

Background: DNA glycosylase NEIL1 initiates prereplicative repair of oxidized DNA.
Results: NEIL1 forms a multiprotein complex with DNA replication proteins via its C-terminal domain (CTD), allowing recruitment at the replication fork. Isolated CTD inhibits this interaction and repair in vitro.
Conclusion: The interactions of NEIL1 are necessary for prereplicative repair.
Significance: The NEIL1 CTD could serve as a target for adjuvant cancer therapy.
Keywords: base excision repair (BER), DNA damage, DNA damage response, DNA polymerase, DNA replication, BERosome, dominant negative inhibition, NEIL1 DNA glycosylase, prereplicative repair
Abstract
The human DNA glycosylase NEIL1 was recently demonstrated to initiate prereplicative base excision repair (BER) of oxidized bases in the replicating genome, thus preventing mutagenic replication. A significant fraction of NEIL1 in cells is present in large cellular complexes containing DNA replication and other repair proteins, as shown by gel filtration. However, how the interaction of NEIL1 affects its recruitment to the replication site for prereplicative repair was not investigated. Here, we show that NEIL1 binarily interacts with the proliferating cell nuclear antigen clamp loader replication factor C, DNA polymerase δ, and DNA ligase I in the absence of DNA via its non-conserved C-terminal domain (CTD); replication factor C interaction results in ∼8-fold stimulation of NEIL1 activity. Disruption of NEIL1 interactions within the BERosome complex, as observed for a NEIL1 deletion mutant (N311) lacking the CTD, not only inhibits complete BER in vitro but also prevents its chromatin association and reduced recruitment at replication foci in S phase cells. This suggests that the interaction of NEIL1 with replication and other BER proteins is required for efficient repair of the replicating genome. Consistently, the CTD polypeptide acts as a dominant negative inhibitor during in vitro repair, and its ectopic expression sensitizes human cells to reactive oxygen species. We conclude that multiple interactions among BER proteins lead to large complexes, which are critical for efficient BER in mammalian cells, and the CTD interaction could be targeted for enhancing drug/radiation sensitivity of tumor cells.
Introduction
A variety of oxidized base lesions in mammalian genomes, induced by endogenous reactive oxygen species, both spontaneously generated during cellular metabolism and generated by several genotoxic agents, including ionizing radiation, are repaired via the base excision repair (BER)4 pathway (1). Many among the mutagenic and cytotoxic oxidized bases contribute to cellular sensitivity to ionizing radiation as well as radiomimetic and other drugs used in cancer therapy. Their repair, which contributes to drug resistance, depends on BER competency of the cells. Thus, various strategies to target the BER pathway are being explored for sensitizing cancer cells as an adjuvant therapy modality with radiation or radiomimetic drugs (2–4).
We recently demonstrated that human DNA glycosylase NEIL1 (Nei-like protein 1) plays a critical role in prereplicative repair of oxidized lesions in S phase cells, a critical process for preventing mutations in the replicating genome (5). NEIL1 is unique among the five oxidized base-specific mammalian DNA glycosylases in the Nth or Nei family for its S phase-specific activation (1). NEIL1 and NEIL2 (in the Nei family, along with the less characterized NEIL3 (6, 7)) are distinct from NTH1 and OGG1 in the Nth family by their ability to act on lesions in single-stranded DNA (ssDNA) substrates (8). NEIL1 functionally interacts with PCNA, FEN-1, and WRN (a RecQ helicase), all of which are involved in DNA replication (10–12). We have previously shown that replication protein A, a mammalian ssDNA-binding protein essential for DNA replication, inhibits DNA glycosylase activity of NEIL1/NEIL2 in vitro with the replication fork-mimicking primer-template DNA substrate (13). This inhibition should be essential in vivo for preventing double strand break formation (5, 13). To further understand the function of NEIL1 in the replicating genome, we recently provided direct experimental evidence for the “cow-catcher” role of NEIL1 in prereplicative repair of oxidized DNA bases in the template strand (5).
We observed that NEIL1 isolated from mammalian cells is present in multiprotein “BERosome” complexes containing DNA replication proteins, and this complex carried out proficient BER in vitro. Furthermore, NEIL1 physically interacted with replication proteins, namely the PCNA clamp loader replication factor C (RFC), replicative DNA polymerase δ (Polδ), and DNA ligase I (LigI) via its disordered C-terminal domain (CTD), which is also the critical region for its interaction with other repair proteins (14, 15). Interestingly, RFC stimulated the DNA glycosylase activity of NEIL1 and NEIL1-initiated complete repair, further supporting the involvement of NEIL1 in replication-associated BER. The inability of a NEIL1 mutant lacking the CTD to associate with chromatin raised the possibility that these replication proteins or some chromatin-associated proteins are essential for recruitment of NEIL1 to the replicating genome. Moreover, isolated CTD significantly inhibited NEIL1-initiated BER both in vitro and in cellulo in a dominant negative manner. These data underscored a unique feature of mammalian BER where repair is regulated via multiprotein interactions within the BERosome and also suggested therapeutic potential of disrupting BERosome as a target to enhance chemo/radiation sensitivity of cancer cells.
Experimental Procedures
Cell Culture, Extract Preparation, and Cell Synchronization
The human embryonic kidney cell line HEK293 was grown in DMEM-high glucose (Life Technologies, Inc.) containing 10% fetal bovine serum (FBS; Sigma), 100 units/ml penicillin, and 100 μg/ml streptomycin mixture (Life Technologies) at 37 °C with 5% CO2. HeLa S3 suspension culture was grown in DMEM-high glucose with 10% FBS and antibiotics.
Whole cell extracts were prepared by lysing the scrape-harvested adherent cells with a buffer containing 50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1% Triton X-100, 0.1 mm EDTA, and protease inhibitor mixture (Roche Applied Science). The extracts were then treated with 500 units/ml each of DNase I and RNase A (Ambion) or benzonase (Novagen) at 37 °C for 30 min and cleared by centrifugation for all experiments to remove nucleic acids. Soluble nuclear and chromatin extracts were prepared using a protocol designed to minimize disruption of in cellulo protein-protein interactions (high salt concentration in the lysis buffers was avoided) as described previously (16). In brief, cells were lysed first in cytoplasmic lysis buffer (10 mm Tris-HCl, pH 8, 0.34 m sucrose, 3 mm CaCl2, 2 mm MgCl2, 0.1 mm EDTA, 1 mm DTT, 0.1% Nonidet P-40, and protease inhibitor), and nuclei were pelleted by centrifugation at 4,000 rpm for 15 min at 4 °C. Nuclei were lysed with nuclear lysis buffer (20 mm HEPES, pH 7.9, 3 mm EDTA, 10% glycerol, 150 mm KOAc, 1.5 mm MgCl2, 0.5% Nonidet P-40, and protease inhibitor). The chromatin pellet was separated from the soluble nuclear fraction by centrifugation at 14,000 rpm for 30 min at 4 °C. Chromatin was then extracted from the pellet by incubating with chromatin lysis buffer (150 mm HEPES, pH 7.9, 1.5 mm MgCl2, 150 mm KOAc, 10% glycerol, and protease inhibitor) supplemented with 0.15 unit/μl benzonase at 37 °C for 30 min followed by centrifugation at 14,000 rpm for 30 min at 4 °C, and the supernatant was collected (16). The extracts were stored at −80 °C.
For synchronization, HEK293 cells were subjected to double-thymidine block as described previously (5, 17). Briefly, ∼40% confluent cells were treated with 10 mm thymidine for 18 h, and then thymidine was removed for 4 h by washing with PBSand adding fresh medium before adding thymidine (10 mm) and incubating for 17 h. Cells were then stimulated to proliferate with fresh media, harvested at various times, and processed for cell cycle analysis as described elsewhere (5).
CRISPR-Cas-mediated Knockout (KO) of NEIL1 in HEK293 Line
The Tet-inducible CRISPR (iCRISPR) strategy was used to knock out NEIL1 in HEK293 cells. Briefly, the single-guide RNA (sgRNA) for the NEIL1 gene (AY257544.1) was designed by screening target sequence with the sgRNA Designer on-line tool (see the Broad Institute Web site). Two high activity sgRNA target sequences were selected targeting exon-2 (sgRNA1, 5′-GTTTGTGAATGAGGCCTGCA-3′; sgRNA2, 5′-TGCAGGGCGCTGGTGTTCGG-3′) and were cloned into lentiviral pLX-sgRNA plasmid (Addgene, catalog no. 50662) at NheI/XhoI sites. HEK293 cells were co-transfected with humanized Cas9 pCW-Cas9 plasmid (Addgene, catalog no. 50661) and customized pLX-sgRNA plasmids using Lipofectamine-2000. Cells stably expressing sgRNA and Cas9 were double-selected with puromycin and blasticidin-S. Cas9 expression was induced by treating the cells to 0.5 μg/ml doxycycline for 96 h. NEIL1 null clones were screened by immunoblotting.
