

Abstract
The method of displaying recombinant proteins on the surface of Saccharomyces cerevisiae via genetic fusion to an abundant cell wall protein, a technology known as yeast surface display, or simply, yeast display, has become a valuable protein engineering tool for a broad spectrum of biotechnology and biomedical applications. This review focuses on the use of yeast display for engineering protein affinity, stability, and enzymatic activity. Strategies and examples for each protein engineering goal are discussed. Additional applications of yeast display are also briefly presented, including protein epitope mapping, identification of protein-protein interactions, and uses of displayed proteins in industry and medicine.
Keywords: Yeast surface display, Protein engineering, Random mutagenesis, DNA shuffling, Affinity maturation, Protein stability engineering, Enzyme engineering
Introduction
The ability to create engineered proteins with enhanced properties, including increased binding affinity, stability, and catalytic activity, has had significant impact on biological research, medicine, and biotechnology. Despite an improved understanding of protein chemistry and folding, it remains challenging to design proteins from first principles. Thus, most strategies rely on combinatorial methods, such as directed evolution, to engineer optimized proteins by applying random or site-directed mutagenesis techniques to generate “libraries” of up to 1014 variants of an individual protein. These protein libraries are then screened in a high-throughput manner to identify amino acid mutations that confer the desired phenotype.
Numerous molecular display platforms have been specifically developed for protein engineering, including tethering libraries of protein variants to ribosomes and mRNA1–5, or to the surface of phage6,7, bacteria8, mammalian9,10, insect11, or yeast12 cells. In the case of cell surface display, each individual cell is transformed with a single vector encoding a protein variant of interest that is genetically fused to a cell-surface anchor protein. The anchor protein contains a signal sequence that directs efficient transport of the fusion protein to the cell surface, where it is immobilized and accessible to the extracellular space. Yeast surface display has become a leading platform for protein engineering due to its collective advantages, including: 1) a eukaryotic expression system capable of incorporating post-translational modifications such as disulfide bond formation, 2) low technical and time requirements relative to other eukaryotic display systems, 3) inclusion of epitope tags, which allows normalization of protein function (e.g., ligand binding) to surface expression and, thus, identification of proteins that both express at high levels and bind with high affinity to a target protein, and 4) compatibility with flow cytometric analysis, which allows quantitative measurements of equilibrium binding constants, dissociation kinetics, stability, and specificity of the displayed proteins without the laborious requirements of soluble protein expression and purification.
This review primarily focuses on the applications of yeast display from a protein engineering perspective, including examples of protein affinity maturation, stability engineering, and enzyme engineering. Other applications of yeast display are briefly reviewed, such as protein epitope mapping, identification of protein-protein interactions, and display of proteins and enzymes on yeast cells for biotechnology and biomedical applications.
Yeast surface display platform
Yeast offer multiple options for cell surface anchor proteins, including Agα1p, Aga2p, Cwp1p, Cwp2p, Tip1p, Flo1p, Sed1p, YCR89w, and Tir113. Fusion of a protein of interest to the C- or N-terminus of an anchor protein typically results in the display of up to 100,000 copies of the fusion protein on the cell surface of Saccharomyces cerevisiae14. The choice of the anchor protein and fusion terminus depends on the protein to be engineered; generally the terminus farthest from the functional portion of the protein should be tethered to the anchor protein to avoid disrupting activity. The most common yeast display system employs fusion of the protein of interest to the C-terminus of the a-agglutinin mating protein Aga2p subunit, a technology pioneered by Boder and Wittrup12 (Figure 1). The yeast surface display construct designed for this system includes two epitope tags: a hemagglutinin (HA) tag between Aga2p and the N-terminus of the protein of interest, and a C-terminal c-myc tag (Figure 1A). Induction of protein expression results in surface display of the fusion protein through disulfide bond formation of Aga2p to the β1,6-glucan-anchored Aga1p domain of a-agglutinin15–17 (Figure 1B). The epitope tags allow quantification of fusion protein expression, and thus normalization of protein function to expression level by flow cytometry using fluorescently labeled antibodies. However, detection of epitope tags yields no information on the fold or function of the protein of interest. Therefore, a ligand or antibody specific for the native fold of the displayed protein must be used to interrogate these properties.
Figure 1.

