

Background: FEN1 removes 5′-flaps during DNA replication and repair.
Results: With increasing flap length, rate of removal decreases, and the rate-determining step changes. Trinucleotide repeat (TNR) flaps are removed slower than mixed sequence flaps of the same length.
Conclusion: FEN1 is biased against long flaps and TNR sequences.
Significance: These results provide a kinetic perspective on the role of FEN1 in DNA replication and repair.
Keywords: base excision repair (BER), DNA enzyme, DNA repair, DNA replication, enzyme kinetics
Abstract
Flap endonuclease 1 (FEN1) is a structure-specific nuclease responsible for removing 5′-flaps formed during Okazaki fragment maturation and long patch base excision repair. In this work, we use rapid quench flow techniques to examine the rates of 5′-flap removal on DNA substrates of varying length and sequence. Of particular interest are flaps containing trinucleotide repeats (TNR), which have been proposed to affect FEN1 activity and cause genetic instability. We report that FEN1 processes substrates containing flaps of 30 nucleotides or fewer at comparable single-turnover rates. However, for flaps longer than 30 nucleotides, FEN1 kinetically discriminates substrates based on flap length and flap sequence. In particular, FEN1 removes flaps containing TNR sequences at a rate slower than mixed sequence flaps of the same length. Furthermore, multiple-turnover kinetic analysis reveals that the rate-determining step of FEN1 switches as a function of flap length from product release to chemistry (or a step prior to chemistry). These results provide a kinetic perspective on the role of FEN1 in DNA replication and repair and contribute to our understanding of FEN1 in mediating genetic instability of TNR sequences.
Introduction
Human flap endonuclease 1 (FEN1)3 is a Mg2+-dependent, structure-specific nuclease responsible for removing the 5′-DNA flaps created by a DNA polymerase during strand displacement synthesis (1, 2). During lagging strand replication, DNA polymerase δ extends RNA-primed segments and displaces a portion of the downstream Okazaki fragment, creating a 5′-flap. FEN1 cleaves at the base of the flap, creating a nick that can be sealed by DNA ligase. In long patch base excision repair, a DNA polymerase also performs strand displacement synthesis and creates a 5′-flap that is removed by FEN1, and the repair is completed by DNA ligase.
In the active site of FEN1, several acidic residues bind two Mg2+ ions, which coordinate the scissile phosphate and activate a nearby water for nucleophilic attack (3–9). Above the active site is a disordered loop that, upon threading of the 5′-flap, orders into a helical arch and is thought to be responsible for maintaining specificity for 5′-flaps (10). The DNA substrate is anchored to the enzyme through a 1-nt 3′-flap, which is bound in a 3′-flap binding pocket adjacent to the active site. The presence of the 3′-flap binding pocket suggests that DNA containing a 3′/5′-double flap is the native substrate for FEN1 (4, 7, 11). Interestingly, these 3′/5′-double flap species arise as part of “flap equilibration” (Fig. 1) and reflect the dynamic nature of FEN1 substrates.
FIGURE 1.
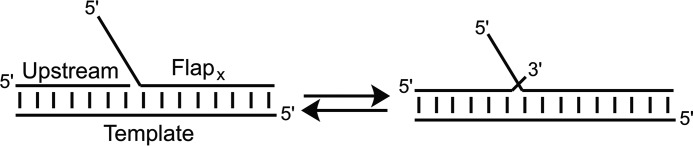
Equilibration of DNA substrates between 5′-single flap and 3′/5′-double flap species. DNA substrates used in this study have a common nucleotide on the 3′-end of the upstream strand and 1 nt into the flap on the FlapX strand, thus allowing the substrate to equilibrate between a 5′-single flap X nt in length and a 3′/5′-double flap. FEN1 will cleave the single flap substrate at the base of the flap and the double flap substrate 1 nt downstream of the base of the 5′-flap, yielding products x nt in length.
FEN1 is capable of binding to and cleaving substrates containing a 5′-single flap, with cleavage occurring at the base of the 5′-flap (12–14). The enzyme, however, prefers substrates with 3′/5′-double flaps and cleaves the DNA 1 nt downstream of the base of the 5′-flap (7, 11, 12). Although the 3′-flap is not necessary for catalysis, its presence increases both single-turnover and multiple-turnover catalytic rates (12). Furthermore, previous kinetic characterizations of FEN1 (10, 12) have shown that the rate of cleavage under single-turnover conditions (kSTO) does not change for 3′/5′-double flap substrates as the 5′-flap varies from 3 to 21 nt in length. There are mixed reports of whether the rate of cleavage under multiple-turnover conditions (kcat) is significantly slower than (12) or comparable to (10) kSTO. Notably, whereas these previous kinetic studies highlight the importance of the 3′-flap, the substrates used constitutively maintain the 3′/5′-double flaps via a mismatch between the template strand and a 1-nt 3′-flap, and thus flap equilibration cannot occur.
Defining the kinetic parameters of FEN1 activity on biological substrates is increasingly important because we and others have proposed that FEN1 contributes to genetic instability when DNA replication or long patch base excision repair is initiated in regions of trinucleotide repeat (TNR) DNA, such as CAG sequences (15–20). Flaps formed by TNR DNA have the ability to fold back upon themselves and form intramolecular secondary structures, which have been demonstrated to hamper FEN1 activity (15, 17, 21–23). If the flap is a substrate for DNA ligase, extra nucleotides can be ligated into the genome, and the TNR tract expands (24). Many neurodegenerative diseases, such as Huntington disease and Friedrich ataxia, are associated with an expanded length of TNR sequences (25), which highlights the importance of defining the enzymatic players involved in this genetic instability.
In this work we report the first kinetic characterization of FEN1 acting on substrates capable of equilibrating between 5′-single flap and 3′/5′-double flap species. We define the effects of flap characteristics on the rate of flap cleavage. With flaps of varying length and sequence context (mixed sequence or TNR), we observe that FEN1 kinetically discriminates shorter flaps from longer flaps and mixed sequence flaps from CAG repeat flaps of comparable length. The results enhance our understanding of the role of FEN1 in DNA replication and repair, as well as the substrate specificity of the enzyme. Further, we elucidate inherent limitations of FEN1 and how those limitations can mediate or promote disease-initiating TNR expansion.
Experimental Procedures
Oligonucleotide Synthesis and Purification
The DNA oligonucleotides used in this study are listed in Table 1 and were synthesized using standard phosphoramidite chemistry on a BioAutomation DNA/RNA synthesizer. The 5′-DMT group was retained on all strands for ease of purification. For strands shorter than 45 nt in length, the strands were purified by HPLC using a Dynamax Microsorb C18 column (10 × 250 mm), with acetonitrile (solvent A) and 30 mm ammonium acetate (solvent B) as mobile phases (gradient: solvent A was increased from 5% to 25% over 25 min; 3.5 ml/min). The 5′-DMT group was removed by incubation in 80% glacial acetic acid for 12 min at room temperature, followed by a second round of HPLC purification (gradient: solvent A was increased from 0% to 15% over 35 min; 3.5 ml/min).
TABLE 1.