Size Exclusion Chromatography
Fractionation of nuclear extract from HEK293 or HeLa cells was performed in a Sephacryl-300 high resolution gel filtration/size exclusion chromatography column (GE Healthcare, catalog no. S-300HR) using ÄKTA pure (GE Healthcare) equipment. A column (16/60, 120-ml capacity) was optimized using standard molecular weight marker mix (catalog no. MWGF1000-1KT) before applying samples. 2 ml of nuclear extract was loaded for each run in a buffer containing 1× PBS, pH 7.4, at a 1.0-ml/min (30-cm/h) flow rate. 2-ml fractions were collected. Column equilibration and cleaning were performed per the manufacturer's recommendations.
Co-immunoprecipitation (Co-IP) Analysis
HEK293 cells were transfected with empty-FLAG vector-, NEIL1(WT)-FLAG-, or NEIL1(N311)-FLAG-expressing plasmids. At 48–56 h post-transfection, cells were harvested and lysed as described (5, 14, 18). The benzonase-treated extracts were immunoprecipitated by rocking for 3 h at 4 °C with FLAG M2 antibody (Ab) cross-linked to agarose beads (Sigma) (12, 13). The beads were collected by centrifugation and washed three times with cold Tris-buffered saline (TBS) containing 0.1% Triton X-100, and NEIL1-FLAG protein was eluted from the beads. The eluted immunocomplexes were separated on a 12% SDS-polyacrylamide gel and immunoblotted using Abs for Polδ, RFC, LigI, and other indicated Abs (Bethyl Laboratories), followed by incubation with the corresponding HRP-conjugated secondary antibodies (Sigma).
Chromatin Immunoprecipitation (ChIP)
ChIP analysis was performed with exponentially growing cells and following the procedure described previously (5, 19). Briefly, cells in 10-cm plates were washed with PBS, cross-linked with 1% formaldehyde, lysed, and sonicated (XL-2000; QSonica LLC). IP was performed in the sonicated cleared lysate with 5 μg of Abs or control IgG (Santa Cruz Biotechnology, Inc.) and Magna ChIP Protein A magnetic beads (Millipore, catalog no. 16-661) overnight. Following IP, the beads were washed, protein-DNA complexes were eluted, and the cross-links were reversed. DNA was purified using standard phenol/chloroform extraction and finally dissolved in 10 mm Tris-HCl (pH 8). The ChIP and 1% of the input DNA were subjected to SYBR Green-based real-time PCR (7500 Real-Time PCR System; Applied Biosystems) with appropriate primers (Table 1) and SYBR Premix Ex Taq (TaKaRa). Data were calculated using the percent input method (see the Life Technologies Web site).
TABLE 1.
Primer oligonucleotide sequence used for quantitative real time PCR amplification of ChIP porducts in Fig. 6C and for DNA damage analysis in Fig. 9C

Fluorescence Microscopy
HEK293 cells transiently expressing NEIL1(WT)-FLAG or NEIL1(N311)-FLAG were cultured in 8-well chamber slides. To check nuclear localization of ectopic NEIL1, the cells were fixed by incubating in 4% paraformaldehyde for 48 h, followed by permeabilization with 0.2% Tween 20 solution in PBS, and then incubated with anti-FLAG-FITC-conjugated Abs (Sigma). The nuclei were counterstained with DAPI. Microscopy was performed on a Nikon Eclipse Ti System at ×125 magnification. Co-localization was visualized by superimposition of green and red images by using Nikon NIS Elements version 3.5 (Nikon Instruments, Tokyo, Japan).
In Situ Proximity Ligation Assay (PLA)
HEK293 cells grown overnight in 16-well chamber slides were fixed with 4% paraformaldehyde, permeabilized with 0.2% Tween 20, and incubated with a rabbit Ab for NEIL1 (9) and one of several DNA replication proteins (mouse monoclonal; as indicated). The PLA was performed using the Duolink PLA kit (OLink Bioscience, catalog no. LNK-92101-KI01) per the manufacturer's instructions. The nuclei were counterstained with DAPI, and the PLA signals were visualized using a fluorescence microscope (Olympus) at ×200 magnification. For BrdU-NEIL1 PLA, HEK293 cells were grown in 60-mm plates and transfected with NEIL1(WT)-FLAG or NEIL1(N311)-FLAG plasmids. After 24 h, the transfected cells were plated in 8-well chamber slides. The next day, log phase cells were pulse-labeled with BrdU (10 μm/10 min), as described previously (5), and PLA was performed with anti-FLAG Ab (Sigma, catalog no. A8592) and anti-BrdU (FITC BrdU flow kit, BD PharmingenTM, catalog no. 559619).
Expression and Purification of Recombinant NEIL1 and DNA Replication Proteins
Recombinant WT NEIL1, NEIL2, PNKP, PCNA, Polδ, RFC, LigI, and FEN-1 were purified as described previously (9, 11, 12, 20–23). The NEIL1-derived peptides 1–288, 1–311 (N311), 1–349, 289–389, 289–349, 312–389, 312–349, and 350–389 were purified as described previously (5, 12, 14, 18), and their purity was confirmed by Coomassie-stained SDS-PAGE.
Generation of TAT Fusion Peptide Expression Plasmids, Protein Purification, and Peptide Transduction in HEK293 Cells
The protein transduction domain from HIV-TAT has been widely utilized to deliver biologically active macromolecules into a variety of cell types. TAT is an 11-aa (YGRKKRRQRRR), Arg-rich peptide, which rapidly translocates into mammalian cells (24–26). We generated NEIL1 CTD (aa 289–389 and aa 350–389) bacterial expression clones in pMA1113 containing a distal EGFP-HA tag-TAT-His10 tag. The pMA1113 backbone plasmid was a generous gift from Susan Ledoux (25, 26).
Recombinant His/TAT fusion NEIL1 CTD peptides were purified to homogeneity from Escherichia coli as described previously (5, 18, 27). Briefly, bacterial extracts containing His-tagged peptides were separated by affinity chromatography on an Ni2+ column, followed by chromatography on a HiTrap-SP column (GE Healthcare).
In Vitro His and GST Affinity Pull-down Assay
His affinity pull-down assays were carried out as described previously (11, 12). Briefly, WT NEIL1, its truncated mutant polypeptides, or NEIL2 (20 pmol) were mixed with His-tagged Polδ or RFC (40 pmol), which was prebound to His-select magnetic nickel-nitrilotriacetic acid (Ni-NTA) beads (Qiagen) and incubated for 1 h at 4 °C with constant rotation in a buffer containing 1× TBS, 5% BSA, and 10% glycerol. After washing the beads with TBS containing 0.1% Triton X-100 and 400 mm NaCl, the bound proteins were eluted with SDS sample dye and assessed for the presence of NEIL1 and NEIL2 by immunoblotting. For affinity pull-down assays using GST-tagged NEIL1 C-terminal peptides, glutathione-Sepharose beads (20 μl) bound with GST alone or GST-tagged NEIL1 C-terminal peptides (40 pmol) were mixed with Polδ, RFC, or LigI (20 pmol). After appropriate washing, the SDS sample buffer-eluted proteins were probed with antibodies for Polδ, RFC, or LigI.
DNA Glycosylase Assay
Strand incision of oxidized base-containing DNA by NEIL1 or NEIL2 was analyzed using 5′-32P-labeled 51-mer oligonucleotide containing 5-OHU at position 26 (12). The substrate (2 pmol) was incubated with NEIL1 (0.2 pmol) alone or in the presence of the indicated amounts of RFC or FEN-1 at 37 °C for 15 min in a 10-μl reaction mixture containing 40 mm HEPES, pH 7.5, 50 mm KCl, 100 μg/ml BSA, and 5% glycerol. After stopping the reaction with formamide dye (80% formamide, 20 mm NaOH, 20 mm EDTA, 0.05% bromphenol blue, and 0.05% xylene cyanol), the products were separated by 20% polyacrylamide gel containing 8 m urea in 1× Tris borate-EDTA buffer, pH 8.4 (12, 21), and the radioactivity was quantitated using a PhosphorImager (Amersham Biosciences) and ImageQuant software.
The kinetic parameters were determined by incubating with increasing amounts of unlabeled substrate (2.5–120 nm) added to the reaction mixture containing 40 fmol of 5′-radiolabeled substrate and 20 nm NEIL1 or NEIL1 plus 5 nm RFC for 4 min at 37 °C. Km, Vmax, and kcat were calculated from the linear range of the reaction by regression analysis using Sigma plot software (12, 14).
BER Assay
Complete BER was analyzed in a reconstituted system containing recombinant proteins (PNKP, Polδ, PCNA, RFC, and LigI). The enzymes were incubated with 2 pmol of damage-containing duplex oligonucleotide or 200 fmol of plasmid substrate (pUC19CPD) as described previously (5). Protein concentrations were optimized for maximum product formation. A 20-μl reaction mixture containing 1 mmol of ATP, 25 μmol of unlabeled dNTPs, and 10 μmol of [α-32P]dNTPs (the concentration of the corresponding cold dNTP was lowered to 5 μm unless otherwise specified) in BER buffer (25 mm HEPES-KOH, pH 7.9, 50 mm KCl, 2 mm MgCl2, 0.5 mm DTT) was incubated for 30 min at 37 °C. Appropriate controls lacking specific enzymes were included. After incubation for 30 min at 37 °C, the plasmid DNA was phenol/chloroform-extracted, ethanol-precipitated, recovered, digested with N.BstNBI, and resolved on a denaturing polyacrylamide gel and analyzed with a PhosphorImager. The duplex oligonucleotide product of the reaction was separated in situ in the denaturing gel.
