

Background: Inhibition of Neisseria gonorrhoeae type II topoisomerases gyrase and TopoIV by the antibacterial spiropyrimidinetrione AZD0914 was investigated.
Results: AZD0914 stabilized the gyrase-DNA complex with double strand DNA cleavage, retaining potency in a fluoroquinolone-resistant mutant, with little inhibition of human type II topoisomerases.
Conclusion: AZD0914 displays mechanistic differences from fluoroquinolones.
Significance: AZD0914 has the potential to combat drug-resistant gonorrhea.
Keywords: antibiotic action, antibiotic resistance, DNA gyrase, DNA topoisomerase, enzyme inhibitor, AZD0914, gonorrhoeae
Abstract
We characterized the inhibition of Neisseria gonorrhoeae type II topoisomerases gyrase and topoisomerase IV by AZD0914 (AZD0914 will be henceforth known as ETX0914 (Entasis Therapeutics)), a novel spiropyrimidinetrione antibacterial compound that is currently in clinical trials for treatment of drug-resistant gonorrhea. AZD0914 has potent bactericidal activity against N. gonorrhoeae, including multidrug-resistant strains and key Gram-positive, fastidious Gram-negative, atypical, and anaerobic bacterial species (Huband, M. D., Bradford, P. A., Otterson, L. G., Basrab, G. S., Giacobe, R. A., Patey, S. A., Kutschke, A. C., Johnstone, M. R., Potter, M. E., Miller, P. F., and Mueller, J. P. (2014) In Vitro Antibacterial Activity of AZD0914: A New Spiropyrimidinetrione DNA Gyrase/Topoisomerase Inhibitor with Potent Activity against Gram-positive, Fastidious Gram-negative, and Atypical Bacteria. Antimicrob. Agents Chemother. 59, 467–474). AZD0914 inhibited DNA biosynthesis preferentially to other macromolecules in Escherichia coli and induced the SOS response to DNA damage in E. coli. AZD0914 stabilized the enzyme-DNA cleaved complex for N. gonorrhoeae gyrase and topoisomerase IV. The potency of AZD0914 for inhibition of supercoiling and the stabilization of cleaved complex by N. gonorrhoeae gyrase increased in a fluoroquinolone-resistant mutant enzyme. When a mutation, conferring mild resistance to AZD0914, was present in the fluoroquinolone-resistant mutant, the potency of ciprofloxacin for inhibition of supercoiling and stabilization of cleaved complex was increased greater than 20-fold. In contrast to ciprofloxacin, religation of the cleaved DNA did not occur in the presence of AZD0914 upon removal of magnesium from the DNA-gyrase-inhibitor complex. AZD0914 had relatively low potency for inhibition of human type II topoisomerases α and β.
Introduction
In 2013 the United States Centers for Disease Control and Prevention classified the threat level associated with the unmet medical need resulting from multidrug-resistant Neisseria gonorrhoeae as urgent (1), and it estimated that at least 800,000 cases of gonorrhea occur per year in the United States alone (2).
Fluoroquinolone antibacterial drugs previously offered an effective treatment option for gonorrhea. Over the last decade, however, development of resistance, first against fluoroquinolones and subsequently against all drugs used for first line treatment, such as cefixime and ceftriaxone (3, 4), demanded the development of novel agents to combat highly resistant N. gonorrhoeae.
Fluoroquinolones, one of the most successful classes of antibiotics on the market (5, 6), target the homologous bacterial type II topoisomerases gyrase and topoisomerase IV (TopoIV).2 Both enzymes are conserved across most bacterial pathogens and are essential for cellular functions, including DNA replication and decatenation. DNA gyrase, a heterotetramer of two subunits, GyrA2-GyrB2, introduces negative supercoils in DNA ahead of the replication fork, thereby relieving torsional strain during replication (6–8). TopoIV, a ParC2-ParE2 heterotetramer, catalyzes decatenation, which is essential for separating linked catenanes of two DNA molecules during replication.
Type II topoisomerases modulate the topology of DNA in eukaryotes (6–11). The human nuclear type II topoisomerases TopoIIα and -β are the targets of inhibitors that have clinical utility for the treatment of cancer (12, 13). Sufficient selectivity by antibacterial drugs for inhibition of the bacterial over human topoisomerases at clinically relevant doses has been achieved, encouraging continued exploration of these enzymes as viable targets for novel antibacterial drugs.
The molecular mechanism of type II topoisomerases is described by a functional model termed the two-gate mechanism (14–16). The catalytic cycle has several stages that can be blocked by inhibitors. Aminocoumarins, such as novobiocin, inhibit gyrase by competing with ATP, thereby blocking the ATPase activity of the GyrB subunit. Fluoroquinolones, such as ciprofloxacin, stabilize the DNA-cleaved gyrase-DNA complex by binding to an interface between DNA, GyrA, and GyrB (17). Recently, a few novel classes of inhibitors have been reported that target bacterial topoisomerase II with modes of inhibition distinct from the fluoroquinolones and aminocoumarins (18, 19).
In this paper, we characterize the mechanism of inhibition of gyrase and TopoIV from N. gonorrhoeae by the novel spiropyrimidinetrione AZD09143 (Fig. 1), which is currently in clinical trials as a treatment for drug-resistant N. gonorrhoeae infections. We examine the effects of ciprofloxacin and AZD0914 resistance mutations on inhibition of N. gonorrhoeae gyrase by these compounds and measure inhibition of human TopoIIα and -β by AZD0914.
FIGURE 1.

Chemical structure of the spiropyrimidinetrione AZD0914.
Experimental Procedures
Materials
Buffers, salts, and routine biochemicals were sourced from Sigma-Aldrich and were of reagent grade or higher purity. Plasmid NTC0109711-U6-shRNA, a derivative of pCR4-TOPO, was used in supercoiling, cleaved complex, and religation assays. Relaxation of the supercoiled form was done as described previously (20). It was obtained in supercoiled form from Nature Technologies (Lincoln, NE). Kinetoplast DNA used in decatenation assays was obtained from Topogen, Inc. (Port Orange, FL). Ciprofloxacin HCl was from MP Biomedicals (Santa Ana, CA). Etoposide and ATP were from Sigma-Aldrich. Human TopoIIα was from Affymetrix (Santa Clara, CA). Human TopoIIβ was supplied by Prof. Caroline A. Austin (University of Newcastle-upon-Tyne).
Chemistry
AZD0914 was synthesized as described by Basarab et al. (21).
Inhibition of Macromolecule Biosynthesis
The procedure was performed according to Hilliard (10), with modifications published previously (22). Escherichia coli was grown at room temperature in cation-adjusted Mueller Hinton Broth 1 (Sigma-Aldrich) in the presence of radiolabeled precursors. As positive controls, rifamycin blocked the incorporation of labeled uridine into RNA, erythromycin blocked labeled valine and leucine incorporation into protein, penicillin G blocked labeled N-acetylglucosamine incorporation into the cell wall, triclosan blocked labeled acetic acid incorporation into fatty acids, and the aminocoumarin novobiocin as well as the fluoroquinolone norfloxacin blocked DNA synthesis.
SOS Induction Assay
The SOS induction assay was performed as described (23).