DNA oligonucleotides used in this study
| Name | Sequence |
|---|---|
| Template | 5′-ATGGGACGTGCTGTCAGTGCACTGTGACCGTCAGGATGACACGC-3′ |
| Upstream | 5′-GCGTGTCATCCTGACGGTCACAGTG-3′ |
| FlapXa | |
| X = 12R | 5′-CAGCAGCAGCAGCACTGACAGCACGTCCCAT-3′ |
| X = 14M | 5′-CTCGATCAGCTACGCACTGACAGCACGTCCCAT-3′ |
| X = 30M | 5′-CGCACTCTCTGTCAGACTCGATCAGCTACGCACTGACAGCACGTCCCAT-3′ |
| X = 30R | 5′-CAGCAGCAGCAGCAGCAGCAGCAGCAGCAGCACTGACAGCACGTCCCAT-3′ |
| X = 45M | 5′-GGTTGTAAATAAGCTCTCACCCTCAGTCAGACTCGATATACAGCGCACTGACAGCACGTCCCAT-3′ |
| X = 45R | 5′-CAGCAGCAGCAGCAGCAGCAGCAGCAGCAGCAGCAGCAGCAGCAGCACTGACAGCACGTCCCAT-3′ |
| X = 60M | 5′-AGATGTCCGTGATTCGGTTGTAAATAAGCTCTCACCCTCAGTCAGACTCGATATACAGCGCACTGACAGCACGTCCCAT-3′ |
| X = 60R | 5′-CAGCAGCAGCAGCAGCAGCAGCAGCAGCAGCAGCAGCAGCAGCAGCAGCAGCAGCAGCAGCACTGACAGCACGTCCCAT-3′ |
a The 5′-flap region is underlined. The length of the flap (nucleotides) in the 5′-single flap species and sequence of the flap (either mixed sequence (M) or CAG repeat (R)) are indicated in the subscript (X).
Strands longer than 45 nt were purified by HPLC using a Polymer Labs Reverse Phase PLRP-S column (4.6 × 250 mm) at 90 °C, with 90% acetonitrile and 100 mm triethylammonium acetate (solvent A) and 1% acetonitrile and 100 mm triethylammonium acetate (solvent B) as mobile phases (gradient: solvent A was increased from 5% to 25% over 25 min; 1.0 ml/min). The 5′-DMT group was removed as described above, followed by another round of HPLC purification (gradient: solvent A was increased from 0% to 25% over 40 min; 1 ml/min). Following HPLC purification, strands were resuspended in 25–50 μl 100% formamide and further purified by 18% denaturing PAGE (33 × 42 cm, 0.8 mm thick), run for ∼2.5 h at 80 W. Regions of the gel containing desired DNA were excised, crushed, and shaken lightly overnight at 37 °C in deionized water. The supernatant was dialyzed against 200 μm sodium phosphate, 1 mm NaCl, pH 7.4 buffer, dried, resuspended in deionized water, and desalted using a Micro Bio-SpinTM 6 column (Bio-Rad). Oligonucleotides were quantitated at 90 °C using the ϵ260 estimated by the nearest neighbor method (26, 27) and a Beckman Coulter DU800 UV-visible spectrophotometer.
Characterization of DNA Substrates by Native Gel Electrophoresis
For each sample, 10 pmol of a single DNA strand (Template strand for lanes 1–3; FlapX for lanes 4–9) were 5′-32P end-labeled using T4 polynucleotide kinase (New England Biolabs) following the manufacturer's protocol. Strands were annealed in ratios of 1:1.5 Template:TemplateComp (lane 2), 1:1.5:1.5 Template:Upstream:Flap0 (lane 3), or 1:1.5:1.5 FlapX:Template:Upstream (lanes 4–9) by heating to 90 °C and slow cooling to room temperature. The strand TemplateComp is the 44-nt complement to the Template strand. Flap0 is FlapX without a 5′-flap. After annealing, nondenaturing dye was added, and the samples were separated on a 12% nondenaturing PAGE gel (19.5 × 16 cm, 0.8 mm thick) at 150 V and 4 °C for 5 h. Products were visualized by phosphorimaging.
DNA Substrate Assembly for Kinetic Experiments
DNA strands containing a 5′-flap (FlapX) were 5′-32P end-labeled. Strands were annealed in 1:1.5:1.75 FlapX:Template:Upstream (12R, 14M, 30M, 30R) or 1:1.5:2 FlapX:Template:Upstream (45M, 45R, 60M, 60R) molar ratio in annealing buffer (30 mm HEPES-KOH, 40 mm KCl, 8 mm MgCl2, pH 7.4).
FEN1 Expression and Purification
Wild-type human FEN1 was overexpressed and purified as previously described (28), with the addition of a Superdex 200 (GE Healthcare Life Sciences) size exclusion column following the cation exchange column. Fractions from the cation exchange column containing the most FEN1 were concentrated with a 15-ml, 30,000 molecular weight cutoff Amicon centrifugal filter to less than 1 ml and loaded onto the size exclusion column pre-equilibrated with buffer A (50 mm HEPES-KOH, 5% (v/v)_glycerol, pH 7.4) plus 50 mm KCl at 4 °C. Protein was eluted from the column with buffer A plus 50 mm KCl in 3-ml fractions. Fractions containing protein (as determined by the addition of Bradford reagent) were analyzed by 4–20% SDS-PAGE, and those containing FEN1 (>90% purity) were concentrated with a 15-ml, 30,000 molecular weight cutoff Amicon centrifugal filter. Concentrated FEN1 was mixed with FEN1 storage buffer (20 mm HEPES-KOH, 50 mm KCl, 1 mm DTT, 50% (v/v) glycerol, pH 7.4) and aliquoted. Each aliquot was flash frozen with liquid nitrogen and stored at −80 °C until use. Total protein concentration was determined using the Bradford assay with bovine γ-globulin standards. The FEN1 preparation was ∼50% active as determined by multiple-turnover kinetics experiments, and all FEN1 concentrations given below are active enzyme concentrations.
FEN1 Single-turnover Kinetic Assays
FEN1 single-turnover kinetic experiments were performed using a Rapid Quench Flow instrument (RQF-3; KinTek Corp.). For each experiment, 10-pmol FlapX strands were 5′-32P-end labeled and annealed as described above and used to generate a 500-μl aliquot of 20 nm DNA substrate in 30 mm HEPES-KOH, 40 mm KCl, 8 mm MgCl2, pH 7.4. A 100–400 nm FEN1 aliquot in FEN1 reaction buffer (30 mm HEPES-KOH, 40 mm KCl, 8 mm MgCl2, 10% (v/v) glycerol, 0.2 mg/ml BSA) was prepared. For reactions with 14M, 30M, 30R, 60M, and 60R substrates, a 100 nm stock of FEN1 was used; reactions with 12R used a 200 nm FEN1 stock; reactions with 45R and 45M used FEN1 stocks of 100 nm, 200 nm, and 400 nm FEN1, one for each replicate. Enzyme concentration was increased to ensure single-turnover conditions; experiments performed at multiple concentrations of FEN1 yielded the same kSTO, indicating that single-turnover conditions were achieved. For each reaction in the RQF, 15 μl of DNA stock and 15 μl of FEN1 stock were loaded into the reaction loops, rapidly mixed, allowed to incubate at 37 °C for the designated time, and then quenched with 85 μl of 100 mm EDTA. Mixing equal volumes of DNA substrate and enzyme by the RQF yielded a final sample of 10 nm DNA substrate and 50–200 nm FEN1 in 30 mm HEPES-KOH, 40 mm KCl, 8 mm MgCl2, 5% (v/v) glycerol, 0.1 mg/ml BSA, pH 7.4. RQF reactions were allowed to proceed for 0.002–3 s (12R, 14M, 30M, 30R, and 45M), 0.002–5 s (45R), 0.002–10 s (60M), or 0.002–90 s (60R) at 37 °C. Once the sample was expelled from the RQF, it was placed at 90 °C for 2 min and then on dry ice until all samples were collected. The negative control in each experiment (labeled 0 on autoradiograms) involved mixing DNA substrate and FEN1 reaction buffer (in the absence of FEN1) for the longest reaction time in the time course in the RQF at 37 °C and was then quenched with 85 μl of 100 mm EDTA, heated to 90 °C for 2 min, and placed on dry ice. After all samples were collected, 50 μl of denaturing dye was added, and samples were separated on a 18% (12R, 14M, 30M, and 30R) or 12% (45M, 45R, 60M, and d60R) denaturing PAGE gel (33 × 42 cm, 0.4 mm thick) for ∼2 h at 80W, and products were visualized by phosphorimaging. Densitometry was used to quantitate the percentage of product formed, and kinetic plots reflect the amount of product formed versus time.
