinatorial dimer formation, association with IκB inhibitors, sequence-specific DNA binding, and finally activation by IKK. We describe each of these regulatory events using the structures and correlated biochemical studies and link that to cellular activities. Herein we show that small sequence differences within the NFκB family members results in structural differences that are often sufficient to restrict their functions.
Personal and historical narratives
I (GG) first came to know about NF-κB as a second year graduate student in 1988 when a friend of mine was writing a proposal on the putative NF-κB inhibitor as part of his qualifying exam. He discussed with me two articles by Sen and Baltimore published in Cell. Since then I closely followed the NF-κB field and was aware of the discoveries of IκB, p105/p50 and p65/RelA. But I have always been interested in RNA-protein complexes, and my own research in Paul Siglar’s laboratory as a postdoctoral fellow focused on structural studies of RNA-protein interaction related to translation. Soon after joining the laboratory, I realized Paul lost his interest in translational regulation. By 1991, the focus of the Siglar lab research had changed from tRNA and phospholipase A2 to G-protein and transcription factors. In late 1992, I had a discussion with Sankar Ghosh, who had recently joined Yale University as an Assistant Professor, about structural study of NF-κB. Sankar and I thought that the structure of the NF-κB:DNA complex would be different than other protein-DNA complexes known by that time, since much larger NF-κB DNA binding domain recognizes short 10 bp DNA target sites. Paul was not too happy about my new move, but allowed me work on NF-κB on a condition. He wanted to develop basal transcriptional assembly complex on archaebacterial system. So I had to work on two projects.
I expressed and purified p50 and p65/RelA homodimers as well as IκBα. I put them into crystallization trials. None of these proteins in their free states even remotely looked crystallizable. I then focused on p50:DNA complexes. I had to take a brute force approach, since most information on NF-κB came from biological experiments but not from detailed biochemical studies, which is necessary to initiate a crystallographic project. I made five different p50 constructs and over fifty DNA duplexes of different lengths and sequences. This approach led to the first structure of a NF-κB dimer, the p50 homodimer, bound to a κB DNA (7). Steve Harrison of Harvard University also reported a similar structure in the same issue of Nature (8). As we expected, the structure was unique at that time, where loops connecting protein secondary structures contributed residues for sequence-specific DNA binding.
I moved to University of California, San Diego, as an Assistant Professor in 1995 and continued to work on the NF-κB system. I focused on elucidation of the structures of p65/RelA homodimer and p50:p65/RelA heterodimer complexed to DNA. Most of the initial complexes bound to p50 or RelA homodimers were done with perfectly palindromic DNA sequences. We used this strategy since κB sites known by that time showed partial half site symmetry and since this allowed us to screen more sequences as just one DNA stand can self anneal to form a duplex. RelA homodimer bound to one such palindromic DNA sequences revealed unusual structural features, where one monomer was out of the DNA register, thus failing to make base-specific contacts (9). This resulted in a large conformational change and sequence non-specific binding by one RelA subunit. We had to struggle to publish these results, as the reviewers thought that the structures were artificial. We now know NF-κB can bind target DNA sites with only half site specificity in vivo. The biological meaning of non-canonical protein-DNA interactions has been beautifully illustrated by the Hofmann group in collaboration with our group (10). Whether conformational variations of different NF-κB:DNA complexes elicit a major source of specificity in gene regulation remains unknown. One important question is if and how single bp alterations in a DNA target site change transcriptional specificity in vivo. The Baltimore group has shown this to be important in two cases (11), but universality of this is yet to be proved. Whether this specificity alteration is caused by conformational change or affinity or binding kinetics or a combination of these is to be determined.
A key component of the NF-κB signaling system is the IκBα:p50:RelA complex. I felt it was necessary to reveal the molecular features of this complex, which might explain why the complete degradation of IκBα is essential to activate NF-κB. It turned out to be an extremely difficult structural project, since flexible segments of both NF-κB and IκBα are involved in the complex formation and non-contacting portions of both molecules had to identified and then removed to obtain crystals. Our plan was to determine the structures of each IκB protein bound to a NF-κB dimer. However, the knowledge of crystallization of the IκBα:NF-κB complex was insufficient to obtain well-ordered crystals for other IκB complexes. After years of trials we got just one crystal of the IκBβ:RelA complex. We were lucky to determine the structure of the complex from this single crystal (12). The IκBβ:RelA complex structure gave us some key insights into its differential biological activity such as the ability of this complex to recognize DNA. To our comfort, it is now known that IκBβ acts as a coactivator of transcription by recruiting RelA:cRel heterodimer to specific κB sites (13).
Although we struggled to obtain more structures of IκB:NF-κB complexes, through collaborative work with Alex Hoffmann, Elizabeth Komives, and Jane Dyson, we set out to thoroughly investigate IκBα. Our work was motivated by the observation of the Baltimore group that in the absence of NF-κB, IκB proteins are highly unstable in vivo (14). The Komives group found IκBα is partially unfolded as a free protein and its unfoldedness led to its short half-life. We found that proteasome-mediated degradation of free IκBα does not require phosphorylation or ubiquitination but is dependent on a flexible C-terminus and particularly positioned hydrophobic residues in the C-terminus. With the Hoffmann group, we showed homeostatic degradation of free IκBα was necessary for proper NF-κB response by stimulus (15). Dyson and Komives elegantly explained how IκBα actively removes NF-κB from the promoter. They showed IκBα associates with the DNA bound NF-κB forming a transient complex (16). These observations give a provocative suggestion that like IκBβ, IκBα may function as a transcriptional activator if it remains bound to the promoters along with NF-κB long enough time to establish an active transcriptional complex. Therefore, a fascinating conclusion of these recent results is that IκB proteins can function both as inhibitors and activators. Perhaps modifications or variable contexts allow them to function differently. In other words, transcriptional co-activation function is not only limited to Bcl3 or IκBζ but also to IκBα, IκBβ and IκBε. Similarly, Bcl3 and IκBζ may also act as transcriptional repressors.
Another challenging but fascinating problems is the mechanism of p100 and p105 processing. Although many articles have been published since the identification of these two molecules, in my view very little is known about how these molecules undergo processing. In particular, processing of p100 is often depicted in cartoons by everyone in the field as induced degradation of the C-terminus of p100 of the p100:RelB complex. However, this cannot be the case. Since half of the NF-κB family members are inhibited by these two atypical inhibitors and at the same time they serve as the precursors of nine out of 15 possible NF-κB dimers, the field should care to know the truth and not just the partial truth. Unfortunately as many scientists believe that these are the questions of the past, it is hard for mechanistic biochemists to convince them, especially when many believe that structures do not contribute much barring their usefulness in an introductory book chapter.
The NF-κB family
The NF-κB family consists of combinatorial dimers of five protomers (Fig. 2). Two of the family members, p50 and p52, are the processed products of NF-κB subunit precursors p105 and p100, respectively. These five polypeptides belong to a family due to homologous sequences near their N-termini referred to as the Rel homology region (RHR). The RHR is roughly 300 residues in length and is responsible for most of the critical functions including subunit association into active NF-κB dimers, NF-κB nuclear localization and DNA binding, and association with IκB inhibitors. These properties allow the family members to be collectively regulated by a set of stimuli. The RHR can be divided into three structural regions: the N-terminal domain (NTD), dimerization domain (DD), and nuclear localization signal (NLS) (Fig. 2). NF-κB exists and functions only as dimers. The DD alone mediates the association of NF-κB subunits to form combinatorial dimers. Together the NTD and DD perform the DNA binding function. The NLS polypeptide is flexible in solution allowing it to adopt different conformations when bound to distinct partners such as IκB or importin-α proteins (17). The DD and NLS region are the primary binding sites for IκB inhibitors. C-terminal to the RHR of RelA, RelB, and c-Rel is a region essential for transcriptional activation potential and, consequently, NF-κB dimers that possess at least one of these subunits function as activators of transcription. This transcriptional activation domain (TAD) region is not conserved between the NF-κB subunits at the amino acid sequence level and is, therefore, defined functionally. In spite of sequence diversity, the TADs are not known to provide functional specificity in vivo. However, more work is required to understand if an NF-κB TAD imparts transcriptional specificity in vivo. Both the NF-κB p50 and p52 subunits lack a C-terminal TAD and instead contain within this region the glycine-rich region (GRR) as remnants of their incomplete proteolytic processing from p105 and p100 precursors. As a consequence of their lack of a C-terminal TAD, NF-κB dimers composed only of p50 and/or p52 subunits fail to activate transcription in vitro or in vivo.
Fig. 2. Members of the NF-κB and IκB protein families.
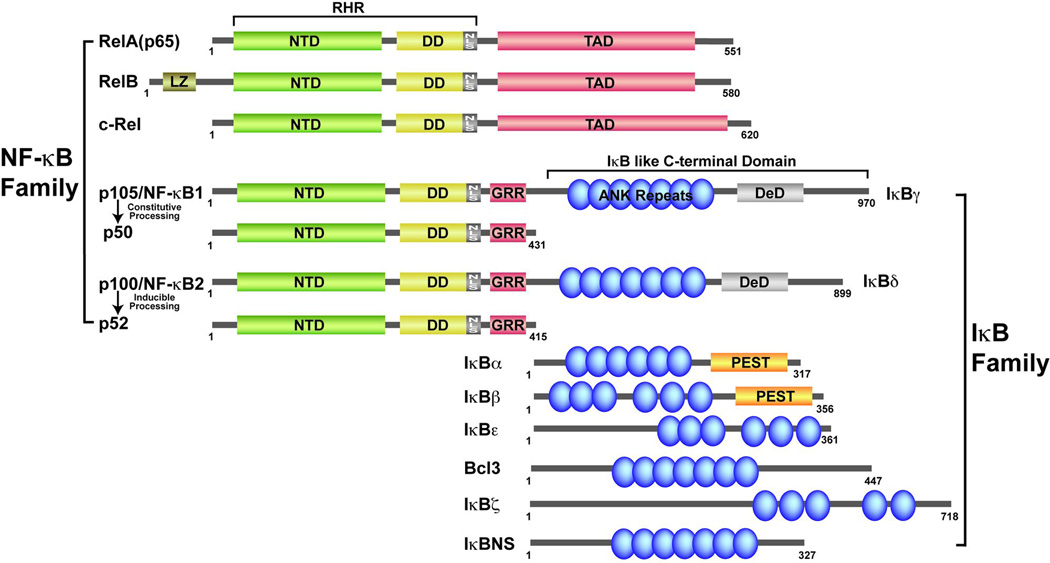
NF-κB family is characterized by having the RHR (Green & Yellow). IκB proteins are characterized by the presence of ankyrin repeat domain (ARD) shown in blue.
NF-κB structure and dimer formation: an overview
The DD of NF-κB was not identified by biochemical means or sequence alignment. Evidence that a small segment within the RHR forms dimer came from the first structure of an NF-κB RHR bound to DNA. The DD consists of approximately 100 amino acids near the C-terminal end of the RHR. Sequence identity and homology within the DD across the family are roughly 20% and 50%, respectively. The molecular structure of the DD has been determined at high resolution by x-ray crystallography for all five NF-κB subunits (18, 19). The NF-κB DD folds into an immunoglobulin-like (Ig-like) fold where two anti-parallel β-sheets form a sandwich (Fig. 3). About 12 to 14 residues from each monomer are involved in dimer formation by making symmetrical (or pseudo-symmetrical for heterodimers) contacts. However, as revealed by alanine scanning mutagenesis in the p50 subunit and subsequent assessment of the p50 homodimer formation, only few of these residues make energetic contributions to dimer stability (20). The x-ray crystal structure of a RelB homodimer DD revealed that it adopts a distinct domain-swapped structure (Fig. 3C).
Fig. 3. Ribbon presentations of the NF-κB RHR and dimmer.

(A) Top, the NF-κB p50 subunit RHR is depicted in green ribbon diagram. Shown in magenta and red are the residues from Loop L1 and L3 that bind DNA. Bottom, sequences of loop L1 and L3 across the family, (•) denotes DNA contacting residues. (B) Ribbon diagram of the NF-κB dimerization domain dimer indicating the secondary elements and the overall tertiary fold. Residues which contribute to the dimer interface are aligned. (C) A similar view of the RelB DD homodimer reveals that the two subunits (dark and light purple) form an intertwined dimer (PDB 1ZK9).
Structures of the NF-κB DD provide insights into the mechanism of preferential dimer formation. These structures have taught us why the p50:RelA heterodimer is more stable than the homodimer of p50 while the RelA homodimer is the weakest of the three. We can now predict why p50 homo- and heterodimers are abundant in cells but the p52 homodimer is observed under specific conditions. A close inspection of these structures reveals that differential selectivity and stability of NF-κB dimers are controlled in two different ways: the first is variation in the amino acid residues that directly contact the other subunit across the dimer interface (Fig. 3B), and the second is variation in surface or core amino acid residues that influence folding stability of the DD. While the first seems obvious, this second class of residues that affects dimerization through an indirect manner is at least as important.