DNA Manipulations and Plasmid Construction
For wild type N. gonorrhoeae gyrase, the following plasmids were used: pJT1330 (N. gonorrhoeae GyrA-TEV-His6), pJT1331 (His6-TEV-N. gonorrhoeae ParE), pJT1337 (His6-TEV-N. gonorrhoeae GyrB), pETite N-His (T7-based expression plasmid), and pETite C-His KAN (T7-based expression plasmid). The primers used were as follows: NgogyrAFor (5′-GAAGGAGATATACATATGACCGACGCAACCATCCGCCAC-3′), NgogyrARev (5′-GTGATGGTGGTGATGATGGGATCCCTGAAAATACAGGTTTTCGCCGCCACCGTTCTCGGCTTCCGGTTCGGT-3′), NgogyrBFor (5′-CATATGCATCATCACCACCATCACGTGGCGGCGAAAACCTGTATTTTCAGGGATCCCATACTGAACAAAAACACGAAGAA-3′), NgogyrBRev (5′-GTGGCGGCCGCTCTATTATGCGTCGATATTTTGCGCAAT-3′), NgoparEFor (5′-CATATGCATCATCACCACCATCACGGTGGCGGCGAAAACCTGTATTTTCAGGGATCCCATGCTAAAAACAACCAATACAGC-3′), and NgoparERev (5′-GTGGCGGCCGCTCTATTAAATATCGAGTTGCGCCGTATC-3′). For DNA gyrase A and B subunits with resistance point mutations, the following plasmids were used: pJT1354 (His6-TEV-N. gonorrhoeae GyrB D429N), pJT1353 (His6-TEV-N. gonorrhoeae GyrB K450T), and pJT1352 (N. gonorrhoeae GyrA (S91F,D95G)-TEV-His6). The primers used were as follows: S91FD95GFor (5′-CACCCCCACGGCGATTTCGCAGTTTACGGCACCATCGTCCGTATGG-3′), S91FD95GRev (5′-CCATACGGACGATGGTGCCGTAAACTGCGAAATCGCCGTGGGGGTG-3′), gyrBK450TFor (5′-GCGATTTTGCCGCTCACCGGTAAAATTTTGAACGTCG-3′), gyrBK450TRev (5′-CGACGTTCAAAATTTTACCGGTGAGCGGCAAAATCGC-3′),gyrBD429NFor (5′-CTCTACCTCGTCGAGGGCAACTCCGCAGGCGGTTCCGCCATGCAG-3′), and gyrBD429NRev (5′-CTGCATGGCGGAACCGCCTGCGGAGTTGCCCTCGACGAGGTAGAG-3′).
Primers for site-directed mutagenesis and PCR DNA amplification were purchased from Eurofins Genomics (Huntsville, AL). Site-directed mutagenesis was performed with the QuikChange II XL site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA) according to the manufacturer's instructions. Plasmid DNA was isolated using the PureYield Plasmid Midiprep System (Promega, Madison, WI). Genomic DNA isolation, plasmid DNA purification, and PCR product purification, were performed using the Wizard Genomic DNA Purification Kit (Promega), PureYield Plasmid Midiprep System (Promega), and QuickStepTM2 PCR Purification Kit (EdgeBio, Gaithersburg, MD), respectively. All PCRs were performed with High Fidelity PCR Master (Roche Applied Science) using reaction conditions specified by the manufacturer. All ligation-independent cloning reactions were performed using the Expresso T7 cloning and expression system (Lucigen, Middleton, WI) according to the manufacturer's instructions using the pETite N-His or pETite C-His KAN vector. DNA sequences of cloned genes were confirmed by sequencing on an ABI PRISM 3100 DNA sequencer (Applied Biosystems, Foster City, CA) using the Big Dye Terminator cycle sequencing kit (Applied Biosystems). Computer analysis of DNA sequences was performed with Sequencher (Gene Codes Corp., Ann Arbor, MI).
Plasmids for expressing either a TEV protease-cleavable N-terminal His6-tagged N. gonorrhoeae GyrB (pJT1337) or ParE (pJT1331) were made by amplifying the gene using the primers NgogyrBFor and NgogyrBRev (gyrB) or NgoparEFor and NgoparERev (parE) from genomic DNA by PCR. The PCR product was purified and cloned into the plasmid pETite N-His by ligation-independent cloning. The cloning reaction was transformed into Hi-Control 10G competent cells (Lucigen). Transformants were selected on LB + 25 μg/ml kanamycin, plasmids were isolated, and inserts were verified by PCR and sequencing.
Plasmid pJT1330 for expressing a TEV protease-cleavable C-terminal His6-tagged N. gonorrhoeae GyrA was constructed by amplifying the gene encoding N. gonorrhoeae GyrA from genomic DNA by PCR using the primers NgogyrAFor and NgogyrARev. The resulting PCR product was spin column-purified and cloned into the plasmid pETite C-His by ligation-independent cloning. The cloning reaction was transformed into Hi-Control 10G competent Cells. Transformants were selected on LB + 25 μg/ml kanamycin, plasmids were isolated, and inserts were verified by PCR and sequencing.
Plasmid pJT1337 was used as a template for site-directed mutagenesis to create D429N and K450T mutations in separate PCRs. The resulting plasmids were designated pJT1354 and pJT1353, respectively. Plasmid pJT1352 for expressing a TEV protease-cleavable C-terminal His6-tagged N. gonorrhoeae GyrA (S91F,D95G) was constructed by mutagenesis using pJT1330 as a template for PCR. Mutagenic PCRs were transformed into XL10-Gold ultracompetent cells (Agilent Technologies). Plasmids were isolated from transformants and verified by sequencing.
Protein Expression and Purification
Protein concentrations were determined by the method of Bradford (24). Purity was confirmed by SDS-PAGE and the correct mass was confirmed by LC-MS. Proteins were stored at −80 °C.
N. gonorrhoeae GyrA
To produce GyrA protein, E. coli Hi-Control BL21(DE3) cells transformed with plasmid pJT1330 were inoculated into LB medium with 25 μg/ml kanamycin at A600 = 0.1, incubated at 37 °C for 3 h, induced with 0.5 mm IPTG at A600 = 0.5, incubated overnight at 18 °C, harvested at A600 2.5 by centrifugation, and frozen at −20 °C. Cell paste from 2 liters of culture expressing GyrA was resuspended in 40 ml of buffer A consisting of 25 mm Tris-HCl (pH 8.0), 0.5 m NaCl, and 5% glycerol, supplemented with one EDTA-free protease inhibitor mixture tablet (Roche Applied Science) and passed through a French press at 4 °C twice at 18,000 p.s.i. The extract was centrifuged at 130,000 × g for 30 min at 4 °C. The supernatant was loaded at a flow rate of 2.0 ml/min onto a 5-ml HiTrap Ni2+ chelating column (GE Healthcare) pre-equilibrated with Buffer A. The column was washed with Buffer A, and the protein was eluted by a linear gradient from 0 to 0.5 m imidazole in Buffer A. Fractions containing GyrA were pooled and dialyzed against 1 liter of Buffer B, consisting of 25 mm HEPES (pH 7.3), 1 mm EDTA, 1 mm DTT, and 5% glycerol. The dialyzed sample was loaded at a flow rate of 2.0 ml/min onto a 20-ml Q-Sepharose HP (HR16/10) column (GE Healthcare) pre-equilibrated with Buffer B. The column was then washed with Buffer B, and the protein was eluted by a linear gradient from 0 to 1 m NaCl in Buffer B. Fractions containing GyrA were pooled and concentrated to 10 ml by an Amicon® Ultracel-10K concentrator (Millipore, Billerica, MA). The yield was 31 mg from 1 liter of cell paste. To obtain GyrA protein carrying point mutations, the same procedure as described for wild type protein was used except that the plasmid carrying the respective point mutations was used for transformation.
N. gonorrhoeae GyrB
To produce protein GyrB, E. coli Hi-Control BL21(DE3) cells transformed with plasmid pJT1337 were inoculated into LB medium with 25 μg/ml kanamycin at A600 = 0.1, incubated at 37 °C for 2 h, induced with 0.5 mm IPTG at A600 = 0.5, incubated for 3 h at 37 °C, harvested at A600 = 1.76 by centrifugation, and frozen at −20 °C. The purification followed the same protocol as described for GyrA. The yield of GyrB was 8.8 mg from 1 liter of cell paste. The protein was stored at −80 °C. To obtain GyrB protein carrying point mutations, the same procedure as described for wild type protein was used, except that the plasmid carrying the respective point mutations was used for transformation.
