

Abstract
Non-small cell lung cancers (NSCLCs) harbor thousands of passenger events that hide genetic drivers. Even highly recurrent events in NSCLC, such as mutations in PTEN, EGFR, KRAS, and ALK, are only detected in, at most, 30% of patients. Thus, many unidentified low-penetrant events are causing a significant portion of lung cancers. To detect low-penetrance drivers of NSCLC a forward genetic screen was performed in mice using the Sleeping Beauty (SB) DNA transposon as a random mutagen to generate lung tumors in a Pten deficient background. SB mutations coupled with Pten deficiency were sufficient to produce lung tumors in 29% of mice. Pten deficiency alone, without SB mutations, resulted in lung tumors in 11% of mice, while the rate in control mice was ~3%. In addition, thyroid cancer and other carcinomas as well as the presence of bronchiolar and alveolar epithelialization in mice deficient for Pten were also identified. Analysis of common transposon insertion sites identified 76 candidate cancer driver genes. These genes are frequently dysregulated in human lung cancers and implicate several signaling pathways. Cullin3 (Cul3), a member of an ubiquitin ligase complex that plays a role in the oxidative stress response pathway, was identified in the screen and evidence demonstrates that Cul3 functions as a tumor suppressor.
Keywords: Sleeping Beauty, Forward Genetic Screen, Cullin3
Introduction
Most lung cancers are caused by smoking, which explains the higher mutation load in lung cancer compared to most other major epithelial cancers(1). Even though individual lung cancers harbor a large number of mutations, comprehensive genetic analyses of these cancers have identified few recurrent drivers, such as activating mutations in EGFR and KRAS, inactivating mutations in TP53, and translocations affecting ALK. Except for TP53, the percentage of tumors with either one of these driver mutations is less than 25%(2,3). Another recurrent event found in non-small cell lung cancers (NSCLC) and small cell lung cancers (SCLC) is activation of the PI3K/AKT pathway (3–5), which is frequently caused by loss of PTEN (6). Because of this confusing abundance of genetic mutations and other genetic anomalies, it is difficult to pinpoint specific drivers in any given patient and many lung cancer genetic drivers have yet to be identified (7).
Mouse models have been used to confirm that these altered proteins result in increased rates of lung cancer. For example, loss of Trp53 in the lungs results in adenocarcinomas after a long latency and, if Rb is simultaneously deleted in the lungs, tumor latency is decreased and the tumor phenotype switches to SCLC (8). If loss of one or both alleles of Pten are introgressed into this model, tumor latency is reduced even further (9). Decreasing Pten protein levels in wildtype mice by introducing a hypomorphic Pten allele also results in lung cancer in 28% of mice (10). Interestingly, lung-specific deletion of Pten in mice did not result in tumors, but when combined with lung-specific activation of KrasG12D, tumor latency was significantly decreased (11).
The aim of our experiments was to discover new genetic drivers of lung cancer by performing forward genetic screens in mice using the SB DNA transposon as a mutagen in lung epithelial cells. We performed one genetic screen on a wild-type background and three additional screens using mice with predisposing mutations in Trp53, p19ARF, and Pten. Contrary to our expectations, the only screen that produced at least twice as many lung tumors in the experimental animals compared to controls was in mice deficient for Pten. We sequenced the tumors that arose in these mice and identified 78 common transposon insertion sites (CISs) and were able to identify 76 candidate cancer genes. Over 85% of these candidate genes have documented alterations in ten or more human lung cancers, and several have been strongly implicated as drivers of lung cancer. The three genes identified with the most transposon insertions were Serinc3, Magi1 and Nckap5. There is evidence that Serinc3 is an oncogene while Magi1 has tumor suppressive activities (12,13). We performed functional tests on another of our candidate cancer genes, Cul3, and found that reducing levels of CUL3 and/or PTEN resulted in cancer phenotypes in human lung cancer cell lines. Furthermore, analysis of gene expression patterns in cells deficient for CUL3, PTEN, or both CUL3 and PTEN suggests this phenotype may be due to alterations in the NRF2 signaling pathway.
Materials and Methods
Mice
Pten floxed mice (PtenLoxp/+) (14) on the C57Bl6/J background were a generous gift of Pier Pandolfi (Memorial Sloan Kettering). Trp53fl/R270H mice [129S4-Trp53tm3Tyj/Nci] strain 01XM3, and p19ARF−/− mice [B6.129-Cdkn2atm1Cjs/Nci] strain 01XG7, were purchased from the National Cancer Institute Mouse Repository. Both Trp53fl/R270H and p19ARF−/− mice were backcrossed > 10 generations to the C57Bl6/J background. Conditional Sleeping Beauty transposase mice (RosaSBaseLsL) (15)on the C57Bl6/J background were a generous gift of Adam Dupuy (University of Iowa). Lung-specific Cre Recombinase mice (Spc-Cre) (16) on the ICR x FVB/n background were a generous gift of Brigid Hogan (Duke University). Spc-Cre mice were backcrossed to C57Bl6/J wild-type mice > 10 generations. T2/Onc15 mice were generated as described (rosa 68) (17) and backcrossed to C57Bl6/J wild-type mice > 10 generations. T2/Onc4 mice were generated on the C57Bl6/J background as described (TG6070) (18). Mice were genotyped using DNA from tail biopsies. PCR protocols and primer sequences are available in Supplemental Data. All mice protocols were approved by the University of Minnesota’s IACUC.
Cells
All cell lines, except HBEC, were obtained from ATCC and the authenticity of these cell lines was verified by short tandem repeat analysis (Johns Hopkins). Human bronchial epithelial cells immortalized with CDK4 and hTERT were provided by John Minna (UT Southwestern). Functional assays were conducted in stage 2 lung cancer cells A549 or H522. 293T human embryonic kidney cells were transfected with Open-Biosystems lentiviral packaging mix with Non-silencing, CUL3 2702, CUL3 32413, or CUL3 351781 shRNA plasmids to produce lentivirus that harbors CUL3 specific shRNA sequence. Cells were transfected according to the Open Biosystems’ lentivirus production protocol. To make stable CUL3 knockdown cells, CUL3 specific shRNA encoding lentivirus was used to transduce A549 or H522 cells. The cells were then grown under puromycin selection in RPMI. A549 cells with stable CUL3 knockdown, or expressing the non-silencing control, were then transfected with SABiosciences SureSilencing shRNA plasmids for human PTEN (catalog number KH00327H) or with the negative shRNA encoding plasmid control. The cells were maintained in RPMI with 1 X Penicillin, Streptomycin, 10% fetal bovine serum, 1 μg/ml Puromycin and 32 μg/ml Hygromycin at 37°C and 5% CO2.
Histopathology and Immunohistochemistry (IHC)
Formalin-fixed tissues were embedded in paraffin, and stained with H&E. IHC for CC10 was performed using a goat anti-mouse C-terminus peptide CC10 polyclonal antibody (Santa Cruz) with detection by a goat horse radish peroxidase (HRP)-Polymer Kit (Biocare) using diaminobenzidine (DAB) (Dako) as the chromogen. IHC for SPC was performed using a rabbit anti-proSP-C polyclonal antibody (Millipore); detection was with a rabbit EnVision™+ HRP-polymer kit (Dako) with DAB as the chromogen. IHC for SB was performed using goat polyclonal anti-SB (R&D Systems). More details are provided in Supplementary Methods.