For all single-turnover time courses, data from the full time course were fit to a monoexponential equation using Kaleidagraph to obtain the single-turnover rate (kSTO). For substrates where multiple product bands were observed, the products were quantitated together, and kSTO reflects the formation of the total population of products. The errors on single-turnover rates represent the standard deviation of three separate experiments. Two-tailed Student's t tests were performed to obtain p values for single-turnover rates for comparing kSTO values; we considered p ≤ 0.01 to be statistically different.
FEN1 Multiple-turnover Kinetic Assays
DNA substrates were assembled as described above. For each time course, a 500-μl aliquot of 100 nm DNA substrate in annealing buffer and a 400-μl aliquot of 4 nm FEN1 in FEN1 reaction buffer were prepared. An RQF instrument was used to mix equal volumes of DNA and FEN1 (final sample concentrations of 50 nm DNA substrate, 2 nm FEN1, 30 mm HEPES-KOH, 40 mm KCl, 8 mm MgCl2, 5% (v/v) glycerol, 0.1 mg/ml BSA, pH 7.4) for 0.002–120 s at 37 °C prior to quenching with 85 μl of 100 mm EDTA. Samples were processed and separated by denaturing PAGE as described above. Products were visualized and quantitated as described above. Kaleidagraph was used to fit data to a burst plus linear phase, or solely linear phase, with the slope of the linear phase equal to kcat × [active enzyme] (29). The active enzyme concentration of ∼50% was determined by extrapolating the linear phase to the y axis. The multiple-turnover catalytic rates (kcat) are from a single data set, and the error reported is the error associated with the fit.
Results
DNA Substrates
DNA substrates containing either mixed sequence or (CAG)n flaps capable of flap equilibration were used to determine the kinetic parameters of FEN1 (Table 1). The substrates are named according to the length of the 5′-flap (as observed in the 5′-single flap species) and the content of the flap, either mixed sequence (M) or CAG repeat (R). Thus a substrate containing a 14-nt mixed sequence flap is here called 14M, and cleavage by FEN1 yields a product 14 nt in length.
Annealing of the template, upstream, and FlapX strands was confirmed by native gel electrophoresis (Fig. 2). Lanes 1, 2, and 3 serve as size controls and represent single-stranded template, duplex, and nicked duplex, respectively. The nicked duplex is a three-stranded substrate that lacks a 5′-flap. Relative to the nicked duplex (lane 3), there is decreased mobility for all samples containing a 5′-flap (lanes 4–9), with longer flaps migrating more slowly than shorter flaps. Furthermore, there is a slight difference in migration of substrates containing mixed sequence and CAG repeat flaps of the same length, most visible for 30R and 30M (lanes 4 and 5, respectively).
FIGURE 2.
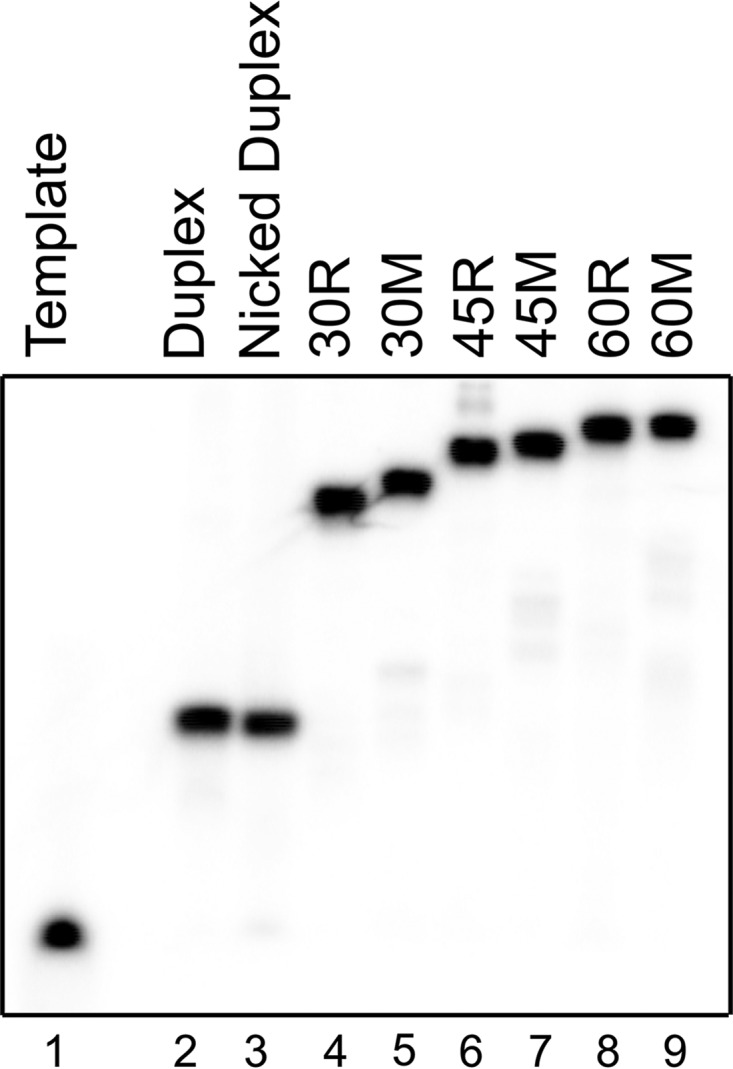
Native gel characterization of DNA substrates. DNA substrates were annealed as described (see “Experimental Procedures”). Single-stranded template DNA (lane 1), duplex DNA (lane 2), and a nicked duplex (lane 3) serve as migration controls. Lanes 4–9 are FEN1 substrates used in kinetic experiments.
Minimal Kinetic Scheme of FEN1
Until recently, the accepted model was that FEN1 recognized the 5′-end of the flap and tracked down the flap until it reached the double-stranded region and cleaved the scissile phosphate (30). However, recent studies suggest that FEN1 binds at the double-stranded/flap junction and threads the flap through the disordered loop, followed by ordering of the loop into the helical arch and cleavage (10, 12, 31). Thus the minimal kinetic scheme shown in Fig. 3 presents the basic steps necessary for the enzymatic cycle of FEN1: binding of FEN1 to the DNA substrate (kon and koff), threading of the 5′-flap through the disordered loop (kthread), ordering of the loop to form the helical arch (korder), chemistry (kchemistry) to cleave the 5′-flap, and product release (krelease).
FIGURE 3.
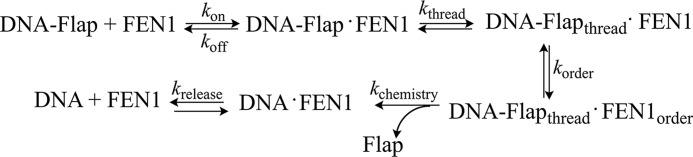
Minimal kinetic scheme of FEN1. FEN1 binds its substrate (kon and koff); the 5′-DNA flap is threaded through the disordered loop (kthread); the disordered loop orders into the helical arch (korder); the flap is cleaved (kchemistry); and the DNA product is released (krelease).
Single-turnover Kinetics of FEN1: Substrates with Flaps of 30 nt or fewer
Single-turnover kinetic time courses were performed such that the concentration of FEN1 exceeded the DNA concentration by at least 5-fold. The enzyme and DNA substrate were mixed and rapidly quenched by use of a RQF instrument. Under single-turnover conditions, the observed rate (kSTO) is representative of the slowest step up to and including chemistry. For substrates containing flaps of 30 nt or fewer (12R, 14M, 30M, and 30R), an accumulation of product is observed, followed by a plateau (Fig. 4A and data not shown). All substrates with flaps of 30 nt or fewer are processed at a rate of kSTO = 30–55 s−1 (Table 2) and are not statistically different.
FIGURE 4.