Regulation of NF-κB dimerization
Although the inter-subunit distances in p50 and RelA homodimers are similar, suggesting overall similarity in their respective mechanisms of dimer formation, fewer hydrogen bonds at the RelA homodimer interface suggested that it would be weaker than the p50 homodimer. Three differences in the amino acids at the dimer interfaces of p50 and RelA homodimer suggest how the resulting dimerization of p50:p50, RelA:RelA, and p50:RelA dimers might be affected. Residues at positions 254 and 267 are an Asp and a Tyr, respectively, in p50 (murine p50 numbering). The equivalent positions are occupied by an Asn and a Phe in RelA. The placement of Asp-Asp and Asn-Asn pairs are unfavorable to the stability of the p50 homodimer and RelA homodimer, respectively (Fig. 3B). This is particularly true in these homodimers since the carbonyl oxygen in both Asp (in p50) and Asn (in RelA) is locked in a fixed position through contact with backbone amide leaving the anionic oxygen in case of Asp or amine in case of Asn to face each other. Indeed, to avoid negative interactions, these functional groups move away from the interface. In the p50:RelA heterodimer, direct hydrogen-bonding contact between the amine and oxygen stabilize the dimer. The Tyr to Phe change accounts for the remaining difference in the number of hydrogen bonds at the dimer interface of p50 and RelA homodimers. The lack of a hydroxyl group on Phe makes RelA homodimer less stable than p50 homodimer, wherein the hydroxyl group of Tyr mediates several hydrogen bonds across the subunit interface.
The third key difference at the dimer interface that influences NF-κB dimer selectivity is the change from a Phe at position 307 in p50 to a Val at the corresponding position of RelA. The aromatic phenylalanine ring in p50 orients itself differently optimizing van der Waals interactions between the beta carbons of Phe from two subunits. This observation explains why alterations of Phe to Ala in p50 did not reduce the strength of the p50 homodimer to any measurable extent. The Val side chains in RelA homodimer position themselves uncomfortably close as they approach one another. Together these differences at least partly explain differential dimer stability.
The remaining amino acid residues at the NF-κB dimer interface are identical across the family. Therefore, the differences in affinity observed between different combinatorial dimers could be explained by the amino acid identity at these three positions only. However, this is not the case. Several inconsistencies, culminating with our observation of an unusual domain-swapped architecture of the RelB homodimer, indicated that non-interfacial amino acid residues also play a vital role in controlling assembly of active NF-κB dimers (19). In RelB, the three critical interfacial residues in question are Asn287, Tyr300, and Ile335. Of these, only Ile is distinct, as equivalent Asn residues are present in RelA and c-Rel and an equivalent Tyr is present in p50 and p52. Mutation of Ile to either Val or Tyr or Phe, the residues present in RelA or p52 or p50, respectively, does not convert RelB into a regular side-by-side NF-κB homodimer. However, alterations of Tyr300 to Phe and Ile335 to Phe (or Tyr or Val) resulted in a side-by-side dimer as in the RelA:RelA homodimer. This finding suggests that the hydrogen bonds mediated by Tyr300 in wt RelB attempt to bring the two subunits close, which forces the Ile side chains to come non-permissibly close together. Is it possible to change the RelB homodimer into a homodimer more like the p50:p50 homodimer with Tyr300 at the center of the interface? If alterations of Asn287 to Asp and Ile335 to Phe as in p50 allows RelB to form a regular dimer, it would suggest variations of residues at only three positions could give a broad range of dimer stability in the NF-κB family (Vu et al., unpublished results).
Affinity of NF-κB dimers
There is no report describing in vitro affinity (stability) of different NF-κB dimers. Stability of the NF-κB p50:RelA heterodimer is relatively higher than the corresponding homodimers as judged by the fact that the heterodimer forms preferentially when p50 and RelA homodimers are mixed together. However, under similar conditions, p50:c-Rel heterodimer formation is not as efficient. This observation suggests that c-Rel homodimer might be more stable than RelA homodimer. This is particularly intriguing, as RelA and c-Rel share greater than 70% sequence identity within their dimerization domains and all inter-subunit contacting residues are identical in both proteins. Despite its high degree of sequence and structural homology to the p50 subunit, the NF-κB p52 homodimer is rarely observed in vivo. Although there exist many explanations for this negative result, it is possible that instability in the p52:p52 homodimer allows for its more stable assembly into the p52:RelA and p52:RelB heterodimers. These observations suggest that small variations in sequence can impact dimerization of two closely related proteins such as RelA and c-Rel or p50 and p52. Supporting these rather qualitative data is more quantitative information on the dimer strengths of five homodimers based on analytical ultracentrifugation (AUC) experiments. Preliminary results have shown that among the five homodimers, p50:p50 and c-Rel:c-Rel homodimers are more stable than RelA:RelA and p52:p52 homodimers. Of these homodimers, p52:p52 homodimer is the weakest and p50:p50 homodimer is the strongest where the difference is nearly two orders of magnitude (Vu & Ghosh, unpublished observation).
Regulation of NF-κB dimerization at a distance
The above observations suggest that residues outside the dimer interface contribute to the stability of the NF-κB dimers. Understanding the role of surface and buried amino acid residues outside of the dimer interface that indirectly influence dimer selectivity and stability is more difficult to imagine. Furthermore, x-ray crystal structures do little to provide a clear explanation. The influence on dimerization of amino acid residues far from the subunit interface is more directly assessed by mutational analysis and measurement of dimerization affinity. In RelA, a Cys at position 216 occupies a position in the core of the DD that is projected opposite to the dimer interface. When this Cys is mutated to an Ala, RelA homodimer stability is significantly reduced. The simplest explanation for this reduced dimer stability is that removal of the sulfhydryl group destabilizes the core structure of DD, which in turn affects the stability of interactions at the dimer interface.
Several amino acid residues located on the surface of RelB opposite to the dimer interface are hydrophobic. These surface-exposed hydrophobic amino acids are unique to RelB among the mammalian NF-κB family. Equivalent residues in other NF-κB subunit structures are polar and are involved in the surface hydrogen bond formation to stabilize the DD structure. Mutation of these residues to polar residues decreases the affinity of RelBDD for dimerization with p52DD. It is likely that the mutant RelB forms a more stable homodimer, thereby decreasing its availability to heterodimer formation with p52. The importance of domain folding stability for NF-κB dimer assembly and subunit dimerization selectivity is further supported by the mutation of Ser at position 319 of RelB. When this surface residue is mutated to Ala, RelB protein stability is dramatically reduced.
Conditional NF-κB dimerization
Some of the NF-κB dimers are rarely observed in vivo, such as RelA:RelB and c-Rel:RelB. It has been reported that phosphorylation of Ser276 of RelA allows the modified protein to form heterodimer with RelB. It is clear from structural studies that phosphorylation of RelA at Ser276 cannot directly affect dimerization, as the amino acid is positioned opposite the dimer interface (Fig. 3B). This observation suggests that phosphorylation alters domain stability in a manner such that RelB is able to associate with RelA. Unpublished results from our laboratory suggest that RelB is largely unfolded in solution at physiological concentrations and that it becomes folded upon dimerization with p50 or p52. We also have found that the introduction of a Glu or an Asp mutation at position 276 to mimic phosphorylation significantly decreases the folding stability of RelA. It is possible that phosphorylation-dependent destabilization of RelA functions to catalyze formation of a stable RelA:RelB heterodimer. Recently, the c-Rel:RelB dimer has also been detected in vivo. It would be interesting to see if these dimers are domain swapped dimers as the RelB homodimer.
A recent study has demonstrated that p52 can heterodimerize with c-Rel. However, this dimer requires phosphorylation of Ser residue 226 of p52 by GSK3β. Ser226 is located within the linker peptide that connects the DD and NTD. It is unclear at this point how phosphorylation would induce the p52:c-Rel heterodimer formation. We suspect that the phosphorylated linker contacts the c-Rel subunit. Such contact by a linker residue is expected not to bind DNA, forming an inhibited dimer. Indeed, the p52:c-Rel heterodimer is inactive. In addition to modification-dependent enforcement, some of the IκB family members may also select NF-κB dimers (discussed later).
The RelB:RelB homodimer forms a domain-swapped artificial dimer whose affinity cannot be measured by AUC (Fig. 3C). We have shown that if RelB is produced alone, it undergoes degradation in part due to its failure to form a stable dimer. How does RelB form dimers with p50 and p52 in vivo? We suggest that RelB and p52 or p50 dimerize during or immediately after their translation. That is, both polypeptides must be synthesized at the same time or be kept unfolded by chaperones until they encounter each other to form the heterodimer.
Recognition of DNA by NF-κB: an overview
κB DNA
NF-κB recognizes 9 to 11 bp (base pairs) long double-stranded DNA elements located within the promoters and enhancers of about 500 known target genes. Early comparisons of the first DNA sequences demonstrated to bind specifically to NF-κB dimers led to the following consensus sequence: 5'-GGGRNWYYCC-3', where R = A or G; N = A, C, G, or T; W = A or T; and Y = C or T. The critical feature of this consensus is the presence of a series of G nucleotides at the 5' ends, while the central portion of the sequence displays greater variation. DNA sequences from gene promoter/enhancer regions that meet this consensus and can be shown to drive NF-κB-dependent reporter gene expression are termed ‘κB DNA’ or ‘κB sites’. Hundreds of such sequences have been confirmed experimentally, and the total number of unconfirmed κB sites detected by computational methods is in the thousands. Many of these newer sites reveal significantly greater variation than allowed by the original consensus κB DNA sequence.
Structures of the NF-κB:DNA complexes
The first two structures of NF-κB:DNA complex were determined in the laboratories of Paul Sigler and Steve Harrison in 1995. It was known that the entire RHR and perhaps a longer protein was needed for NF-κB’s DNA binding. By that time, DNA-binding domains (DBDs) of most transcription factors was found be much smaller in size, but most of these DBDs recognized a longer DNA response elements than 10-bp. It was therefore presumed that the structure of the NF-κB:DNA would be unique. The structure of NF-κB was indeed unique at the time of its determination by virtue of the fact that all of the contacts with DNA were mediated by amino acids on loops connecting β-strands (Figs 3A and 4). Three-dimensional structures of several NF-κB RHR dimers in complex with diverse κB DNAs are now known. These structures have provided important insights into the DNA recognition mechanism of NF-κB (7–10, 21–30). In general, the κB DNA is pseudo-symmetric, and each NF-κB monomer binds to one DNA half site (Figs 4 and 5). The loops in each NTD (loop L1 and L2) recognize a flanking region of DNA half-site from the major groove side, the linker (loop L3) and the loops form the dimerization domain then consume the rest of the major groove at the center. Since the minor groove is very narrow in all NF-κB:DNA complexes, it appears that residues from the loops encircles the DNA. The arrangement of the NF-κB dimer about the major groove of one entire turn of DNA gives rise to a global structure that is reminiscent of a butterfly with a DNA ‘body’ and a pair RHR ‘wings’.
Fig. 4. Ribbon structure diagram of the NF-κB p50:RelA heterodimer in complex with κB DNA.

The assembled RHR of the p50 (green) and RelA (red) subunits viewed orthogonal to their vertical axis of 2-fold pseudo-symmetry (left); and rotated 120° about the vertical axis (right) to show the interaction of p50 subunit loop L1 (magenta) and L3 (blue) with DNA bases through the major groove.
Fig. 5. NF-κB recognition of consensus κB DNA.

(A) Schematic representation of base-specific contacts mediated by NF-κB p50 (green) and RelA (red) subunits and HIV-κB DNA observed in the x-ray crystal structure. Amino acid numbering comes from the murine sequences. (B) Examples of κB DNAs in the natural target genes with different length and variable half-side.
Amino acid side chains from the immunoglobulin-like NTD of each NF-κB RHR mediate all of the DNA base-specific contacts. One of the striking features of these complexes is conformational variability. The NTD translates and/or rotates as it encounters different DNA sequences. Even a single bp difference can cause large conformational change. Bi-lobal architecture of the RHR where the NTD is linked to the DD by a 10-residue linker makes it possible for the NTD to rotate/translate with respect to the DD. The NF-κB dimer interface is maintained upon κB DNA binding and multiple additional non-specific DNA backbone interactions are made by the NTD and DD. The C-terminal NLS polypeptide is disordered when it is included in the NF-κB RHR constructs used for x-ray crystal structure determination. The p50 and p52 subunits optimally contact a 5 bp half site, whereas the RelA, c-Rel, and RelB subunits contact a 4 bp half site. With the central bp at the pseudo-dyad axis, p50 and p52 homodimers prefer an 11 bp κB DNA. The p50 (or p52) heterodimers with RelA, c-Rel, or RelB bind to 10 bp κB DNA, whereas the homodimers of RelA and c-Rel or RelA:c-Rel heterodimer prefer 9 bp κB DNA. These are however ideal cases, and as we shall see later, this recognition rule is not stringently followed by the dimers in vivo.
NF-κB recognition of consensus κB DNA at the 5'-end
Six conserved amino acids of p50 and p52 directly contact bases within κB DNA. In p50 (murine numbering), these residues are Arg54, Arg56, Tyr57, Glu60, His64, and Lys241 (Fig. 5A). The two Arg, the Tyr, and the Glu are invariant in all NF-κB subunits. His64 and Lys241 are an Ala and an Arg, respectively, in RelA, c-Rel, and RelB. His64 (His62 in p52) directly contacts the 5' G. Ala at this position in c-Rel, RelA, and RelB gives rise to differences in the half site length preferred by these two classes of NF-κB subunits, as the Ala cannot compensate for the loss of this base-specific contact. As a consequence, the NF-κB p50 and p52 subunits prefer a 5' half site that begins 5'-GGG and is 5 bp in length while the other subunits (RelA, RelB, or c-Rel) bind preferentially to 4 bp half sites that begin 5'-GG. A central bp, which is nearly always A:T, is not contacted by either subunit, suggesting that homodimers of p50 or p52 would bind optimally to an 11 bp κB DNA (two 5 bp half sites and a central A:T bp) while RelA, RelB, and c-Rel prefer 9 bp κB DNA. This also perfectly explains the original observation of NF-κB p50:RelA heterodimer bound to the 10 bp κB DNA from the enhancer of the Ig κ light chain gene (31).