Estimation of Oxidized Base/Single Strand Break Repair by End Point PCR-based Quantitation
Nuclear genome-specific semiquantitative PCR assays of long DNA fragments for measuring DNA damage were performed as described earlier (28, 29) using LongAmp TaqDNA polymerase (New England Biolabs) to amplify a 10-kb region of genomic DNA, using appropriate primer sequences (Table 1). Preliminary assays were carried out to ensure the linearity of PCR amplification with respect to the number of cycles and DNA concentration. Unrepaired oxidized bases in DNA were measured by digestion with Fpg/endonuclease III to generate strand breaks before PCR analysis (29).
Clonogenic Cell Survival Assay
Log phase HEK293 cultures were transfected with FLAG-NEIL1(289–389) expression plasmid or the vector control. 24 h after transfection, cells were trypsinized and transferred to 60-mm dishes (400 cells/dish). 24 h post-transfection, the cells were treated with glucose oxidase (0–100 ng/ml) for 15 min. After allowing the cells to grow in fresh medium for 10 days, the colonies were counted after staining with crystal violet to calculate the surviving fraction (14, 18).
Results
NEIL1 Forms BER-proficient Multiprotein Complexes with DNA Replication Proteins in Human Cells
We have previously shown by co-IP analysis that NEIL1 associates with proteins involved in DNA replication machinery for prereplicative repair of the template strand via long patch BER (LP-BER) (5). To test whether NEIL1 associated with these replication proteins in a large repair-competent replication complex, we size-fractionated nuclear extracts (precleared with benzonase to remove nucleic acids) from HEK293 cells in a Sephacryl 300 gel filtration column. Immunoblotting of the eluates showed that about one-third of NEIL1 was in a large megadalton fraction (Fig. 1A), which also contained replication proteins PCNA, RFC, Polδ, LigI, FEN-1, and replication protein A. Another one-third of NEIL1, eluted in a ∼400–500-kDa fraction, mostly contained single-nucleotide BER proteins (data not shown). Nuclear extracts from HeLa S cells also showed a similar multiprotein complex containing NEIL1 (data not shown). Interestingly, NEIL1 in the large size fractions contained multiple lower migrating bands, presumably representing post-translational modifications. Whereas NEIL1 is acetylated in the S phase cells, a detailed account of these modifications will be the topic of a future paper.5 Using a previously described BER assay (5, 18), we show here that this complex fraction, containing NEIL1 and replication proteins, was BER-proficient in vitro (Fig. 1B). Furthermore, OGG1 eluted distinctly from NEIL1 in gel filtration (Fig. 1A), which led us to test whether the complexes contained specific components required for downstream repair steps. For example, NEIL1 immunocomplexcontained PNKP but not APE1 (Fig. 1C), whereas OGG1 immunocomplex contained APE1 but not PNKP (Fig. 1D). PNKP is required to remove the NEIL1 βδ-elimination product 3′P at the strand break, and APE1 removes the OGG1 β-elimination product 3′dRP or cleaves the AP site, mostly generated by OGG1 due to its weak lyase activity (21, 30). The presence of BER activity in the large complex supported our model in which preformed repair complexes in mammalian cells are responsible for endogenous oxidized base repair, which led to the present study to characterize the NEIL1-containing prereplicative repair complexes.
FIGURE 1.

NEIL1 forms a specific BER-proficient multiprotein complex with DNA replication proteins in human cells that contains PNKP but not APE1. A, HEK293 cell nuclear extracts were precleared with benzonase and fractionated on a Sephacryl 300 gel filtration column. A significant fraction of NEIL1 elutes as megadalton-size complex (>2000 kDa), which contains DNA replication proteins. B, complete LP-BER assay was performed with 5′-biotinylated duplex oligonucleotide substrate (51 nucleotides long) with 5-OHU at position 26. Sephacryl fractions 44 and 45, containing NEIL1 and replication proteins, showed repair activity. In contrast, fraction 80, mostly containing uncomplexed NEIL1 and a small amount of PNKP and Polβ, generated unligated repair products. Complete BER was achieved with fraction 62 when supplemented with recombinant PNKP, Polβ, and DNA ligase IIIα (LigIIIα). C and D, co-IP of FLAG-NEIL1 from HEK293 cells contained PNKP but not APE1; conversely, co-IP of FLAG-OGG1 contained APE1 but not PNKP, suggesting that NEIL1 and OGG1 form unique BER complexes containing essential downstream repair proteins. E–G, CRISPR/Cas-mediated NEIL1 KO in HEK293 cells (E) does not affect RFC in cellulo association with PCNA, Polδ, or LigI, as analyzed by RFC co-IP (F) and PLA (G).
To test whether the loss of NEIL1 affects the formation of replication complex in HEK 293 cells, we examined in cellulo association of RFC with other replication proteins in CRISPR/Cas-mediated NEIL1 null/KO HEK293 cells. A CRISPR/Cas9 NEIL1 KO HEK293 cell line (Fig. 1E) was generated as described under “Experimental Procedures.” As we had expected, based on nonessentiality of a DNA repair protein in DNA replication, loss of NEIL1 did not significantly affect RFC interaction with PCNA, FEN-1, Polδ, or LigI, as analyzed by RFC co-immunoprecipitation (Fig. 1F) and PLA (Fig. 1G). These data suggest that whereas NEIL1 stably interacts with replication proteins and is present in a replication protein complex in human cells for prereplicative damage recognition and repair, it would not contribute significantly to the stability of the replication complex. It is more likely that NEIL1, due to its affinity for most replication proteins, is recruited to the preassembled replication complex. In any event, although the spatial organization of these dynamic complexes remains to be investigated, we asked whether NEIL1 stably associates with individual DNA replication proteins in a pairwise fashion.
NEIL1 Directly Interacts with Replication Proteins via Its CTD
After establishing that NEIL1 associated with DNA replication proteins in human cells (5) and that these proteins could be isolated as a NEIL1-containing multiprotein complex (Fig. 1), we examined whether NEIL1 directly interacts with DNA replication proteins RFC, Polδ, and LigI in vitro. These experiments were performed using His and GST fusion recombinant proteins via affinity pull-down assays. Immunoblotting revealed that the His-RFC-bound Ni-NTA beads also contained both NEIL1 and NEIL2 (Fig. 2A (a)). We further mapped the region of the NEIL1 CTD that interacted with RFC by a GST pull-down assay using GST-tagged NEIL1 truncated polypeptides containing aa residues 312–389, 312–349, and 350–389 (Fig. 2A (b)). Positive interactions were observed for residues 312–389 and 312–349 but not for the peptide corresponding to residues 350–389, indicating that the RFC binding interface is localized within residues 312–349. Untagged GST served as the control.
FIGURE 2.

Pairwise interaction of NEIL1 with DNA replication proteins in vitro via CTD. A, interaction with RFC. a, in vitro His tag affinity analysis with recombinant proteins shows that both NEIL1 and NEIL2 directly interact with His-RFC. b, GST-NEIL1 CTD fragments with aa 312–389 and 312–349 but not aa 350–389 co-elute with RFC. GST alone was used as the control. B, interaction with Polδ. a, full-length NEIL1 and its CTD peptide aa 289–389 but not aa 1–288 polypeptide were co-eluted with His-Polδ. WT NEIL2 also co-eluted with His-Polδ. b, as with RFC, Polδ was co-eluted with GST-NEIL1 CTD peptides aa 312–389 and 312–349 but not with aa 350–389. c, Far-Western analysis of Polδ shows its interaction with NEIL1(WT) and the polypeptide aa 1–349 but not the aa 1–311 peptide. FEN1 served as a positive control. d, reverse Far-Western analysis (Polδ on the membrane with four subunits) shows interaction of NEIL1 with p66 and p50 subunits. Polβ served as the positive control. C, interaction with LigI. a, Far-Western analysis of NEIL1 CTD peptides indicates that LigI binding requires aa 289–349 in the NEIL1 CTD. b, co-elution of LigI with GST-NEIL1 fragments aa 289–389 and 289–349 but not aa 312–389 or 312–349. D, the interaction mapping of NEIL1 domains with RFC, Polδ, and LigI shows distinct but overlapping binding residues within the CTD.