Schematic representation of yeast surface display. (A) The protein of interest is flanked by two epitope tags: a 9-amino acid hemagglutinin antigen (HA) tag and a 10-amino acid c-myc tag, and is fused to the C-terminus of the a-agglutinin Aga2p subunit. (B) Protein display on the yeast cell surface. Following translation, the 69-amino acid Aga2p subunit associates with 725-amino acid a-agglutinin Aga1p subunit via two disulfide bonds. The fusion protein is subsequently secreted to the extracellular space where Aga1p is anchored to the cell wall via a β1,6-glucan covalent linkage. As a result, the protein of interest is displayed on the cell surface where it is accessible by soluble ligands. Functional display of the protein of interest (shown here as a scFv130 modified from PDB 1X9Q using the UCSF Chimera package131) can be detected by a fluorescently labeled antibody or ligand (red star) specific to the native fold. The epitope tags are used to normalize protein function to surface expression level through either labeled anti-HA or anti-c-myc antibodies (green stars). These features allow flow cytometric sorting of a heterogeneous mixture of yeast cells, each displaying up to 100,000 copies of a single protein variant, based on the biophysical and biochemical properties of the displayed proteins.
Protein engineering applications using yeast display involve expression of a protein library, which is generated from the underlying genetic material that codes for the protein variants. This connection is known as a genotype-phenotype linkage and must be maintained throughout the protein engineering process so that the desired protein variants can be identified by sequencing following library screening. To create a protein library, diverse genetic material, which can be obtained directly from organisms or generated by mutagenic PCR, is transformed into yeast cells and induction of expression through a GAL promoter leads to surface display of the protein variants. The resulting library can then be screened using flow cytometric sorting (also known as fluorescence-activated cell sorting, or FACS) to isolate yeast displaying proteins with the desired properties. For this purpose, yeast are incubated with fluorescent probes that differentially label the cells based on the biochemical and biophysical properties of their displayed protein (e.g., affinity, stability, specificity, etc.). The yeast cells are then passed single-file through the fluidics stream of a FACS instrument, which analyses and sorts them based on cell size, granularity, and fluorescence measurements. Detailed protocols for library creation and screening have been well described in the field14,18–21.
Phage display and ribosome display techniques take advantage of panning-based methods to efficiently screen libraries as large as 1012–1014 variants. In comparison, yeast display offers lower throughput due to limitations in yeast transformation efficiency and current cell screening technology. The upper limit of library sizes that can currently be screened by FACS is ~108–109 yeast cells, determined by the maximum sampling rate (~50,000 cells per second) of the leading flow cytometry instruments. Moreover, libraries are typically sampled by FACS at higher coverage (e.g., 10x) to increase the probability of sampling each variant at least once. Thus, to increase throughput, yeast-displayed libraries of greater than 108 variants can first be screened using bead-based magnetic-activated cell sorting (MACS) to reduce the library diversity before screening with FACS22,23. In this method, magnetic beads are coated with a soluble target of interest, for example, an antibody or ligand. The beads are then incubated with the yeast library, and yeast displaying non-binding protein variants are removed by washing, after which the yeast binding the desired target are eluted and recovered. This method is advantageous for removing truncated, misfolded, and weak affinity proteins from the library, thereby reducing library diversity to a size that is amenable to quantitative screening by FACS. Finally, while differences between human and yeast glycosylation patterns have typically not prevented functional display of glycosylated human proteins, yeast strains that express human glycosylation machinery have been engineered, and similar technology could potentially be developed for yeast display applications if human glycosylation is desired (see 24–26 for reviews).
Engineering proteins for increased affinity
The affinity a protein has for its binding partner is a key parameter that often regulates the biological function of the bound complex. High binding affinity is a desired characteristic of proteins used for research, therapeutic, and diagnostic applications, and thus multiple strategies for increasing protein affinity (termed “affinity maturation”) have been developed, with the most common involving directed evolution and molecular display technologies. Over the last decade, yeast display has become a leading platform for affinity maturation; in addition to the aforementioned advantages, yeast display can discriminate between proteins with only 2-fold differences in affinity21,27, further illustrating the sensitivity of this approach.