Transposon insertion analysis
Detailed methods are available in supplementary materials. Briefly, LM- PCR was performed on DNA isolated from tumors. PCR amplicons were sequenced using the Illumina GAIIx sequencing platform. Sequences were mapped to the genome and CISs were identified using the TAPDANCE bioinformatics pipeline.
Western blotting
Cells were lysed in standard RIPA buffer and loaded onto 12.5% SDS-PAGE gels. Lysates were transferred to PVDF membranes and blocked in 5% milk TBS-T. Primary antibodies used: anti-CUL3 (Bethyl Labs A301-109A), anti-PTEN (138G6) (Cell Signaling #9559S), anti-NRF2 (D1C9) (Cell Signaling #8882S), anti-KEAP1(D1G10) (Cell Signaling #7705S) and anti-GAPDH (14C10) (Cell Signaling #2118L). Goat-anti-rabbit IgG-HRP was used to detect primary antibodies (Santa Cruz Biotechnology SC-2004). HRP was detected using SuperSignal® West Femto Maximum Sensitivity Substrate (Thermo Scientific product # 34096) and visualized using a FluorChemE imager (Protein Simple). Densitometry was measured using ImageJ software.
Cell proliferation assay
Cell proliferation was measured using an MTS assay for 5–6 days (Cell Titer 96® Aqueous One Solution Cell Proliferation Assay, Promega). Cells (3,500) were plated in 96-well plates and OD490/650 levels were measured after 1 hour using a spectrophotometer (Epoch BioTek) Data were analyzed using Graphpad Prism.
Colony formation assay
Cells were grown in between layers of 0.5% Sea Plaque Low Melt Agarose (Lonza Cat. # 50101) in RPMI supplemented with 10% FBS, penicillin, streptomycin, and antibiotic selection for 3 weeks. Colonies were fixed, stained with crystal violet, and imaged. Colony counts were analyzed using ImageJ software.
Bead Array
RNA was isolated from A549, A549-CUL3-KD, A549-PTEN-KD and A549 CUL3-PTEN-DKD cells using Trizol (Life Technologies, Cat # 15596-026) following manufacturer’s protocol. RNA was analyzed using the Human HT-12v4 bead array (Illumina, Cat # BD-103-0204). Differentially expressed genes were defined by pair-wise comparisons using rank invariant normalization following the gene expression workflow from Partek Genomics Suite software. All Bead Array data has been deposited in the Gene Expression Omnibus database (GSE68869).
Superoxide detection assay
Cells were seeded at equal numbers in 6-well plates and incubated for 24 hours. Cells were washed in PBS and then incubated with media containing 5μM MitoSOX reagent (Molecular Probes, Cat #M36008) for 3 hours. A minimum of 20 brightfield and red fluorescent images were taken for each well at 20x magnification using an inverted fluorescent microscope (Nikon Eclipse TE200). Images were analyzed using ImageJ. Cells were counted in each brightfield image. Red fluorescence was measured using the ImageJ integrated density measurement function after setting the maximum and minimum pixel intensities to 30 and 90, respectively. Average integrated density/cell was calculated for each well. Statistical differences were determined using the student T-test.
Results
SB Mutagenesis drives lung tumor formation
We performed four forward genetic screens in mice to identify genes involved in lung cancer. Our screen in wild-type mice consisted of the following three alleles: 1) A conditional SB11 transposase allele (Rosa26-LsL-SB11)(15,19); 2) a Cre-recombinase cDNA driven by the Surfactant Protein-C promoter (Spc-Cre)(16); and 3) a concatamer of oncogenic transposons (T2/Onc)(17,18) (Fig. S1a–c). In these mice, SB mutagenesis occurs in lung epithelial cells because SB11 transposase enzyme is activated by the lung-specific Cre recombinase (Fig. S2). Control mice consisted of littermates with two of the three alleles. We also conducted three additional screens using the same three alleles introgressed into mice with the following predisposing alleles: 1) p19ARF−/−, 2) p53floxed-R270H, and 3) PtenLoxp/Loxp (Fig. S1d–f). These mice are referred to as p19, p53, and Pten. The p19ARF−/− allele is a germline knockout. The initial report describing these mice found that roughly 30% of these mice develop tumors within six months and the majority of these tumors are blood tumors or sarcomas (20). The p53floxed-R270H allele is a dominant negative p53 allele that is not expressed in cells unless Cre recombinase is present. Therefore, these mice are heterozygous for p53, except in Cre expressing cells. In the absence of Cre, these mice develop tumors due to p53 heterozygosity; while in animals expressing a germline Cre the tumor spectrum changes slightly to favor carcinomas (21). PtenLoxP/LoxP mice lose expression of Pten in cells expressing Cre recombinase and this allele been used to study brain and prostate cancer (14,22).
The experimental genotype, number of mice, lung tumor penetrance and median survival in the four screens are displayed in Table 1. Potential neoplasms in other tissues were also detected at necropsy (Table S1), which may have resulted from leaky or non-lung-specific expression of the Spc-Cre transgene. Lung tumor penetrance in the wild-type, p19 and p53 screens was not substantially higher in the experimental groups compared to the control groups, so we did not analyze these tumors. Instead we focused on tumors arising in the Pten predisposed background, as there was a large difference in penetrance between all controls (6%) and experimental animals (29%).
Table 1.
Lung tumor penetrance and median survival in four SB screens
| Screen | Genotype | Exp mice with lung tumors % (N) | Median survival of exp mice (days) | Ctrl mice with lung tumors % (N) | Median survival of ctrl mice (days) |
|---|---|---|---|---|---|
| wild-type | Rosa26-LsL-SB11 x T2/Onc x Spc-Cre | 17% (53) | NA* | 12 % (93) | NA* |
| p19 | Rosa26-LsL-SB11 x T2/Onc x Spc-Cre x p19ARF−/− | 6% (99) | 303 | 8% (100) | 326 |
| p53 | Rosa26-LsL-SB11 x T2/Onc x Spc-Cre x p53+/floxedR270H | 22% (116) | 419 | 15 % (87) | 443 |
| Pten | Rosa26-LsL-SB11 x T2/Onc x Spc-Cre x Ptenfloxed/floxed | 29% (62) | 320 | 6 % (94) | 415 |
Median survival of wild-type mice was not determined because the majority lived longer than the endpoint of 1.5 years
The Pten cohort consisted of five different genotypes (Table 2). All animals were homozygous for the Ptenloxp knock-in allele and contained the conditional SB11 transposase. Because SB transposon mobilization in some cases is biased towards re-insertion into the donor chromosome we used two strains of T2/Onc transgenic mice. In one strain, there were roughly 25 copies of the transposon linked as a concatamer and resident on chromosome 15 (T2/Onc(15)) (17), while the second strain contained approximately 215 copies of the transposon as a concatamer on chromosome 4 (T2/Onc(4)) (18). The difference in lung tumor penetrance between these two experimental lines was 23% for T2/Onc(4) and 37% for T2/Onc(15). Of the three control groups, the two with wild-type Pten had a tumor penetrance of 3% for T2/Onc(4) and 4% for T2/Onc(15), while the control group with loss of Pten had an intermediate penetrance of 11% (Table 2). Interestingly, we expected a higher penetrance in the experimental group with the larger number of transposons (T2/Onc(4)), but this group actually had a lower penetrance than the low-copy group (T2/Onc(15)). It is possible that the higher number of transposon insertions was detrimental to cell survival, and thus resulted in a lower tumor penetrance. Survival correlated with the presence or absence of Pten, as the Pten−/− control group became moribund at the same rate as the experimental group (Pten−/− and SB mutagenesis) (Fig. 1). Upon histological examination, both the Pten−/− control group and the experimental group had extensive bronchiolar epithelial hyperplasia, which may be the reason why morbidity was similar between these two groups. There was no difference in survival between the mice carrying the different transposon alleles (Fig. S3).