We next examined whether NEIL1 (or NEIL2) binarily interacted with Polδ. Both NEIL1 and NEIL2 co-eluted with His-Polδ bound to Ni-NTA-agarose beads, indicating a direct interaction between these proteins (Fig. 2B). Interaction mapping analysis by His or GST pull-down assays showed that NEIL1 fragments of aa 289–389, 312–389, and 312–349 bound to Polδ, but aa 1–288 and 350–389 fragments did not (Fig. 2B, a and b). Together, these data suggested that aa 312–349 in the NEIL1 CTD served as the minimal peptide for interaction with Polδ. This was also confirmed by a Far-Western analysis showing a positive interaction with both NEIL1(WT) and NEIL1(N349) but not with NEIL1(N311) peptides (Fig. 2B (c)). A reverse Far-Western analysis with Polδ subunits separated in SDS-PAGE revealed interaction of NEIL1 with both the p66 and p50 subunits (Fig. 2B (d)).
Similarly, we confirmed in vitro binding of NEIL1 with LigI and mapped the interacting peptide to residues 289–349 in NEIL1 by Far-Western (Fig. 2C (a)) and GST co-elution analysis (Fig. 2C (b)). In addition, we have previously shown pairwise interaction of NEIL1 with PCNA, FEN-1, and replication protein A (11–13). Together, these data show that NEIL1 directly interacts with most DNA replication proteins to form a large multiprotein prereplicative repair complex, which was stabilized by pairwise interactions with individual replication proteins via the NEIL1 CTD. Although it is intriguing that a BER-initiating enzyme like NEIL1 or NEIL2 interacted with most replication/downstream repair proteins, including RFC and LigI, the biological role of such large complexes where the components directly interact with each other warrants further investigation.
NEIL1 CTD Is Required for Its Complex Formation with DNA Replication Proteins in Cells
Extending our in vitro interaction mapping studies, we used co-IP to demonstrate that FLAG-NEIL1(WT) but not the FLAG-NEIL1(N311) mutant, which lacked the CTD, interacted with RFC, Polδ, LigI, and FEN-1 (Fig. 3A). PCNA binding was comparable with WT and N311 NEIL1, as reported previously (11), because PCNA binding requires residues 289–311. Involvement of the NEIL1 CTD in the in cellulo association of NEIL1 with DNA replication proteins was further confirmed in situ by PLA (Fig. 3B). Strong interaction was observed between NEIL1(WT) and Polδ, RFC, PCNA, FEN1, and LigI, indicated by a significant number (>10/nuclei) of PLA foci in the nucleus. The number of foci were significantly reduced for the N311 mutant, nearly 3-fold less than with WT NEIL1 (Fig. 3, B and C). Taken together, these data indicate that NEIL1 exists in complexes with the replication proteins in vivo using a minimal peptide in its CTD. We previously observed that NEIL2 interacted with replication proteins at a weaker level than NEIL1 in human cells (5).
FIGURE 3.

Loss of CTD abrogates in cellulo association of NEIL1 with DNA replication proteins. A, FLAG-IP from FLAG-NEIL1(N311)-expressing HEK293 cells shows a significantly reduced level of LigI, RFC, and Polδ, but not PCNA, compared with that from FLAG-NEIL1(WT)-expressing cells. PCNA binding requires residues 289–312, as shown previously (11). B, PLA of WT versus N311 mutant NEIL1-FLAG with replication proteins confirms that CTD is required for binding. C, quantitation of PLA foci from 25 or more cells. Error bars, S.E.
RFC Enhances NEIL1 DNA Glycosylase Activity and NEIL1-initiated BER in Vitro
We tested whether the base excision/strand incision activity of NEIL is affected by RFC. In vitro activity assays were performed as described using purified NEIL1(WT), NEIL1(N311), and NEIL2. RFC significantly stimulated (∼8-fold) NEIL1 to incise the primer-template substrate at the damaged base site in a concentration-dependent manner (Fig. 4A). Activity of neither NEIL2 nor NEIL1(N311) (which lacked the RFC binding domain) was affected. Furthermore, stimulation of NEIL1 activity by RFC was 2-fold higher with the primer-template substrate than with the duplex substrate (Fig. 4B).
FIGURE 4.

Activation of NEIL1 but not NEIL2 by RFC via pairwise interaction. A, 5-OHU lesion excision/lyase activity of NEILs with 5′-32P-labeled primer-template oligonucleotide substrate shows that RFC stimulates WT NEIL1 by ∼8-fold but does not affect N311 mutant lacking the CTD. NEIL2 activity was also not affected. B, RFC stimulated NEIL1 activity with duplex oligonucleotide substrate, albeit to a lesser extent compared with the primer-template substrate. C, kinetic parameters reveal RFC stimulation of NEIL1 activity primarily by increasing its turnover (∼5-fold increase in Kcat), although a ∼2.5-fold decrease in Km suggested an increase in substrate affinity as well. D–E, RFC activates NEIL1-initiated LP-BER (lanes 5 and 6) with DNA replication proteins. Repair was carried out with pUC19CPD plasmid substrate containing a single 5-OHU lesion (D) (5, 14). The LP-BER reaction was optimized for linear dose dependence for the NEIL1 level (lanes 7 and 8). Error bars, S.E.
Analysis of the kinetic parameters (Fig. 4C) of NEIL1 activity indicated that RFC increased both the substrate affinity (2–3-fold reduction in Km) and product release (∼4-fold increase in kcat) of NEIL.
We reconstituted NEIL1-initiated LP-BER in vitro (characterized by incorporation of [32P]dGMP at the second nucleotide position from the lesion) using purified Polδ, PCNA, RFC, FEN-1, LigI, and PNKP with a 5-OHU lesion-containing plasmid (Fig. 4D). As expected, the presence of all LP-BER factors, including NEIL1, generated completely repaired product (Fig. 4E, lane 4). Because RFC stimulated NEIL1 glycosylase activity via direct interaction, we examined whether RFC stimulated complete repair. A dose-dependent enhancement of LP-BER was observed with increasing RFC amount (Fig. 4E, lanes 5 and 6). As expected, NEIL1 was rate-limiting (Fig. 4E, lanes 1, 7, and 8).
Functional stimulation of NEIL1, but not NEIL2, by RFC was intriguing, because both NEIL1 and NEIL2 physically interact with RFC. We previously made a similar observation with PCNA (11). To test whether this was a common feature of the interaction of NEILs with replication proteins, we then analyzed the impact of FEN-1 binding on NEIL2 activity. FEN-1, which activated NEIL1 in vitro (12) (Fig. 5B, lane 6) did not affect NEIL2 (Fig. 5B, lanes 2–5), although direct physical interaction was observed with both NEIL1 and NEIL2 (Fig. 5A). Interestingly, the binding affinity, measured by the dissociation constant (Kd) of FEN-1 for NEIL1, was 3-fold higher than that for NEIL2 (Fig. 5, C and D). This differential affinity may explain the differential functional association of replication proteins with the two NEIL glycosylases, which was consistent with our model of the back-up role for NEIL2 in BER during replication (5). Furthermore, unlike RFC, Polδ and LigI did not affect NEIL1 activity in vitro (data not shown).
FIGURE 5.

Pairwise interaction and differential stimulation of NEILs with FEN-1. A, Far-Western analysis and His tag co-elution of NEIL1 and NEIL2 with WT FEN1 and its C-terminal domain (aa 328–380). B, BER activity with 5-OHU-containing bubble oligonucleotide (33) shows that FEN-1 stimulates NEIL1 but not NEIL2. C and D, binding (Kd) measurement using fluorescence spectroscopy (12, 14) showed 3-fold reduced affinity of the FEN-1 CTD (aa 328–380) for NEIL2 relative to NEIL1. The FEN-1 CTD lacking Trp or Tyr residues has a negligible contribution to the fluorescence. Error bars, S.E.
Deletion of CTD Impairs NEIL1 Recruitment at Replication Foci
We have previously shown the cell cycle phase-specific recruitment of NEIL1 to chromatin along with DNA replication proteins (5). To test the role of the CTD in NEIL1 chromatin recruitment, we first confirmed comparable expression of ectopic NEIL1(WT) versus NEIL1(N311) in HEK293 cells (Fig. 6A). Although the N311 mutant and WT NEIL1 showed comparable nuclear localization, as indicated by immunofluorescence microscopy (Fig. 6B) and immunoblotting of the nuclear fraction (Fig. 6A), the N311 mutant was absent in the chromatin fraction, unlike WT NEIL1 (Fig. 6A). This suggested that the CTD is required for NEIL1 to associate with chromatin. To test this quantitatively in S phase cells, we performed ChIP analysis on FLAG-tagged NEIL1(WT)- versus NEIL1(N311)-expressing cells with a FLAG antibody. Quantitative ChIP analysis showed significantly higher binding of NEIL1(WT) relative to the N311 mutant at the same representative sequences in S phase cells, but not in the G1/S boundary cells (Fig. 6C).
FIGURE 6.