A general strategy of affinity maturation using yeast display involves creation of a library on the order of 107–109 protein variants by random mutagenesis, followed by display of these variants on the surface of yeast as fusions to the Aga2p cell wall protein (Figure 2). Subsequently, two main strategies are used to label the yeast-displayed library prior to sorting by FACS. In the first approach, library members are screened based on their equilibrium dissociation constants (KD)27,28. The yeast-displayed library is incubated with the soluble binding partner (ligand) at a concentration ~5–10-fold greater than the expected KD of the highest affinity variants (typically near the KD of the wild-type interaction), and binding is allowed to reach equilibrium. This method requires at least a 10-fold excess of ligand relative to the number of yeast-displayed protein variants in the binding reaction. If a lower ratio is used, binding may significantly alter the concentration of free ligand in solution, which would result in ligand depletion and non-compliance with rules that govern equilibrium binding isotherms. The second approach uses kinetic competition and screens yeast-displayed variants based on their dissociation rate constants (koff)12,29,30. The library is incubated with a saturating concentration of fluorescently labeled ligand, washed, and then either incubated with 100-fold excess of unlabeled ligand, if available, or incubated in a sufficiently large volume of buffer to prevent rebinding of the labeled ligand after dissociation. This method is primarily used when evolving variants of a protein with a strong starting affinity (KD < 1 nM) that would require inconveniently large incubation volumes to meet the 10-fold excess ligand requirement of the former method, or when the dissociation kinetics are the most important functional characteristic of the desired protein. Regardless of which labeling method is used, high-affinity variants are selected using FACS to isolate cells that exhibit high levels of binding for a given amount of cell surface expression, as measured by a fluorescent antibody against the C-terminal epitope tag and a fluorescently labeled soluble ligand, respectively (Figure 1).
Figure 2.
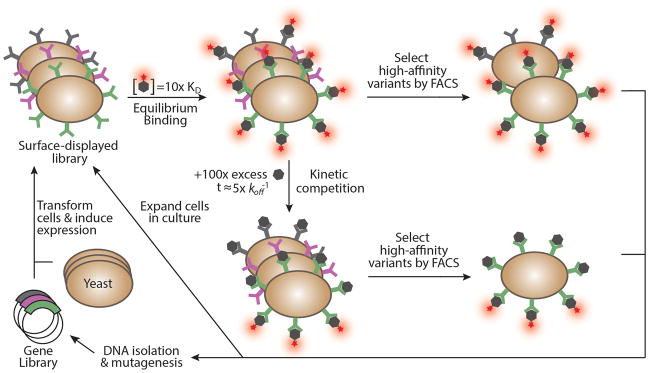
Isolating high-affinity protein variants from a yeast-displayed library by FACS. Following transformation of yeast cells with a gene library and induction of surface expression, two main strategies are used to differentially label the displayed library prior to screening: 1) an equilibrium binding strategy where the library is incubated with a ligand concentration 5–10-times greater than the expected KD value of the highest affinity variant, resulting in near saturation of tight binding variants and partial labeling of weaker affinity variants at equilibrium, and 2) a kinetic binding strategy where the library is incubated with ligand as described for the equilibrium binding strategy, but unbound ligand is removed by washing and the library is then either incubated with a 100-fold excess of unlabeled ligand, or incubated in a sufficiently large volume of buffer to prevent rebinding of dissociated ligand. During this second incubation step, the excess unlabeled ligand or large incubation volume prevents dissociated labeled ligands from rebinding. Proteins are thus differentiated based on their dissociation rate constants (koff), with variants having the slowest koff retaining the largest percentage of pre-bound labeled ligand. Addition of a fluorescently-labeled anti-epitope tag antibody (not shown) permits normalization of yeast surface expression levels with binding, allowing the highest affinity variants to be isolated by FACS. Sorted pools of yeast clones can be expanded in culture for either analysis or a subsequent round of sorting, or DNA from these clones can be isolated, subjected to mutagenesis, and used to transform a new batch of yeast for further directed protein evolution. Components of the yeast display platform, including Aga1p, Aga2p, HA and c-myc epitope tags, and detection antibodies depicted in Figure 1, are omitted for clarity.