Table 2.
Lung tumor penetrance in Pten screen
| Group | Genotype | Mice with tumors % (N) |
|---|---|---|
| Ctrl 1 | Ptenloxp/loxp x Rosa26-LsL-SB11 x T2/Onc(4) | 3% (31) |
| Ctrl 2 | Ptenloxp/loxp x Rosa26-LsL-SB11 x T2/Onc(15) | 4% (26) |
| Ctrl 3 | Ptenloxp/loxp x Rosa26-LsL-SB11 x Spc-Cre | 11% (37) |
| Exp 1 | Ptenloxp/loxp x Rosa26-LsL-SB11 x T2/Onc(4) x Spc-Cre | 23% (35) |
| Exp 2 | Ptenloxp/loxp x Rosa26-LsL-SB11 x T2/Onc(15) x Spc-Cre | 37% (27) |
Figure 1. Kaplan Meier three groups.
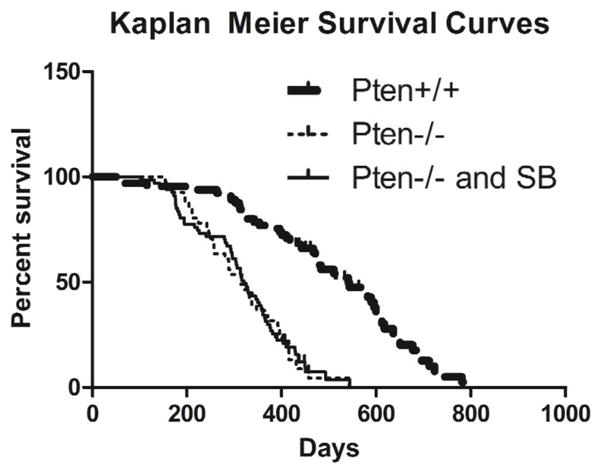
Kaplan Meier Survival curves for two control groups (Pten+/+ and Pten−/−) and the experimental group (Pten−/− and SB). (Log-rank Mantel-Cox test p-values < 0.0001 for Pten−/− vs. Pten+/+ and SB vs. Pten+/+).
Due to the small size of the tumors that developed (Fig. S4), in most cases the entire tumor was excised and used for extracting DNA. In those cases where microscopic examination of lung lesions was performed, a spectrum of lesions was observed including bronchiolar and alveolar epithelial hyperplasia attributable to proliferation of club cells, primary pulmonary adenoma and adenocarcinomas (Fig. 2). The diagnosis of pulmonary adenomas and adenocarcinomas was based on the criteria recommended by the Mouse Models of Human Cancers Consortium (23), and expression of prosurfactant protein C (SPC) and/or club cell 10-kDa protein (CC10) detected by immunohistochemistry (IHC) (Fig. 2). These tumors fit in the broad category of human non-small cell lung carcinomas. In addition to primary pulmonary lesions, several mice had metastatic adenocarcinomas in the lung of diverse primary origins (e.g., thyroid, mammary, preputial etc., or of undetermined origin) (Fig. S5). CIS data was generated from single, discrete tumors that had a gross appearance similar to those lesions diagnosed as primary pulmonary adenomas or adenocarcinomas; however, we cannot completely exclude the possibility that our CIS data may include some data that was derived from one or more metastatic tumors.
Figure 2. Lung tumor histology.

Representative photomicrographs demonstrating histological findings. Lung sections show bronchiolar and alveolar epithelial hyperplasia (Panel A, H&E) attributable to club cell hyperplasia (Panel B, hyperplastic cells express CC10); Panel C (H&E) shows an adenoma that expresses CC10 (Panel D). Lung section shows an adenocarcinoma (Panel E, H&E) that expresses prosurfactant protein C (Panel F). IgG serum control staining for CC10 and proSP-C are shown in panels G and H, respectively (See supplementary materials and methods for more details). Bar sizes in panels A–H are 100, 100, 250, 50, 500, 50, 50 and 50 microns, respectively.
CIS analysis identifies candidate lung cancer genes
To identify the genes driving tumorigenesis in our mice we performed linker-mediated PCR (LM-PCR) using DNA isolated from 23 lung tumors from 13 mice. We then subjected the LM-PCR amplicons to high-throughput sequencing and analyzed the sequences using the TAPDANCE bioinformatics pipeline that we developed (24). Over 18 million sequences were assigned to the 23 tumors based on the barcodes attached during the LM-PCR process. Of these, six million mapped to non-repeat regions in the mouse genome. Combining redundant sequences and insertions that occurred within 100 base pairs of each other reduced this number to 178,028 unique 100 bp regions, or an average of ~7,740 regions per tumor. The depth of sequence reads using the Illumina platform allowed us to filter regions based on the number of sequence reads that mapped to the region. We reasoned that regions with only one or a few reads could either be artifacts or only present in a minority of cells, while regions with a larger number of reads were more likely to be present in a majority of tumor cells (25). We set a read threshold of 0.01% of total reads mapping in a single tumor for each region. Out of the 178,028 non-redundant regions, 27,077 unique regions met the threshold (Table S2).
In previous screens we noted that transposon insertions mapping to the donor chromosome, where the original transposon concatamer was located, constituted up to half of all the mapped transposon insertions, a phenomenon referred to as “local hopping”. In this experiment we generated experimental mice using either the T2/Onc(4) or the T2/Onc(15) concatamers, which reside on chromosome 4 and 15, respectively. In this study, we did not see as large of a bias of insertions, with only 11% of insertions in chromosome 4 with T2/Onc(4) and 18% of insertions in chromosome 15 with T2/Onc(15). Based on the distribution of TA dinucleotides in the genome, the preferred SB integration site, we would expect 6% in chromosome 4 and 4% in chromosome 15. Nevertheless, to avoid this bias when determining CIS loci, we did not include insertions in these chromosomes in the analysis. After eliminating these insertions we identified 78 CISs, or regions of the mouse genome interrupted by transposon insertions at a rate higher than expected. Manual analysis of each CIS region allowed us to identify 76 genes most likely to be affected in these regions, as three of the regions were all in the same gene, Magi1 (Table S3). Based on the pattern of transposon insertions we could also predict whether the genes acquired loss- or gain-of function mutations (See supplemental methods). The majority (66%) appeared to be loss-of-function mutations, while 12% had a pattern suggestive of gain-of-function mutations. Of the 76 mouse candidate genes, 75 had human orthologs (Table S3).