Loss of CTD abolishes recruitment of NEIL1 to chromatin and replication fork. A, immunoblotting of whole cell extracts, soluble nuclear fraction, and chromatin fraction from HEK293 cells transiently expressing FLAG-NEIL1(WT) or FLAG-N311 shows an absence of the latter in chromatin. B, immunofluorescence staining with FITC-conjugated anti-FLAG antibody shows comparable nuclear localization of N311 and WT NEIL1. C, real-time ChIP-PCR analysis at arbitrary genome sequences (RARβ2 or CDKN1A promoter regions) in double-thymidine block-synchronized HEK293 cells at the G1/S boundary versus S phase shows a requirement of CTD for S phase-specific recruitment of NEIL1 to chromatin. D, log phase HEK293 cells ectopically expressing WT NEIL1-FLAG or N311-FLAG were pulse-labeled with BrdU. PLA was performed with anti-FLAG and anti-BrdU Ab. A significant number of PLA foci in WT NEIL1-expressing cells indicates its recruitment at the replication fork/foci. An absence of PLA foci in N311-expressing cells confirms the requirement of the CTD. 25 cells were analyzed for quantitation. Error bars, S.E.
Next, we utilized PLA to examine the role of the CTD in NEIL1 recruitment at the replication fork after pulse-labeling the HEK293 cells with BrdU. BrdU after pulse-labeling for a short time (>10 min) is observed in discrete foci and was used to identify replication forks (5, 31, 32). Here, we developed a novel approach using PLA with BrdU to confirm localization of NEIL1 at the replication forks. A significant number of PLA foci were observed for FLAG/BrdU in FLAG-NEIL1(WT)-expressing cells, but no PLA foci were observed in cells expressing FLAG-NEIL1(N311) (Fig. 6D). Taken together, these results confirm that the CTD is essential for the recruitment of NEIL1 to replication forks.
The NEIL1 CTD Is Required for Efficient BER with Replication Proteins
We previously showed that the CTD, although dispensable for NEIL1 excision activity, contributed cellular resistance to oxidative stress via its interactions with single-nucleotide BER proteins (18). To determine the impact of the CTD on the prereplicative repair function of NEIL1, we examined the effect of CTD deletion on LP-BER involving DNA replication proteins. The repair reaction was reconstituted with a 5-OHU-containing plasmid substrate. LP-BER, represented by labeled, second nucleotide incorporation from the lesion site in a plasmid substrate (Fig. 4D), showed that repair initiated by NEIL1(N311) was significantly less efficient relative to NEIL1(WT) or NEIL1(1–349), which contained the interaction domain (Fig. 7A). No repair was observed with NEIL1(1–288), which is known to be inactive (18, 30). Similar observations were made using a linear duplex substrate (Fig. 7B). As shown previously (18), the DNA glycosylase activity of NEIL1(WT), NEIL1(1–311), and NEIL1(1–349) were comparable, whereas NEIL1(1–288) was inactive (Fig. 7C). Together, these results showed that the NEIL1 CTD, which is dispensable for its glycosylase activity, is crucial for its interactions with replication proteins, resulting in efficient repair of oxidized bases.
FIGURE 7.
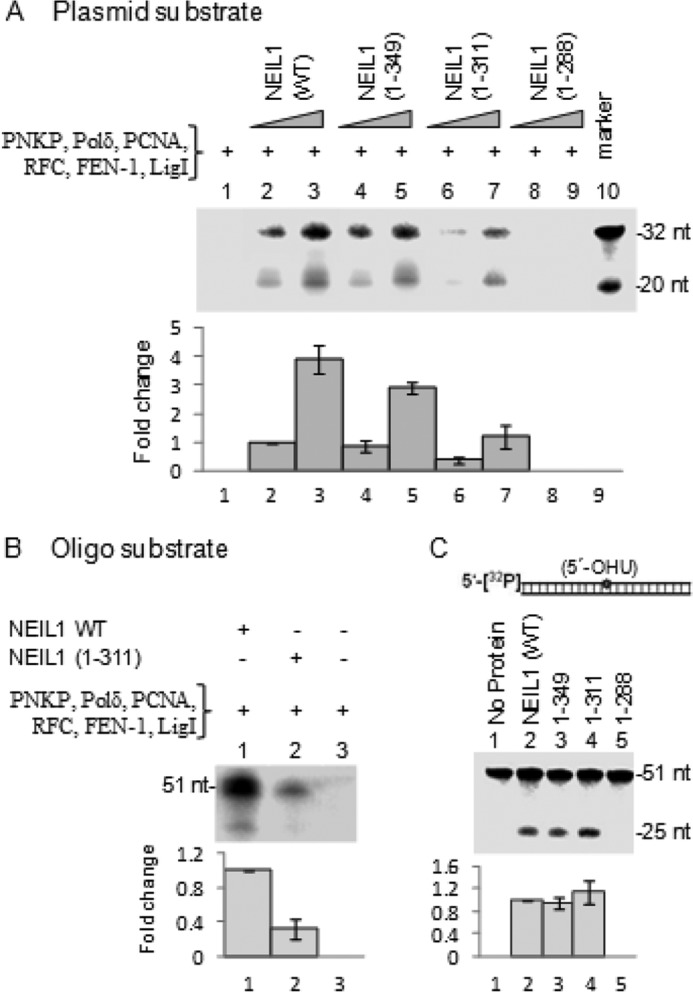
The CTD is dispensable for NEIL1 base excision/AP lyase activity but is required for optimum repair activity involving replication proteins. A, complete LP-BER assay with pUC19CPD as in Fig. 4D, using recombinant proteins, shows proficient repair with WT NEIL1 but not with the nested CTD deletion polypeptides aa 1–349 and 1–311. The 1–288 mutant was inactive, as shown previously (30). B, similar results were obtained with linear duplex oligonucleotide substrate for WT NEIL1 and N311 mutant. C, base excision/AP lyase activity was comparable for WT NEIL1, 1–349, and 1–311. The 1–288 polypeptide was inactive. Error bars, S.E.
Ectopic CTD Peptide Acts as a Dominant Negative Inhibitor of BER
The present study and our previous observations cumulatively demonstrated that mammalian BER is carried out by repair complexes guided by repair-initiating glycosylases, including NEIL1. NEIL1 utilizes its disordered CTD for most of its interactions. Hence, we tested whether ectopic CTD could inhibit BER by inhibiting NEIL1-BERosome formation. When a recombinant CTD peptide was added in vitro, in a LP-BER reaction initiated by NEIL1 and replication proteins using a 5-OHU-containing duplex substrate (Fig. 8A), a dose-dependent inhibition of repair was observed (Fig. 8B, lanes 2–5). The non-interacting peptide, aa 350–389, did not inhibit NEIL1-initiated BER (Fig. 8B, lanes 7 and 8). Complete BER initiated with FLAG-NEIL1 immunocomplex from HEK293 cells was similarly reduced in the presence of the purified CTD peptide (Fig. 8B, lanes 9 and 10).
FIGURE 8.

Isolated CTD peptide of NEIL1 dominant negatively inhibits BER in vitro. A, 5-OHU-containing duplex oligonucleotide, end-biotinylated, used in the LP-BER assay. B, dose-dependent inhibition of LP-BER activity by CTD (aa 312–389; lanes 1–5). The non-interacting CTD (aa 350–389) peptide does not affect BER (lanes 6–8). LP-BER activity of FLAG-NEIL1 IP was inhibited by recombinant CTD peptide (lanes 9 and 10). AU, arbitrary units. Error bars, S.E.
The CTD Peptide Sensitizes HEK293 Cells to Reactive Oxygen Species by Impairing BER
We then examined the effect of ectopic CTD at the cellular level. We generated TAT fusion NEIL1 CTD peptides, aa 289–389 and aa 350–389, also with a GFP tag. NEIL1 aa 289–389 contained the common interaction domain, whereas aa 350–389 was outside of the interaction region and served as a control. Transduction of TAT fusion peptides in HEK293 cells was confirmed by green fluorescence (Fig. 9B). The cells were then exposed to 75 ng/ml glucose oxidase (reactive oxygen species source), grown for 4–5 h to allow time for repair, and then harvested to isolate the genomic DNA. Quantitative PCR analysis of genomic DNA following Fpg and EndoIII treatment (to excise unrepaired oxidized base lesions) showed that unrepaired oxidized base damage was significantly higher in cells transduced with TAT-NEIL1 aa 289–389 (Fig. 9C). Similar results were observed in cells expressing FLAG-NEIL1 aa 289–389 compared with those expressing empty vector or non-interacting peptide (aa 350–389) (Fig. 9C).
FIGURE 9.

Ectopic introduction of CTD peptide sensitizes cells to oxidative stress by inhibiting BERosome formation. A, flow diagram depicting the protocol to measure genome damage/repair after transduction of TAT/GFP-fused CTD peptides or transfection of FLAG-CTD plasmid, which was confirmed by immunofluorescence or FITC-conjugated anti-FLAG antibody (B). C, estimation of oxidized base lesions by long amplicon PCR. Glucose oxidase (GO)-induced DNA damage (measured at 0.5 and 5 h after glucose oxidase treatment) in HEK293 cells was measured by long amplicon PCR of Fpg/EndoVIII-treated genomic DNA. Persistent damage was identified at 5 h in CTD fragment 289–389-expressing cells but not in 350–389-expressing cells. D, TAT-CTD peptide transduction reduced RFC association with PCNA, Polδ, or LigI as analyzed by PLA. Quantitation is shown as a histogram. E, a clonogenic cell survival assay shows reduced survival of glucose oxidase-treated HEK293 cells expressing FLAG-NEIL1(289–389) as compared with vector control or non-interacting peptide (aa 350–389). Error bars, S.E.