The central A:T bp serves as a point of reference in studying base-specific interactions between NF-κB subunits and κB DNA. The 5' G that is contacted by His64 of the p50 subunit occupies the position ±5 bp from this origin. The G:C bp at positions ±4 and ±3 are contacted similarly by each of the NF-κB subunits. The two invariant Arg (Arg56 and Arg54 in murine p50) make direct contact with these two G bases and the invariant Glu contacts the paired C at the ±3 position. Recognition of both nucleotide bases at this position suggests a more important role of the G:C bp at position ±3 than either the G:C bp at the ±5 and ±4 positions. This explains the observation that at least one of the half sites contains the G:C bp at this position in all κB DNA known to date.
NF-κB binding to consensus κB DNA at the inner positions
Base pairs at positions ±2 and ±1 in κB DNA exhibit more variability in sequence than the peripheral bases. In the crystal structure of the NF-κB p50:RelA heterodimer in complex with κB DNA from the Ig κ light chain gene, an Arg residue contained within the linker region that joins the NTD and DD in the RelA subunits crosses over and contacts the T of an A:T bp at position +2 (Fig. 5A). An identical Arg is present in c-Rel. An analogous Lys residue at the corresponding position in p50 and p52 can interact with both T in an A:T bp or G from a G:C bp at the same position. Base pairs at position ±1 in κB DNA do not participate in any contacts with either RelA and c-Rel. However, the Lys residue within the interdomain linker of p50 and p52 can mediate contacts at this position dependent on the DNA sequence. The corresponding residue in RelB is also a Lys (Lys274 in murine RelB). However, rather than DNA contacting this Lys, it projects inward to make an ion pair with Asp272. This observation suggests that RelB subunits might tolerate more sequence diversity at the inner positions of its κB DNA targets.
An invariant Tyr of loop L1 (Tyr57 in murine p50 and Tyr36 in murine RelA) participates in stacking interactions with bases at both ±1 and ±2 of the same strand. This stacking is favored by the presence of two successive T bases, as their exocyclic 5-methyl groups favor the interaction. Although a Phe at the same position could substitute for Tyr and maintain these stacking interactions, Tyr also participates in hydrogen bonds through its hydroxyl group making Tyr an absolutely required residue for DNA recognition and binding. Either two C bases or any combination of T and C can also be accommodated at these positions, but an A or G at either position is unfavorable. The critical role played by this invariant Tyr is illustrated by the overrepresentation of the sequence AAATT or AATTT at the central 5 positions of the κB sequences recognized by RelA and c-Rel homodimers. It is likely that these Tyr base stacking interactions toward the center of the 9 bp κB sites preferred by RelA and c-Rel compensate for the fact that these NF-κB subunits contact fewer flanking GC bp.
Stabilization of NF-κB:DNA complexes
The interaction of proteins can significantly alter binding affinity of NF-κB:DNA complexes. This can be true even if the protein binding is distal from the NF-κB:DNA interface. Both protein-DNA and protein-protein interactions are interdependent. This means that assembly of NF-κB into larger transcription complexes can be affected by subtle changes in DNA conformation. This point is illustrated by two loops, one from the DD and the other from the NTD, which play particularly important roles. The βf-βg loop of the NF-κB DD projects toward κB DNA but does not directly contact it (Fig. 3A). Two conserved acidic residues (Asp267 and Glu269 in chicken c-Rel) are located within this loop and reside near the DNA in the complex between c-Rel homodimer and the IL2-CDRE κB DNA complex. These residues would be expected to repel DNA and weaken binding (24). However, these negative charges are neutralized by an Arg in loop L1 (Fig. 3A). Loop L1 is the same loop that contributes 5 of the 6 base-contacting residues. Loop L1 can be divided into three parts: N-terminal front, N-terminal back, and C-terminal flexible part. The C-terminal portion of loop L1 is flexible and can contact the DNA backbone of nucleotides flanking the κB sequence. The N-terminal front and back end forms a rigid core structure that remain unchanged both in DNA-bound and -unbound forms. The surface residues projected from the front portion contribute the DNA base-contacting residues. An Arg on the back surface of the N-terminal portion contacts the acidic residues of the βf-βg loop. Interestingly, not all NF-κB:DNA complex crystallographic structures show this protein-protein interaction. We suggest that DNA conformation differences play a role in dictating RHR interdomain interactions. In the case of oncogenic v-Rel, a viral homologue of c-Rel, two core residues within the rigid part of Loop L1 are mutated. These two residues are at least partly responsible for altered DNA binding profiles by v-Rel as compared to c-Rel (32). Finally, these two loops also undergo modification, which also appears to regulate NF-κB DNA recognition as discussed below.
RelA and c-Rel homodimers bind classical κB sites such as Ig-κB or IFNβ-κB with lower affinity than their heterodimers with p50. Since historically κB DNA is used to detect NF-κB activation in electromagnetic shift assay (EMSA), low DNA binding activity of the RelA homodimer made people believe RelA had little or no DNA binding activity even when it acts as a heterodimer with the p50 subunit. Therefore, until the late 1990s, it was thought that RelA homodimer binding to DNA was artificial and that RelA homodimers had no physiological function.
κB DNA dynamics
One of the intriguing regulatory aspects of protein-DNA recognition involves DNA dynamics. A major question in the NF-κB field is the role of κB sequence specificty in gene regulation. Even a single bp variation within the κB site can impact gene regulation. A single nucleotide variation can affect direct contact with NF-κB or indirectly affect binding by DNA dynamics change due to the sequence change or both. For instance, it is known that the dynamics of the TA base step is different than the AT or AA base step. DNA dynamics and conformational differences are often difficult to measure accurately from these rather simple sequence alterations. However, it can be inferred by measuring differences in affinity and/or kinetics of binding in solution, and possibly through the elucidation of structures of complexes with mutant DNA. Although the dynamics of DNA due to a TA base step might be essential for specific recognition in some protein-DNA complexes, the TA base step is rarely observed in κB sequences. Although both GGAATTTCC and GGAAATTCC sequences are optimum targets for the RelA or c-Rel homodimers from the standpoint of hydrogen-bonding contacts, the GGAATATCC and GGAAAATCC sequences are not ideal κB sequences. Although these two DNAs should make identical contact with NF-κB, the presence of an A in the second half site is detrimental but when it is present next to T, the effect becomes significantly worse. The TA step is expected to induce dynamics of the neighboring bp, resulting in a reduced binding affinity. Therefore, GGAAAATCC can be more common than GGAATATCC in spite of the fact that the central bp (underlined nucleotide) does not contact protein.
Molecular dynamics simulations of the IL-2-κB (AGAAATTCC) site embedded within a 22-mer duplex DNA over a long time scale (1-µs) has revealed striking structural transitions (33). At around 0.7 µs of simulation, the central A:T bp becomes completely sheared, and the bases undergo cross-stand stacking. This phenomenon leads to the flipping of neighboring thymine at position –1, leaving its paired adenine free. These structural changes might be linked to sequence. For example, interruption of an otherwise AT-rich central sequence by a G:C bp may not result in similar structural changes. In all, these studies on free κB DNA strongly suggest that sequence-dependent DNA conformations play a vital role in NF-κB recognition. Moreover, sequence variations in the flanking regions can also affect the conformation of the κB DNA in such a way that NF-κB binding affinity is altered. We have observed that the overall conformations of all NF-κB:DNA complexes studied thus far exhibit unexpected differences. Nuclear magnetic resonance (NMR) analysis of both wildtype and mutant human immunodeficiency virus (HIV) κB DNA sequences showed some key differences in their conformations. Although the base changes were introduced at the 5′-junction in the mutant κB DNAs, differences in conformation within the κB DNA core were observed. These mutant HIV κB DNA sequences used in the NMR studies showed reduced NF-κB binding suggesting that the flanking sequence negatively affects DNA binding by changing the preferred phosphate conformation (34, 35).
The presence of a G:C or C:G bp at the central position is fairly uncommon among κB sites, although this position does not directly contact protein. A G:C or C:G bp could also potentially affect the conformation/ dynamics of the neighboring sites. For instance, the narrow width of the minor groove at the center, a universal feature of A:T/T:A centered κB sites observed to date, might be difficult to achieve with a G:C/C:G bp at the center. The X-ray structure of a free κB DNA allowed for comparison of the structural transitions of the same DNA upon binding to a RelA:RelA homodimer (36). The sequence of the central part of the DNA was identical to another DNA sequence whose X-ray structure is available. The features of the central AAATTT sequence in both DNA sequences are similar with a small roll angle, and consequently, these DNAs are closer to ideal B form. RelA induces a smooth bending around the central AT-rich sequence. Moreover, the free κB DNA exhibits a wider minor grove and deeper major groove. NF-κB binding results in narrowing of the minor groove and widening of the major groove where direct protein contacts are observed. We suggest that RelA and c-Rel homodimers would discriminate a κB site with G/C-centric κB sites. The case might be different for p50 and p52 homodimers. Because greater flexibility of the Lys side chain (Lys241 in p50) instead of an Arg side chain (Arg187 in RelA) will allow it make contact with diverse sequences at the inner core.
Non-classical κB sites and transcriptional regulation
It is not clear from structural analysis alone why A:T is by far the preferred central bp (position 0). It is likely that the presence of this base pair is necessary to convey DNA bending and/or dynamic characteristics necessary for optimum NF-κB:DNA complex formation. However, it is possible that most of the κB DNA identified are heavily biased due to the traditional experimental set up. These experiments have selectively looked for early response sites through tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), or lipopolysaccharide (LPS), which preferentially activates the p50:RelA heterodimer. This dimer prefers the canonical sites described above. However, recent experiments have identified κB sites that deviates significantly from the classical sites. Indeed, having only one good half site may allow binding of a dimer to a site as long as the other half site does not contain bps are facilitate rapid dissociation of the complex.
Post-translational modification and NF-κB:DNA complex regulation
A recent report has shown that monomethylation of RelA at Lys37 in response to induction by TNF-α and IL-1 is required for the expression of a subset of NF-κB target genes (37). The methylated form of RelA displays extended gene activation as a result of prolonged DNA binding by RelA. Although a detailed mechanism to explain how modification of this residue might affect DNA binding is lacking, its position within loop L1 suggests that the effect is likely indirect through altering the conformation the residues that directly contact DNA. It is important to mention here that some of the DNA contacting residues from loop L1 contact one another further stabilizing the loop L1 conformation and allowing them all to act as a unit. In p50, Glu60 bridges Arg54 and Arg56 and together they contact DNA as a structured module. The stability and utility of this folded polypeptide structure was illustrated when it was found to be exploited by RNA in selection binding experiments (38). In RelA and c-Rel, a similar Glu brings together one of the two Arg residues form the loop L1 and the Arg from the interdomain linker. These cooperative interactions between the amino acids side chains not only maintain a properly oriented conformation of the functional groups primed to contact DNA but also contribute to differential base pair selectivity. Hence modification of lysine could affect orientation of these residues as well as residues that are involved in contact with residues at loop βf-βg (discussed earlier).
Cells expressing a RelA Ser276Ala mutant show dramatically reduced transcriptional activity. This Ser has been shown to undergo phosphorylation by two different kinases, MSK and protein kinase A (PKA), and this post-translational modification is essential for RelA transcriptional activity (39–42). Although the RelA Ser276Ala appears to bind DNA, defects in DNA binding affinity cannot be completely ruled out in light of the importance of other residues in the same loop in the protein-DNA complex formation. Phosphorylation at position 276 has also been shown to be required for coactivator recruitment. Therefore, in addition to affinity modulation, phosphorylation may play a role in changing chromatin dynamics through acetylation activity of CBP/p300.
Rules of NF-κB:DNA complex formation in vivo
One aspect of NF-κB regulation that has not yet been adequately addressed experimentally is the potential for exchange of subunits between active NF-κB dimers in the nucleus. The available structural and biochemical data suggest that this is likely to occur, especially in light of the relatively low dimerization affinity exhibited by many of the NF-κB dimer combinations and the preference of different κB DNA half sites toward specific NF-κB subunits. Functionally, it seems reasonable that such an exchange might coincide with the transition of a gene promoter from a repressed to an activated state. Homodimers of p50 and p52 are present in the nucleus of uninduced cells. Rapid mobilization of additional NF-κB subunits to the nucleus in response to IκB degradation could then lead to the replacement of repressive NF-κB with dimers that possess inherent transcription activation potential. It is not known whether the repressive p50 and p52 homodimers are directly exchanged with p50:RelA or other activating homo- and heterodimers or, alternatively, if the individual p50 or p52 subunit monomers can exchange with a RelA monomer on κB DNA. Interestingly, analysis of the mice lacking the gene encoding IκBNS revealed a significant decrease in IL-2 production. ChIP assays on T cells revealed that IκBNS co-localizes with p50 homodimer at κB DNA and remains associated even after the p50 is dissociated (43). Therefore, it is possible that nuclear NF-κB subunit exchange on κB DNA falls under the purview of the nuclear IκB proteins.