FEN1 processing of substrates containing flaps of 30 nt under single-turnover conditions. A, representative time courses of FEN1 acting upon 30M (filled squares) and 30R (open squares). B, representative autoradiograms depicting cleavage of 30M (top panel) and 30R (bottom panel) under single-turnover conditions. The 0 reaction time is a negative control (see “Experimental Procedures”). Experimental conditions were 10 nm DNA substrate, 50 nm FEN1, 30 mm HEPES-KOH, 40 mm KCl, 8 mm MgCl2, 5% (v/v) glycerol, 0.1 mg/ml BSA, pH 7.4. Reactions were performed at 37 °C using an RQF instrument and quenched with EDTA.
TABLE 2.
Kinetic parameters of FEN1 acting on flap-containing substrates
| Flap length (nt) |
kSTOa |
kburstb |
kcatb |
|||
|---|---|---|---|---|---|---|
| Mixed sequence | (CAG)n | Mixed sequence | (CAG)n | Mixed sequence | (CAG)n | |
| s−1 | s−1 | s−1 | ||||
| 12 | NDc | 54 ± 8 | NDc | 21 ± 6 | NDc | 4.4 ± 0.2 |
| 14 | 33 ± 6 | NDc | 42 ± 10 | NDc | 2.9 ± 0.1 | NDc |
| 30 | 49 ± 9 | 36 ± 9 | 36 ± 8 | 33 ± 18 | 2.0 ± 0.1 | 3.4 ± 0.1 |
| 45 | 12 ± 1 | 0.95 ± 0.20 | 6.4 ± 3.0 | NAd | 1.7 ± 0.1 | 0.40 ± 0.01 |
| 60 | 3.0 ± 0.7 | 0.104 ± 0.004 | NAd | NAd | 1.43 ± 0.04 | 0.071 ± 0.001 |
a Measured at 37 °C under single-turnover conditions.
b Measured at 37 °C under multiple-turnover conditions.
c ND, not determined.
d NA, not applicable because no burst phase was observed.
It is worthy of note that although 30M and 30R are indistinguishable by kSTO, the autoradiograms indicate that the two substrates are processed differently by FEN1 (Fig. 4B). The autoradiogram of 30M depicts a single product corresponding to the cleaved 30-nt flap. Similar to 30M, both 12R and 14M substrates yield only a single product band (data not shown). Cleavage of 30R, however, yields the 30-nt cleaved flap, as well as several smaller products fewer than 30 nt in length, producing a ladder-type pattern of cleavage. Because the smaller and full-length products accumulate at similar rates, the reported value for kSTO reflects formation of this total population of products.
Single-turnover Kinetics of FEN1: Substrates with Flaps Longer than 30 nt
There is a clear decrease in kSTO when the flap is lengthened beyond 30 nt, and this change is depicted graphically for the mixed sequence flaps in Fig. 5A and much more dramatically for the CAG repeat flaps in Fig. 5B. The 45M and 45R substrates are cleaved more slowly than the shorter flap substrates, with kSTO of 12 and 0.95 s−1, respectively. When the flap is increased to 60 nt, kSTO is decreased further still. Interestingly, for flaps shorter than 30 nt, there was no difference in kSTO for the mixed sequence and CAG repeat flaps; however, for flaps greater than 30 nt, FEN1 kinetically discriminates between the two sequence contexts with the repetitive DNA flap removed at a slower rate (Fig. 6 for 45-nt flaps; Fig. 7 for 60 nt flaps).
FIGURE 5.
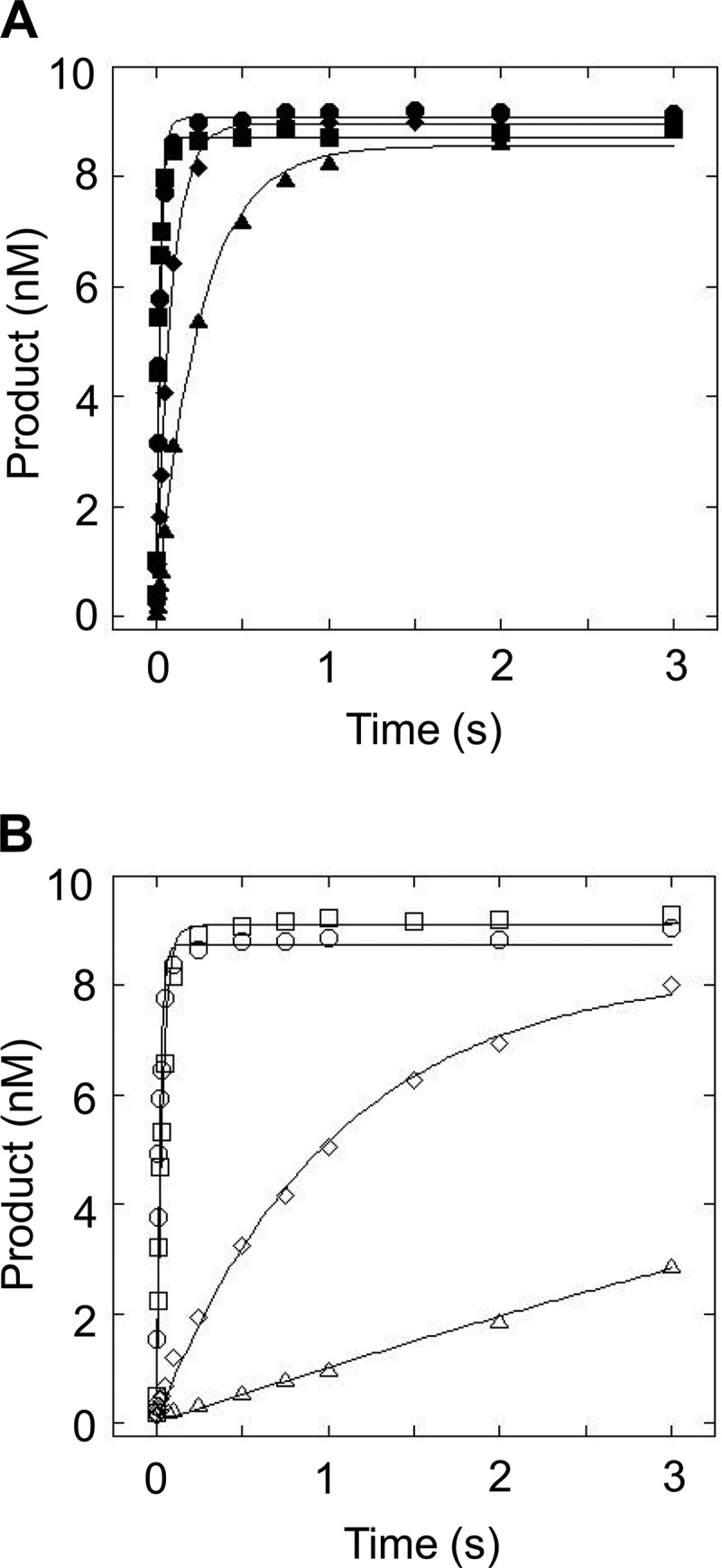
Single-turnover time course of FEN1 acting upon mixed sequence and TNR flap substrates. A, comparison of mixed sequence flap substrates shows 14M (filled circles), 30M (filled squares), 45M (filled diamonds), and 60M (filled triangles). B, comparison of CAG repeat flap substrates depicts 12R (open circles), 30R (open squares), 45R (open diamonds), and 60R (open triangles). Experimental conditions were 10 nm DNA substrate, 50–200 nm FEN1 (see “Experimental Procedures” for details), 30 mm HEPES-KOH, 40 mm KCl, 8 mm MgCl2, 5% (v/v) glycerol, 0.1 mg/ml BSA, pH 7.4. Reactions were performed at 37 °C using an RQF instrument and quenched with EDTA.
FIGURE 6.