Finally, we tested whether ectopic CTD affects NEIL1 in cellulo association with replication proteins. Fig. 9D shows significant (>5-fold) reduction in the number of PLA foci for NEIL1 pair with RFC, Polδ, or LigI. Moreover, the TAT-NEIL1 aa 289–389 peptide strongly inhibited cell survival after glucose oxidase treatment, as measured by a clonogenic assay (Fig. 9E). Together, these studies confirmed that the NEIL1 CTD peptide could inhibit in cellulo BER in a dominant negative manner by inhibiting NEIL1-BERosome formation and sensitizes cells via accumulation of unrepaired oxidized DNA bases.
Discussion
The minimal BER reaction in vitro using naked DNA substrate was previously shown to require only four enzymes, including the repair-initiating DNA glycosylase (1). However, subsequent studies indicated the involvement of more than 20 additional polypeptides in mammalian BER comprising several subpathways. These proteins include scaffolds, damage sensors, RNA/DNA binding proteins, and proteins involved in DNA transcription and replication (5, 12, 14, 33, 34). We recently demonstrated that cross-talk of BER proteins in the NEIL-initiated subpathway and DNA replication machinery ensures repair of mutagenic oxidized lesions prior to DNA replication in S phase cells (5).
Excision repair pathways, including BER and nucleotide excision repair, are commonly believed to comprise multistep, sequential hand-off reactions (35, 36). BER steps include damaged base recognition, its excision and strand scission at the damage site, end-trimming at DNA breaks, and finally repair synthesis followed by nick ligation (1). During hand-off repair, individual steps would require sequential recruitment of specific components.
Whereas the minimal pathway for BER may be operative in prokaryotes (and possibly other primitive organisms), recent observations by us and others suggest that mammalian BER is far more complex and more highly regulated, which is supported by the following observations. (a) Several accessary and scaffold proteins are involved in BER, many of which stably interact with most essential BER proteins (34). (b) BER co-opts proteins involved in DNA replication, transcription, and stress response, presumably to select specific repair subpathways (5, 33, 34). (c) Multiple pairwise interactions are common among the BER components, often involving BER-initiating DNA glycosylases, which even interact with DNA ligase, the last enzyme in the repair pathway (18, 30). (d) Finally, these interactions may involve common interaction interfaces of the repair-initiating enzyme, namely DNA glycosylase or APE1, most of which contain a disordered, non-conserved terminal peptide segment, absent in the E. coli ortholog (1, 15, 34, 37). These studies raise the issue of the intracellular state of mammalian BER components. Specifically, are they present as individual entities or in preformed complexes? In the latter scenario, such complexes would remain bound to the DNA segment containing a substrate lesion until completion of repair. Alternatively, they could be recruited and handed off sequentially as individual repair proteins.
Thus far, our studies have provided support for the presence of preformed BER complexes that carry out sequential reaction steps. We observed that nearly half of NEIL1 in the nuclear extracts from HeLa and HEK293 cells existed in multiprotein complexes, which also contained significant levels of DNA replication proteins. These complexes, separated by size fractionation after removal of DNA, were proficient in carrying out complete BER in vitro. Although multiprotein complexes for DNA repair were characterized previously, particularly for nucleotide excision repair (35, 36, 38), our studies for the first time demonstrated the existence of a BERosome in human cells, which included replication proteins needed for the replication-associated repair subpathway. Surprisingly, NEIL1 interacted directly with most of the proteins present in the complex in a DNA-independent manner, often resulting in its activity enhancement (12, 14). In this report, we have shown marked stimulation of NEIL1 by RFC, an unexpected observation considering that the role of RFC was postulated to be as the PCNA clamp loader. However, contrary to this previous notion, recent studies have indicated the key role of RFC in coordinating LP-BER, especially in regulating FEN-1 and LigI functions (39). RFC is also required for unloading of PCNA after completion of repair, necessitating its presence in the BER complex throughout the repair process (40, 41). Furthermore, PCNA and FEN-1 also activate NEIL1, whereas replication protein A inhibits NEIL1 activity, all of which require direct protein-protein interaction (5, 13). As mentioned previously, the NEIL1-BERosome complex exists in the absence of DNA, suggesting that the entire preformed complex is recruited to or formed on the chromatin to ensure complete BER at the damaged sites.
As observed earlier for single-nucleotide BER, the disordered CTD (aa 289–349) of NEIL1 also interacted with replication proteins. Involvement of a relatively short peptide domain in NEIL1 in its interactions with as many as 20 partner proteins suggests the formation of dynamic complexes where NEIL1 appears to act as a “hub protein” (37). This is consistent with our cow-catcher prereplicative BER model, where NEIL1, as part of the replication complex, provides lesion surveillance to prevent mutagenic replication (5, 34). Such dynamic complexes have also been shown to exist in other cellular processes, including transcription (42–44). Constitutive association of WT NEIL1 with chromatin, which is abrogated by deleting its CTD, further raised the possibility that the preformed multiprotein complex, which is stabilized by binary interactions involving this disordered CTD, is essential for the stable association of NEIL1 with chromatin. Our recent unpublished observations6 suggest that chromatin association of BER complexes, including that of NEIL1, may also involve their interaction with key chromatin factors, including histones, histone chaperones, etc., which is regulated via post-translational modifications.
How such multiprotein complexes overcome structural or steric constraints remains unexplored because of the lack of appropriate experimental tools for in cellulo analysis. It is likely that the intrinsic disorder in the interaction peptide provides the flexible surface needed for allosteric changes to modulate interactions among the components within the complex. Furthermore, pairwise binding among the partner proteins would exponentially increase the stability of large functional complexes in cells. As a proof of concept, using recombinant NEIL1, XRCC1, and Polβ, which were previously shown to interact in a pairwise fashion (30), we showed by size fractionation of the mixture of these proteins that although the binary complexes are not highly stable, the presence of all three proteins facilitates a significant amount of a stable ternary complex (>50% of total protein; Fig. 10).
FIGURE 10.
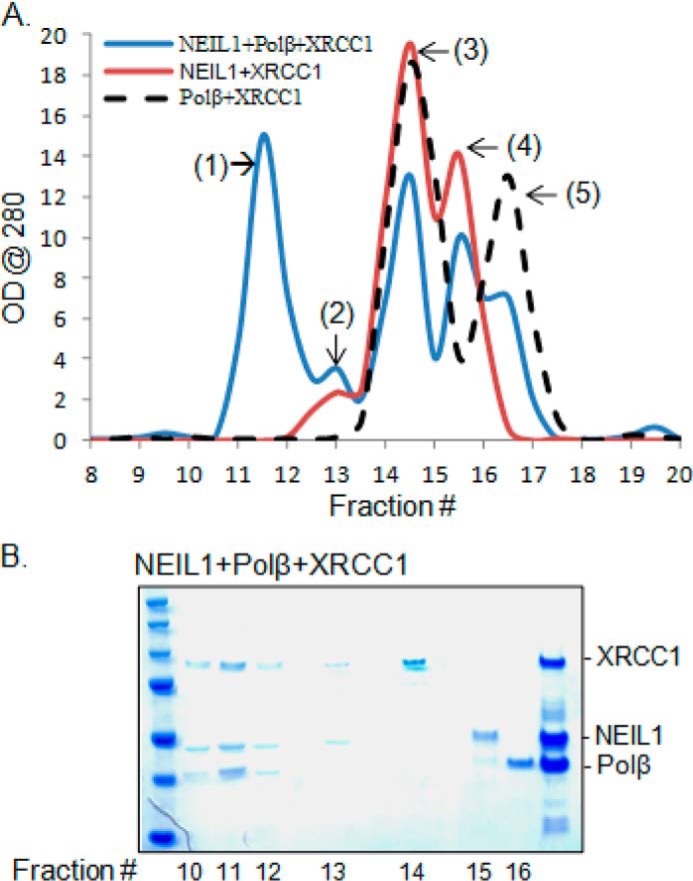
Pairwise interaction between NEIL1, XRCC1, and Polβ stabilizes the ternary complex. A, analytical gel filtration chromatography profile of NEIL1 + XRCC1, Polβ + XRCC1, or NEIL1 + XRCC1 + Polβ. Whereas the two-protein mixtures formed only a negligible fraction of stable complex (peak 2), a significant portion of the mixture of three proteins exists in ternary complex (peak 1). B, Coomassie staining of the peak fractions confirms the ternary complex formation. Peaks 3, 4, and 5 represent monomeric XRCC1, NEIL1, and Polb, respectively.