The IκB Family
There are eight known IκB family proteins (Fig. 2). They belong to a family by virtue of the presence of ankyrin repeat domain (ARD) that is funcitonally linked to the NF-κB dimers. The ankyrin repeat is a roughly 33 amino acid tandem helical repeat motif that appears in multiple copies in numerous proteins (44, 45). Using this domain, IκB proteins can form biological complex with at least one NF-κB dimers. These proteins are IκBα, IκBβ, IκBε, IκBγ (NF-κB1/p105), IκBδ (NF-κB2/p100), Bcl3, IκBζ and IκBNS. Three of these IκB proteins, IκBα, IκBβ, and IκBε, mediate classical IκB activities as they relate to NF-κB binding in the cytoplasm and phosphorylation-dependent proteasome-mediated degradation in response to induction.These classical IκB proteins contain six ankyrin repeats. The ARD is flanked by sequences that are predicted to be unstructured. The classical IκB proteins preferentially bind to NF-κB dimers that contain at least one RelA or c-Rel subunit.The N-terminal flexible regions in IκBα, IκBβ, and IκBε contain two serine residues within the consensus sequence DSGXXS that are sites for phosphorylation by the IKKβ subunit. Once phosphorylated, this N-terminal region serves as the recognition site for the E3 receptor β-TrCP for IκBα polyubiquitination (Reviewed in Kanarek & Ben-Neriah, this volume). Ubiquitinated IκBα is degraded by the 26S proteasome (46). The importance of this N-terminal region for the inducible proteolysis of classical IκB proteins is illustrated by the fact that an IκBα molecule with its two IKK phosphorylation sites mutated to Ala functions as a ‘super-repressor’ of NF-κB activation through the canonical NF-κB activation pathway (47). C-terminal to the ARD is a structurally flexible region rich in the amino acids proline, glutamic acid, serine, and threonine (PEST). This so-called PEST region is common to many proteins that exhibit rapid turnover in the cell (48) .
The unprocessed NF-κB1/p105 and NF-κB2/p100 also act as inhibitors of NF-κB (49, 50). However, unlike the oligomerization exhibited by IκBα, IκBβ, and IκBε, where a monomeric IκB assembles with a single NF-κB dimer to form an inactive complex, p100 and p105 assemble into larger complexes wherein they integrate two inhibitor molecules and at least two NF-κB molecules (51). p105 dimer binds RelA, c-Rel, and its own processed form p50. p100 prefers RelB and p52 for binding and to a lesser extent RelA. The complete degradation of p100 releases different NF-κB subunits, which then could initiate physiologic programs that are distinct from those regulated by p105 and the classical IκB inhibitors.
Three additional ARD-containing polypeptides have been shown to participate in NF-κB regulation. These are Bcl3, IκBζ, and IκBNS. Of these, Bcl3 is the most well-studied because of its early discovery (52, 53). Though they are grouped with other IκB proteins because of their structural similarity and abilities to bind NF-κB dimers, Bcl3, IκBζ, and IκBNS exhibit significant differences from classical IκB proteins (54–57). First of all, they display binding specificity toward p50 and p52 homodimers. Each of the three proteins migrates to the nucleus when over-expressed in cells, leading to their classification as ‘nuclear IκB proteins’. However, more recent experiments show that activities of these proteins are also regulated at least in part by the nuclear localization and half-life. In the nucleus, it appears that these proteins play regulatory roles that may include chromatin rearrangement, NF-κB dimer exchange, and co-activation of specific NF-κB target gene expression. IκBζ, for example, was shown in mice knockout studies to be necessary for the NF-κB-induced activation of the inflammatory cytokine IL-6 in response to LPS treatment of peritoneal macrophages (58). The functional regulation of Bcl3 is particularly complicated as it has been shown that both the phosphorylation status and partner selection dictate whether Bcl3 might act as a co-activator, a co-repressor, or an inhibitor (59).
IκB structure and regulation of NF-κB: an overview
IκB proteins control induction of NF-κB in a stimulus-specific manner. In particular, the rapid and transient activation of NF-κB that is required to mount immune and inflammatory responses is mediated by the degradation of classical IκB proteins, IκBα, IκBβ, and IκBε, and the subsequent transcriptional upregulation of response genes. The non-classical IκB proteins, p105 and p100, induce a more prolonged gene activation program, as these IκB proteins are co-opted for a slow degradation processes. Finally, atypical IκB proteins, Bcl3, IκBζ, and IκBNS, regulate NF-κB activity at the level of transcription.
IκBα binding to the NF-κB p50:RelA heterodimer
The primary function of IκBα is to inhibit the DNA binding activity of NF-κB. It does so by biasing the steady-state level of IκB:NF-κB complexes more in the cytoplasm rather than in the nucleus. Inhibited IκB:NF-κB complex is posed for rapid activation of NF-κB via fast IκBα degradation. The X-ray crystal structures of IκBα bound to the NF-κB p50:RelA heterodimer have provided insight into how IκBα inhibits NF-κB activity (60, 61). The modular binding surface can be divided into three distinct segments. First is the rigid body interaction between the ARD of IκBα and the p50:RelA dimer platform (Fig. 6A).This interaction interface, which is mediated primarily by Van der Waals interactions, accounts for the greatest amount of buried surface area in the complex. The second mode of interaction is mediated by C-terminal NLS polypeptide region beyond the DD of RelA and the first two ankyrin repeats of the IκBα ARD. The C-terminal extended portion, which is flexible in its unbound state, binds to IκBα by forming two helices that mediate specific ion-pair and hydrophobic interactions between conserved amino acid side chains from both proteins. Indeed, the complementary interactions at this site are responsible for the majority of the binding energy of the complex. The third mode of interaction involves the C-terminal PEST region of IκBα, which binds the NTD of the RelA subunit through dynamic long-range electrostatic interactions. This interaction converts the RelA subunit NTD into a conformation relative to the DD that is distinct from that observed when RelA binds DNA (discussed in the next section). The structures explain why binding of NF-κB to IκBα inhibits its ability to bind to DNA. In addition, they suggest that IκBα conceals the NLS of RelA explaining the primarily cytoplasmic localization of the IκBα:p50:RelA complex.
Fig. 6. Ribbon diagrams of IκB: NF-κB complexes.

(A) X-ray crystal structure of IκBα in complex with NF-κB p50:RelA heterodimer (PDB 1NFI). The helical ankyrin repeat domain (ARD) of IκBα is depicted in gold; the NF-κB p50 and RelA subunits are in green and red, respectively. RelA NTD is contacting the PEST sequence of IκBα. (B) X-ray crystal structure of IκBβ in complex with NF-κB RelA homodimer (PDB 1K3Z). IκBβARD is depicted in grey.
Regulation of IκBα:NF-κB complex formation
Biophysical analysis of free IκBα in solution revealed that only the first four ankyrin repeats adopt a stable folded structure, while the two remaining C-terminal repeats and the contiguous PEST sequence remain mostly unfolded (62). This is rare for ARD-containing proteins, most of which display high folding stability in solution (63). Upon binding to NF-κB, however, the six ankyrin repeats of IκBα stack as a continuous folded domain (64). These observations suggest that as the disordered NLS polypeptide region of RelA adopts an ordered structure upon binding to the more stable ankyrin repeats 1 and 2, the binding energy allows folding of the last two repeats which in turn are involved in binding the DD of the p50:RelA heterodimer. Ferreiro et al. (65) have introduced mutations in IκBα that do not directly contact NF-κB but converts non-consensus repeats 6 to a more consensus repeat. As expected, these mutants are better folded. However, these pre-folded mutants showed reduced binding to NF-κB (65, 66). Kinetic analysis revealed that an increased rate of dissociation of the pre-folded mutants was responsible for the observed decrease in NF-κB binding. Therefore, by coupling folding and binding, IκBα significantly decreases its rate of dissociation from NF-κB resulting in a high affinity protein-protein interaction (KD in the high pM range). These observations, based on structural studies and biophysical characterization, serve to explain why IκBα must be actively removed via the 26S proteasome-mediated proteolysis in order to supply free NF-κB for inducible gene expression. They also suggest that deviations from consensus ankyrin repeat sequence endow IκBα with its signature NF-κB binding and regulatory properties.
The IκBβ:RelA homodimer complex
The X-ray crystal structure of the IκBβ:RelA homodimer complex revealed similar modes of interaction between IκB and NF-κB proteins (12) (Fig. 6B). However, there are clear differences. First, interactions between the ARD and an NLS are nearly identical. Interestingly, the NLS of the second RelA subunit also appears to interact weakly. Although IκBβ alone was shown to partially protect this second RelA subunit NLS polypeptide from proteolysis with limiting amounts of protease in vitro, it appears as if the IκBβ requires some other component to stabilize its complex with RelA homodimer. Second, the last ankyrin repeat (6th) of IκBβ appeared to be less intimately involved in the NF-κB binding as compared with the similar region of IκBα in the IκBα:NF-κB complex structure. This observation may explain why the C-terminal PEST sequence in IκBβ is not critical for interaction with the N-terminal domain of RelA as it appears to be positioned away from the protein-protein interface. Third, IκBβ contains a unique insertion of 42 amino acids in length located between ankyrin repeats 3 and 4. This insert, the majority of which is disordered in the x-ray structure, is projected into solution far from the protein-protein interface. It is likely that the insert is used for other purposes such as binding to other factors.
The IκBβ:RelA complex also provides intriguing insights into how IκBβ might bind and regulate activity of the NF-κB c-Rel homodimer. It was previously reported that IκBβ interacts with c-Rel in a phosphorylation-dependent manner, whereby two Ser in the IκBβ PEST sequence (Ser313 and Ser315 in murine IκBβ) must be phosphorylated (67). This suggests the intriguing possibility of two distinct modes of NF-κB inhibition by IκBβ: one that relies primarily on interactions between the IκBβ ARD and RelA DD and NLS polypeptide and another that involves the phosphorylated IκBβ PEST and c-Rel. The requirement of PEST phosphorylation for IκBβ:c-Rel homodimer complex formation further suggests that the c-Rel NTD may play a role in IκBβ complex formation in a manner analogous to interactions between the analogous NTD of RelA and IκBα in the IκBα:NF-κB complex structure. Alternative binding modes by IκBβ could explain why IκBβ is not a good inhibitor of DNA binding by the RelA homodimer as the unphosphorylated IκBβ PEST does not engage the RelA NTD. In contrast, a PEST-phosphorylated IκBβ might be able to inhibit DNA binding by the c-Rel homodimer. In contrast to IκBα, which can readily dissociate RelA homodimer or p50:RelA heterodimer from their complexes with target DNA, IκBβ appears to be unable to carry out this function. Future experiments will determine whether IκBβ is capable of stripping c-Rel from κB DNA. Finally, it has been observed that deletion of the insert between ankyrin repeats 3 and 4 of IκBβ reduces its affinity for c-Rel homodimer. κB-Ras, a small GTPase, was shown to be involved in IκBβ-mediated inhibition of NF-κB and might interact with IκBβ:NF-κB complexes through this inter-repeat loop (68, 69).
NF-κB regulation by IκBε
IκBε was originally reported to inhibit homodimers of RelA and c-Rel (70–72). However, recent work also suggested that IκBε negative feedback regulates p50:RelA to dampen IκBα-mediated oscillations (73). Significant differences in domain architecture between IκBε and other classical IκB proteins include the relative absence of acidic amino acid residues within the C-terminal PEST region and an extended N-terminus. These differences may allow IκBε to use these peripheral regions to specifically recognize features unique to RelA or c-Rel homodimers. More structural and in vitro biochemical studies are required in order to gain mechanistic insight into how IκBε regulates NF-κB activity.
Non-classical IκB protein- p105 and p100
The paradigm of NF-κB regulation in the cytoplasm for the better part of the past 25 years has relied upon stimulus-dependent rapid degradation of IκBα followed by nuclear translocation of the NF-κB p50:RelA heterodimer. Recently, it has become increasingly clear that the NF-κB precursors p105 and p100 are important IκB inhibitors. The ability of p105 to function as an IκB molecule was demonstrated previously (74, 75). Furthermore, the biological significance of the inhibitory activities of both p105 and p100 have been evident for many years since mouse studies revealed that chromosomal deletion of their C-terminal ARD leads to severe misregulation of NF-κB (76, 77). However, as p105 and p100 also function as the immature precursors of the NF-κB subunits p50 and p52, respectively, dissecting the specific consequences on NF-κB regulation due to modification or disruption of these proteins has been a challenge. Recent experiments have established that two copies of p100 and p105 can assemble wherein diverse NF-κB subunits are bound (51). The p105 protein binds and inhibits RelA, c-Rel, and its own processed product p50. p100, in contrast, binds and inhibits all NF-κB subunits. In addition to more relaxed NF-κB binding specificity, multiple NF-κB subunits can remain associated in a single p100 or p105 inhibitory complex. That is, different NF-κB subunits can be released through degradation of these inhibitors. Both p100 and p105 are degraded with slower kinetics than the classical IκB proteins for different reasons. Slower degradation of p100 is due to the slower activation kinetics of kinases (NIK/IKK1) essential for p100 phosphorylation. Although p105 phosphorylation and degradation follow similar pathways as IκBα degradation, IKK2 phosphorylates IκBα rapidly, while it phosphorylates p105 with slow kinetics (Reviewed in Gantke et al., this volume). The observed differences in structural arrangement, binding specificity, and degradation kinetics exhibited by the non-classical inhibitors result in distinct kinetic profiles of NF-κB activation, identity of activated NF-κB subunits, and post-induction repression as compared to the classical inhibitors. Since over half of cellular RelA and c-Rel and all of RelB are bound to p100 and p105 in the steady state of most cells, they impact profoundly on NF-κB -mediated cellular regulations (78, 79). The unique physiological consequences of NF-κB regulation by p105 and p100 are just beginning to be determined (80, 81).