FEN1 processing of substrates containing flaps of 45 nt under single-turnover conditions. A, representative single-turnover time courses of FEN1 acting upon 45M (filled diamonds) and 45R (open diamonds). B, representative autoradiograms depicting cleavage of 45M (top panel) and 45R (bottom panel). The 0 reaction time is a negative control (see “Experimental Procedures”). Experimental conditions were 10 nm DNA substrate, 50–200 nm FEN1 (see “Experimental Procedures” for details), 30 mm HEPES-KOH, 40 mm KCl, 8 mm MgCl2, 5% (v/v) glycerol, 0.1 mg/ml BSA, pH 7.4. Reactions were performed at 37 °C using an RQF instrument and quenched with EDTA.
FIGURE 7.

FEN1 processing of substrates containing flaps of 60 nt under single-turnover conditions. A, representative time courses of FEN1 acting upon 60M (filled triangles) and 60R (open triangles). B, representative autoradiograms depicting cleavage of 60M (top panel) and 60R (bottom panel). The 0 reaction time is a negative control (see “Experimental Procedures”). The zoomed in box to the right of 60R depicts the ladder-type pattern of cleavage. Experimental conditions were 10 nm DNA substrate, 50 nm FEN1, 30 mm HEPES-KOH, 40 mm KCl, 8 mm MgCl2, 5% (v/v) glycerol, 0.1 mg/ml BSA, pH 7.4. Reactions were performed at 37 °C using an RQF instrument and quenched with EDTA.
Similar to the shorter flaps, the 45M and 60M substrates yield a single product corresponding to cleavage of the full flap (Fig. 6B, top panel; Fig. 7B, top panel). However, the 45R substrate displays a ladder-like cleavage pattern, similar to that observed for 30R (Fig. 6B, bottom panel). The 60R substrate is different still and yields products corresponding to cleavage of the full flap, the ladder pattern of cleavage observed in 30R and 45R, and significantly shorter products that are fewer than 20 nt in length (Fig. 7B, bottom panel).
Multiple-turnover Kinetics of FEN1
Multiple-turnover kinetic time courses were performed such that the concentration of DNA greatly exceeded the concentration of FEN1. If the rate-determining step occurs after chemistry, FEN1 should exhibit burst kinetics under multiple-turnover conditions, with a rapid accumulation of product followed by a linear phase. For substrates with flaps of 30 nt or fewer, FEN1 demonstrates burst kinetics (Fig. 8A and data not shown), and for each substrate, the rate of the burst phase (kburst) is comparable to kSTO (Table 2). For these shorter flap substrates, we obtained multiple-turnover catalytic rates of kcat = 2–4.5 s−1. The 45M substrate exhibits relatively small differences in rates between the burst and linear phases, manifested as a less well defined burst for 45M (filled diamonds) relative to 30M (filled squares), as seen in Fig. 8A. Interestingly, the 45R, 60M, and 60R substrates do not exhibit burst kinetics, and rather solely a linear phase is observed with kcat of 0.40, 1.43, and 0.071 s−1, respectively (Fig. 8B and data not shown).
FIGURE 8.
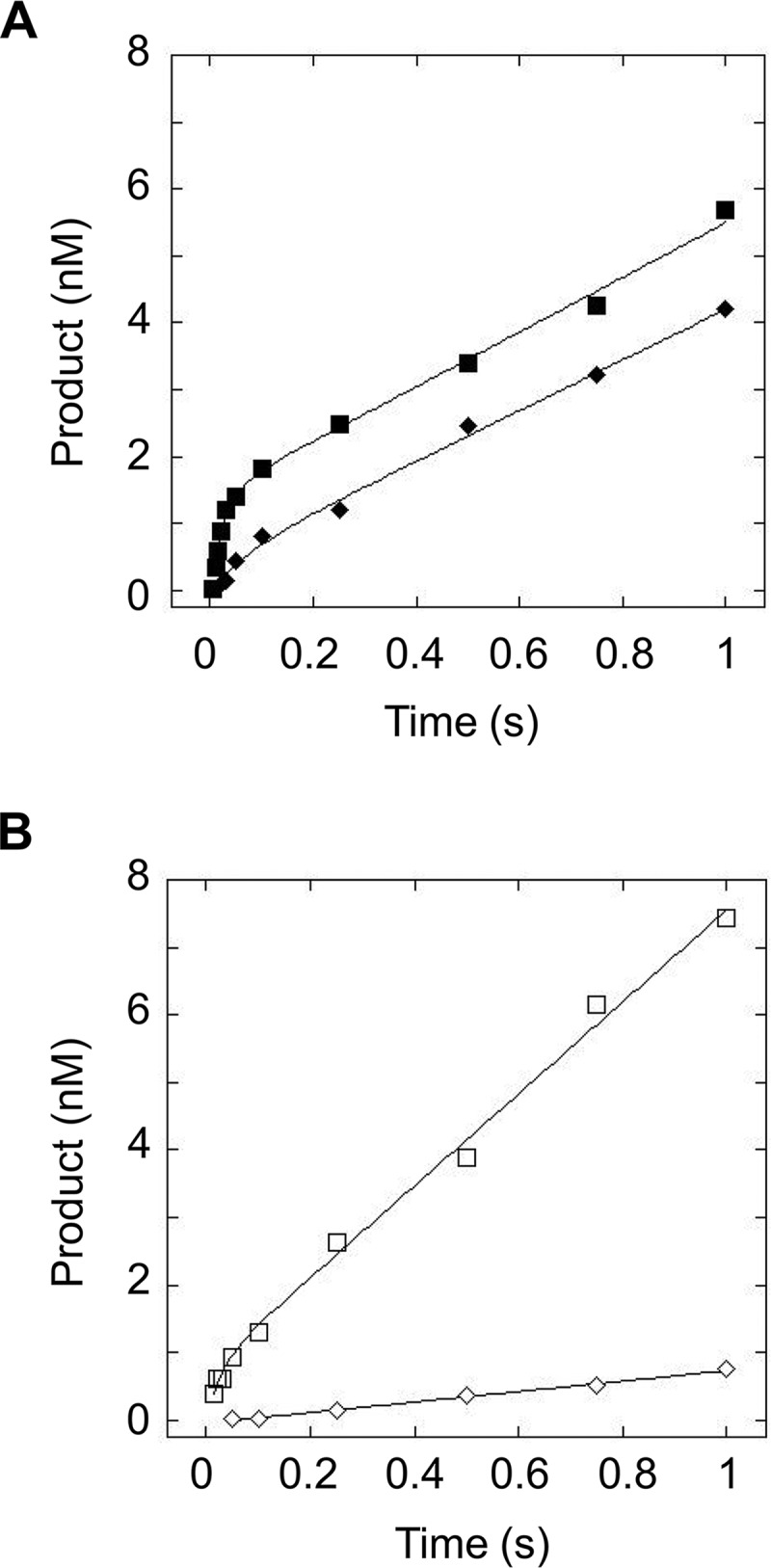
Multiple-turnover time course of FEN1 acting upon substrates with flaps of 30 and 45 nt in length. A, multiple-turnover time course comparing cleavage of 30M (filled squares) and 45M (filled diamonds). B, multiple-turnover time course comparing cleavage of 30R (open squares) and 45R (open diamonds). Experimental conditions were 50 nm DNA substrate, 2 nm FEN1, 30 mm HEPES-KOH, 40 mm KCl, 8 mm MgCl2, 5% (v/v) glycerol, 0.1 mg/ml BSA, pH 7.4. Reactions were performed at 37 °C using an RQF instrument and quenched with EDTA.