N. gonorrhoeae ParC
The ParC gene from N. gonorrhoeae was codon-optimized for expression in E. coli and custom-synthesized with a TEV protease cleavage recognition sequence and C-terminal His6 purification tag (BlueSky Bioscience, Worcester, MA). The optimized gene was cloned into pET-24a(+) (Novagen Biosciences, Madison, WI) using XbaI and XhoI restriction sites to create plasmid pNG055.
To produce ParC protein, E. coli BL21(DE3) cells transformed with plasmid pNG055 were inoculated into LB medium with 25 μg/ml kanamycin at A600 = 0.1, incubated at 30 °C for 3.5 h, induced with 0.5 mm IPTG at A600 = 0.6, incubated for 3.5 h at 30 °C, harvested at A600 = 1.7 by centrifugation, and frozen at −20 °C. Cell paste from 2 liters of culture expressing ParC was extracted and subjected to Ni2+-chelating chromatography as for GyrA, except that the flow-through fractions containing ParC were pooled and concentrated to 5 ml by an Amicon® Ultracel-10K concentrator. The concentrated sample was loaded onto a 120-ml Superdex 200 (16/60) column (GE Healthcare Life Sciences) pre-equilibrated with Buffer C, consisting of 25 mm HEPES (pH 7.3), 0.5 m NaCl, 1 mm EDTA, 1 mm DTT, and 5% glycerol. Fractions containing ParC were pooled and concentrated to by an Amicon® Ultracel-10K concentrator. The yield was 25 mg from 1 liter of cell paste.
N. gonorrhoeae ParE
To produce ParE protein, E. coli Rosetta (DE3) cells transformed with plasmid pJT1331 were inoculated into LB medium with 25 μg/ml kanamycin at A600 0.1, incubated at 30 °C for 3.5 h, induced with 0.5 mm IPTG at A600 0.6, incubated for 3 h at 30 °C, harvested at A600 1.7 by centrifugation, and frozen at −20 °C. The purification followed the same protocol as described for GyrA. The yield of ParE was 21 mg from 1 liter of cell paste.
Enzyme Assays
All assays were conducted at room temperature, ∼21 °C. Ciprofloxacin and AZD0914 were dissolved in dimethyl sulfoxide (DMSO), which contributed 1% of the final volume for all assays. Because mutant enzymes displayed differences in specific activity of up to 10-fold, the concentrations of individual protein subunits were adjusted accordingly. To arrive at final assay conditions, each subunit was varied with the other in limiting quantity. Reconstitutions to form active enzyme were performed as described previously (20). Final enzyme concentrations are described such that the limiting subunit concentration is presumed to define one-half of the reconstituted enzyme. These concentrations and the ratio of subunits used in assays are given in Table 1. The composition of TopoIV used in the decatenation and cleaved complex assays were 2 and 20 nm, respectively, with a 1:2 ratio of ParC/ParE.
TABLE 1.
Gyrase reconstitution conditions
Ratio (A:B) is the GyrA to GyrB ratio used in the respective experiment; SC, supercoiling assay; CC, cleaved complex assay; nanomolar final enzyme concentrations are such that limiting subunit concentration is presumed to define one-half of the reconstituted enzyme.
| GyrA | GyrB | Ratio (A:B) | SC | CC |
|---|---|---|---|---|
| nm | nm | |||
| WT | WT | 1:2 | 2 | |
| WT | WT | 1:8 | 20a | |
| WT | D429N | 1:4 | 2 | 40 |
| WT | K450T | 1:8 | 5 | 50 |
| S91F,D95G | WT | 1:2 | 10 | 50 |
| S91F,D95G | K450T | 2:3 | 20 | 50 |
a Religation assay used the same conditions.
Gel Analysis and Quantitation
All assays were evaluated by gel electrophoresis. Gel dimensions were 12 cm wide × 13.5 cm long × 0.7 cm thick. Samples (25 μl) were loaded into wells of 1% agarose gels buffered with 40 mm Tris, 20 mm acetic acid, and 1 mm EDTA at pH 8.4 and run for 18–20 h at 30 V or 3 h at 60 V. For all gel assays except decatenation, 1 μg/ml ethidium bromide was included in the agarose gel running buffer. DNA bands for decatenation assays were visualized by staining with ethidium bromide (5 μg/ml in TAE (Tris base, acetic acid, and EDTA)) after electrophoresis. The DNA was quantified by AlphaEase software (Genetic Technologies) using digital images acquired during UV transillumination. For supercoiling and decatenation reactions, percentage of inhibition was determined directly from band intensities of supercoiled and unlinked DNA bands, referenced to control reactions containing no inhibitor. For cleaved complex and religation assays, background was subtracted based on control wells without inhibitor.
DNA supercoiling assays were carried out using a modification of the method of Mizuuchi (25). Assays were conducted in a 30-μl volume in buffer composed of 35 mm Tris-HCl (pH 7.5), 1.8 mm spermidine, 8 mm MgCl2, 24 mm KCl, 6.5% (w/v) glycerol, 0.005% Brij-35, 2 mm dithiothreitol, 400 ng of relaxed DNA plasmid, and 1 mm ATP. Reactions were initiated by the addition of enzyme and quenched by the addition of 6 μl of 0.5 m EDTA after 30 min, followed by 4 μl of DNA loading dye consisting of 40% sucrose, 100 mm Tris-HCl (pH 7.5), 1 mm EDTA-NaOH (pH 8), and 0.5 mg/ml bromphenol blue. Negative control wells were treated with the quench solution prior to initiation with enzyme.
DNA decatenation assays were performed in 30 μl at room temperature and contained 20 mm Tris-HCl (pH 8.0), 50 mm ammonium acetate, 5 mm dithiothreitol, 8 mm MgCl2, 0.5 mm EDTA, 5% (w/v) glycerol, 0.005% (w/v) Brij-35, 200 ng of kinetoplast DNA, and 1 mm ATP. Reactions were initiated with 2 nm TopoIV and allowed to react for 60 min prior to quenching and processing as described for DNA supercoiling. Negative controls were performed as for supercoiling assays.
For supercoiling and decatenation reactions, the amounts of product present in the negative and positive control reactions were used to define DNA band intensities for 100% (MIN) and 0% (MAX) inhibition, respectively. The percentage of inhibition at each inhibitor concentration was calculated with Equation 1,
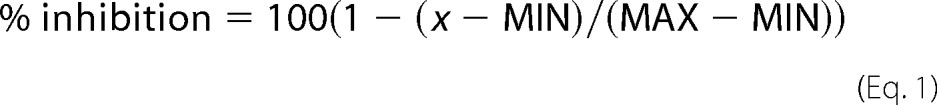 |
where x is the band intensity for the particular reaction. The half-maximal inhibitory concentration (IC50) was obtained by non-linear least squares regression of the percentage of inhibition data with Equation 2,
where n is the Hill coefficient, and [I] is the inhibitor concentration.
Cleaved complex assays for gyrase and TopoIV were conducted similarly to the method described previously (26) with minor modifications to make conditions consistent with the supercoiling and decatenation assays described above. Reactions were performed in 30 μl under the same conditions used for supercoiling or decatenation, respectively, with the exception that enzyme concentrations were increased, as listed in Table 1. TopoIV assays used 400 ng of relaxed plasmid in place of kinetoplast DNA. Reaction times were 30 and 60 min for gyrase and TopoIV, respectively, after which 6 μl of 1% SDS containing 0.5 mg/ml proteinase K was added as a quench. Solutions were gently mixed and incubated at 37 °C for 30 min. DNA loading dye (4 μl) was added, and samples of 30 μl were analyzed by gel electrophoresis.