Post-induction repression of NF-κB by IκB
Most NF-κB activating signals, such as TNF-α and IL-1, lead to the elevated expression of the NF-κB-inducible IκB proteins IκBα, IκBε, p105, and p100. The newly synthesized inhibitors can then function to repress NF-κB activity. IκBα-mediated NF-κB repression activity has been studied extensively. Free IκBα enters the nucleus where it is capable of binding and disrupting NF-κB:DNA complexes (82) and forming new IκBα:NF-κB complexes. Continuous signaling however degrades IκBα of newly formed IκBα:NF-κB complexes sending a second wave of free NF-κB. Indeed, prolong stimulation can results in NF-κB activation in periodic phases (14, 83). Computational modeling of IκB:NF-κB regulation using a systems biology approach has correctly predicted the temporal control of NF-κB in response to several stimuli. Since RelA can activate other IκB inhibitors, then in principle, newly synthesized IκB molecules should bind RelA and other NF-κB liberated during stimulation. Indeed, newly synthesized p100 (IκBδ) has been shown to trap p50:RelA NF-κB during the later stages of induction to provide negative feedback inhibition (84). This newly synthesized IκBδ inhibitory complex can subsequently become the target for non-canonical signaling. Therefore, NF-κB signaling pathways are intricately intertwined and susceptible to alteration as cells respond continuously to their environment (85). However, the mechanism by which IκBα and p100 squelch NF-κB might be different. IκBα actively destabilizes and dissociates NF-κB:DNA complexes. NMR studies have revealed that using the acidic PEST tail sequence IκBα makes contact with NF-κB residues that are present in the vicinity of DNA. Indeed, a transient ternary complex has been shown to form before full dissociation of the NF-κB:DNA complex and formation of the new IκBα:NF-κB complex (Fig. 7A). It would be important to test if p100 can associate with p50:RelA using a similar strategy.
Fig. 7. A cartoon model of the IκB:NF-κB:κB DNA ternary complex formation.

(A) Unstable ternary complex of IκBα:(p50:RelA):DNA. Addition of κB DNA causes the dissociation of the IκBα PEST sequence and ANK 5–6 from NF-κB. (B) Bcl3:p52 homodimer forms a relatively stable ternary complex with κB DNA comparing to IκBα.
IκB as enforcer of NF-κB dimer formation
The IκBα:NF-κB complex crystal structure hints that IκB proteins might influence NF-κB dimer formation. IκBα sits atop the p50:RelA heterodimer interface forming a ternary interface, suggesting that IκBα could function to further stabilize the NF-κB dimer (Fig. 6A). Since IκBα binding affinity is much higher than NF-κB dimerization affinity (KIκB:NF-κB <0.1 nM compared to Kdim~0.1 µM), IκBα must bring together two NF-κB subunits at concentration much lower than the Kdim. One possible functional advantage of this IκB-mediated NF-κB dimer stabilization is that different IκB proteins could catalyze the assembly of otherwise rare/weak NF-κB dimers. For example, as mentioned previously, the free NF-κB p52 homodimer has not been observed in vivo. However, the p52 homodimer bound to the nuclear IκB protein Bcl3 has been detected. This finding suggests that, although p52 preferably forms heterodimers with RelB, interaction with a specific IκB molecule can induce the formation the p52 homodimer.
Two recent reports have shown that IκBβ acts as an co-activator of transcription by preferentially interacting with the c-Rel:RelA heterodimer or RelA:RelA homodimer and recruiting them to NF-κB target promoters. Both c-Rel and RelA form preferential heterodimers with p50. They also form homodimers. Therefore, when all four subunits are present in similar amounts distribution of all possible dimer would depend on the dimer affinities. Such distribution can alter in the presence of IκB proteins if they exhibit preferential dimer association. We suggest that all IκB proteins effectively alter the distribution of cellular NF-κB dimers by virtue of their differential affinities towards different dimers.
IκB at the DNA sites
Three of the IκB proteins, Bcl3, IκBζ, and IκBNS, act as transcriptional regulators by directly associating with the NF-κB:DNA complexes. While Bcl3 and IκBζ have been shown to act both as co-repressors and co-activators of transcription, IκBNS is known only for its co-repression activity. Bcl3 acts as a co-activator of p52 homodimer and shown to activate the expression of Cyclin D1, P-selectin, and Skp2 genes. We suggested that part of the Bcl3 co-activator function might come from its ability to induce the formation of p52 homodimer in addition to its ability to recruit histone acetyl transferases such as the Tip 60 complex. How Bcl3 also represses transcription is a subject of debate. One thought is that Bcl3 inhibits DNA binding like the classical IκB proteins or it binds to a subclass of NF-κB:DNA complexes and functions as a co-repressor as IκBNS. IκBζ acts as a co-activator of p50 homodimer. Genes such as IL-6, β-defensin, neutrophil gelatinase-associated lipolactin (NGAL), and TNF-γ are known to be activated by the p50:IκBζ complex. They are particularly important in innate immunity response. Like Bcl3, IκBζ provides the transcriptional activation domain in its N-terminus, possibly to recruit histone acetyl transferases.
Although the classical IκB proteins, IκBα, IκBβ, and IκBε, are famously known for their NF-κB inhibitory activity, two of these proteins also function as transcriptional coregulators of NF-κB. It is possible that all three of them might act both as an activator or an inhibitor of NF-κB under specific cellular conditions. The first convincing evidence that the classical IκB protein can also act as a transcription activation has been described only recently, where it was shown that IκBβ activates transcription through association with either RelA homodimer or c-Rel:RelA heterodimer by binding to κB sites present in the promoters of IL-1β or TNF-α (13, 86). It is not difficult to envision from the three dimensional structure of IκBβ:RelA complex that this binary complex can associate with κB DNA forming the ternary complex. Intriguingly, promoter association of IκBα was also reported, and it was shown IκBα binding is essential for Hes transcriptional repression (87). As discussed earlier, in vitro experiments also revealed a quarternary complex between IκBα:p50:RelA:DNA. An intriguing hypothesis would be that while IκBα eventually removes NF-κB p50:RelA heterodimer from some promoters, it can remain associated with the same or different NF-κB dimers for a ‘long enough’ time such that it effectively functions as a co-activator. That is, promoter directed half-life of IκBα would dictate inhibition versus activation. A similar ternary complex is also expected to be formed by IκBε.
Kinetics of IκB degradation: steady state and inducible
An important aspect of NF-κB activation emerged from the structures of IκB proteins. H/D exchange and NMR studies have revealed that IκBα is a weakly folded protein. As a consequence, IκBα undergoes rapid degradation in its ‘free’ form. Degradation of IκBα requires proteasome but not IKK-mediated phopshorylation or ubiquitination. Free 20S proteasome or, more likely, 20S bound to regulatory REG complex targets the C-terminus of IκBα for rapid degradation. The feature of this degradation mechanism, which is referred to as Ub-independent degradation, is the presence of multiple degrons within the PEST, 6th and 5th ankyrin repeats. These degrons are recognized by the 20S:REG proteasome complex. A Ub-independent degron contains a patch or patches of hydrophobic residues within an extended unfolded region. This C-terminal degron of IκBα is concealed when it is bound to NF-κB. Therefore, Ub-independent degradation of IκBα in IκBα:NF-κB complexes is non-operational. The N-terminal degradation signal however remains exposed both in the free and NF-κB bound form of IκBα. The N-terminal degradation signal uses phosphorylation (at Ser32 and Ser36) and ubiquitination (Lys21 and Lys22) for IκBα degradation. Existence of a rapid degradation of free IκBα became evident in NF-κB deficient mouse embryonic fibroblasts. Levels of all three classical IκB proteins are extremely low in these cells, which suggested low protein stability was at least partly responsible. Since that observation a series of experiments dictated from modeling clarified the steady state regulation of IκB-NF-κB. In resting cells, low levels of nuclear NF-κB allows continuous synthesis of IκBα which undergoes continuous degradation primarily by Ub-independent pathway such that free IκBα does not build up in the cell to dampen NF-κB activation upon stimulation. This free IκBα pool is not entirely wasted, as it captures excess free NF-κB that arise from continuous degradation of bound IκBα due to low level of cellular IKK activity. Homeostatic control of nuclear NF-κB through continuous synthesis and degradation of free and bound IκBα is essential for cell physiology as many of essential NF-κB target genes must be expressed at a steady level to properly maintain several cellular functions such as cell survival (Fig. 8).
Fig. 8. A model of the degradation pathways controlling IκBα in basal and stimulated cells.
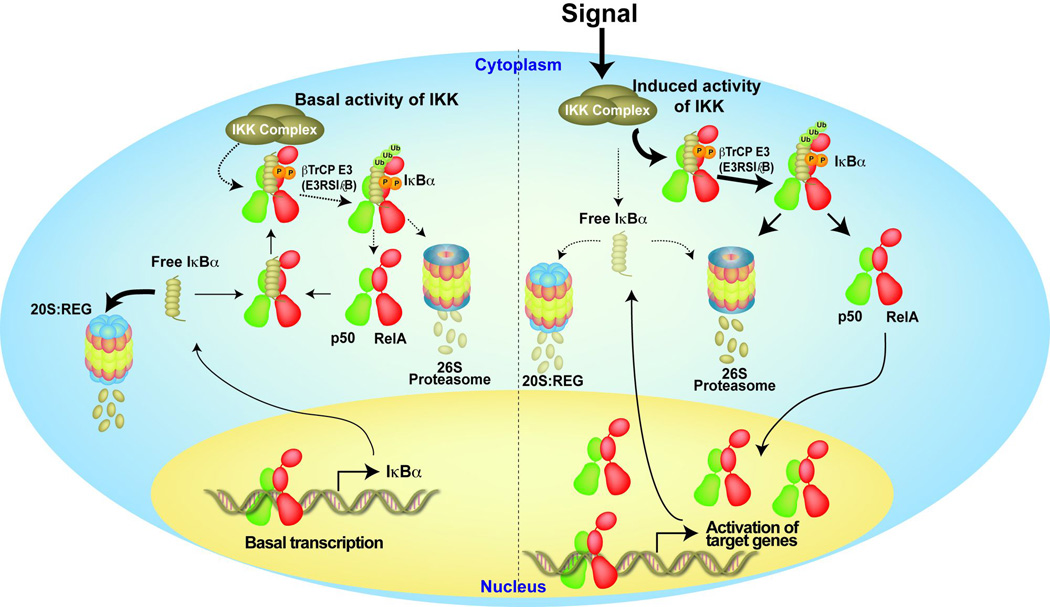
In the resting cells (left), IκBα is continuously synthesized and degraded in IKK and Ub-independent pathway (this pathway is dominant as denoted by the bold arrow), and at the same time, NF-κB bound IκBα is also degraded by the basal IKK activity. Upon stimulation (right), the IKK activity is increased (shown by bold arrow) such that most IκBα is rapidly degraded and leads to the NF-κB activation. Free IκBα is being continuously degraded to allow for the rapid and robust NF-κB activation.
The IKK Family
The IKK family is a distinct family of protein kinases in having an approximately 300-residues long homologous segment located at the center flanked by a ubiquitin-like domain (UBL) and the protein kinase core domain (KD) at the N-terminus and a flexible adapter binding segment in the C-terminus (Fig. 9A). Four kinases constitute the IKK family; IKK1 (also known as IKKα), IKK2 (IKKβ), IKK3 (IKKε), and TBK1. IKK1 and IKK2 can form homodimers and heterodimers. Similarly, IKK3 and TBK1 also form homodimers and heterodimers (88). Thus, the IKK family can be divided into two sub-families. Although IKK3 and TBK1 participate in NF-κB activation pathway, they are not directly involved in IκB phosphorylation leading to its degradation, which is the primary function of IKK1 and IKK2. We shall confine our discussion to IKK1 and IKK2 only.
Fig. 9. The IKK family.

(A) A schematic representation of IKK family proteins. Ribbon diagrams of (B) NEMO coiled coil domain (CC) in complex with IKK2 NEMO binding domain (NBD) (PDB 3BRV); NEMO zinc finger domain (PDB 2JVX); NEMO CC in complex with linear di-Ub (PDB 2ZVO); and NEMO CC in complex with ks-vFLIP (PDB 3CL3); and (C) IKK2 dimer (PDB 3QA8).
Prototypical IKK consists of three subunits, IKK1, IKK2, and NEMO (also known as IKKγ). NEMO is an adapter molecule that shows higher affinity for IKK2 than IKK1. Whereas the trimeric IKK is most abundant, both IKK1 and IKK2 homodimers also exist in cells. IKK2 is primarily responsible for activation of NF-κB through signal-dependent IκBα phosphorylation. Different stimuli regulate IKK2 activity by distinct but overlapping mechanisms, which include K63-linked ubiquitination of membrane proximal adapter proteins and activation of upstream kinases such as NIK, TAK1, MEKK3, and p38. NEMO is primarily a coiled coil (CC) protein with occasional interruptions ending with a Zn finger motif at the extreme C-terminus (Fig. 9A). The N-terminus of NEMO interacts with the C-terminus of IKK2 and the central CC region interacts with K63 poly-Ub chain. Thus NEMO links IKK2 to the upstream signal (89). There are a few other proteins such as ABIN2, Optineurin, and ELKS structurally similar to NEMO and act in the NF-κB activation pathway. However, the role of these proteins in the regulation of IKK activation and function has not been as well characterized as that of NEMO (90).
Several upstream kinases are known to activate IKK2. TAK1 is the best characterized of these kinases (91, 92). TAK1 remains inactive in resting cells bound to an adapter protein TAB2. K63-linked poly-Ub chain interacts with TAB2, which clusters TAK1 in close proximity leading to the activation of TAK1 through trans-auto-phosphorylation. Active TAK1 then phosphorylates IKK2 at two Ser (positions 177 and 181) located within its activation loop rendering its function as an active kinase. Mutations of both these activation loop Ser to Ala block the kinase activity while phosphomimetic Glu substitutions at these positions render IKK2 constitutively active (5, 93).