Candidate cancer genes are dysregulated in human lung cancers
To determine the relevance to human cancer we analyzed the overlap of the 75 human CIS orthologs with various lists of human cancer genes. The current version of the Cancer Gene Census lists 522 functionally validated cancer genes (26).. Nine of the CIS human orthologs overlapped with these documented cancer genes, an overlap that would not be expected by chance (p < 0.0001) (Table S4). The majority of cancer genes reported in the census are linked to hematopoietic cancers. To determine the specific relevance to lung cancer we compared our CIS list to two major sources documenting somatic lung cancer mutations, The Cancer Genome Atlas (TCGA) and the Catalog of Somatic Mutations in Cancer (COSMIC). The vast majority of all annotated genes have a documented somatic mutation in human lung cancer based on these two sources (> 18,000 genes in COSMIC and > 17,000 genes in TCGA). If we limit our analysis to genes with at least 10 reported mutations (COSMIC = 7,209 and TCGA = 5,513), we find a significant overlap with our CIS genes (COSMIC overlap = 52 [p-value 5.3e-8], TCGA overlap = 44 [p-value 3.5e-11], Table S4, Supplementary Methods).
Lung cancer has one of the highest mutation rates among all types of cancer making it difficult to discern cancer-associated genetic mutations from background mutations. Factors including heterogeneity between cancer patients and differences in mutation rates within the genome due to transcription-coupled repair (27) and DNA replication timing(28) also confound analyses. Two recent large-scale genomic analyses of human lung squamous cell carcinoma (SCC) (3) and adenocarcinoma (AC)(2) published lists of significant amplifications/deletions, mutations, and genomic rearrangements based on exome and whole genome sequencing. These analyses used algorithms that took into account some of the confounding factors. Our list of CIS human orthologs were significantly enriched in all three categories of genes determined in those studies to be possible drivers based on amps/dels, mutations, and rearrangements (Table S4 [p-values from 0.001 to 0.018]).
Pathway analysis
We used Ingenuity Pathway Analysis (Ingenuity Systems, www.ingenuity.com) to analyze canonical pathways enriched in our list of 75 CIS genes. As might be expected, the canonical pathway identified with the lowest p-value was Molecular Mechanisms of Cancer (Table S5). This association was based on seven genes (GAB2, TAB2, CREBBP, CTNNA1, RBPJ, SMAD4, and GSK3B). Other canonical signaling pathways that were associated with these genes included the NFkB, RANK, Integrin, and NGF pathways.
Our mouse model generates cancers by creating multiple random mutations in a single cell, as every cell contains either ~25 (T2/Onc(15)) or ~215 (T2/Onc(4)) transposons that can be mobilized to create mutations. We reasoned that the cancers arising in these mice are likely generated from multiple independent transposon insertions, but the frequency of each of these mutations in the different mice might not reach the level of significance required for classification as a CIS. To find combinations of insertions that may be cooperating to cause tumorigenesis we adapted an algorithm called frequent itemset mining (29,30). The algorithm identifies combinations of insertions that frequently co-occur in multiple tumors. These groups of genes can reach statistical significance, even though they do not individually reach significance as a CIS. All transposon insertions were associated with the nearest annotated gene and frequent itemset mining was used to determine sets of genes co-occurring in tumors from at least three different mice (Supplemental Methods). The 10 sets of genes with -Log (p-value) >5 are listed in Table S6. All of the sets were coordinately mutated in three separate mice and half of the sets consist of three genes, while the other half consisted of four genes. There were 23 unique genes included in the 10 gene sets, and only 6 of these reached significance as an individual CIS. The majority of these genes are associated with cancer and we propose that the mutations caused by the transposon insertions in or near these sets work together to cause tumorigenesis.
Cul3 functions as a tumor suppressor gene in human lung cancer cells
In addition to demonstrating a strong correlation between our CIS list of candidate driver genes and genes frequently disrupted in human lung cancer it is important to demonstrate that these genes functionally act as driver genes. To this end we choose to analyze one of the candidate drivers discovered in our screen, Cullin3 (Cul3). We selected Cul3 for several reasons. First, TCGA analysis of lung squamous cell carcinomas identified the oxidative response pathway, specifically KEAP1/CUL3/NFE2L2, as a potential driver in this cancer (3). Second, CUL3 was mutated in 10 human lung adenocarcinomas as reported in the COSMIC database (31). Finally, we wanted to validate a “middle of the list” gene and Cul3 was identified in 25% of the mice in our screen. Approximately half of our CIS genes were identified in a larger percentage, and half in a smaller percentage of mice. Based on the location and orientation of the transposon insertions in the Cul3 mutant tumors (Fig. S6) we predict that the transposon is causing a loss-of-function mutation. In humans, the protein product of CUL3 is a core scaffolding protein in an E3 ubiquitin ligase complex that includes KEAP1 and RBX1. Known substrates of this complex include NRF2 (gene name NFE2L2), IKBKB and other proteins (32,33). This complex is important in regulating oxidative stress and the recent TCGA analysis of lung SCC indicates that members of the complex may be disrupted in a significant percentage of these lung tumors (3).
In support of our hypothesis that CUL3 is a tumor suppressor, a trend towards reduced levels of CUL3 protein was seen in a series of cancer cell lines compared to a normal human bronchiolar epithelial cell line (HBEC) (34) (Fig. 3). Two of the cell lines analyzed were from patients with stage II cancer (A549 and H522) and one line was from a patient with stage IV cancer (H2030). The mutational status of A549 and H522 has been analyzed by whole genome sequencing (35). CUL3, PTEN and RBX1 were wild-type in both cell lines, while A549 had a non-synonymous mutation in KEAP1. Surprisingly, we found that further reduction of CUL3 levels in these lines using shRNA resulted in an increased growth rate compared to control lines (Fig. 4 and Fig. 5, p-value day 5 < 0.05 both graphs). Knockdown of CUL3 in the stage IV line, H2030 did not have a significant effect on growth (data not shown). Because our mouse model was predisposed to cancer using a conditional Pten knockout allele, we generated A549 cell lines stably expressing shRNA targeting CUL3 alone, PTEN alone, or both CUL3 and PTEN (Fig. S7). Consistent with our hypothesis that loss of CUL3 cooperates with loss of PTEN, the double knockdown cells grew faster than the control line or either of the single knockdown lines (Fig. 5, p-value day 5 < 0.01). To test the role of CUL3 and PTEN in anchorage-independent growth we grew the cell lines in soft agar. Similar to the proliferation assay, the CUL3/PTEN double-knockdown line formed the most colonies in soft agar compared to the single knockdown lines and the control line, although the difference between the CUL3 single and the double-knockdown was not significant (Fig. 5).
Figure 3. CUL3 protein levels are reduced in cancer cell lines.