Assays probing religation of gyrase-cleaved complex used a modification of a published method (27). Assays were conducted in 30 μl with ciprofloxacin and AZD0914 included at 40 μm (>20-fold excess over cleaved complex IC50 values). The cleaved complex was first formed in a 20-μl volume with all reaction components as described for the gyrase-cleaved complex assay, but at a 1.5-fold higher concentration to account for a subsequent dilution, and incubated 30 min. Religation was then induced by the addition of 10 μl of EDTA and sodium sulfate solutions, to variably buffer the free Mg2+ concentration and balance the ionic strength at ∼0.53 m for all conditions. Religation reactions were incubated for 45 min, quenched by the addition of 3 μl of 2% SDS and 1 mg/ml proteinase K, and incubated for 30 min at 37 °C. DNA loading dye (4 μl) was added to each reaction. DNA products were quantified by gel electrophoresis. The concentrations of EDTA and sodium sulfate in the final 30-μl volume were as follows: 104 and 0 mm, 81 and 42 mm, 58 and 83 mm, 35 and 125 mm, 23 and 146 mm, 12 and 166 mm, 6 and 176 mm, and 3 and 177 mm.
The concentration of free Mg2+ for each condition in the religation assays was calculated from the indicated concentrations of total Mg2+ (8 mm; [M]0 and EDTA ([L]0) using Equation 3.
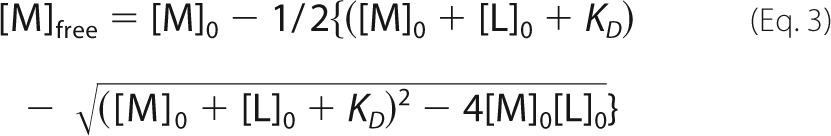 |
The value of KD, representing the pH-adjusted apparent affinity of EDTA for Mg2+, was taken as 2.5 μm (28). Two protonation constants for EDTA and the Mg2+-chelate stability constant (log K) were 10.19, 6.13, and 8.96, respectively (29). Ionic strength contributions to the buffer made by Mg2+ EDTA speciation were determined for each solution by calculating the concentration of free EDTA using Equation 4.
At pH 7.5, the protonation equilibrium of unliganded EDTA is 0.01:0.945:0.045 for the tetra-, tri-, and dianionic species (28). Finally, the excess ionic strength (I) contributed by EDTA for each condition was calculated with Equation 5.
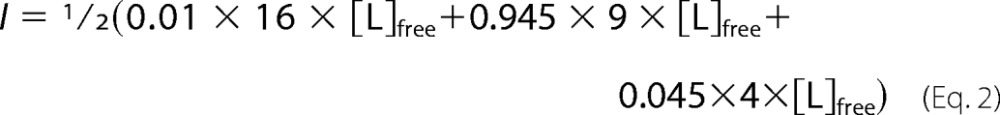 |
Human Topoisomerase Supercoiling Assay
The potency of compounds for inhibiting human TopoIIα and -β, expressed as IC50, was determined with a fluorescence anisotropy-based assay as described (30, 31).
Results
Mechanism of Action of AZD0914
The effect of AZD0914 on macromolecular biosynthesis pathways was investigated by tracking the incorporation of radioactively labeled precursors into actively growing E. coli cells (22). AZD0914 inhibited DNA biosynthesis with an IC50 of 0.037 μg/ml for [3H]thymidine incorporation. The IC50 for RNA biosynthesis was 4.3 μg/ml. The IC50 values for protein, cell wall, and fatty acid biosynthesis were >256 μg/ml. This result is consistent with inhibition of DNA biosynthesis being the primary mechanism of action of AZD0914, as expected for a topoisomerase inhibitor.
SOS Induction in E. coli
When E. coli senses DNA damage or interference with DNA replication, a range of physiological changes known as the SOS response occurs (32–34). Induction of the E. coli SOS response was monitored using an E. coli ΔtolC biosensor strain containing a transcriptional fusion between the recA promoter and GFP on a low copy plasmid. Fig. 2 depicts the SOS response induced after exposure to the aminocoumarin novobiocin, the fluoroquinolone ciproloxacin, and AZD0914. Novobiocin, a slow killing antibiotic that interferes with DNA gyrase by competing with ATP binding, induced a relatively low level, concentration-dependent SOS response. Ciprofloxacin, a rapidly bactericidal fluoroquinolone antibiotic, induced a high level SOS response. AZD0914, which is also rapidly bactericidal (35), induced a high level response similar to that observed for ciprofloxacin at concentrations below its MIC.
FIGURE 2.

Dose-dependent induction of SOS-inducible recA promoter measured as GFP-mediated fluorescence 4 h after challenge with ampicillin, ciprofloxacin, novobiocin, or AZD0914. Data are graphed as an average of 3 samples. Error bars, S.D. The increase in induction is relative to a control that contained 1% DMSO but no compound. The dose is shown in multiples of the MIC for each compound.
Supercoiling and Decatenation
AZD0914 and ciprofloxacin were compared with respect to their inhibitory potencies toward N. gonorrhoeae gyrase-catalyzed supercoiling and N. gonorrhoeae TopoIV-catalyzed decatenation (Fig. 3). Ciprofloxacin was slightly more potent than AZD0914 for gyrase inhibition (1.1 versus 6.3 μm IC50, respectively) and TopoIV inhibition (6.3 versus 19 μm IC50), respectively. For both compounds, inhibition of gyrase was more potent than inhibition of TopoIV.
FIGURE 3.

Inhibition of gyrase-catalyzed supercoiling and TopoIV-catalyzed decatenation by AZD0914 and ciprofloxacin. A, example of supercoiling reactions catalyzed by gyrase in the presence of the indicated concentrations of ciprofloxacin or AZD0914. Wells a and b were used as controls: a, no enzyme; b, enzyme, no compound. B, example of decatenation reactions catalyzed by TopoIV in the presence of the indicated concentrations of ciprofloxacin or AZD0914. Wells a and b were used as controls: a, no enzyme; b, enzyme, no compound. The IC50 and S.E. values for supercoiling and decatenation (in μm) were determined by a global fit of n independent data sets (for AZD0914, IC50 gyrase = 6.3 ± 0.2 (n = 4) and IC50 TopoIV = 19 ± 2 (n = 6); for ciprofloxacin, IC50 gyrase = 1.1 ± 0.2 (n = 4) and IC50 TopoIV = 7 ± 1 (n = 7)).
Cleaved Complex Assay
Both ciprofloxacin and AZD0914 stabilized double strand-broken DNA bound to gyrase and TopoIV (cleaved complex). Whereas the compound concentrations required to produce 50% cleaved complex were similar for both compounds with gyrase (1.9 and 1.7 μm CC50 for ciprofloxacin and AZD0914, respectively), ciprofloxacin was more potent than AZD0914 with TopoIV (0.065 versus 0.5 μm CC50, respectively) (Fig. 4).
FIGURE 4.

Stabilization of cleaved complex for gyrase and TopoIV by AZD0914 and ciprofloxacin. A, example of stabilization of cleaved complex with gyrase in the presence of the indicated concentrations of ciprofloxacin or AZD0914. Wells a and b were used as controls: a, no enzyme; b, enzyme, no compound. B, example of stabilization of cleaved complex with TopoIV in the presence of the indicated concentrations of ciprofloxacin or AZD0914. Wells a and b were used as controls: a, no enzyme; b, enzyme, no compound. N, single strand-nicked DNA; L, linearized, double strand-cleaved DNA; C, intact, closed circular DNA. The CC50 and S.E. values for cleaved complex stabilization (in μm) were determined by a global fit of n independent data sets (for AZD0914, CC50 gyrase = 1.7 ± 0.3 (n = 8) and CC50 TopoIV = 0.5 ± 0.1 (n = 7); for ciprofloxacin, CC50 gyrase = 1.9 ± 0.7 (n = 7) and CC50 TopoIV = 0.065 ± 0.002 (n = 5)).