IKK1 defines the non-canonical NF-κB activation pathway, wherein association with NF-κB inducing kinase (NIK) it regulates p100 processing into p52 (94, 95, reviewed in Sun, this volume). It is thought that NIK phosphorylates the activation Ser of IKK1 leading to its activation. However, both NIK and IKK1 target p100 for phosphorylation and subsequent processing of p100 by the proteasome (96).
The IKK structure: an overview
The three-dimensional structure of the physiological IKK complex has remained elusive. Three-dimensional structures of different segments of NEMO and a large fragment of IKK2 have been elucidated recently (97–101). These structures offer insights into the mechanisms of NEMO dimerization, NEMO binding to IKK2, NEMO interactions with poly-Ub chain, and finally, IKK2 dimerization (Fig. 9B, C). However, these structures so far are insufficient to create a model of IKK2-NEMO assembly or the regulation of IKK2 activation.
Structures of NEMO sub-domains
Secondary structure predictions suggest that NEMO is a mostly helical protein with two signature coiled coil (CC) elements and a leucine zipper motif in the middle are flanked by a helical dimerization domain near the N-terminal end and a zinc (Zn)-finger motif at the C-terminus. Structures of several NEMO fragments, as free or bound to their partners, are now known. Barring the Zn finger motif rest of the protein might adopt a continuous coiled coil structure (Fig. 9A). Full length recombinant NEMO elutes as a high MW (~600 kDa) oligomer in size exclusion chromatography. The N-terminal IKK binding domain of NEMO (residues 40 to 130 or 196) has been shown to exist in multiple oligomeric states including monomeric and dimeric states. This N-terminal dimerization domain of NEMO interacts with a peptide spanning residues 703 to 743 near the C-terminus of IKK2, which is known as the NEMO binding domain (NBD). In solution NEMO:IKK2 minimal complex exists as a 2:2 complex. The X-ray structure of this minimal complex appeared as an imperfect parallel four-helix bundle where the NEMO CC dimer symmetrically interacts with two NBD helices (100) (Fig. 9B). Affinity of this complex is reasonably high (IC50 ~25 nM). One of the intriguing features of this complex is that Ser68 of NEMO undergoes phosphorylation upon TNF-α induction, which has shown to destabilize the complex (102). This observation suggests that induced phosphorylation of NEMO may have two consequences: it may signal termination of IKK activation or induce dissociation of the steady state IKK2:NEMO complex resulting in reorganization of the complex essential during signaling (90). That is, IKK2:NEMO may assemble in two or more possible ways. The X-ray structure of the central CC-LZ domain spanning residues 250 to 350 is also known both free and bound to a linear di-Ub molecule (98, 101). The CC-LZ domain of NEMO also adopts a CC structure. Two di-Ub molecules interact with the NEMO CC dimer where each NEMO protomer interacts with both di-Ub molecules. However, a recent study has shown only one di-Ub binds to the NEMO CC dimer with high affinity and that only at high di-Ub concentration the second molecule assembles. Therefore, it is unclear if NEMO contacts two di-Ub molecules or just one in vivo.
The structure of NEMO spanning residues192–250 bound to ks-vFLIP further reveals that this segment too adopts CC structure (99). In essence, there is no specific dimerization domain in NEMO. Rather, the entire length of NEMO, with the exception of the C-terminus, is able to form a CC structure either alone or perhaps in association with partner protein. The CC regions are not perfect, punctuated with several disruptions with residues out of CC register. Often these breaks within the CC regions serve as receptor sites for interacting partners. Various CC regions of NEMO can bind to different activators or inhibitors in a sequence specific manner, which is further aided by the flexible nature of the CC region adapting to the need for stable binding. The C-terminus of NEMO (residues 388–419) is a CCHC-type Zn-finger motif. The solution structure of this Zn-finger has been determined by multidimensional NMR spectroscopy (97). Its structure adopts the familiar fold of a Zn-finger motif (Fig. 9B). Furthermore, this motif has been characterized as an Ub-binding motif required for NF-κB signaling in response to TNF-α (103).
Structure of IKK2
The x-ray crystal structure of IKK2 from Xenopus oocyte has recently been determined (104) (Fig. 9C). In this structure, the C-terminal segment NBD is absent. The most important known function of the deleted region is its ability to bind NEMO as described above. Since the structure of the NBD bound to NEMO is known, at least from a structural perspective, all the important parts of NEMO and IKK2 are now known. While both the KD and ULD folds into expected conformations, the most striking feature of the IKK structure is its elongated helical structure of the C-terminal segment encompassing residues ~290 to 669. The signature leucine zipper (LZ) and helix loop helix (HLH) domains do not form separate structures but are embedded within the helical domain. This long segment forms a three-helix bundle, where one of the long helices has two interruptions. Since the resolution of the structure is low, it needs to be seen if these interruptions are functional signatures or densities of these segments are not clear at this resolution. The KD and the ULD interacts with each other and with the HSD at the base and the middle, respectively, of the HSD forming a tri-modular architecture. The HSDs of two molecules associate with each other forming the IKK2 dimer, thus the helical domain is also the dimerization domain of IKK2, and hence we will refer this domain as helical scaffold dimerization domain (HSDD).
The dimer interface of the HSD is discontinuous. Two subunits meet at both ends of the helical domain. The interface in one end is mostly hydrophobic in nature, whereas in the other end it is mostly hydrophilic. Surprisingly, mutations that disrupt the subunit interface do not abolish IκBα-specific catalytic activity of IKK. This is contrary to previous observations, which showed dimerization is essential for IKK activity (105, 106). These previously characterized hydrophobic residues (L465 and M472; all in human IKK2 numbering) are located within the core of the helical domain helping the core to properly pack. It is possible that those mutations altered the structure of IKK2 significantly indirectly affecting kinase activity. Mutations of residues that abolish both catalytic activity and dimerization are therefore structural. Dimerization-defective mutants that apparently do not alter structure fail to interact with NEMO (such as Leu654Glu) (104). This is another surprise, since the NEMO-binding domain of IKK2 (NBD) is spatially separated from the IKK2 dimerization domain. Moreover, interaction between NEMO and IKK2 is stable. Therefore, it is unclear how mutations within the IKK2 dimerization domain distal to the NEMO binding domain abolish the IKK:NEMO binding.
IKK activation mechanism
Have these structures have taught us anything about signal-induced IKK2 activation, or can we propose a structural model of an active IKK2 complex that explain results obtained from cellular experiments? The key players in rendering IKK2 activity are NEMO, poly-Ub chain (K63 or linear), TAK1/TAB1/2 complex, and IKK2. While a structural model of the IKK2:NEMO:poly-Ub:TAB2:TAK1 complex can be proposed showing how IKK2-bound NEMO can recruit TAB2:TAK1 complex, this model may not properly explain results on IKK activation accumulated over several years. Biochemical experiments showed massive shifts of the IKK2 complex into high molecular weight (MW) upon induction. The catalytic TAB1/2:TAK1 complex recruitment to the IK2:NEMO complex does not explain the shift to high MW. Binding to the IκB:NF-κB complex is also not relevant, as in induced cells, IκBα is degraded to free NF-κB. It is likely that significant structural reorganization occur that might result in a IKK2:NEMO complex with different architecture and hence a shift in MW. Cross-linking results showed that four IKK molecules are assembled with four NEMO proteins in the activated complex (107). However, the stoichiometry of IKK and NEMO in the complex in basal state is not known. Both NEMO and IKK2 are apparently capable of adopting various structures since both contain extensive helical structures. These elongated helical domains are highly flexible and malleable to alter without input of much energy. We are tempted to speculate that plastic nature of elongated helical scaffolds allows IKK2 and NEMO to assemble them in different ways before and after stimulation. Poly-Ub chains might trigger the differential IKK2:NEMO assembly after stimulation. Structure of the holo-IKK2:NEMO complexes both in their active and inactive states are required to justify this model. While we await new structures of these complexes, structural frameworks of IKK2 and NEMO sub-domains presently known will serve as valuable guidance to design experiments to investigate signal dependent activation of IKK2.
Regulation of IκB phosphorylation
Active IKK complex recognizes and phosphorylates IκB proteins at specific sites. It is clear that the phosphorylation sites of classical IκB proteins are accessible to IKK when they are bound to NF-κB. Although active IKK phosphorylates unbound IκBα in cells (15), NF-κB bound IκBα is a better substrate for phosphorylation (106). A recent report has shown that the long HSD of IKK2 interacts with the ARD of IκBα (104). Since the face of the ARD with the fingers interact with the NF-κB dimer, it is likely that the helical face of ARD opposite to the finger is contacted by IKK HSD in a ternary complex arrangement. However, how NEMO participates in this multiprotein assembly remains an unresolved issue. Phosphorylation of free IκBβ by IKK occurs at a slower rate compared to that of IκBα, although the rate of IκBβ phosphorylation remains on the time scale of minutes. Small variations in the amino acid sequences at their respective phosphorylation sites account for the observed differences in the rates of IκBβ and IκBα phosphorylation by IKK2 (108). Structural and biochemical studies indicate that phosphorylation sites of IκBβ remain accessible to IKK in the IκBβ:NF-κB complex, and it is reasonable to assume that the same is true for IκBε (12). Since it is clear that other parts of IκB also participate in the kinase recognition, sequence differences in the ARD among these IκB proteins also explains differential phosphorylation kinetics.
The state of the IKK phosphorylation sites in p100 and p105 are unknown when the inhibitor proteins are multimerized. The recent structure of the C-terminal IκBδ domain of p100 have revealed that in the oligomeric p100 complex C-terminal phosphorylation sites would remain free for kinase binding and phosphorylation. It is expected that p105 oligomerization would not engage the C-terminal region where the IKK2 phosphorylation sites are located. Therefore, differences in phosphorylation kinetics might arise from differential recognition modes as described above. Differences in the degradation kinetics of IκB may also arise from changes in the levels of ubiquitination, recognition by the proteasome, and unfolding of the IκB proteins prior to degradation. Detailed in vitro and cellular experiments to test these hypotheses are required to improve our understanding of NF-κB regulation arising from kinetic control of IκB degradation.
Perspectives: conclusions and outstanding questions
It is becoming increasingly clear that the roles of NF-κB, IκB, and IKK are extensive. However, we know little about the mechanism of how these factors function in a very fundamental away. The primary reason is very little input on structural and correlated in vitro biochemistry. The most glaring failure is in our knowledge of how IKK becomes active. It took only five years to determine the structure of an NF-κB:DNA complex after the first NF-κB gene was discovered. In just seven years after the cloning of IκBα, the structure of IκB bound to NF-κB was reported. However, it took 14 years to obtain the structure of a catalytic IKK molecule after the fist gene was cloned. This is in spite of the fact that at least 10–20-fold more scientists worked to obtain the IKK structure. It is nearly impossible to propose a rational model of IKK2 activates in vivo using the current structural information. The next challenge is to determine the IKK2:IKK2:NEMO or IKK1:IKK2:NEMO complexes. These structures will certainly provide insights into if NEMO induced IKK2 structure or how NEMO is poised to integrate other adapters to activate IKK. What is the function of IKK1 in IKK1:IKK2: NEMO complex? Why does IKK1 specifically interact with NIK? Associated with the structural studies, there must be in vitro mechanistic studies using enzyme kinetics or other strategies.
Another long-standing question is the mechanism of p105 and p100 processing. Why is p100 processed efficiently upon stimulation, whereas p105 processing is constitutive? Since both these molecules are upregulated by NF-κB and processing occur in all known cell types, it is reasonable to assume that small variations in sequences between the two precursors dictate this outcome. Since half of NF-κB is inhibited by p105 and p100 and all of p50 and p52 are generated from this processing, we must know the biochemical regulation underlying the differences between processing and inhibition. The role of IκB in transcription activation or inhibition at DNA sites is another fascinating area of investigation. Is there a role for differential phosphorylation in the outcome of co-activator versus co-repressor function?
In the area of NF-κB dimerization, new dimers are now being detected. Are these dimers abundant, forming under a variety of cellular conditions, or do they only form conditionally? Are these dimers formed as regular side-by-side dimer as in p50:RelA, or is a different structural strategy used for formation? Finally, does a κB site restrict NF-κB dimer specificity? Although in a few cases the relationship between a κB site and NF-κB dimer has been established, it is not clear if this will be a rule for all κB sites.
Acknowledgements
We acknowledge Tom Huxford for reading the manuscript. The work in the G. Ghosh lab has been supported by grants from NIH (GM085490, AI064326 and CA141722).
Footnotes
The authors declare no conflicts of interest.