Discussion
In this work we establish that the kinetic behavior of FEN1 is modulated by both flap length and sequence on substrates capable of equilibrating between 5′-single flap and 3′/5′-double flap species. For substrates with flaps of 30 nt or fewer, regardless of sequence, kSTO is the same within error. Importantly, previous studies examined binding of FEN1 to 5–53-nt mixed sequence flaps and established that the KD is unchanged over this length regime (31). This prior work along with our own characterizations establish that the conditions of our single-turnover experiments are such that the concentration of FEN1 is well above the KD, and binding to substrate does not limit kSTO. Thus the kSTO values reported in Table 2 correspond to either kthread, korder, or kchemistry. For these shorter flap substrates, kSTO likely corresponds to kchemistry; however, these experiments cannot rule out that kthread or korder influence kSTO. The values we obtained for kSTO for flaps of 30 nt or fewer are comparable to those reported previously for a poly(dT) flap of 5 nt (12) and mixed sequence flaps of 3 or 21 nt (10). Because these previous studies used substrates in which the flaps cannot equilibrate, these results suggest that for flaps shorter than 30 nt in length, the ability of the flaps to equilibrate does not influence kSTO. This conclusion is supported by our observation that kSTO of a 14-nt mixed sequence flap substrate that constitutively maintains the 3′/5′-double flap is comparable to the kSTO of 14M (data not shown).
The observation of burst kinetics in our multiple-turnover experiments for substrates with flaps of 30 nt or fewer indicates that for these shorter flaps, a step after chemistry is rate-limiting, in this case product release. This observation of burst kinetics and the reported rates are consistent with a previous kinetic characterization of FEN1 on a poly(dT) flap of 5 nt (12).
In this study, we report the first kinetic characterization of substrates containing flaps longer than 21 nt in length. Our results demonstrate that several notable changes occur when the flap length is increased beyond 30 nt: 1) kSTO decreases as a function of length; 2) FEN1 kinetically discriminates between mixed sequence and CAG repeat flaps; and 3) the rate-determining step of the enzymatic cycle changes.
We will first discuss the length dependence of kSTO for mixed sequence flaps. Mixed sequence flaps longer than 30 nt are likely not formed in vivo (32, 33), and thus this drop in kSTO would not have biological implications. Nevertheless, it has been shown that substrates with longer flaps dissociate from FEN1 less readily than those with shorter flaps because of a slower rate of unthreading of the flap (31). Therefore, we propose that kthread is slowed significantly for mixed sequence flaps longer than 30 nt and drives the observed decrease in kSTO.
Whereas flaps longer than 30 nt are likely not formed in vivo for mixed sequences, such longer flaps are biologically relevant for TNR sequences because of well documented strand slippage displacement synthesis activity by polymerases, where slippage can occur on the newly synthesized strand and/or the template strand (34–38). Indeed, reconstitution of base excision repair in vitro has demonstrated that strand displacement synthesis by DNA polymerases leads to significant expansion of TNR sequences (16, 17). Importantly, we observe that whereas shorter flaps are processed comparably regardless of sequence, at 45 nt FEN1 kinetically discriminates between mixed sequence and CAG repeat flaps and is biased such that the repeat-containing flaps are removed at a slower rate. Previous studies observed a decrease in activity of FEN1 on substrates containing TNR flaps 15–60 nt in length; however, these studies did not report rates (15, 17, 21, 23).
The kinetic bias we observe may derive from differences in processing of the CAG repeat flap at numerous steps in the enzymatic cycle: binding, threading, ordering, or chemistry. Regardless of the step(s) affected, the differences observed for the CAG repeat sequences are likely due to secondary structure in the flap. Indeed, the migratory differences for mixed sequence and CAG flaps observed on a native gel indicate that the TNR flap adopts a secondary structure. Previous work from our and other labs showed that CAG oligonucleotides are capable of forming secondary structure in vitro (39–45) and in vivo (46), specifically stem-loop hairpins.
Binding of FEN1 may be influenced if the flap forms a secondary structure. Furthermore, it has been shown that FEN1 can nonproductively bind to substrates containing flaps that it cannot thread and thus cannot cleave (10, 31). The disordered loop is large enough to accommodate threading of double-stranded DNA, as well as other secondary flap modifications (10, 13, 21), which need not be resolved prior to or in the process of threading (10), suggesting that a CAG hairpin can be threaded. Nevertheless, the structure may sterically clash with the disordered loop, decreasing the rate at which the flap is threaded. We also considered that the kinetic discrimination between mixed sequence and CAG repeat flaps could be the result of a decrease in korder (10). Crystal structures of FEN1, as well as its homologues, indicate that the helical arch is capable of accommodating only single-stranded DNA once it has ordered (4–9), and substrates with flap modifications close to the base of the flap are processed very poorly, if at all (13). It is thus thought that there must be a few nucleotides near the base of the flap that are single-stranded for ordering to occur (4, 10, 13, 47); therefore, secondary structure at the base of the flap, as might occur in a CAG stem-loop hairpin, may impede ordering of the loop.
It is also possible that a decreased kchemistry is responsible for the kinetic discrimination between the CAG repeat flap substrates and their mixed sequence counterparts. The structured CAG flap may be situated in the active site differently than an unstructured flap, thus changing orientation of or decreasing accessibility to the scissile phosphate (10, 48, 49). The scissile phosphate, furthermore, is not always at the base of the flap for CAG repeat substrates, as evidenced by the ladder-type cleavage patterns observed for 30R, 45R, and 60R. The predominate product is cleavage of the full flap and suggests that hairpin(s) formed are sufficiently disordered at the base and/or the hairpin is spatially separated from the base by a sufficient number of nucleotides to allow for ordering around a single-stranded region. The smaller products may arise from different binding orientations of FEN1 to a single hairpin species or, more likely, from a population of alternate hairpin species where FEN1 binding positions an alternate phosphate in the active site. For the former situation, Fig. 9 depicts the several places along the 3′-end of the flap that FEN1 could bind and cleave to produce the ladder pattern seen in 30R, 45R, and 60R. We hypothesize that FEN1 may have difficulty recognizing the base of the flap if a hairpin forms, resulting in multiple binding orientations. Furthermore, the amount of ladder pattern cleavage decreases with increasing TNR flap length, which is likely due to increased energy requirements to unpair the base of the hairpin. The interpretation that the 30R, 45R, and 60R flaps form hairpins is bolstered by the observation that 12R, which contains a (CAG)4 flap that is likely too short to maintain secondary structure, yields only the full-length cleavage product and no ladder-type cleavage.
FIGURE 9.
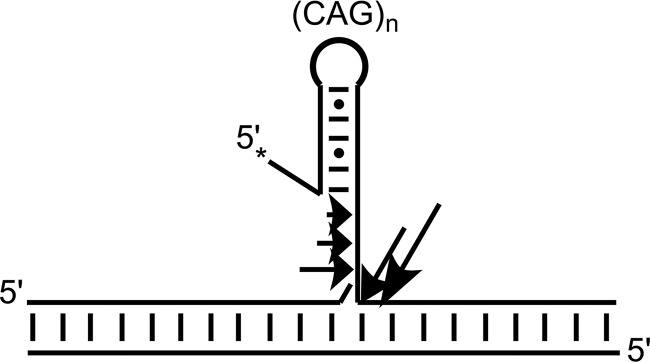
Schematic showing FEN1 cleavage of a CAG repeat substrate to produce a ladder pattern of cleavage. FEN1 may bind in several places on a hairpin-containing substrate, cleaving the backbone at each location. Arrows indicate possible sites of binding and cleavage on a DNA substrate where the relative sizes of arrows correspond to amounts of cleavage product. The asterisk indicates the location of 5′-32P radiolabel.