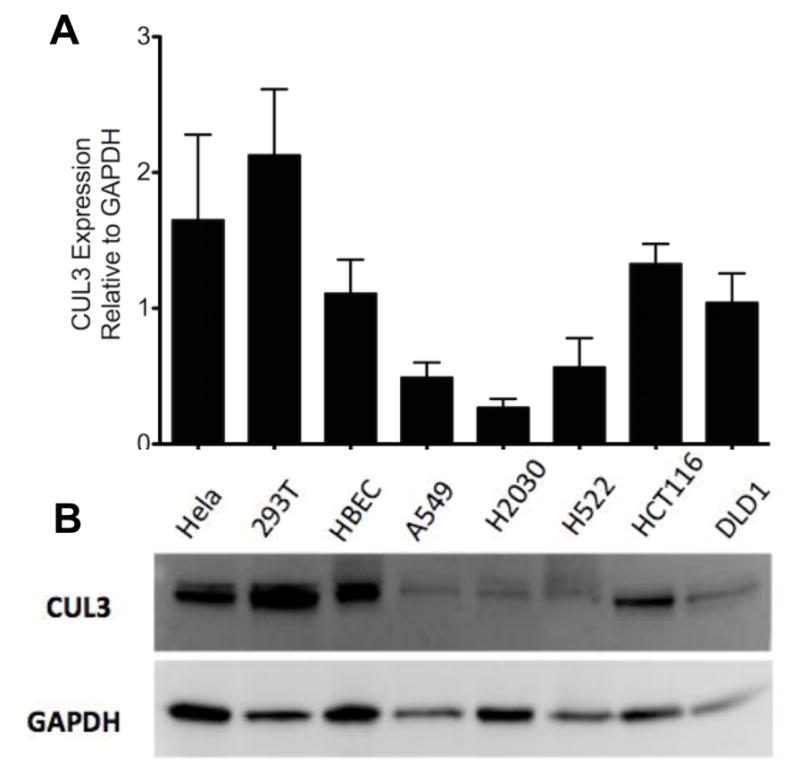
A decreasing trend is seen in CUL3 protein levels in human lung cancer cell lines A549, H2030, H522 compared to human bronchial epithelial cells. A) Western blot for Cul3 expression in human cell lines. Hela = cervical cancer, 293T = kidney, HBEC = immortalized bronchial epithelial, A549 and H522 = stage 2 lung adenocarcinoma, H2030 = stage 4 lung adenocarcinoma, HCT116 and DLD1 = colorectal cancer. B) Quantification of western blots analyzing Cul3 expression from 3 separate cell lysates. (t-test P-values: HBEC vs. A549 = 0.12, HBEC vs. H2030 = 0.07, HBEC vs. H522 = 0.18)
Figure 4. Knockdown of CUL3 causes increased growth in lung cancer cell lines.

CUL3 knock down in H522 cells increases cell proliferation and anchorage-independent cell growth. A) Western blot indicating that CUL3 is knocked down in H522 cells. B) Cell proliferation assay showing that CUL3 knockdown cells proliferate more rapidly than the non-silencing negative control (t-test day 5 p-value < 0.05). C) Images of colonies growing in soft agar in Non-silencing (left panel) and CUL3 knockdown (right panel) H522 cells. D) Quantification of 12 replicates of the soft agar assay indicating that CUL3 knockdown leads to increased colony formation in soft agar (t-test p-value < 0.05).
Figure 5. Double knockdown of CUL3 and PTEN results in increased growth.

Knockdown of CUL3 and PTEN in A549 cells increases cell proliferation and anchorage-independent growth. A) Western blot showing knockdown of PTEN, CUL3, or PTEN and CUL3 in A549 cells. B) Soft Agar Assay shows that knocking down both PTEN and CUL3 in A549 cells leads to increased colony formation compared to controls. In addition, the single CUL3 knockdown formed more colonies than the PTEN knockdown (t-test p-value ** ≤ .01, p-value *** ≤ .001). C) MTS based cell proliferation assay indicates that knocking down both CUL3 and PTEN leads to increased cell growth compared to knocking down CUL3 or PTEN or the negative control. CUL3 knock down also led to increased growth compared to knocking down PTEN or the negative control (t-test p-values day 5 < 0.01, all comparisons).
Loss of CUL3 and PTEN activates the NRF2 signaling pathway
We measured changes in gene expression in the A549 cell strains with reduced levels of CUL3, PTEN, or both CUL3 and PTEN using the Illumina BeadArray platform. Interestingly, knockdown of PTEN resulted in changes in expression of only 11 genes (fold change > 2.0, FDR < 0.05), while knockdown of CUL3 caused changes in 87 genes, with the majority (59) being downregulated (Table S7). Knockdown of both CUL3 and PTEN resulted in changes in expression of 120 genes, also with a majority (76) being downregulated.
Knockdown of CUL3 could affect many proteins regulated by ubiquitination because CUL3 is a scaffolding component of BTB-CUL3-RBX1 ubiquitin ligase complexes. The BTB protein functions as the targeting protein in this complex and CUL3 can theoretically bind to over 50 different BTB proteins, making it difficult to predict the effect of a knockdown (36). One of the best characterized CUL3 partners is KEAP1, and the KEAP1-CUL3-RBX1 complex is known to ubiquitinate the transcription factor NRF2 (gene name NFE2L2) (37). In accordance with this role, we would predict that loss of CUL3 would lead to increased transcriptional activity of NRF2. Chorley, et al., identified 232 NRF2 target genes based on ChIP-Seq analysis of cells treated with sulforaphane, which induces NRF2-mediated antioxidant activity (38). Seven of these genes were differentially expressed in CUL3/PTEN double knockdown cells compared to controls (significant by Fisher’s Exact Test P < 0.005). The same group identified 18 genes differentially expressed (> 2-fold) in cells treated with sulforaphane compared to controls, and 7 of these genes are also differentially expressed in CUL3/PTEN double knockdown cells (significant by Fisher’s Exact Test P < 6.0e-11) (Table S8 and Supplementary Methods). These genes include the well-known NRF2 targets NQO1 and HMOX1. Surprisingly, NQO1 and HMOX1, along with GCLM and SQ5TM1 were upregulated 2-fold only in the CUL3/PTEN double knockdown cells, and not in the CUL3 knockdown cells, suggesting loss of PTEN indirectly enhances NRF2 signaling caused by loss of CUL3. Notably, when comparing the CUL3/PTEN double knockdown gene expression changes to those identified in either PTEN or CUL3 single knockdown cells, 48 of the 120 genes that changed more than 2-fold in the CUL3/PTEN double knockdown cells were not identified with a 2-fold change in CUL3 or PTEN single knockdown cells. The two highest upregulated genes were OSGIN1 and HMOX1, both of which are activated by the oxidative stress response (39,40).
Further support that loss of PTEN and CUL3 synergize to activate NRF2 signaling comes from Ingenuity Pathway Analysis (Ingenuity Systems, www.ingenuity.com). The canonical pathway identified by IPA with the lowest p-value using the CUL3/PTEN gene list was the NRF2-mediated Oxidative Stress Response pathway, while this pathway was not in the top 50 significant canonical pathways in the CUL3 list (Table S9 & S10).
To understand the mechanism of synergy between CUL3 and PTEN we identified genes that were discordantly regulated when CUL3, PTEN, or CUL3/PTEN were knocked down compared to control cells. Only one gene had a greater than 2-fold change in all three comparisons that was discordant. The gene, DKK1, is a secreted inhibitor of Wnt signaling and DKK1 down regulation is associated with poor prognosis in lung cancer(41,42). DKK1 was increased over 3-fold when PTEN was knocked down, but in CUL3 or CUL3/PTEN knockdown cells, DKK1 mRNA was decreased over 3-fold.