Religation of Double Strand DNA in the Cleaved Complex
Double strand-cleaved DNA in the DNA-N. gonorrhoeae gyrase complex stabilized by ciprofloxacin can be religated if Mg2+ is removed from the complex (36). It has been proposed that this effect is due to the requirement for Mg2+ in the enzyme-fluoroquinolone complex formation (37, 38). In this experiment, the stabilized cleaved complex between DNA gyrase, inhibitor, and DNA is preformed by preincubation with the respective inhibitor, ciprofloxacin or AZD0914, as described above. Subsequently, EDTA additions to the preformed cleaved complex were chosen to deplete the free Mg2+ to concentrations between 0.1 μm and 1.7 mm. As expected, the cleaved complex formed in the presence of ciprofloxacin religated over a range of free Mg2+ concentrations between 0.1 and 100 μm with an optimal concentration of 0.4–1 μm (Fig. 5). At these concentrations, the magnesium involved in stabilizing ciprofloxacin is probably dissociating, and therefore ciprofloxacin dissociates from the complex, thus allowing religation of the DNA. Reduced religation at lower Mg2+ concentrations may be due to removal of Mg2+ from a second, high affinity site required for religation. In contrast to the results with ciprofloxacin, very little religation was observed over the entire range of Mg2+ concentrations tested for cleaved DNA complex with N. gonorrhoeae gyrase that was induced by AZD0914. A hypothetical model for the binding of AZD0914 to DNA gyrase where AZD0914, in contrast to fluoroquinolones, lacks the chelating interaction with Mg2 was recently proposed by Alm et al. (39) for DNA gyrase. In contrast to these observations for DNA gyrase, religation of the cleaved complex formed with TopoIV by both ciprofloxacin and AZD0914 occurred more completely at Mg2+ concentrations between 0.2 and 100 μm.
FIGURE 5.

Effect of magnesium depletion on the religation reaction of cleaved DNA-gyrase or DNA-TopoIV complex formed in the presence of AZD0914 or ciprofloxacin, respectively. A, example of religation reaction of DNA bound to gyrase inhibited by AZD0914 or ciprofloxacin, respectively. Final concentrations of free Mg2+ are indicated above each well. Wells a and b were used as controls: a, no enzyme; b, enzyme, no compound. The plots below the gel images show nicked (N), linearized (L), and closed circular (C) DNA as a function of free Mg2+ concentration. AZD0914 blocks religation, and ciprofloxacin does not. B, example of religation reaction of DNA bound to TopoIV inhibited by AZD0914 or ciprofloxacin, respectively. Final concentrations of free Mg2+ are indicated above each well. Wells a and b were used as controls: a, no enzyme; b, enzyme, no compound. The plots to the right show nicked, linearized, and closed circular DNA at increasing amounts of MgCl2. Religation occurs both in the presence of AZD0914 and ciprofloxacin.
Effect of Resistance Mutations on Gyrase Inhibition by Ciprofloxacin and AZD0914
We prepared the N. gonorrhoeae GyrA subunit having both of two well characterized point mutations (S91F,D95G) in the quinolone resistance-determining region conferring resistance to fluoroquinolones (40–42), as well as separate N. gonorrhoeae GyrB subunits with one of two point mutations (K450T or D429N) from strains resistant to AZD0914 (39). These mutant subunits were reconstituted with either the wild type or mutated form of the other gyrase subunit. The effects of ciprofloxacin and AZD0914 on supercoiling and cleaved complex stabilization by the mutant enzymes were compared with wild type DNA gyrase (Table 2). All mutant forms had to be reconstituted at higher mutant subunit stoichiometries, as indicated in Table 1, to achieve comparable activities.
TABLE 2.
Effects of ciprofloxacin and AZD0914 on supercoiling and cleaved complex stabilization by mutant enzymes compared with wild type DNA gyrase
| Gyrase |
Supercoiling IC50 |
Cleavage complex CC50 |
MIC |
||||
|---|---|---|---|---|---|---|---|
| GyrA | GyrB | CIP | AZD0914 | CIP | AZD0914 | CIP | AZD0914 |
| μm | μm | μg/ml | |||||
| GyrA (WT) | GyrB (WT) | 1.1 ± 0.2 | 6.3 ± 0.2 | 1.9 ± 0.7 | 1.7 ± 0.3 | 0.008a | 0.125 |
| GyrA (S91F,D95G) | GyrB (WT) | >500 | 2.0 ± 0.2 | >500 | 1 ± 0.4 | 16 | 0.125 |
| GyrA (WT) | GyrB (K450T) | 1 ± 0.3 | 13 ± 3 | 0.3 ± 0.01 | 6 ± 1 | 0.001 | 1 |
| GyrA (WT) | GyrB (D429N) | 1 ± 0.3 | 6 ± 1 | 3 ± 0.4 | 12 ± 0.4 | NDb | ND |
| GyrA (S91F,D95G) | GyrB (K450T) | 24 ± 0.4 | 19 ± 5 | 19 ± 1 | 12 ± 3 | 0.25 | 1 |
a MIC values are taken from Alm (43). The MIC value shown for GyrA (S91F,D95G) GyrB (K450T) is from a strain possessing GyrA (S91F,D95A) GyrB (K450T).
b ND, not determined.
For gyrase with GyrA mutations conferring resistance to ciprofloxacin, the potencies of ciprofloxacin for inhibition of supercoiling (IC50) and stabilization of the cleaved complex (CC50) were more than 20-fold decreased (>500 for mutant versus 1–2 μm for wild type). AZD0914, however, showed a slightly enhanced inhibition of supercoiling (6 μm for wild type versus 2 μm for the GyrA mutant) and cleaved complex stabilization (1.7 μm for wild type versus 1 μm for the GyrA mutant).
Point mutations in the GyrB subunit conferring resistance to AZD0914 did not change the potency of ciprofloxacin with respect to supercoiling inhibition. Whereas the D429N mutation had no significant effect on the potency of cleaved complex stabilization by ciprofloxacin, the K450T mutation increased the potency by 6-fold. The D429N mutation had no effect on the supercoiling IC50 of AZD0914, and the K450T mutation elevated it only 2-fold. In contrast, both mutations substantially elevated the CC50 of AZD0914.
Surprisingly, when the K450T mutant GyrB subunit was reconstituted with the mutant GyrA subunit, the potencies for supercoiling inhibition and cleaved complex stabilization by ciprofloxacin were enhanced by more than 20-fold from >500 μm with the GyrA-only mutant to 24 μm for supercoiling and 19 μm for cleaved complex stabilization. In contrast, this enhancement of potency was not seen for AZD0914.
Selectivity for Bacterial Topoisomerases
Compared with its inhibition of N. gonorrhoeae gyrase, AZD0914 was a relatively weak inhibitor of the ATP-dependent supercoiled DNA relaxation activity of both human topoisomerase IIα and IIβ (IC50 = >400 and 79 μm, respectively). These potencies are comparable with those of ciprofloxacin (IC50 = 110 and 111 μm, respectively). In comparison, the anticancer drug etoposide, which targets human TopoII, showed more potent inhibition for both isozymes (IC50 = 11 and 13 μm, respectively) (Table 3).
TABLE 3.
Inhibition of human topoisomerases by AZD0914, ciprofloxacin and etoposide
| Compound | IC50 (mean ± S.D.) |
|
|---|---|---|
| Topoisomerase IIα | Topoisomerase IIβ | |
| μm | ||
| AZD0914 | >400 (n = 6) | 79 ± 3 (n = 3) |
| Ciprofloxacin | 110 ± 25 (n = 3) | 111 ± 15 (n = 3) |
| Etoposide | 11 ± 2 (n = 5) | 13 ± 2 (n = 3) |
Discussion
AZD0914 preferentially inhibits DNA biosynthesis over biosynthesis of other key macromolecules in sensitive bacteria. This is consistent with inhibition of type II topoisomerases by this compound. The large SOS response caused by AZD0914 (Fig. 2) was similar to that seen with the fluoroquinolone ciprofloxacin, consistent with the observation that AZD0914, like ciprofloxacin, is a type II topoisomerase poison (i.e. it causes double strand DNA breaks due to cleaved complex stabilization). Inhibition of gyrase or topoisomerase IV by an ATP-competitive inhibitor, such as novobiocin, which does not cause direct DNA damage, resulted in a much smaller SOS response.