It is noteworthy that the pattern of FEN1 cleavage for 60R varies from that previously reported for a substrate containing a (CAG)20 flap; the predominate product in this previous study was not cleavage of the full flap, as we observed, but rather very small products (17). We attribute this difference to a variation in substrate design, where in the previous work the majority of the 5′-(CAG)20 flap could equilibrate to generate a large 3′-flap and a small 5′-flap. The shorter products could be the result of cleavage of the small 5′-flap or from a 5′-overhang of a CAG stem-loop hairpin. Furthermore, the significantly shorter products (<20 nt) we observe for 60R form only on long time scales and may arise from FEN1 activity on a previously cleaved flap. Although it remains unclear which step in the enzymatic cycle is responsible for the dramatic decrease in kSTO, we have observed that FEN1 exhibits a flap length-dependent decrease in single-turnover rate for both mixed sequence and CAG repeat flaps. Our results also demonstrate that there is a threshold flap length at which the rate-determining step changes. For all flaps shorter than 30 nt, burst kinetics are observed in multiple-turnover experiments, and thus the rate-determining step occurs after chemistry and corresponds to product release. For flaps of 60 nt, the lack of a burst under multiple-turnover conditions indicates that chemistry or a step prior to chemistry is rate-limiting. Therefore, the rate-determining step of FEN1 changes as a function of increased flap length. Indeed, for substrates that do not exhibit burst behavior, kSTO and kcat likely represent the same kinetic step. The threshold flap length that represents the transition point for the rate-determining step is between 45 and 60 nt for mixed sequence flaps and for CAG repeat flaps is between 30 and 45 nt, and notably is shorter than mixed sequence flaps. We also recognize that there is a difference in the burst amplitudes of 30M and 45M, which likely arises because of additional kinetic complexity in processing flaps of increasing length, suggesting that the transition point for the rate-determining step is closer to 45 nt than 60 nt. Nevertheless, the specific threshold length may be different in vivo, because of the cellular environment and presence of other proteins, but we wish to emphasize that there exists a flap length where the rate-determining step of FEN1 changes.
In summary, we demonstrate a length and sequence context effect on kSTO for FEN1. Furthermore, the rate-determining step for FEN1 changes as a function of flap length, for substrates containing both mixed sequence and CAG repeat flaps. A change in the rate-determining step can have significant implications, especially for events requiring coordination of numerous enzymes, such as DNA replication and repair. We want to emphasize the bias FEN1 displays against TNR flaps. Depending on the length of the flap, FEN1 may not be able to remove the flap on a biologically relevant timescale. Indeed, if flap cleavage is sufficiently slow, ligase competes with FEN1 to incorporate the uncleaved flap into the genome, thus expanding the TNR tract (24). These findings underscore the importance of defining kinetic parameters for enzymes and highlight the functional implications that changes in those parameters can have.
Author Contributions
M. E. T., K. B., J. H., and S. D. designed the study and wrote the paper. M. E. T. performed and analyzed the experiments. K. B. and J. H. provided technical assistance. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgment
We thank Dr. David M. Wilson III (NIA, National Institutes of Health) for providing the expression plasmid for FEN1.
This work was supported, in whole or in part, by National Institutes of Health Grant R01ES019296. The authors declare that they have no conflicts of interest with the contents of this article.
- FEN1
- flap endonuclease 1
- nt
- nucleotide
- RQF
- rapid quench flow
- TNR
- trinucleotide repeat
- DMT
- dimethoxytrityl.
References
- 1. Balakrishnan L., Bambara R. A. (2013) Flap endonuclease 1. Annu. Rev. Biochem. 82, 119–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Liu Y., Kao H.-I., Bambara R. A. (2004) Flap endonuclease 1: a central component of DNA metabolism, Ann. Rev. Biochem. 73, 589–615 [DOI] [PubMed] [Google Scholar]
- 3. Tock M. R., Frary E., Sayers J. R., Grasby J. A. (2003) Dynamic evidence for metal ion catalysis in the reaction mediated by a flap endonuclease. EMBO J. 22, 995–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tsutakawa S. E., Classen S., Chapados B. R., Arvai A. S., Finger L. D., Guenther G., Tomlinson C. G., Thompson P., Sarker A. H., Shen B., Cooper P. K., Grasby J. A., Tainer J. A. (2011) Human flap endonuclease structures, DNA double-base flipping, and a unified understanding of the FEN1 superfamily. Cell 145, 198–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mueser T. C., Nossal N. G., Hyde C. C. (1996) Structure of bacteriophage T4 RNase H, a 5′ to 3′ RNA-DNA and DNA-DNA exonuclease with sequence similarity to the RAD2 family of eukaryotic proteins. Cell 85, 1101–1112 [DOI] [PubMed] [Google Scholar]
- 6. Ceska T. A., Sayers J. R., Stier G., Suck D. (1996) A helical arch allowing single-stranded DNA to thread through T5 5′-exonuclease. Nature 382, 90–93 [DOI] [PubMed] [Google Scholar]
- 7. Chapados B. R., Hosfield D. J., Han S., Qiu J., Yelent B., Shen B., Tainer J. (2004) Structural basis for FEN-1 substrate specificity and PCNA-mediated activation in DNA replication and repair. Cell 116, 39–50 [DOI] [PubMed] [Google Scholar]
- 8. Sakurai S., Kitano K., Yamaguchi H., Hamada K., Okada K., Fukuda K., Uchida M., Ohtsuka E., Morioka H., Hakoshima T. (2005) Structural basis for recruitment of human flap endonuclease 1 to PCNA. EMBO J. 24, 683–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Devos J. M., Tomanicek S. J., Jones C. E., Nossal N. G., Mueser T. C. (2007) Crystal structure of bacteriophage T4 5′ nuclease in complex with a branched DNA reveals how FEN-1 family nucleases bind their substrates. J. Biol. Chem. 282, 31713–31724 [DOI] [PubMed] [Google Scholar]
- 10. Patel N., Atack J. M., Finger L. D., Exell J. C., Thompson P., Tsutakawa S., Tainer J. A., Williams D. M., Grasby J. A. (2012) Flap endonucleases pass 5′-flaps through a flexible arch using a disorder-thread-order mechanism to confer specificity for free 5′-ends. Nucleic Acids Res. 40, 4507–4519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kao H.-I., Henricksen L. A., Liu Y., Bambara R. A. (2002) Cleavage specificity of saccharomyces cerevisiae flap endonuclease 1 suggests a double-flap structure as the cellular substrate. J. Biol. Chem. 277, 14379–14389 [DOI] [PubMed] [Google Scholar]
- 12. Finger L. D., Blanchard M. S., Theimer C. A., Sengerová B., Singh P., Chavez V., Liu F., Grasby J. A., Shen B. (2009) The 3′-flap pocket of human flap endonuclease 1 is critical for substrate binding and catalysis. J. Biol. Chem. 284, 22184–22194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bornarth C. J., Ranalli T. A., Henricksen L. A., Wahl A. F., Bambara R. A. (1999) Effect of flap modifications on human FEN1 cleavage. Biochemistry 38, 13347–13354 [DOI] [PubMed] [Google Scholar]
- 14. Balakrishnan L., Brandt P. D., Lindsey-Boltz L. A., Sancar A., Bambara R. A. (2009) Long patch base excision repair proceeds via coordinated stimulation of the multienzyme DNA repair complex. J. Biol. Chem. 284, 15158–15172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Spiro C., Pelletier R., Rolfsmeier M. L., Dixon M. J., Lahue R. S., Gupta G., Park M. S., Chen X., Mariappan S. V., McMurray C. T. (1999) Inhibition of FEN-1 processing by DNA secondary structure at trinucleotide repeats. Mol. Cell 4, 1079–1085 [DOI] [PubMed] [Google Scholar]