Following each round of cell sorting, two paths can be taken: 1) the selected cells can be amplified in culture and sorted again to further reduce the library diversity to a smaller subset of clones with the highest affinity, or 2) DNA from the selected cells can be extracted and subjected to an another round of mutagenesis (e.g. random mutagenesis or DNA shuffling31) to introduce additional diversity or combine potentially favorable mutations, followed by display of the new library and another round of cell sorting. The latter approach is termed “directed evolution”, and incorporates multiple rounds of mutagenesis and library screening to iteratively evolve proteins with the desired binding characteristics. After the library size is reduced to a smaller pool of high-affinity proteins, the concentration of soluble ligand can be lowered (while still avoiding ligand depletion) to adjust the resolution between the weaker and tighter binding proteins and further aid in selection of variants with the highest affinity. Typically, multiple rounds of mutagenesis and/or library sorting are applied to isolate high-affinity variants with equilibrium dissociation constants in the low nanomolar to picomolar range.
Following library sorting, individual variants can be quantitatively analyzed for their binding properties while still tethered to the yeast cell surface27,28. This is a major advantage of yeast display technology as it allows fast, quantitative comparisons of binding properties without the need for laborious soluble expression and purification of each protein. For this purpose, the enriched library pool is amplified by culturing and is then plated to isolate individual yeast clones that each express a single variant. These individual clones are induced for cell surface expression and tested for binding to varying concentrations of ligand typically ranging from 10-fold above to 10-fold below the expected KD of the displayed variant. Equilibrium binding constants or dissociation kinetics are then determined by flow cytometry27,28. Importantly, many studies have demonstrated that the KD of a protein-protein binding interaction measured on the surface of yeast is essentially equal to the KD measured using soluble proteins32.
Using the general strategies outlined above numerous proteins have been engineered with enhanced affinities for their binding partners, including T cell receptors with greater than 100-fold enhanced affinity for a peptide/MHC ligand33, epidermal growth factor (EGF) with 30-fold enhanced affinity for the EGF receptor34, interleukin-2 (IL-2) with up to 30-fold enhanced affinity for the IL-2 receptor alpha subunit35,36, leptins with up to 60-fold enhanced affinity for the leptin receptor37, an Axl receptor variant with 12-fold enhanced affinity for it’s ligand Gas6 (final KD = 2.7 pM)38, and a signal-regulatory protein α (SIRPα) variant with ~50,000-fold enhanced affinity for CD47 (final KD = 11.1 pM)39,40. Yeast display has also been applied to engineer and affinity mature numerous antibodies, including antibodies against cholera toxin41, FITC22, HIV-1 gp12042, hemagglutinin surface glycoprotein of the H1N1 virus43, HER2/neu44, T cell receptors28, TNF-α45, and a host of other targets (see reference 46 for a review). In a classical demonstration of the technological capabilities afforded by yeast display, a fluorescein binding antibody was engineered with a KD equal to 48 fM and a dissociation rate greater than 1,000-fold lower than the parent antibody30, representing one of the strongest protein binding interactions ever engineered and among the strongest found in nature.
Yeast display technology has also been used to engineer novel non-antibody protein binders against targets of interest. These so-called alternative scaffold proteins have been chosen for protein engineering applications based on positive attributes including stability, amenability to mutation, ease of expression and purification, and binding epitope surface area. Typically, the amino acid sequence of a contiguous solvent exposed region of the scaffold is randomized to generate a “naïve” library, and the library is displayed on the surface of yeast and screened for binding to a target protein using FACS47,48. A number of novel ligands have been engineered using this strategy, including: cysteine knot peptides (knottins) that bind to various integrins with KD values in the picomolar to nanomolar range47,49–53, or that inhibit human matriptase-1 with picomolar to nanomolar inhibition constants (Ki)54; human fibronectin 10th type III domain scaffold55 variants that bind to a variety of protein targets18,56,57, such as lysozyme, with KD values in the nanomolar to picomolar range29,32; green fluorescent protein variants that bind streptavidin-phycoerythrin, biotin-phycoerythrin, glyceraldehyde 3-phosphate dehydrogenase, and a neurotrophin receptor with KD values of 70 nM, 190 nM, 18 nM, and 3.2 nM, respectively58; human kringle domain variants that bind death receptor 4 (DR4), DR5, or TNF-α with KD values of 680 nM, 172 nM, and 29 nM, respectively59; and Sso7d protein variants from the hyperthermophilic archaeon Sulfolobus solfataricus that bind fluorescein, a peptide fragment from β-catenin, hen egg lysozyme, streptavidin, and chicken and mouse immunoglobulins with KD values in the nanomolar to micromolar range60.