References
- 1.Ghosh S, May MJ, Kopp EB. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 2.Gilmore TD. Introduction to NF-kappaB: players, pathways, perspectives. Oncogene. 2006;25:6680–6684. doi: 10.1038/sj.onc.1209954. [DOI] [PubMed] [Google Scholar]
- 3.Whiteside ST, Israel A. I kappa B proteins: structure, function and regulation. Semin Cancer Biol. 1997;8:75–82. doi: 10.1006/scbi.1997.0058. [DOI] [PubMed] [Google Scholar]
- 4.Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu Rev Immunol. 2000;18:621–663. doi: 10.1146/annurev.immunol.18.1.621. [DOI] [PubMed] [Google Scholar]
- 5.Chen ZJ. Ubiquitin signalling in the NF-kappaB pathway. Nat Cell Biol. 2005;7:758–765. doi: 10.1038/ncb0805-758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pahl HL. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene. 1999;18:6853–6866. doi: 10.1038/sj.onc.1203239. [DOI] [PubMed] [Google Scholar]
- 7.Ghosh G, van Duyne G, Ghosh S, Sigler PB. Structure of NF-kappa B p50 homodimer bound to a kappa B site. Nature. 1995;373:303–310. doi: 10.1038/373303a0. [DOI] [PubMed] [Google Scholar]
- 8.Muller CW, Rey FA, Sodeoka M, Verdine GL, Harrison SC. Structure of the NF-kappa B p50 homodimer bound to DNA. Nature. 1995;373:311–317. doi: 10.1038/373311a0. [DOI] [PubMed] [Google Scholar]
- 9.Chen YQ, Ghosh S, Ghosh G. A novel DNA recognition mode by the NF-kappa B p65 homodimer. Nat Struct Biol. 1998;5:67–73. doi: 10.1038/nsb0198-67. [DOI] [PubMed] [Google Scholar]
- 10.Cheng CS, et al. The specificity of innate immune responses is enforced by repression of interferon response elements by NF-kappaB p50. Sci Signal. 4 doi: 10.1126/scisignal.2001501. ra11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leung TH, Hoffmann A, Baltimore D. One nucleotide in a kappaB site can determine cofactor specificity for NF-kappaB dimers. Cell. 2004;118:453–464. doi: 10.1016/j.cell.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 12.Malek S, Huang DB, Huxford T, Ghosh S, Ghosh G. X-ray crystal structure of an IkappaBbeta x NF-kappaB p65 homodimer complex. J Biol Chem. 2003;278:23094–23100. doi: 10.1074/jbc.M301022200. [DOI] [PubMed] [Google Scholar]
- 13.Rao P, et al. IkappaBbeta acts to inhibit and activate gene expression during the inflammatory response. Nature. 466:1115–1119. doi: 10.1038/nature09283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoffmann A, Levchenko A, Scott ML, Baltimore D. The IkappaB-NF-kappaB signaling module: temporal control and selective gene activation. Science. 2002;298:1241–1245. doi: 10.1126/science.1071914. [DOI] [PubMed] [Google Scholar]
- 15.Mathes E, O’Dea EL, Hoffmann A, Ghosh G. NF-kappaB dictates the degradation pathway of IkappaBalpha. Embo J. 2008;27:1357–1367. doi: 10.1038/emboj.2008.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sue SC, Alverdi V, Komives EA, Dyson HJ. Detection of a ternary complex of NF-kappaB and IkappaBalpha with DNA provides insights into how IkappaBalpha removes NF-kappaB from transcription sites. Proc Natl Acad Sci U S A. 108:1367–1372. doi: 10.1073/pnas.1014323108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Conti E, Kuriyan J. Crystallographic analysis of the specific yet versatile recognition of distinct nuclear localization signals by karyopherin alpha. Structure. 2000;8:329–338. doi: 10.1016/s0969-2126(00)00107-6. [DOI] [PubMed] [Google Scholar]
- 18.Huang DB, Huxford T, Chen YQ, Ghosh G. The role of DNA in the mechanism of NFkappaB dimer formation: crystal structures of the dimerization domains of the p50 and p65 subunits. Structure. 1997;5:1427–1436. doi: 10.1016/s0969-2126(97)00293-1. [DOI] [PubMed] [Google Scholar]
- 19.Huang DB, Vu D, Ghosh G. NF-kappaB RelB forms an intertwined homodimer. Structure. 2005;13:1365–1373. doi: 10.1016/j.str.2005.06.018. [DOI] [PubMed] [Google Scholar]
- 20.Sengchanthalangsy LL, Datta S, Huang DB, Anderson E, Braswell EH, Ghosh G. Characterization of the dimer interface of transcription factor NFkappaB p50 homodimer. J Mol Biol. 1999;289:1029–1040. doi: 10.1006/jmbi.1999.2823. [DOI] [PubMed] [Google Scholar]
- 21.Cramer P, Larson CJ, Verdine GL, Muller CW. Structure of the human NF-kappaB p52 homodimer-DNA complex at 2.1 A resolution. Embo J. 1997;16:7078–7090. doi: 10.1093/emboj/16.23.7078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen FE, Huang DB, Chen YQ, Ghosh G. Crystal structure of p50/p65 heterodimer of transcription factor NF-kappaB bound to DNA. Nature. 1998;391:410–413. doi: 10.1038/34956. [DOI] [PubMed] [Google Scholar]
- 23.Chen YQ, Sengchanthalangsy LL, Hackett A, Ghosh G. NF-kappaB p65 (RelA) homodimer uses distinct mechanisms to recognize DNA targets. Structure. 2000;8:419–428. doi: 10.1016/s0969-2126(00)00123-4. [DOI] [PubMed] [Google Scholar]
- 24.Huang DB, Chen YQ, Ruetsche M, Phelps CB, Ghosh G. X-ray crystal structure of proto-oncogene product c-Rel bound to the CD28 response element of IL-2. Structure. 2001;9:669–678. doi: 10.1016/s0969-2126(01)00635-9. [DOI] [PubMed] [Google Scholar]
- 25.Chen-Park FE, Huang DB, Noro B, Thanos D, Ghosh G. The kappa B DNA sequence from the HIV long terminal repeat functions as an allosteric regulator of HIV transcription. J Biol Chem. 2002;277:24701–24708. doi: 10.1074/jbc.M200007200. [DOI] [PubMed] [Google Scholar]
- 26.Berkowitz B, Huang DB, Chen-Park FE, Sigler PB, Ghosh G. The x-ray crystal structure of the NF-kappa B p50.p65 heterodimer bound to the interferon beta -kappa B site. J Biol Chem. 2002;277:24694–24700. doi: 10.1074/jbc.M200006200. [DOI] [PubMed] [Google Scholar]
- 27.Escalante CR, Shen L, Thanos D, Aggarwal AK. Structure of NF-kappaB p50/p65 heterodimer bound to the PRDII DNA element from the interferon-beta promoter. Structure. 2002;10:383–391. doi: 10.1016/s0969-2126(02)00723-2. [DOI] [PubMed] [Google Scholar]
- 28.Moorthy AK, Huang DB, Wang VY, Vu D, Ghosh G. X-ray structure of a NF-kappaB p50/RelB/DNA complex reveals assembly of multiple dimers on tandem kappaB sites. J Mol Biol. 2007;373:723–734. doi: 10.1016/j.jmb.2007.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Panne D, Maniatis T, Harrison SC. An atomic model of the interferon-beta enhanceosome. Cell. 2007;129:1111–1123. doi: 10.1016/j.cell.2007.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fusco AJ, Huang DB, Miller D, Wang VY, Vu D, Ghosh G. NF-kappaB p52:RelB heterodimer recognizes two classes of kappaB sites with two distinct modes. EMBO Rep. 2009;10:152–159. doi: 10.1038/embor.2008.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sen R, Baltimore D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell. 1986;46:705–716. [PubMed] [Google Scholar]
- 32.Phelps CB, Ghosh G. Discreet mutations from c-Rel to v-Rel alter kappaB DNA recognition, IkappaBalpha binding, and dimerization: implications for v-Rel oncogenicity. Oncogene. 2004;23:1229–1238. doi: 10.1038/sj.onc.1207242. [DOI] [PubMed] [Google Scholar]
- 33.Mura C, McCammon JA. Molecular dynamics of a kappaB DNA element: base flipping via cross-strand intercalative stacking in a microsecond-scale simulation. Nucleic Acids Res. 2008;36:4941–4955. doi: 10.1093/nar/gkn473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tisne C, Hartmann B, Delepierre M. NF-kappa B binding mechanism: a nuclear magnetic resonance and modeling study of a GGG --> CTC mutation. Biochemistry. 1999;38:3883–3894. doi: 10.1021/bi982402d. [DOI] [PubMed] [Google Scholar]
- 35.Tisne C, Hantz E, Hartmann B, Delepierre M. Solution structure of a non-palindromic 16 base-pair DNA related to the HIV-1 kappa B site: evidence for BI-BII equilibrium inducing a global dynamic curvature of the duplex. J Mol Biol. 1998;279:127–142. doi: 10.1006/jmbi.1998.1757. [DOI] [PubMed] [Google Scholar]
- 36.Huang DB, Phelps CB, Fusco AJ, Ghosh G. Crystal structure of a free kappaB DNA: insights into DNA recognition by transcription factor NF-kappaB. J Mol Biol. 2005;346:147–160. doi: 10.1016/j.jmb.2004.11.042. [DOI] [PubMed] [Google Scholar]
- 37.Ea CK, Baltimore D. Regulation of NF-kappaB activity through lysine monomethylation of p65. Proc Natl Acad Sci U S A. 2009;106:18972–18977. doi: 10.1073/pnas.0910439106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang DB, Vu D, Cassiday LA, Zimmerman JM, Maher LJ, 3rd, Ghosh G. Crystal structure of NF-kappaB (p50)2 complexed to a high-affinity RNA aptamer. Proc Natl Acad Sci U S A. 2003;100:9268–9273. doi: 10.1073/pnas.1632011100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhong H, Voll RE, Ghosh S. Phosphorylation of NF-kappa B p65 by PKA stimulates transcriptional activity by promoting a novel bivalent interaction with the coactivator CBP/p300. Mol Cell. 1998;1:661–671. doi: 10.1016/s1097-2765(00)80066-0. [DOI] [PubMed] [Google Scholar]
- 40.Vermeulen L, De Wilde G, Van Damme P, Vanden Berghe W, Haegeman G. Transcriptional activation of the NF-kappaB p65 subunit by mitogen- and stress-activated protein kinase-1 (MSK1) Embo J. 2003;22:1313–1324. doi: 10.1093/emboj/cdg139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dong J, Jimi E, Zhong H, Hayden MS, Ghosh S. Repression of gene expression by unphosphorylated NF-kappaB p65 through epigenetic mechanisms. Genes Dev. 2008;22:1159–1173. doi: 10.1101/gad.1657408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reber L, Vermeulen L, Haegeman G, Frossard N. Ser276 phosphorylation of NF-kB p65 by MSK1 controls SCF expression in inflammation. PLoS One. 2009;4:e4393. doi: 10.1371/journal.pone.0004393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Touma M, et al. Functional role for I kappa BNS in T cell cytokine regulation as revealed by targeted gene disruption. J Immunol. 2007;179:1681–1692. doi: 10.4049/jimmunol.179.3.1681. [DOI] [PubMed] [Google Scholar]
- 44.Bork P. Hundreds of ankyrin-like repeats in functionally diverse proteins: mobile modules that cross phyla horizontally? Proteins. 1993;17:363–374. doi: 10.1002/prot.340170405. [DOI] [PubMed] [Google Scholar]
- 45.Gaudet R. A primer on ankyrin repeat function in TRP channels and beyond. Mol Biosyst. 2008;4:372–379. doi: 10.1039/b801481g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yaron A, et al. Identification of the receptor component of the IkappaBalpha-ubiquitin ligase. Nature. 1998;396:590–594. doi: 10.1038/25159. [DOI] [PubMed] [Google Scholar]
- 47.Muenchen HJ, Lin DL, Walsh MA, Keller ET, Pienta KJ. Tumor necrosis factor-alpha-induced apoptosis in prostate cancer cells through inhibition of nuclear factor-kappaB by an IkappaBalpha “super-repressor”. Clin Cancer Res. 2000;6:1969–1977. [PubMed] [Google Scholar]
- 48.Rogers S, Wells R, Rechsteiner M. Amino acid sequences common to rapidly degraded proteins: the PEST hypothesis. Science. 1986;234:364–368. doi: 10.1126/science.2876518. [DOI] [PubMed] [Google Scholar]
- 49.Hatada EN, Naumann M, Scheidereit C. Common structural constituents confer I kappa B activity to NF-kappa B p105 and I kappa B/MAD-3. Embo J. 1993;12:2781–2788. doi: 10.1002/j.1460-2075.1993.tb05939.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Naumann M, Nieters A, Hatada EN, Scheidereit C. NF-kappa B precursor p100 inhibits nuclear translocation and DNA binding of NF-kappa B/rel-factors. Oncogene. 1993;8:2275–2281. [PubMed] [Google Scholar]
- 51.Savinova OV, Hoffmann A, Ghosh G. The Nfkb1 and Nfkb2 proteins p105 and p100 function as the core of high-molecular-weight heterogeneous complexes. Mol Cell. 2009;34:591–602. doi: 10.1016/j.molcel.2009.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ohno H, Takimoto G, McKeithan TW. The candidate proto-oncogene bcl-3 is related to genes implicated in cell lineage determination and cell cycle control. Cell. 1990;60:991–997. doi: 10.1016/0092-8674(90)90347-h. [DOI] [PubMed] [Google Scholar]
- 53.Hatada EN, et al. The ankyrin repeat domains of the NF-kappa B precursor p105 and the protooncogene bcl-3 act as specific inhibitors of NF-kappa B DNA binding. Proc Natl Acad Sci U S A. 1992;89:2489–2493. doi: 10.1073/pnas.89.6.2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kitamura H, Kanehira K, Okita K, Morimatsu M, Saito M. MAIL, a novel nuclear IkappaB protein that potentiates LPS-induced IL-6 production. FEBS Lett. 2000;485:53–56. doi: 10.1016/s0014-5793(00)02185-2. [DOI] [PubMed] [Google Scholar]