To measure the functional effect of CUL3 and PTEN in regulating oxidative stress, we incubated A549 control and knockdown cells with MitoSOX Red, a dye that fluoresces in response to levels of superoxide. As predicted, A549 cells with reduced levels of CUL3 and/or PTEN had significantly less fluorescence than control cells (Figure 6, p-value < 0.005). Although there was no significant difference between the three knockdown lines, in three replicate experiments the CUL3 single knockdown A549 cells consistently had the lowest amount of fluorescence. These results suggest that reduced levels of PTEN and/or CUL3 results in decreased production of superoxide, which could be protective for cancer cells.
Figure 6. Knockdown of CUL3 and PTEN results in decreased levels of superoxide.

Knockdown of CUL3 and/or PTEN in A549 cells results in reduced superoxide production. Representative brightfield (left) and red fluorescent (right, converted to grayscale) images of (A, B) A549 control cells and (C,D) A549 CUL3/PTEN knockdown cells treated with MitoSox. E) Graph of average MitoSox fluorescence/field of view/cell comparing control cells to PTEN, CUL3, and CUL3/PTEN knockdown A549 cells.
Discussion
It is likely that multiple agents targeting several pathways will be required to extend the survival of lung cancer patients. For this strategy to work new targets must be identified and validated. To this end, we have developed a method of identifying novel drivers of lung cancer using SB mutagenesis in forward genetic screens in mice. Using this method we were able to identify 76 potential lung cancer drivers. A caveat to any mouse study is that the lung tumors generated may not be genetically or physiologically equivalent to human tumors. Unfortunately, identifying driver genes in human lung cancer is particularly problematic due to the large number of background passenger mutations. If our forward genetic screen in mice were identifying bona fide drivers of human lung cancer we would expect to find the orthologous genes mutated or altered to a significant extent in human lung cancer. Analysis of large-scale genomic datasets on human lung cancer indicate that almost 90% of the orthologous human genes we identified in our mouse screen are mutated or present in genomic regions of amplification, deletion or rearrangement in a significant portion of human lung cancers (Table S4). This suggests that these genes are likely drivers of human lung cancer and warrant further investigation as potential targets for therapy.
The first screen we performed was on a wild-type background and although we saw a slight increase in lung tumor penetrance over controls (17% vs. 12%), we believe that the SB system did not provide enough mutations per cell to actively accelerate lung tumorigenesis. To overcome this we decided to use mice with common predisposing mutations for lung cancer. Interestingly, we found the highest lung tumor penetrance in mice with conditional ablation of Pten in the lungs, while mice with a conditional dominant negative Trp53 or a germline loss of p19ARF had lung tumor rates similar to controls. It is possible that p19 and p53 mice would have developed more lung tumors if they survived longer (Table 1), but instead became moribund due to other neoplasms caused by Trp53 heterozygosity or germline loss of p19ARF, respectively (Table S1). This suggests that tumor initiation is accelerated by loss of Pten while Trp53 mutations are probably not required for the early stages of cancer formation. In previous screens using the same approach in the intestines and the liver, tumor latency was shorter than in our study, which could be related to a slower turnover rate and lower absolute numbers of cells being targeted using the Spc promoter (46).
Three genes were identified in our screen in more than 30% of mice: Magi1, Nckap5, and Serinc3, MAGI1 is a scaffold protein that plays a role in maintaining tight junctions in epithelial cells. There is evidence that MAGI1 may be playing a role in human lung cancer, as there are more than 10 patient samples with mutations in MAGI1 in both the COSMIC database and TCGA datasets (Table S4). Furthermore, whole genome sequencing revealed a genomic rearrangement in MAGI1 in a human lung cancer (2). SERINC3 belongs to a family of proteins that have the ability to incorporate serine into lipid membranes. Based on the pattern of transposon insertions in our screen we predict that Serinc3 is being activated in the mouse tumors (Table S3). Supporting the hypothesis that SERINC3 is an oncogene, overexpression in rat fibroblast cells increased their resistance to apoptosis and enhanced their tumorigenicity in a xenograft model (12). Similar to MAGI1, SERINC3 is mutated in more than 10 lung cancer patients in COSMIC (Table S4) and was found at a rearranged genomic locus (2). Relatively little is known about the SH3-domain containing NCKAP5, except that mutations are found in patients with melanoma (47). Based on the results of our mouse screen, the known biochemical function of these proteins and their presence in human tumor genomes, we believe further study is warranted.
New approaches are being developed to mine transposon insertion data to find combinations of genes that cooperate in tumorigenesis (24,48,49). We used a form of frequent itemset mining to identify sets of genes that are recurrently mutated in more than one mouse tumor (50). Using this method we identified 10 sets of genes that were mutated in three different mice (−Log(p-value) > 5.0, Table S6). There were 20 annotated protein-coding genes identified in these ten gene sets and all 20 are associated with cancer (www.ingenuity.com) while 15 have been functionally identified as tumor suppressors or oncogenes (CADM2, CDH8, CNTNAP5 CPXCR1, FOXA2, GRB14, LRLP1B, PSME4, ROCK1, SERINC3, SOCS6, SVIL, TCF4 and WNK1). Interestingly nine out of ten of the gene sets included at least one member with a demonstrated functional role in cell adhesion (Svil, Cntnap5a, Cadm2, Cdh8, Flrt2, Foxa2). In addition to cell adhesion, the majority of these gene sets include a gene that protects from apoptosis and enhances tumorigenicity (Serinc3 (12)). Each gene set also typically contained a gene that functions in a signaling pathway known to be important in cancer. Examples include, Socs6 regulating STAT signaling, Magi3 regulating PTEN signaling, Wnk1 regulating WNT signaling and Svil regulating p53 signaling. To prove these genes function cooperatively will require simultaneous alterations in their expression levels. Once validated, the findings could be used to design combinations of targeted therapies attacking the cooperating pathways.
To functionally validate one of our candidate CIS genes we used shRNA to decrease CUL3 expression in stage II lung cancer cell lines and demonstrated that these cells grew faster and were able to form more colonies in soft agar than control cells (Figs. 4 & 5). We analyzed gene expression patterns in A549 human lung cancer cell lines with CUL3, PTEN, or both CUL3 and PTEN knocked down. This analysis indicated that the NRF2 oxidative stress pathway is activated in CUL3/PTEN double knockdown cells, which could explain how these cancer cells are able to withstand the strong oxidative stress generated in cancer. We also measured the effect of knocking down CUL3 and PTEN in the human lung cancer cell line and noted a significant decrease in the production of reactive oxygen species in cells with reduced CUL3 and PTEN. Our results indicate that loss of CUL3 and PTEN may synergize to produce some phenotypes, including proliferation (Fig. 5C) and oxidative stress gene expression regulation, while other phenotypes, including anchorage independent growth (Fig. 5B) and superoxide production (Fig. 6) do not demonstrate a synergistic effect in the double knockdown.
In conclusion, this study has identified a large number of potential genetic drivers of lung cancer. Furthermore, we have shown that loss of CUL3 and PTEN may be driving tumorigenesis by activating the NRF2 oxidative stress pathway.
Supplementary Material
Implications.