- 16. Kovtun I. V., Liu Y., Bjoras M., Klungland A., Wilson S. H., McMurray C. T. (2007) OGG1 initiates age-dependent CAG trinucleotide expansion in somatic cells. Nature 447, 447–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liu Y., Prasad R., Beard W. A., Hou E. W., Horton J. K., McMurray C. T., Wilson S. H. (2009) Coordination between polymerase β and FEN1 can modulate CAG repeat expansion. J. Biol. Chem. 284, 28352–28366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jarem D. A., Wilson N. R., Schermerhorn K. M., Delaney S. (2011) Incidence and persistence of 8-oxo-7,8-dihydroguanine within a hairpin intermediate exacerbates a toxic oxidation cycle associated with trinucleotide repeat expansion. DNA Repair 10, 887–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu Y., Wilson S. H. (2012) DNA base excision repair: a mechanism of trinucleotide repeat expansion. Trends Biochem. Sci. 37, 162–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kim J. C., Mirkin S. M. (2013) The balancing act of DNA repeat expansions. Curr. Opin. Genet. Dev. 23, 280–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Henricksen L. A., Tom S., Liu Y., Bambara R. A. (2000) Inhibition of flap endonuclease 1 by flap secondary structure and relevance to repeat sequence expansion. J. Biol. Chem. 275, 16420–16427 [DOI] [PubMed] [Google Scholar]
- 22. Xu M., Gabison J., Liu Y. (2013) Trinucleotide repeat deletion via a unique hairpin bypass by DNA polymerase β and alternate flap cleavage by flap endonuclease 1. Nucleic Acids Res. 41, 1684–1697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xu M., Lai Y., Torner J., Zhang Y., Zhang Z., Liu Y. (2014) Base excision repair of oxidative DNA damage coupled with removal of a CAG repeat hairpin attenuates trinucleotide repeat expansion. Nucleic Acids Res. 42, 3675–3691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Henricksen L. A., Veeraraghavan J., Chafin D. R., Bambara R. A. (2002) DNA ligase I competes with FEN1 to expand repetitive DNA sequences in vitro. J. Biol. Chem. 277, 22361–22369 [DOI] [PubMed] [Google Scholar]
- 25. Andrew S. E., Goldberg Y. P., Kremer B., Telenius H., Theilmann J., Adam S., Starr E., Squitieri F., Lin B., Kalchman M. A. (1993) The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington's disease. Nat. Genet. 4, 398–403 [DOI] [PubMed] [Google Scholar]
- 26. Cantor C. R., Warshaw M. M., Shapiro H. (1970) Oligonucleotide interactions. III. Circular dichroism studies of the conformation of deoxyoligonucleolides. Biopolymers 9, 1059–1077 [DOI] [PubMed] [Google Scholar]
- 27. Cavaluzzi M. J., Borer P. N. (2004) Revised UV extinction coefficients for nucleoside-5′-monophosphates and unpaired DNA and RNA. Nucleic Acids Res. 32, e13–e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lee B.-I., Wilson D. M., 3rd (1999) The RAD2 domain of human exonuclease 1 exhibits 5′ to 3′ exonuclease and flap structure-specific endonuclease activities. J. Biol. Chem. 274, 37763–37769 [DOI] [PubMed] [Google Scholar]
- 29. Leipold M. D., Workman H., Muller J. G., Burrows C. J., David S. S. (2003) Recognition and removal of oxidized guanines in duplex DNA by the base excision repair enzymes hOGG1, yOGG1, and yOGG2. Biochemistry 42, 11373–11381 [DOI] [PubMed] [Google Scholar]
- 30. Murante R. S., Rust L., Bambara R. A. (1995) Calf 5′ to 3′ exo/endonuclease must slide from a 5′ end of the substrate to perform structure-specific cleavage. J. Biol. Chem. 270, 30377–30383 [DOI] [PubMed] [Google Scholar]
- 31. Gloor J. W., Balakrishnan L., Bambara R. A. (2010) Flap endonuclease 1 mechanism analysis indicates flap base binding prior to threading. J. Biol. Chem. 285, 34922–34931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ayyagari R., Gomes X. V., Gordenin D. A., Burgers P. M. (2003) Okazaki fragment maturation in yeast: I. distribution of functions between FEN1 and Dna2. J. Biol. Chem. 278, 1618–1625 [DOI] [PubMed] [Google Scholar]
- 33. Garg P., Stith C. M., Sabouri N., Johansson E., Burgers P. M. (2004) Idling by DNA polymerase δ maintains a ligatable nick during lagging-strand DNA replication. Genes Dev. 18, 2764–2773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Petruska J., Hartenstine M. J., Goodman M. F. (1998) Analysis of strand slippage in DNA polymerase expansions of CAG/CTG triplet repeats associated with neurodegenerative disease. J. Biol. Chem. 273, 5204–5210 [DOI] [PubMed] [Google Scholar]
- 35. Hartenstine M. J., Goodman M. F., Petruska J. (2002) Weak strand displacement activity enables human DNA polymerase β to expand CAG/CTG triplet repeats at strand breaks. J. Biol. Chem. 277, 41379–41389 [DOI] [PubMed] [Google Scholar]
- 36. Chan N. L., Guo J., Zhang T., Mao G., Hou C., Yuan F., Huang J., Zhang Y., Wu J., Gu L., Li G.-M. (2013) Coordinated processing of 3′ slipped (CAG)n/(CTG)n hairpins by DNA polymerases β and δ preferentially induces repeat expansions. J. Biol. Chem. 288, 15015–15022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Canceill D., Viguera E., Ehrlich S. D. (1999) Replication slippage of different DNA polymerases is inversely related to their strand displacement efficiency. J. Biol. Chem. 274, 27481–27490 [DOI] [PubMed] [Google Scholar]
- 38. Ruggiero B. L., Topal M. D. (2004) Triplet repeat expansion generated by DNA slippage is suppressed by human flap endonuclease 1. J. Biol. Chem. 279, 23088–23097 [DOI] [PubMed] [Google Scholar]
- 39. Jarem D. A., Wilson N. R., Delaney S. (2009) Structure-dependent DNA damage and repair in a trinucleotide repeat sequence. Biochemistry 48, 6655–6663 [DOI] [PubMed] [Google Scholar]
- 40. Jarem D. A., Delaney S. (2011) Premutation huntingtin allele adopts a non-B conformation and contains a hot spot for DNA damage. Biochem. Biophys. Res. Commun. 416, 146–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Volle C. B., Jarem D. A., Delaney S. (2012) Trinucleotide repeat DNA alters structure to minimize the thermodynamic impact of 8-oxo-7,8-dihydroguanine. Biochemistry 51, 52–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Figueroa A. A., Cattie D., Delaney S. (2011) Structure of even/odd trinucleotide repeat sequences modulates persistence of non-B conformations and conversion to duplex. Biochemistry 50, 4441–4450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gacy A. M., Goellner G., Juranić N., Macura S., McMurray C. T. (1995) Trinucleotide repeats that expand in human disease form hairpin structures in vitro. Cell 81, 533–540 [DOI] [PubMed] [Google Scholar]
- 44. Mitas M. (1997) Trinucleotide repeats associated with human disease. Nucleic Acids Res. 25, 2245–2254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Paiva A. M., Sheardy R. D. (2004) Influence of sequence context and length on the structure and stability of triplet repeat DNA oligomers. Biochemistry 43, 14218–14227 [DOI] [PubMed] [Google Scholar]
- 46. Liu G., Chen X., Bissler J. J., Sinden R. R., Leffak M. (2010) Replication-dependent instability at (CTG)·(CAG) repeat hairpins in human cells. Nat. Chem. Biol. 6, 652–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Allawi H. T., Kaiser M. W., Onufriev A. V., Ma W.-P., Brogaard A. E., Case D. A., Neri B. P., Lyamichev V. I. (2003) Modeling of flap endonuclease interactions with DNA substrate. J. Mol. Biol. 328, 537–554 [DOI] [PubMed] [Google Scholar]
- 48. Beddows A., Patel N., Finger L. D., Atack J. M., Williams D. M., Grasby J. A. (2012) Interstrand disulfide crosslinking of DNA bases supports a double nucleotide unpairing mechanism for flap endonucleases. Chem. Commun. (Camb.) 48, 8895–8897 [DOI] [PubMed] [Google Scholar]
- 49. Finger L. D., Patel N., Beddows A., Ma L., Exell J. C., Jardine E., Jones A. C., Grasby J. A. (2013) Observation of unpaired substrate DNA in the flap endonuclease-1 active site. Nucleic Acids Res. 41, 9839–9847 [DOI] [PMC free article] [PubMed] [Google Scholar]