To investigate the biological importance of multiprotein BER complexes containing NEIL1, we compared the repair efficiency of NEIL1(WT) versus NEIL1(N311), which lacks the common interface for interaction with other repair proteins and thus does not allow active complex formation. Although the base excision/strand incision activities of NEIL1(WT) and NEIL1(N311) are comparable (the CTD is dispensable for NEIL1 DNA glycosylase activity), complete BER was significantly lower with the N311 mutant relative to the WT protein. Together, these results suggest that repair by multiprotein complexes is much more efficient than that by non-complexed proteins. This could be particularly relevant in higher eukaryotes with chromatinized DNA, where the impact of chromatin dynamics on BER is still largely unknown. Although the basic chemistry of BER is conserved from E. coli to mammals, its regulation is unique to higher eukaryotes, including the mammals.
Finally, the in vivo relevance of the BERosome was confirmed by the novel observation of BER inhibition by the NEIL1 CTD. Our finding that this common interaction peptide functions as a dominant negative inhibitor of BER both in vitro and in cellulo, by disrupting BERosome formation, could have significant implications in cancer therapy. Similar interaction-inhibiting peptides have been extensively used to modulate cellular outcomes with clinical relevance. Examples include a peptide corresponding to the interaction domain of NFRF (NF-κβ-repressing factor) that inhibits NFRF-NF-κβ p65 interaction and consequent IL-8 gene activation (45), and a STAT3-derived phosphopeptide that inhibits its dimerization and transcriptional activities (46, 47). Our study providing the proof of concept of inhibiting in vivo BER by disrupting complex formation could be exploited to enhance tumor cell susceptibility to drugs.
Author Contributions
M. L. H. and S. M. conceived and coordinated the study and wrote the paper with contributions from P. M. H., A. D., S. S., A. E. T., I. B., G.-M. L., and T. K. H. P. M. H. purified proteins and performed the experiments shown in Figs. 1 (A and C), 2, 3A, 9, and 10. A. D. performed the experiments in Figs. 1G, 3, 6B, and 9D. S. S. performed the experiments in Fig. 6 (A and C). J. M. generated the CRISPR/Cas-mediated NEIL1 null cell line and performed the experiments in Fig. 1 (E and F). S. A. provided technical assistance and contributed to the manuscript preparation. T. K. H., A. E. T., and G.-M. L. provided reagents. I. B. assisted in microscopy. M. L. H. designed all of the experiments and performed the experiments shown in Figs. 1 (A and B), 3B, 4, 5 (B–D), 7, 8, 9, and 10, and analyzed data. All of the authors approved the final version of the manuscript.
Acknowledgments
We thank Prof. Susan Ledoux (University of South Alabama) for providing the TAT expression plasmid. We also thank Drs. D. W. Bolen and Luis Holthauzen (Department of Biochemistry and Molecular Biology) for the use of the Biophysical Core Facility at the University of Texas Medical Branch at Galveston. We utilized the Imaging Core at Houston Methodist Research Institute for fluorescence microscopy. We thank Drs. C. Theriot and A. Das and V. Borra for various assistance.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 NS088645 (to M. L. H.), R01 CA158910, R01 GM105090, and P01 CA092584 (to S. M.), R01 NS073976 (to T. K. H.), R01 ES018948 (to I. B.), R01 GM57479 (to A. E. T), and R01 CA167181 (to G. M. L). This work was also supported by Muscular Dystrophy Association Grant MDA 294842, ALS Association Grant 15-IIP-204), and Alzheimer's Association Grant NIRG-12-242135) (to M. L. H.). The authors declare that they have no conflicts of interest with the contents of this article.
K. K. Bhakat, S. Sengupta, P. M. Hegde, M. L. Hegde, and S. Mitra, unpublished observations.
S. Sengupta, C. Yang, M. L. Hegde, and S. Mitra, unpublished observations.
- BER
- base excision repair
- Ab
- antibody
- FEN-1
- flap endonuclease 1
- LigI
- DNA ligase I
- PNKP
- polynucleotide kinase 3′-phosphatase
- Polβ
- DNA polymerase β
- Polδ
- DNA polymerase δ
- RFC
- replication factor C
- CTD
- C-terminal domain
- sgRNA
- single-guide RNA
- IP
- immunoprecipitation
- PLA
- proximity ligation assay
- aa
- amino acid(s)
- 5-OHU
- 5-hydroxyuracil
- PCNA
- proliferating cell nuclear antigen
- LP-BER
- long patch BER
- Ni-NTA
- nickel-nitrilotriacetic acid.
References
- 1. Hegde M. L., Hazra T. K., Mitra S. (2008) Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Res. 18, 27–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Adhikari S., Choudhury S., Mitra P. S., Dubash J. J., Sajankila S. P., Roy R. (2008) Targeting base excision repair for chemosensitization. Anticancer Agents Med. Chem. 8, 351–357 [DOI] [PubMed] [Google Scholar]
- 3. Begg A. C., Stewart F. A., Vens C. (2011) Strategies to improve radiotherapy with targeted drugs. Nat. Rev. Cancer 11, 239–253 [DOI] [PubMed] [Google Scholar]
- 4. Bulgar A. D., Snell M., Donze J. R., Kirkland E. B., Li L., Yang S., Xu Y., Gerson S. L., Liu L. (2010) Targeting base excision repair suggests a new therapeutic strategy of fludarabine for the treatment of chronic lymphocytic leukemia. Leukemia 24, 1795–1799 [DOI] [PubMed] [Google Scholar]
- 5. Hegde M. L., Hegde P. M., Bellot L. J., Mandal S. M., Hazra T. K., Li G. M., Boldogh I., Tomkinson A. E., Mitra S. (2013) Prereplicative repair of oxidized bases in the human genome is mediated by NEIL1 DNA glycosylase together with replication proteins. Proc. Natl. Acad. Sci. U.S.A. 110, E3090–3099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liu M., Bandaru V., Bond J. P., Jaruga P., Zhao X., Christov P. P., Burrows C. J., Rizzo C. J., Dizdaroglu M., Wallace S. S. (2010) The mouse ortholog of NEIL3 is a functional DNA glycosylase in vitro and in vivo. Proc. Natl. Acad. Sci. U.S.A. 107, 4925–4930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Liu M., Doublié S., Wallace S. S. (2013) Neil3, the final frontier for the DNA glycosylases that recognize oxidative damage. Mutat. Res. 743, 4–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dou H., Mitra S., Hazra T. K. (2003) Repair of oxidized bases in DNA bubble structures by human DNA glycosylases NEIL1 and NEIL2. J. Biol. Chem. 278, 49679–49684 [DOI] [PubMed] [Google Scholar]
- 9. Hazra T. K., Izumi T., Boldogh I., Imhoff B., Kow Y. W., Jaruga P., Dizdaroglu M., Mitra S. (2002) Identification and characterization of a human DNA glycosylase for repair of modified bases in oxidatively damaged DNA. Proc. Natl. Acad. Sci. U.S.A. 99, 3523–3528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Das A., Boldogh I., Lee J. W., Harrigan J. A., Hegde M. L., Piotrowski J., de Souza Pinto N., Ramos W., Greenberg M. M., Hazra T. K., Mitra S., Bohr V. A. (2007) The human Werner syndrome protein stimulates repair of oxidative DNA base damage by the DNA glycosylase NEIL1. J. Biol. Chem. 282, 26591–26602 [DOI] [PubMed] [Google Scholar]
- 11. Dou H., Theriot C. A., Das A., Hegde M. L., Matsumoto Y., Boldogh I., Hazra T. K., Bhakat K. K., Mitra S. (2008) Interaction of the human DNA glycosylase NEIL1 with proliferating cell nuclear antigen: the potential for replication-associated repair of oxidized bases in mammalian genomes. J. Biol. Chem. 283, 3130–3140 [DOI] [PubMed] [Google Scholar]
- 12. Hegde M. L., Theriot C. A., Das A., Hegde P. M., Guo Z., Gary R. K., Hazra T. K., Shen B., Mitra S. (2008) Physical and functional interaction between human oxidized base-specific DNA glycosylase NEIL1 and flap endonuclease 1. J. Biol. Chem. 283, 27028–27037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Theriot C. A., Hegde M. L., Hazra T. K., Mitra S. (2010) RPA physically interacts with the human DNA glycosylase NEIL1 to regulate excision of oxidative DNA base damage in primer-template structures. DNA Repair 9, 643–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hegde M. L., Banerjee S., Hegde P. M., Bellot L. J., Hazra T. K., Boldogh I., Mitra S. (2012) Enhancement of NEIL1 protein-initiated oxidized DNA base excision repair by heterogeneous nuclear ribonucleoprotein U (hnRNP-U) via direct interaction. J. Biol. Chem. 287, 34202–34211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hegde M. L., Tsutakawa S. E., Hegde P. M., Holthauzen L. M., Li J., Oezguen N., Hilser V. J., Tainer J. A., Mitra S. (2013) The disordered C-terminal domain of human DNA glycosylase NEIL1 contributes to its stability via intramolecular interactions. J. Mol. Biol. 425, 2359–2371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Aygün O., Svejstrup J., Liu Y. (2008) A RECQ5-RNA polymerase II association identified by targeted proteomic analysis of human chromatin. Proc. Natl. Acad. Sci. U.S.A. 105, 8580–8584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chiu R. W., Baril E. F. (1975) Nuclear DNA polymerases and the HeLa cell cycle. J. Biol. Chem. 250, 7951–7957 [PubMed] [Google Scholar]