In N. gonorrhoeae, the rise of resistance to fluoroquinolones makes it imperative to identify new drugs, like AZD0914, that are able to maintain activity against resistant strains. Fluoroquinolone resistance resulting from mutations in the target type II topoisomerases has been attributed to a common mechanism, namely reduced affinity of inhibitor binding because of disruption of a “water-metal ion bridge” between the compound and GyrA or ParC (43, 44). It has been proposed that by minimizing the requirement for this bridge for binding affinity, as seen with quinazolinediones, compounds binding in the same region of these enzymes could overcome this resistance mechanism (45). Although there is as yet no x-ray structural confirmation of the binding site of AZD0914 or related spiropyrimidinetriones, based on mapping resistance mutations in GyrB, Alm and co-workers (39) proposed that AZD0914 occupies the same pocket in gyrase as fluoroquinolones. However, they propose that AZD0914 does not engage the non-catalytic magnesium ion coordinated through GyrA but rather interacts differently, through residues in GyrB that do not involve a chelating interaction with Mg2+. The lack of a water-metal ion bridge to GyrA in the binding of AZD0914 with gyrase, therefore, is a potential explanation for the ability of this compound to maintain activity in fluoroquinolone-resistant strains.
One important feature to define in a new class of inhibitors that act as topoisomerase poisons is selectivity over human type II topoisomerases (Table 2). AZD0914 exhibited little inhibition of human TopoIIα (IC50 > 400 μm versus 110 μm for ciprofloxacin) and weak inhibition of human TopoIIβ (IC50 = 79 μm). The latter IC50 was comparable with that of ciprofloxacin (111 μm), which is well tolerated even after long term clinical administration (46). Thus, it is expected that AZD0914 should not exhibit significant mechanism-based toxicity in humans. Indeed, AZD0914 showed no in vitro mammalian genotoxicity and no measurable toxicity in an in vivo rat micronucleus assay (21).
A comparison of the MIC value of AZD0914 for N. gonorrhoeae (0.25 μg/ml = 0.5 μm) (4) with the observed IC50 and CC50 values for gyrase and TopoIV (IC50 = 6.3 and 19 μm, respectively, and CC50 = 1.7 and 0.5 μm, respectively (Figs. 3 and 4) reveals that the CC50 values are of a similar magnitude as the MIC90 value. This is a similar finding as with fluoroquinolones in E. coli and is explained by the acutely toxic nature of the DNA lesions created by fluoroquinolone action. However, in E. coli, fluoroquinolones display a potency asymmetry, with gyrase inhibited at lower concentrations than TopoIV (47). First-step mutants to fluoroquinolones in E. coli appear exclusively in gyrase and, in combination with the observed lower CC50 values for gyrase, serve to implicate gyrase as the primary fluoroquinolone target in E. coli. In N. gonorrhoeae, mutational studies also implicate gyrase as the primary fluoroquinolone target (41, 48), but surprisingly, in our enzyme assays, the CC50 values for ciprofloxacin were asymmetrical, with the TopoIV CC50 being 29-fold more potent than the CC50 for gyrase. This observation suggests that N. gonorrhoeae may be less susceptible to poisoning of TopoIV as opposed to gyrase. In this regard, the observation with fluoroquinolones in E. coli that TopoIV-mediated cell killing is slower than gyrase-mediated killing (49) may be relevant.
With AZD0914, first step and second step resistance mutations appear in GyrB, implicating gyrase as the primary target, despite a balanced biochemical profile between gyrase and TopoIV as measured in IC50 and CC50 assays, consistent with the hypothesis that poisoning of TopoIV in N. gonorrhoeae may not be as lethal as poisoning of gyrase. This hypothesis could be corroborated with future experiments employing strains engineered with single GyrB and ParE mutations or further with a recent technique that quantifies the amount of inhibitor-bound topoisomerase complexes in cells (50).
Both ciprofloxacin and AZD0914 stabilized the cleaved complex, but to differentiate between the mode of inhibition of AZD0914 and ciprofloxacin, we examined the potential of each drug to interfere with religation of double strand-nicked DNA bound to the topoisomerase tetramer. Investigators working on Staphylococcus aureus and E. coli topoisomerases demonstrated that ciprofloxacin inhibits the religation reaction and that Mg2+ stabilizes the interaction of fluoroquinolones with DNA (27, 36, 51). This interaction is maintained in the DNA topoisomerase-fluoroquinolone complex via the 3-carboxyl group on the fluoroquinolone and an aspartate/glutamate and a serine residue in helix IV of the GyrA subunit (38, 52). To compare ciprofloxacin and AZD0914, we first accumulated cleaved complex and then removed Mg2+ by the addition of EDTA. For the ciprofloxacin-induced DNA-gyrase cleaved complex, religation of the cleaved complex was observed at free Mg2+ concentrations between 0.2 and 100 μm. In contrast, very little religation was observed for the complex induced by AZD0914 at all concentrations of Mg2+ tested. This indicates that Mg2+ is not critically involved in the binding of AZD0914 to N. gonorrhoeae DNA gyrase and is a clear differentiation between the binding modes of AZD0914 and ciprofloxacin (Fig. 5A). This differentiation is further supported by the observation that there is no cross-resistance between ciprofloxacin-resistant N. gonorrhoeae strains and AZD0914 (35, 39). No religation occurred below 0.2 μm Mg2+, probably due to the removal of the Mg2+ from the catalytic site on DNA gyrase. This binding site must have a tighter affinity, and its occupation by Mg2+ may be required for the ligation reaction.
When the same experiment was conducted with TopoIV from N. gonorrhoeae, the cleaved complexes stabilized by both AZD0914 and ciprofloxacin religated when Mg2+ was removed (Fig. 5B). This is in contrast to the observation that the cleaved complex formed with E. coli or S. aureus TopoIV in the presence of AZD0914 cannot religate (data not shown). The result for the N. gonorrhoeae TopoIV enzyme is therefore unusual and may reflect a different mode of binding to TopoIV that involves Mg2+.
The AZD0914 mode of inhibition was further differentiated from that of ciprofloxacin by comparing the ability of both compounds to interact with wild type gyrase as well as previously described mutant versions of gyrase that confer resistance to either ciprofloxacin or AZD0914 (39) (Table 3). We studied the catalytic and inhibition properties of gyrase enzymes containing resistance mutations at loci in GyrA (Ser-91 and Asp-95) that disrupt the fluoroquinolone water-metal ion bridge and two loci for AZD0914 resistance in GyrB (Asp-429 and Lys-450). In the reconstituted fluoroquinolone-resistant gyrase, ciprofloxacin IC50 and concentrations where 50% cleaved complex is observed (CC50) were increased >250-fold from the wild type enzyme, whereas AZD0914 IC50 and CC50 values were unchanged. This result biochemically confirms why AZD0914 maintains activity in fluoroquinolone-resistant strains. The reconstituted gyrase with AZD0914-resistant mutations in GyrB displayed no differences in IC50 values to AZD0914 compared with wild type but did display 3–6-fold elevation in CC50, which agrees with the moderate MIC elevation (∼8-fold) conferred by the GyrB D429N and K450T mutations. Interestingly, the AZD0914-resistant gyrase containing the K450T GyrB mutation displayed a 6-fold lowered CC50 for ciprofloxacin, which is reflected in the 8-fold reduced MIC that GyrB K450T confers in a wild type background. Similarly, the gyrase tetramer carrying resistance mutations against both ciprofloxacin and AZD0914 (GyrA S91F D95G and GyrB K450T) also displayed a lower CC50 for ciprofloxacin compared with the GyrA tetramer that carried just the ciprofloxacin resistance mutations S91F and D95G. These biochemical findings corroborate that the K450T mutation, while conferring resistance to AZD0914, also conveys enhanced susceptibility to ciprofloxacin. They also further reinforce that although ciprofloxacin and AZD0914 both target DNA gyrase, they employ binding modes that are clearly distinct from each other. The observation that resistance against binding of AZD0914 sensitizes binding of ciprofloxacin to gyrase raises the question of whether concomitant dosing of ciprofloxacin and AZD0914 could prevent the emergence of resistant N. gonorrhoeae strains.