This study identifies many novel candidate genetic drivers of lung cancer and demonstrates that CUL3 acts as a tumor suppressor by regulating oxidative stress.
Acknowledgments
Financial Support:
TKS was supported by grants from the National Institutes of Health, National Cancer Institute (5R00CA151672-04 and P30-CA77598), a grant from the American Cancer Society (PF-06-282-01-MGO), and startup funds from the University of Minnesota. CD was supported by an NCI Cancer Biology Training Grant (5-T32-CA009138-37 and 2-T32-CA009138-36) and an NIGMS intramural research and career development award (3-K12-GM074628-03-S1). SRL was supported by the University of Minnesota’s Interdisciplinary Doctoral Fellowship and Doctoral Dissertation Fellowship. MS was supported by a grant from the NSF (IIS-134415).
We thank Blake Jacobsen and Robert Kratzke for sharing reagents and protocol suggestions. We thank Ameeta Kelekar and Eric Hanse for technical assistance measuring reactive oxygen species. This work was possible due to the generous help from the Minnesota Supercomputing Institute, the University of Minnesota Genomics Center, and the University of Minnesota Comparative Pathology Shared Resource.
Footnotes
Conflicts of Interest
The authors do not declare any conflicts of interest.
Authors contributions
Conception and design (CD, DAL, TKS), development of methodology (CD, CJ, MW, RB, SL, MS, ALS, TKS), acquisition of data (CD, CJ, MW, RAB, JB, RK, BN, LF, TKS), analysis and interpretation of the data (CD, SL, MGO’S, SL, MS, AS, KATS, TKS), and writing of manuscript (CD, TKS)
References
- 1.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339(6127):1546–58. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012;150(6):1107–20. doi: 10.1016/j.cell.2012.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hammerman PS, Lawrence MS, Voet D, Jing R, Cibulskis K, Sivachenko A, et al. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012 doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bellacosa A, Kumar CC, Di Cristofano A, Testa JR. Activation of AKT kinases in cancer: implications for therapeutic targeting. Advances in cancer research. 2005;94:29–86. doi: 10.1016/S0065-230X(05)94002-5. [DOI] [PubMed] [Google Scholar]
- 5.Walls M, Baxi SM, Mehta PP, Liu KK, Zhu J, Estrella H, et al. Targeting small cell lung cancer harboring PIK3CA mutation with a selective oral PI3K inhibitor PF-4989216. Clin Cancer Res. 2014;20(3):631–43. doi: 10.1158/1078-0432.CCR-13-1663. [DOI] [PubMed] [Google Scholar]
- 6.Cumberbatch M, Tang X, Beran G, Eckersley S, Wang X, Ellston RP, et al. Identification of a subset of human non-small cell lung cancer patients with high PI3Kbeta and low PTEN expression, more prevalent in squamous cell carcinoma. Clin Cancer Res. 2014;20(3):595–603. doi: 10.1158/1078-0432.CCR-13-1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rosell R, Bivona TG, Karachaliou N. Genetics and biomarkers in personalisation of lung cancer treatment. Lancet. 2013;382(9893):720–31. doi: 10.1016/S0140-6736(13)61715-8. [DOI] [PubMed] [Google Scholar]
- 8.Meuwissen R, Linn SC, Linnoila RI, Zevenhoven J, Mooi WJ, Berns A. Induction of small cell lung cancer by somatic inactivation of both Trp53 and Rb1 in a conditional mouse model. Cancer Cell. 2003;4(3):181–9. doi: 10.1016/s1535-6108(03)00220-4. [DOI] [PubMed] [Google Scholar]
- 9.Cui M, Augert A, Rongione M, Conkrite K, Parazzoli S, Nikitin AY, et al. PTEN is a Potent Suppressor of Small Cell Lung Cancer. Mol Cancer Res. 2014 doi: 10.1158/1541-7786.MCR-13-0554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alimonti A, Carracedo A, Clohessy JG, Trotman LC, Nardella C, Egia A, et al. Subtle variations in Pten dose determine cancer susceptibility. Nat Genet. 2010;42(5):454–8. doi: 10.1038/ng.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iwanaga K, Yang Y, Raso MG, Ma L, Hanna AE, Thilaganathan N, et al. Pten inactivation accelerates oncogenic K-ras-initiated tumorigenesis in a mouse model of lung cancer. Cancer Res. 2008;68(4):1119–27. doi: 10.1158/0008-5472.CAN-07-3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bossolasco M, Veillette F, Bertrand R, Mes-Masson AM. Human TDE1, a TDE1/TMS family member, inhibits apoptosis in vitro and stimulates in vivo tumorigenesis. Oncogene. 2006;25(33):4549–58. doi: 10.1038/sj.onc.1209488. [DOI] [PubMed] [Google Scholar]
- 13.Laura RP, Ross S, Koeppen H, Lasky LA. MAGI-1: a widely expressed, alternatively spliced tight junction protein. Exp Cell Res. 2002;275(2):155–70. doi: 10.1006/excr.2002.5475. [DOI] [PubMed] [Google Scholar]
- 14.Trotman LC, Niki M, Dotan ZA, Koutcher JA, Di Cristofano A, Xiao A, et al. Pten dose dictates cancer progression in the prostate. PLoS biology. 2003;1(3):E59. doi: 10.1371/journal.pbio.0000059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dupuy AJ, Rogers LM, Kim J, Nannapaneni K, Starr TK, Liu P, et al. A modified sleeping beauty transposon system that can be used to model a wide variety of human cancers in mice. Cancer Res. 2009;69(20):8150–6. doi: 10.1158/0008-5472.CAN-09-1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Okubo T, Knoepfler PS, Eisenman RN, Hogan BL. Nmyc plays an essential role during lung development as a dosage-sensitive regulator of progenitor cell proliferation and differentiation. Development. 2005;132(6):1363–74. doi: 10.1242/dev.01678. [DOI] [PubMed] [Google Scholar]
- 17.Collier LS, Carlson CM, Ravimohan S, Dupuy AJ, Largaespada DA. Cancer gene discovery using Sleeping Beauty transposon-based somatic mutagenesis in the mouse. Nature. 2005;436(7048):272–6. doi: 10.1038/nature03681. [DOI] [PubMed] [Google Scholar]
- 18.Dupuy AJ, Akagi K, Largaespada DA, Copeland NG, Jenkins NA. Mammalian mutagenesis using a highly mobile somatic Sleeping Beauty transposon system. Nature. 2005;436(7048):221–6. doi: 10.1038/nature03691. [DOI] [PubMed] [Google Scholar]
- 19.Starr TK, Allaei R, Silverstein KA, Staggs RA, Sarver AL, Bergemann TL, et al. A Transposon-Based Genetic Screen in Mice Identifies Genes Altered in Colorectal Cancer. Science. 2009;323(5922):1747–50. doi: 10.1126/science.1163040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kamijo T, Zindy F, Roussel MF, Quelle DE, Downing JR, Ashmun RA, et al. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell. 1997;91(5):649–59. doi: 10.1016/s0092-8674(00)80452-3. [DOI] [PubMed] [Google Scholar]
- 21.Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, et al. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell. 2004;119(6):847–60. doi: 10.1016/j.cell.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 22.Xiao A, Yin C, Yang C, Di Cristofano A, Pandolfi PP, Van Dyke T. Somatic induction of Pten loss in a preclinical astrocytoma model reveals major roles in disease progression and avenues for target discovery and validation. Cancer Res. 2005;65(12):5172–80. doi: 10.1158/0008-5472.CAN-04-3902. [DOI] [PubMed] [Google Scholar]