- 55.Yamazaki S, Muta T, Takeshige K. A novel IkappaB protein, IkappaB-zeta, induced by proinflammatory stimuli, negatively regulates nuclear factor-kappaB in the nuclei. J Biol Chem. 2001;276:27657–27662. doi: 10.1074/jbc.M103426200. [DOI] [PubMed] [Google Scholar]
- 56.Haruta H, Kato A, Todokoro K. Isolation of a novel interleukin-1-inducible nuclear protein bearing ankyrin-repeat motifs. J Biol Chem. 2001;276:12485–12488. doi: 10.1074/jbc.C100075200. [DOI] [PubMed] [Google Scholar]
- 57.Fiorini E, et al. Peptide-induced negative selection of thymocytes activates transcription of an NF-kappaB inhibitor. Mol Cell. 2002;9:637–648. doi: 10.1016/s1097-2765(02)00469-0. [DOI] [PubMed] [Google Scholar]
- 58.Yamamoto M, et al. Regulation of Toll/IL-1-receptor-mediated gene expression by the inducible nuclear protein IkappaBzeta. Nature. 2004;430:218–222. doi: 10.1038/nature02738. [DOI] [PubMed] [Google Scholar]
- 59.Palmer S, Chen YH. Bcl-3, a multifaceted modulator of NF-kappaB-mediated gene transcription. Immunol Res. 2008;42:210–218. doi: 10.1007/s12026-008-8075-4. [DOI] [PubMed] [Google Scholar]
- 60.Huxford T, Huang DB, Malek S, Ghosh G. The crystal structure of the IkappaBalpha/NF-kappaB complex reveals mechanisms of NF-kappaB inactivation. Cell. 1998;95:759–770. doi: 10.1016/s0092-8674(00)81699-2. [DOI] [PubMed] [Google Scholar]
- 61.Jacobs MD, Harrison SC. Structure of an IkappaBalpha/NF-kappaB complex. Cell. 1998;95:749–758. doi: 10.1016/s0092-8674(00)81698-0. [DOI] [PubMed] [Google Scholar]
- 62.Croy CH, Bergqvist S, Huxford T, Ghosh G, Komives EA. Biophysical characterization of the free IkappaBalpha ankyrin repeat domain in solution. Protein Sci. 2004;13:1767–1777. doi: 10.1110/ps.04731004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kohl A, Binz HK, Forrer P, Stumpp MT, Pluckthun A, Grutter MG. Designed to be stable: crystal structure of a consensus ankyrin repeat protein. Proc Natl Acad Sci U S A. 2003;100:1700–1705. doi: 10.1073/pnas.0337680100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bergqvist S, Croy CH, Kjaergaard M, Huxford T, Ghosh G, Komives EA. Thermodynamics reveal that helix four in the NLS of NF-kappaB p65 anchors IkappaBalpha, forming a very stable complex. J Mol Biol. 2006;360:421–434. doi: 10.1016/j.jmb.2006.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ferreiro DU, Cervantes CF, Truhlar SM, Cho SS, Wolynes PG, Komives EA. Stabilizing IkappaBalpha by "consensus" design. J Mol Biol. 2007;365:1201–1216. doi: 10.1016/j.jmb.2006.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Truhlar SM, Mathes E, Cervantes CF, Ghosh G, Komives EA. Pre-folding IkappaBalpha alters control of NF-kappaB signaling. J Mol Biol. 2008;380:67–82. doi: 10.1016/j.jmb.2008.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Thompson JE, Phillips RJ, Erdjument-Bromage H, Tempst P, Ghosh S. I kappa B-beta regulates the persistent response in a biphasic activation of NF-kappa B. Cell. 1995;80:573–582. doi: 10.1016/0092-8674(95)90511-1. [DOI] [PubMed] [Google Scholar]
- 68.Fenwick C, et al. A subclass of Ras proteins that regulate the degradation of IkappaB. Science. 2000;287:869–873. doi: 10.1126/science.287.5454.869. [DOI] [PubMed] [Google Scholar]
- 69.Chen Y, Vallee S, Wu J, Vu D, Sondek J, Ghosh G. Inhibition of NF-kappaB activity by IkappaBbeta in association with kappaB-Ras. Mol Cell Biol. 2004;24:3048–3056. doi: 10.1128/MCB.24.7.3048-3056.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li Z, Nabel GJ. A new member of the I kappaB protein family, I kappaB epsilon, inhibits RelA (p65)-mediated NF-kappaB transcription. Mol Cell Biol. 1997;17:6184–6190. doi: 10.1128/mcb.17.10.6184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Whiteside ST, Epinat JC, Rice NR, Israel A. I kappa B epsilon, a novel member of the I kappa B family, controls RelA and cRel NF-kappa B activity. Embo J. 1997;16:1413–1426. doi: 10.1093/emboj/16.6.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Simeonidis S, Liang S, Chen G, Thanos D. Cloning and functional characterization of mouse IkappaBepsilon. Proc Natl Acad Sci U S A. 1997;94:14372–14377. doi: 10.1073/pnas.94.26.14372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kearns JD, Basak S, Werner SL, Huang CS, Hoffmann A. IkappaBepsilon provides negative feedback to control NF-kappaB oscillations, signaling dynamics, and inflammatory gene expression. J Cell Biol. 2006;173:659–664. doi: 10.1083/jcb.200510155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mercurio F, Didonato J, Rosette C, Karin M. Molecular cloning and characterization of a novel Rel/NF-kappa B family member displaying structural and functional homology to NF-kappa B p50/p105. DNA Cell Biol. 1992;11:523–537. doi: 10.1089/dna.1992.11.523. [DOI] [PubMed] [Google Scholar]
- 75.Rice NR, MacKichan ML, Israel A. The precursor of NF-kappa B p50 has I kappa B-like functions. Cell. 1992;71:243–253. doi: 10.1016/0092-8674(92)90353-e. [DOI] [PubMed] [Google Scholar]
- 76.Ishikawa H, Carrasco D, Claudio E, Ryseck RP, Bravo R. Gastric hyperplasia and increased proliferative responses of lymphocytes in mice lacking the COOH-terminal ankyrin domain of NF-kappaB2. J Exp Med. 1997;186:999–1014. doi: 10.1084/jem.186.7.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ishikawa H, Claudio E, Dambach D, Raventos-Suarez C, Ryan C, Bravo R. Chronic inflammation and susceptibility to bacterial infections in mice lacking the polypeptide (p)105 precursor (NF-kappaB1) but expressing p50. J Exp Med. 1998;187:985–996. doi: 10.1084/jem.187.7.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tergaonkar V, Correa RG, Ikawa M, Verma IM. Distinct roles of IkappaB proteins in regulating constitutive NF-kappaB activity. Nat Cell Biol. 2005;7:921–923. doi: 10.1038/ncb1296. [DOI] [PubMed] [Google Scholar]
- 79.Basak S, et al. A fourth IkappaB protein within the NF-kappaB signaling module. Cell. 2007;128:369–381. doi: 10.1016/j.cell.2006.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sriskantharajah S, et al. Proteolysis of NF-kappaB1 p105 is essential for T cell antigen receptor-induced proliferation. Nat Immunol. 2009;10:38–47. doi: 10.1038/ni.1685. [DOI] [PubMed] [Google Scholar]
- 81.Chang M, Lee AJ, Fitzpatrick L, Zhang M, Sun SC. NF-kappa B1 p105 regulates T cell homeostasis and prevents chronic inflammation. J Immunol. 2009;182:3131–3138. doi: 10.4049/jimmunol.0803637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bergqvist S, Alverdi V, Mengel B, Hoffmann A, Ghosh G, Komives EA. Kinetic enhancement of NF-kappaBxDNA dissociation by IkappaBalpha. Proc Natl Acad Sci U S A. 2009;106:19328–19333. doi: 10.1073/pnas.0908797106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kearns JD, Hoffmann A. Integrating computational and biochemical studies to explore mechanisms in NF-{kappa}B signaling. J Biol Chem. 2009;284:5439–5443. doi: 10.1074/jbc.R800008200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shih VF, Kearns JD, Basak S, Savinova OV, Ghosh G, Hoffmann A. Kinetic control of negative feedback regulators of NF-kappaB/RelA determines their pathogen- and cytokine-receptor signaling specificity. Proc Natl Acad Sci U S A. 2009;106:9619–9624. doi: 10.1073/pnas.0812367106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Basak S, Shih VF, Hoffmann A. Generation and activation of multiple dimeric transcription factors within the NF-kappaB signaling system. Mol Cell Biol. 2008;28:3139–3150. doi: 10.1128/MCB.01469-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Scheibel M, et al. IkappaBbeta is an essential co-activator for LPS-induced IL-1beta transcription in vivo. J Exp Med. 207:2621–2630. doi: 10.1084/jem.20100864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Aguilera C, Hoya-Arias R, Haegeman G, Espinosa L, Bigas A. Recruitment of IkappaBalpha to the hes1 promoter is associated with transcriptional repression. Proc Natl Acad Sci U S A. 2004;101:16537–16542. doi: 10.1073/pnas.0404429101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hacker H, Karin M. Regulation and function of IKK and IKK-related kinases. Sci STKE. 2006;2006 doi: 10.1126/stke.3572006re13. re13. [DOI] [PubMed] [Google Scholar]
- 89.Krappmann D, Scheidereit C. A pervasive role of ubiquitin conjugation in activation and termination of IkappaB kinase pathways. EMBO Rep. 2005;6:321–326. doi: 10.1038/sj.embor.7400380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Israel A. The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harb Perspect Biol. 2:a000158. doi: 10.1101/cshperspect.a000158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Xia ZP, et al. Direct activation of protein kinases by unanchored polyubiquitin chains. Nature. 2009;461:114–119. doi: 10.1038/nature08247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 93.Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature. 2001;412:346–351. doi: 10.1038/35085597. [DOI] [PubMed] [Google Scholar]
- 94.Senftleben U, et al. Activation by IKKalpha of a second, evolutionary conserved, NF-kappa B signaling pathway. Science. 2001;293:1495–1499. doi: 10.1126/science.1062677. [DOI] [PubMed] [Google Scholar]
- 95.Xiao G, Harhaj EW, Sun SC. NF-kappaB-inducing kinase regulates the processing of NF-kappaB2 p100. Mol Cell. 2001;7:401–409. doi: 10.1016/s1097-2765(01)00187-3. [DOI] [PubMed] [Google Scholar]
- 96.Xiao G, Fong A, Sun SC. Induction of p100 processing by NF-kappaB-inducing kinase involves docking IkappaB kinase alpha (IKKalpha) to p100 and IKKalpha-mediated phosphorylation. J Biol Chem. 2004;279:30099–30105. doi: 10.1074/jbc.M401428200. [DOI] [PubMed] [Google Scholar]
- 97.Cordier F, Vinolo E, Veron M, Delepierre M, Agou F. Solution structure of NEMO zinc finger and impact of an anhidrotic ectodermal dysplasia with immunodeficiency-related point mutation. J Mol Biol. 2008;377:1419–1432. doi: 10.1016/j.jmb.2008.01.048. [DOI] [PubMed] [Google Scholar]
- 98.Lo YC, et al. Structural basis for recognition of diubiquitins by NEMO. Mol Cell. 2009;33:602–615. doi: 10.1016/j.molcel.2009.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bagneris C, et al. Crystal structure of a vFlip-IKKgamma complex: insights into viral activation of the IKK signalosome. Mol Cell. 2008;30:620–631. doi: 10.1016/j.molcel.2008.04.029. [DOI] [PubMed] [Google Scholar]
- 100.Rushe M, et al. Structure of a NEMO/IKK-associating domain reveals architecture of the interaction site. Structure. 2008;16:798–808. doi: 10.1016/j.str.2008.02.012. [DOI] [PubMed] [Google Scholar]
- 101.Rahighi S, et al. Specific recognition of linear ubiquitin chains by NEMO is important for NF-kappaB activation. Cell. 2009;136:1098–1109. doi: 10.1016/j.cell.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 102.Palkowitsch L, Leidner J, Ghosh S, Marienfeld RB. Phosphorylation of serine 68 in the IkappaB kinase (IKK)-binding domain of NEMO interferes with the structure of the IKK complex and tumor necrosis factor-alpha-induced NF-kappaB activity. J Biol Chem. 2008;283:76–86. doi: 10.1074/jbc.M708856200. [DOI] [PubMed] [Google Scholar]
- 103.Cordier F, Grubisha O, Traincard F, Veron M, Delepierre M, Agou F. The zinc finger of NEMO is a functional ubiquitin-binding domain. J Biol Chem. 2009;284:2902–2907. doi: 10.1074/jbc.M806655200. [DOI] [PubMed] [Google Scholar]
- 104.Xu G, et al. Crystal structure of inhibitor of kappaB kinase beta. Nature. 472:325–330. doi: 10.1038/nature09853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tang ED, Inohara N, Wang CY, Nunez G, Guan KL. Roles for homotypic interactions and transautophosphorylation in IkappaB kinase beta IKKbeta) activation [corrected] J Biol Chem. 2003;278:38566–38570. doi: 10.1074/jbc.M304374200. [DOI] [PubMed] [Google Scholar]
- 106.Zandi E, Chen Y, Karin M. Direct phosphorylation of IkappaB by IKKalpha and IKKbeta: discrimination between free and NF-kappaB-bound substrate. Science. 1998;281:1360–1363. doi: 10.1126/science.281.5381.1360. [DOI] [PubMed] [Google Scholar]
- 107.Krappmann D, et al. The IkappaB kinase complex and NF-kappaB act as master regulators of lipopolysaccharide-induced gene expression and control subordinate activation of AP-1. Mol Cell Biol. 2004;24:6488–6500. doi: 10.1128/MCB.24.14.6488-6500.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wu C, Ghosh S. Differential phosphorylation of the signal-responsive domain of I kappa B alpha and I kappa B beta by I kappa B kinases. J Biol Chem. 2003;278:31980–31987. doi: 10.1074/jbc.M304278200. [DOI] [PubMed] [Google Scholar]