Author Contributions
G. K. conceived the studies and wrote the paper. T. P. and B. A. conducted the laboratory work and data evaluation for gel assays. D. E. E. and A. B. S. analyzed experiments and contributed to writing of the paper. G. S. B. produced the compound. P. D. and J. M. analyzed experiments. J. F. and S. D. M. performed and designed the SOS experiments, N. G. purified the proteins. S. S. performed the mode of action experiments, and J. T. cloned the constructs for protein expression. G. K. W. contributed to writing of the paper and performed the calculations for the EDTA titrations.
The authors declare that they have no conflicts of interest with the contents of this article.
AZD0914 will be henceforth known as ETX0914 (Entasis Therapeutics).
- TopoIV
- topoisomerase IV
- TopoII
- topoisomerase II
- CC50
- potency for stabilization of cleaved complex
- MIC
- minimum inhibitory concentration
- TEV
- tobacco etch virus
- IPTG
- isopropyl 1-thio-β-d-galactopyranoside.
References
- 1. Centers for Disease Control and Prevention (2013) Antibiotic Resistance Threats in the United States, 2013, United States Department of Health and Human Resources, Washington, D. C. [Google Scholar]
- 2. Centers for Disease Control and Prevention (2013) Neisseria gonorrhoeae infection frequency in the U.S.A., United States Department of Health and Human Resources, Washington, D. C. [Google Scholar]
- 3. Lowndes C. (2014) Update from the Health Protection Agency: Update on Resistance of Neisseria gonorrhoeae to Antimicrobials, Health Protection Agency, London [Google Scholar]
- 4. Jacobsson S., Golparian D., Alm R. A., Huband M., Mueller J., Jensen J. S., Ohnishi M., Unemo M. (2014) High in vitro activity of the novel spiropyrimidinetrione AZD0914, a DNA gyrase inhibitor, against multidrug-resistant Neisseria gonorrhoeae isolates suggests a new effective option for oral treatment of gonorrhea. Antimicrob. Agents Chemother. 58, 5585–5588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Emami S., Shafiee A., Foroumadi A. (2006) Structural features of new quinolones and relationship to antibacterial activity against Gram-positive bacteria. Mini Rev. Med. Chem. 6, 375–386 [DOI] [PubMed] [Google Scholar]
- 6. Mitscher L. A. (2005) Bacterial topoisomerase inhibitors: quinolone and pyridone antibacterial agents. Chem. Rev. 105, 559–592 [DOI] [PubMed] [Google Scholar]
- 7. Maxwell A. (1997) DNA gyrase as a drug target. Trends Microbiol. 5, 102–109 [DOI] [PubMed] [Google Scholar]
- 8. Maxwell A. (1999) DNA gyrase as a drug target. Biochem. Soc. Trans. 27, 48–53 [DOI] [PubMed] [Google Scholar]
- 9. Gellert M., Mizuuchi K., O'Dea M. H., Itoh T., Tomizawa J. I. (1977) Nalidixic acid resistance: a second genetic character involved in DNA gyrase activity. Proc. Natl. Acad. Sci. U.S.A. 74, 4772–4776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hilliard J. J., Goldschmidt R. M., Licata L., Baum E. Z., Bush K. (1999) Multiple mechanisms of action for inhibitors of histidine protein kinases from bacterial two-component systems. Antimicrob. Agents Chemother 43, 1693–1699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Maxwell A., Lawson D. M. (2003) The ATP-binding site of type II topoisomerases as a target for antibacterial drugs. Curr. Top. Med. Chem. 3, 283–303 [DOI] [PubMed] [Google Scholar]
- 12. Chu D. T., Hallas R., Clement J. J., Alder J., McDonald E., Plattner J. J. (1992) Synthesis and antitumour activities of quinolone antineoplastic agents. Drugs Exp. Clin. Res. 18, 275–282 [PubMed] [Google Scholar]
- 13. Hoshino K., Sato K., Une T., Osada Y. (1989) Inhibitory effects of quinolones on DNA gyrase of Escherichia coli and topoisomerase II of fetal calf thymus. Antimicrob. Agents Chemother. 33, 1816–1818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Roca J., Berger J. M., Harrison S. C., Wang J. C. (1996) DNA transport by a type II topoisomerase: direct evidence for a two-gate mechanism. Proc. Natl. Acad. Sci. U.S.A. 93, 4057–4062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Roca J., Wang J. C. (1992) The capture of a DNA double helix by an ATP-dependent protein clamp: a key step in DNA transport by type II DNA topoisomerases. Cell 71, 833–840 [DOI] [PubMed] [Google Scholar]
- 16. Roca J., Wang J. C. (1994) DNA transport by a type II DNA topoisomerase: evidence in favor of a two-gate mechanism. Cell 77, 609–616 [DOI] [PubMed] [Google Scholar]
- 17. Laponogov I., Sohi M. K., Veselkov D. A., Pan X. S., Sawhney R., Thompson A. W., McAuley K. E., Fisher L. M., Sanderson M. R. (2009) Structural insight into the quinolone-DNA cleavage complex of type IIA topoisomerases. Nat. Struct. Mol. Biol. 16, 667–669 [DOI] [PubMed] [Google Scholar]
- 18. Ehmann D. E., Lahiri S. D. (2014) Novel compounds targeting bacterial DNA topoisomerase/DNA gyrase. Curr. Opin. Pharmacol. 18, 76–83 [DOI] [PubMed] [Google Scholar]
- 19. Wiener J. J., Gomez L., Venkatesan H., Santillán A., Jr., Allison B. D., Schwarz K. L., Shinde S., Tang L., Hack M. D., Morrow B. J., Motley S. T., Goldschmidt R. M., Shaw K. J., Jones T. K., Grice C. A. (2007) Tetrahydroindazole inhibitors of bacterial type II topoisomerases. Part 2: SAR development and potency against multidrug-resistant strains. Bioorg. Med. Chem. Lett. 17, 2718–2722 [DOI] [PubMed] [Google Scholar]
- 20. Shapiro A., Jahic H., Prasad S., Ehmann D., Thresher J., Gao N., Hajec L. (2010) A homogeneous, high-throughput fluorescence anisotropy-based DNA supercoiling assay. J. Biomol. Screen 15, 1088–1098 [DOI] [PubMed] [Google Scholar]
- 21. Basarab G. S., Brassil P., Doig P., Galullo V., Haimes H. B., Kern G., Kutschke A., McNulty J., Schuck V. J., Stone G., Gowravaram M. (2014) Novel DNA gyrase inhibiting spiropyrimidinetriones with a benzisoxazole scaffold: SAR and in vivo characterization. J. Med. Chem. 57, 9078–9095 [DOI] [PubMed] [Google Scholar]
- 22. Patrzykat A., Friedrich C. L., Zhang L., Mendoza V., Hancock R. E. (2002) Sublethal concentrations of pleurocidin-derived antimicrobial peptides inhibit macromolecular synthesis in Escherichia coli. Antimicrob. Agents Chemother. 46, 605–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fan J., de Jonge B. L., MacCormack K., Sriram S., McLaughlin R. E., Plant H., Preston M., Fleming P. R., Albert R., Foulk M., Mills S. D. (2014) A novel high-throughput cell-based assay aimed at identifying inhibitors of DNA metabolism in bacteria. Antimicrob. Agents Chemother. 58, 7264–7272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 25. Mizuuchi K., Mizuuchi M., Craigie R. (1984) The mechanism of transposition of bacteriophage μ. Cold Spring Harb. Symp. Quant. Biol. 49, 835–838 [DOI] [PubMed] [Google Scholar]
- 26. Gellert M., Fisher L. M., O'Dea M. H. (1979) DNA gyrase: purification and catalytic properties of a fragment of gyrase B protein. Proc. Natl. Acad. Sci. U.S.A. 76, 6289–6293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pierrat O. A., Maxwell A. (2003) The action of the bacterial toxin microcin B17. Insight into the cleavage-religation reaction of DNA gyrase. J. Biol. Chem. 278, 35016–35023 [DOI] [PubMed] [Google Scholar]