- 18. Hegde M. L., Hegde P. M., Arijit D., Boldogh I., Mitra S. (2012) Human DNA glycosylase NEIL1's interactions with downstream repair proteins is critical for efficient repair of oxidized DNA base damage and enhanced cell survival. Biomolecules 2, 564–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sengupta S., Mantha A. K., Mitra S., Bhakat K. K. (2011) Human AP endonuclease (APE1/Ref-1) and its acetylation regulate YB-1-p300 recruitment and RNA polymerase II loading in the drug-induced activation of multidrug resistance gene MDR1. Oncogene 30, 482–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hazra T. K., Kow Y. W., Hatahet Z., Imhoff B., Boldogh I., Mokkapati S. K., Mitra S., Izumi T. (2002) Identification and characterization of a novel human DNA glycosylase for repair of cytosine-derived lesions. J. Biol. Chem. 277, 30417–30420 [DOI] [PubMed] [Google Scholar]
- 21. Hill J. W., Hazra T. K., Izumi T., Mitra S. (2001) Stimulation of human 8-oxoguanine-DNA glycosylase by AP-endonuclease: potential coordination of the initial steps in base excision repair. Nucleic Acids Res. 29, 430–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Taylor R. M., Whitehouse C. J., Caldecott K. W. (2000) The DNA ligase III zinc finger stimulates binding to DNA secondary structure and promotes end joining. Nucleic Acids Res. 28, 3558–3563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Prasad R., Beard W. A., Strauss P. R., Wilson S. H. (1998) Human DNA polymerase β deoxyribose phosphate lyase: substrate specificity and catalytic mechanism. J. Biol. Chem. 273, 15263–15270 [DOI] [PubMed] [Google Scholar]
- 24. Nischan N., Herce H. D., Natale F., Bohlke N., Budisa N., Cardoso M. C., Hackenberger C. P. (2015) Covalent attachment of cyclic TAT peptides to GFP results in protein delivery into live cells with immediate bioavailability. Angew. Chem. Int. Ed. Engl. 54, 1950–1953 [DOI] [PubMed] [Google Scholar]
- 25. Shokolenko I. N., Alexeyev M. F., LeDoux S. P., Wilson G. L. (2005) TAT-mediated protein transduction and targeted delivery of fusion proteins into mitochondria of breast cancer cells. DNA Repair 4, 511–518 [DOI] [PubMed] [Google Scholar]
- 26. Koczor C. A., Snyder J. W., Shokolenko I. N., Dobson A. W., Wilson G. L., Ledoux S. P. (2009) Targeting repair proteins to the mitochondria of mammalian cells through stable transfection, transient transfection, viral transduction, and TAT-mediated protein transduction. Methods Mol. Biol. 554, 233–249 [DOI] [PubMed] [Google Scholar]
- 27. Hegde M. L., Hegde P. M., Holthauzen L. M., Hazra T. K., Rao K. S., Mitra S. (2010) Specific Inhibition of NEIL-initiated repair of oxidized base damage in human genome by copper and iron: potential etiological linkage to neurodegenerative diseases. J. Biol. Chem. 285, 28812–28825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Santos J. H., Meyer J. N., Mandavilli B. S., Van Houten B. (2006) Quantitative PCR-based measurement of nuclear and mitochondrial DNA damage and repair in mammalian cells. Methods Mol. Biol. 314, 183–199 [DOI] [PubMed] [Google Scholar]
- 29. Mandal S. M., Hegde M. L., Chatterjee A., Hegde P. M., Szczesny B., Banerjee D., Boldogh I., Gao R., Falkenberg M., Gustafsson C. M., Sarkar P. S., Hazra T. K. (2012) Role of human DNA glycosylase Nei-like 2 (NEIL2) and single strand break repair protein polynucleotide kinase 3′-phosphatase in maintenance of mitochondrial genome. J. Biol. Chem. 287, 2819–2829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wiederhold L., Leppard J. B., Kedar P., Karimi-Busheri F., Rasouli-Nia A., Weinfeld M., Tomkinson A. E., Izumi T., Prasad R., Wilson S. H., Mitra S., Hazra T. K. (2004) AP endonuclease-independent DNA base excision repair in human cells. Mol. Cell 15, 209–220 [DOI] [PubMed] [Google Scholar]
- 31. Nakamura H., Morita T., Sato C. (1986) Structural organizations of replicon domains during DNA synthetic phase in the mammalian nucleus. Exp. Cell Res. 165, 291–297 [DOI] [PubMed] [Google Scholar]
- 32. Mills A. D., Blow J. J., White J. G., Amos W. B., Wilcock D., Laskey R. A. (1989) Replication occurs at discrete foci spaced throughout nuclei replicating in vitro. J. Cell Sci. 94, 471–477 [DOI] [PubMed] [Google Scholar]
- 33. Banerjee D., Mandal S. M., Das A., Hegde M. L., Das S., Bhakat K. K., Boldogh I., Sarkar P. S., Mitra S., Hazra T. K. (2011) Preferential repair of oxidized base damage in the transcribed genes of mammalian cells. J. Biol. Chem. 286, 6006–6016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dutta A., Yang C., Sengupta S., Mitra S., Hegde M. L. (2015) New paradigms in the repair of oxidative damage in human genome: mechanisms ensuring repair of mutagenic base lesions during replication and involvement of accessory proteins. Cell Mol. Life Sci. 72, 1679–1698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Araújo S. J., Wood R. D. (1999) Protein complexes in nucleotide excision repair. Mutat. Res. 435, 23–33 [DOI] [PubMed] [Google Scholar]
- 36. He Z., Ingles C. J. (1997) Isolation of human complexes proficient in nucleotide excision repair. Nucleic Acids Res. 25, 1136–1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hegde M. L., Hazra T. K., Mitra S. (2010) Functions of disordered regions in mammalian early base excision repair proteins. Cell. Mol. Life Sci. 67, 3573–3587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rodriguez K., Talamantez J., Huang W., Reed S. H., Wang Z., Chen L., Feaver W. J., Friedberg E. C., Tomkinson A. E. (1998) Affinity purification and partial characterization of a yeast multiprotein complex for nucleotide excision repair using histidine-tagged Rad14 protein. J. Biol. Chem. 273, 34180–34189 [DOI] [PubMed] [Google Scholar]
- 39. Fortini P., Dogliotti E. (2007) Base damage and single-strand break repair: mechanisms and functional significance of short- and long-patch repair subpathways. DNA Repair 6, 398–409 [DOI] [PubMed] [Google Scholar]
- 40. Montecucco A., Rossi R., Levin D. S., Gary R., Park M. S., Motycka T. A., Ciarrocchi G., Villa A., Biamonti G., Tomkinson A. E. (1998) DNA ligase I is recruited to sites of DNA replication by an interaction with proliferating cell nuclear antigen: identification of a common targeting mechanism for the assembly of replication factories. EMBO J. 17, 3786–3795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hashiguchi K., Matsumoto Y., Yasui A. (2007) Recruitment of DNA repair synthesis machinery to sites of DNA damage/repair in living human cells. Nucleic Acids Res. 35, 2913–2923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Feaver W. J., Svejstrup J. Q., Bardwell L., Bardwell A. J., Buratowski S., Gulyas K. D., Donahue T. F., Friedberg E. C., Kornberg R. D. (1993) Dual roles of a multiprotein complex from S. cerevisiae in transcription and DNA repair. Cell 75, 1379–1387 [DOI] [PubMed] [Google Scholar]
- 43. Geertsema H. J., van Oijen A. M. (2013) A single-molecule view of DNA replication: the dynamic nature of multi-protein complexes revealed. Curr. Opin. Struct. Biol. 23, 788–793 [DOI] [PubMed] [Google Scholar]
- 44. Luijsterburg M. S., von Bornstaedt G., Gourdin A. M., Politi A. Z., Moné M. J., Warmerdam D. O., Goedhart J., Vermeulen W., van Driel R., Höfer T. (2010) Stochastic and reversible assembly of a multiprotein DNA repair complex ensures accurate target site recognition and efficient repair. J. Cell Biol. 189, 445–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bartels M., Schweda A. T., Dreikhausen U., Frank R., Resch K., Beil W., Nourbakhsh M. (2007) Peptide-mediated disruption of NFκB/NRF interaction inhibits IL-8 gene activation by IL-1 or Helicobacter pylori. J. Immunol. 179, 7605–7613 [DOI] [PubMed] [Google Scholar]
- 46. Turkson J., Ryan D., Kim J. S., Zhang Y., Chen Z., Haura E., Laudano A., Sebti S., Hamilton A. D., Jove R. (2001) Phosphotyrosyl peptides block Stat3-mediated DNA binding activity, gene regulation, and cell transformation. J. Biol. Chem. 276, 45443–45455 [DOI] [PubMed] [Google Scholar]
- 47. Furqan M., Akinleye A., Mukhi N., Mittal V., Chen Y., Liu D. (2013) STAT inhibitors for cancer therapy. J. Hematol. Oncol. 6, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]