- 23.Nikitin AY, Alcaraz A, Anver MR, Bronson RT, Cardiff RD, Dixon D, et al. Classification of proliferative pulmonary lesions of the mouse: recommendations of the mouse models of human cancers consortium. Cancer Res. 2004;64(7):2307–16. doi: 10.1158/0008-5472.can-03-3376. [DOI] [PubMed] [Google Scholar]
- 24.Sarver AL, Erdman J, Starr T, Largaespada DA, Silverstein KA. TAPDANCE: An Automated tool to identify and annotate Transposon insertion CISs and associations between CISs from next generation sequence data. BMC Bioinformatics. 2012;13(1):154. doi: 10.1186/1471-2105-13-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Friedel RH, Friedel CC, Bonfert T, Shi R, Rad R, Soriano P. Clonal Expansion Analysis of Transposon Insertions by High-Throughput Sequencing Identifies Candidate Cancer Genes in a PiggyBac Mutagenesis Screen. PLoS One. 2013;8(8):e72338. doi: 10.1371/journal.pone.0072338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, et al. A census of human cancer genes. Nat Rev Cancer. 2004;4(3):177–83. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fousteri M, Mullenders LH. Transcription-coupled nucleotide excision repair in mammalian cells: molecular mechanisms and biological effects. Cell Res. 2008;18(1):73–84. doi: 10.1038/cr.2008.6. [DOI] [PubMed] [Google Scholar]
- 28.Stamatoyannopoulos JA, Adzhubei I, Thurman RE, Kryukov GV, Mirkin SM, Sunyaev SR. Human mutation rate associated with DNA replication timing. Nat Genet. 2009;41(4):393–5. doi: 10.1038/ng.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Agrawal R, Imieliński T, Swami A. Database mining: A performance perspective. Knowledge and Data Engineering, IEEE Transactions on. 1993;5(6):914–25. [Google Scholar]
- 30.Agrawal R, Imieliński T, Swami A. Mining association rules between sets of items in large databases. ACM; 1993. pp. 207–16. [Google Scholar]
- 31.Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic acids research. 2011;39(Database issue):D945–50. doi: 10.1093/nar/gkq929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T, et al. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol. 2004;24(16):7130–9. doi: 10.1128/MCB.24.16.7130-7139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thu KL, Pikor LA, Chari R, Wilson IM, Macaulay CE, English JC, et al. Genetic disruption of KEAP1/CUL3 E3 ubiquitin ligase complex components is a key mechanism of NF-kappaB pathway activation in lung cancer. J Thorac Oncol. 2011;6(9):1521–9. doi: 10.1097/JTO.0b013e3182289479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ramirez RD, Sheridan S, Girard L, Sato M, Kim Y, Pollack J, et al. Immortalization of human bronchial epithelial cells in the absence of viral oncoproteins. Cancer Res. 2004;64(24):9027–34. doi: 10.1158/0008-5472.CAN-04-3703. [DOI] [PubMed] [Google Scholar]
- 35.Liu J, Lee W, Jiang Z, Chen Z, Jhunjhunwala S, Haverty PM, et al. Genome and transcriptome sequencing of lung cancers reveal diverse mutational and splicing events. Genome Res. 2012;22(12):2315–27. doi: 10.1101/gr.140988.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Prag S, Adams JC. Molecular phylogeny of the kelch-repeat superfamily reveals an expansion of BTB/kelch proteins in animals. BMC Bioinformatics. 2003;4:42. doi: 10.1186/1471-2105-4-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jaramillo MC, Zhang DD. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 2013;27(20):2179–91. doi: 10.1101/gad.225680.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chorley BN, Campbell MR, Wang X, Karaca M, Sambandan D, Bangura F, et al. Identification of novel NRF2-regulated genes by ChIP-Seq: influence on retinoid X receptor alpha. Nucleic Acids Res. 2012;40(15):7416–29. doi: 10.1093/nar/gks409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hu J, Yao H, Gan F, Tokarski A, Wang Y. Interaction of OKL38 and p53 in regulating mitochondrial structure and function. PLoS One. 2012;7(8):e43362. doi: 10.1371/journal.pone.0043362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ade N, Leon F, Pallardy M, Peiffer JL, Kerdine-Romer S, Tissier MH, et al. HMOX1 and NQO1 genes are upregulated in response to contact sensitizers in dendritic cells and THP-1 cell line: role of the Keap1/Nrf2 pathway. Toxicol Sci. 2009;107(2):451–60. doi: 10.1093/toxsci/kfn243. [DOI] [PubMed] [Google Scholar]
- 41.Rachner TD, Gobel A, Benad-Mehner P, Hofbauer LC, Rauner M. Dickkopf-1 as a mediator and novel target in malignant bone disease. Cancer Lett. 2014;346(2):172–7. doi: 10.1016/j.canlet.2014.01.010. [DOI] [PubMed] [Google Scholar]
- 42.Stewart DJ. Wnt signaling pathway in non-small cell lung cancer. J Natl Cancer Inst. 2014;106(1):djt356. doi: 10.1093/jnci/djt356. [DOI] [PubMed] [Google Scholar]
- 43.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350(21):2129–39. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 44.Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304(5676):1497–500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 45.Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, et al. Acquired Resistance of Lung Adenocarcinomas to Gefitinib or Erlotinib Is Associated with a Second Mutation in the EGFR Kinase Domain. PLoS Med. 2005;2(3):e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barkauskas CE, Cronce MJ, Rackley CR, Bowie EJ, Keene DR, Stripp BR, et al. Type 2 alveolar cells are stem cells in adult lung. J Clin Invest. 2013;123(7):3025–36. doi: 10.1172/JCI68782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wagle N, Van Allen EM, Treacy DJ, Frederick DT, Cooper ZA, Taylor-Weiner A, et al. MAP kinase pathway alterations in BRAF-mutant melanoma patients with acquired resistance to combined RAF/MEK inhibition. Cancer Discov. 2014;4(1):61–8. doi: 10.1158/2159-8290.CD-13-0631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.de Ridder J, Kool J, Uren A, Bot J, Wessels L, Reinders M. Co-occurrence analysis of insertional mutagenesis data reveals cooperating oncogenes. Bioinformatics. 2007;23(13):i133–41. doi: 10.1093/bioinformatics/btm202. [DOI] [PubMed] [Google Scholar]
- 49.Bergemann TL, Starr TK, Yu H, Steinbach M, Erdmann J, Chen Y, et al. New methods for finding common insertion sites and co-occurring common insertion sites in transposon- and virus-based genetic screens. Nucleic acids research. 2012;40(9):3822–33. doi: 10.1093/nar/gkr1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Been RA, Linden MA, Hager CJ, DeCoursin KJ, Abrahante JE, Landman SR, et al. Genetic signature of histiocytic sarcoma revealed by a sleeping beauty transposon genetic screen in mice. PLoS One. 2014;9(5):e97280. doi: 10.1371/journal.pone.0097280. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.