- 28. Schwarzenbach G., Flaschka H., Irving H. M. N. H. (1969) Complexometric Titrations, 2nd Ed., Methuen, London, UK [Google Scholar]
- 29. Martell A. E., Smith R. M. (2001) NIST Critical Stability Constants of Metal Complexes, National Institute of Standards and Technology, Gaithersburg, MD [Google Scholar]
- 30. Shapiro A. B. (2013) A high-throughput-compatible, fluorescence anisotropy-based assay for ATP-dependent supercoiled DNA relaxation by human topoisomerase IIα. Biochem. Pharmacol. 85, 1269–1277 [DOI] [PubMed] [Google Scholar]
- 31. Shapiro A. B., Austin C. A. (2014) A high-throughput fluorescence anisotropy-based assay for human topoisomerase II β-catalyzed ATP-dependent supercoiled DNA relaxation. Anal. Biochem. 448, 23–29 [DOI] [PubMed] [Google Scholar]
- 32. Schlacher K., Cox M. M., Woodgate R., Goodman M. F. (2006) RecA acts in trans to allow replication of damaged DNA by DNA polymerase V. Nature 442, 883–887 [DOI] [PubMed] [Google Scholar]
- 33. Schlacher K., Pham P., Cox M. M., Goodman M. F. (2006) Roles of DNA polymerase V and RecA protein in SOS damage-induced mutation. Chem. Rev. 106, 406–419 [DOI] [PubMed] [Google Scholar]
- 34. Janion C. (2008) Inducible SOS response system of DNA repair and mutagenesis in Escherichia coli. Int. J. Biol. Sci. 4, 338–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Huband M. D., Bradford P. A., Otterson L. G., Basarab G. S., Kutschke A. C., Giacobbe R. A., Patey S. A., Alm R. A., Johnstone M. R., Potter M. E., Miller P. F., Mueller J. P. (2015) In vitro antibacterial activity of AZD0914, a new spiropyrimidinetrione DNA gyrase/topoisomerase inhibitor with potent activity against Gram-positive, fastidious Gram-Negative, and atypical bacteria. Antimicrob. Agents Chemother. 59, 467–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Palù G., Valisena S., Ciarrocchi G., Gatto B., Palumbo M. (1992) Quinolone binding to DNA is mediated by magnesium ions. Proc. Natl. Acad. Sci. U.S.A. 89, 9671–9675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Aldred K. J., Breland E. J., McPherson S. A., Turnbough C. L., Jr., Kerns R. J., Osheroff N. (2014) Bacillus anthracis GrlAV96A topoisomerase IV, a quinolone resistance mutation that does not affect the water-metal ion bridge. Antimicrob. Agents Chemother. 58, 7182–7187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Aldred K. J., McPherson S. A., Turnbough C. L., Jr., Kerns R. J., Osheroff N. (2013) Topoisomerase IV-quinolone interactions are mediated through a water-metal ion bridge: mechanistic basis of quinolone resistance. Nucleic Acids Res. 41, 4628–4639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Alm R. A., Lahiri S. D., Kutschke A., Otterson L. G., McLaughlin R. E., Whiteaker J. D., Lewis L. A., Su X., Huband M. D., Gardner H., Mueller J. P. (2015) Characterization of the novel DNA gyrase inhibitor AZD0914: low resistance potential and lack of cross-resistance in Neisseria gonorrhoeae. Antimicrob. Agents Chemother. 59, 1478–1486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Endimiani A., Guilarte Y. N., Tinguely R., Hirzberger L., Selvini S., Lupo A., Hauser C., Furrer H. (2014) Characterization of Neisseria gonorrhoeae isolates detected in Switzerland (1998–2012): emergence of multidrug-resistant clones less susceptible to cephalosporins. BMC Infect. Dis. 14, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Belland R. J., Morrison S. G., Ison C., Huang W. M. (1994) Neisseria gonorrhoeae acquires mutations in analogous regions of gyrA and parC in fluoroquinolone-resistant isolates. Mol. Microbiol. 14, 371–380 [DOI] [PubMed] [Google Scholar]
- 42. Deguchi T., Yasuda M., Nakano M., Ozeki S., Kanematsu E., Fukuda H., Maeda S., Saito I., Kawada Y. (1997) Comparison of in vitro antimicrobial activity of AM-1155 with those of tosufloxacin and fleroxacin against clinical isolates of Neisseria gonorrhoeae harboring quinolone resistance alterations in GyrA and ParC. Chemotherapy 43, 239–244 [DOI] [PubMed] [Google Scholar]
- 43. Aldred K. J., Kerns R. J., Osheroff N. (2014) Mechanism of quinolone action and resistance. Biochemistry 53, 1565–1574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wohlkonig A., Chan P. F., Fosberry A. P., Homes P., Huang J., Kranz M., Leydon V. R., Miles T. J., Pearson N. D., Perera R. L., Shillings A. J., Gwynn M. N., Bax B. D. (2010) Structural basis of quinolone inhibition of type IIA topoisomerases and target-mediated resistance. Nat. Struct. Mol. Biol. 17, 1152–1153 [DOI] [PubMed] [Google Scholar]
- 45. Aldred K. J., Schwanz H. A., Li G., McPherson S. A., Turnbough C. L., Jr., Kerns R. J., Osheroff N. (2013) Overcoming target-mediated quinolone resistance in topoisomerase IV by introducing metal-ion-independent drug-enzyme interactions. ACS Chem. Biol. 8, 2660–2668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Segev S., Yaniv I., Haverstock D., Reinhart H. (1999) Safety of long-term therapy with ciprofloxacin: data analysis of controlled clinical trials and review. Clin. Infect. Dis. 28, 299–308 [DOI] [PubMed] [Google Scholar]
- 47. Cambau E., Matrat S., Pan X. S., Roth Dit Bettoni R., Corbel C., Aubry A., Lascols C., Driot J. Y., Fisher L. M. (2009) Target specificity of the new fluoroquinolone besifloxacin in Streptococcus pneumoniae, Staphylococcus aureus, and Escherichia coli. J. Antimicrob. Chemother. 63, 443–450 [DOI] [PubMed] [Google Scholar]
- 48. Shultz T. R., Tapsall J. W., White P. A. (2001) Correlation of in vitro susceptibilities to newer quinolones of naturally occurring quinolone-resistant Neisseria gonorrhoeae strains with changes in GyrA and ParC. Antimicrob. Agents Chemother. 45, 734–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Khodursky A. B., Cozzarelli N. R. (1998) The mechanism of inhibition of topoisomerase IV by quinolone antibacterials. J. Biol. Chem. 273, 27668–27677 [DOI] [PubMed] [Google Scholar]
- 50. Aedo S., Tse-Dinh Y. C. (2012) Isolation and quantitation of topoisomerase complexes accumulated on Escherichia coli chromosomal DNA. Antimicrob. Agents Chemother. 56, 5458–5464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Anderson V. E., Zaniewski R. P., Kaczmarek F. S., Gootz T. D., Osheroff N. (1999) Quinolones inhibit DNA religation mediated by Staphylococcus aureus topoisomerase IV: changes in drug mechanism across evolutionary boundaries. J. Biol. Chem. 274, 35927–35932 [DOI] [PubMed] [Google Scholar]
- 52. Aldred K. J., McPherson S. A., Wang P., Kerns R. J., Graves D. E., Turnbough C. L., Jr., Osheroff N. (2012) Drug interactions with Bacillus anthracis topoisomerase IV: biochemical basis for quinolone action and resistance. Biochemistry 51, 370–381 [DOI] [PMC free article] [PubMed] [Google Scholar]