In many of the scaffold examples described above, the screening strategy identified a number of high affinity ligands that bound to different epitopes of the target protein. Novel ligands have also been engineered to bind to a specific epitope of the target protein by first selecting all library variants that bind to a wild-type target protein, and then screening the selected pool of binders for variants that do not bind to an epitope-altered form of the target protein. As a recent example, a dengue virus-neutralizing antibody was engineered using yeast display by selecting antibodies from a library that bound to the wild-type viral envelope protein domain III, but not to a form of the target protein with a specific epitope mutated61. Importantly, this strategy is contingent on proper design of the mutant target epitope used for library screening. First, the target epitope must be sufficiently mutated such that ligands that bind to the wild-type epitope will not bind to the mutated form. Second, the mutation(s) must only affect the structure of the protein at the site of the target epitope and must not affect the global fold of the protein, as screening for epitope-specific binders using a completely misfolded mutant competitor would be futile.
Engineering proteins for increased stability
The stability of a protein generally refers to its ability to resist thermal and chemical denaturation and proteolytic degradation. High stability is a desired characteristic of proteins that are used for research, industrial, and therapeutic applications, and translates to longer shelf-life, duration of activity, and in vivo activity. As with binding affinity, thermal stability can be analyzed while a protein variant is still tethered to the yeast cell surface, allowing for rapid, quantitative measurement of half-maximal denaturation (TM) values. Three general strategies have been applied to engineer proteins with increased stability (Figure 3). In each approach, a library on the order of 107–109 protein variants is generated by random mutagenesis and displayed on the surface of yeast as a fusion to the Aga2p cell wall protein.
Figure 3.

Isolating high-stability protein variants from a yeast-displayed library by FACS. (A) Screening of stable protein variants based on their level of surface expression. Transformation of yeast with a mutant gene library generally results in display of properly folded and truncated protein variants that range in expression level, which can be used as a proxy for protein stability62–64. Cells are labeled with a fluorescent antibody (green star) against the c-myc epitope tag (purple box) and a ligand (red star) specific to the native fold of the displayed protein, and cells expressing the highest levels of properly folded variants are selected by FACS. (B) Screening of stable protein variants based on their ability to resist irreversible thermal denaturation. Following heat incubation and sorting of yeast displaying stable protein variants, viable cells can be expanded in culture for additional screening and/or analysis, whereas DNA from nonviable cells must be isolated and amplified for analysis or an additional round of yeast transformation and screening. Point mutations are shown as red circles, and colors indicating protein identity match those shown in Figure 1. The displayed protein depicted here is a scFv130 modified from PDB 1X9Q using the UCSF Chimera package131.
The first strategy for stability engineering exploits a correlation between the yeast surface expression levels of properly folded proteins and their thermal stability62–64 (Figure 3A). For example, a library of single-chain T-cell receptor (scTCR) variants was expressed on the yeast surface and enriched for cells displaying the highest levels of properly folded protein as determined by binding to a conformationally specific antibody65. When individual protein mutants from this enriched pool of yeast were recombinantly expressed in soluble form and assayed, the most stable scTCR variant retained 80% activity after incubation at 50°C for 30 minutes, whereas the parent scTCR protein retained less than 10% activity under the same conditions. In another example, yeast surface display and library screening were used to identify an epidermal growth factor receptor (EGFR) mutant with a TM of 61.0 ± 1.3°C compared to a TM of 52.5 ± 0.7°C for wild-type EGFR66. Similarly, yeast display was used to identify a single-chain class II major histocompatibility complex protein (scDR1αβ) with a TM of 73.3 ± 1.8°C, whereas display of the properly folded wild-type scDR1αβ protein was barely detectable67. This general strategy has been applied to enhance the stability of numerous other proteins and is reviewed in detail elsewhere68.
Despite the successes described above, using surface expression level as a proxy for protein stability may be better suited for proteins with low inherent thermal stabilities. The correlation between expression level and protein stability is due, in part, to the quality control process that occurs in the endoplasmic reticulum (ER) during protein synthesis and post-translational processing. The ER quality control mechanism ensures efficient export of properly folded proteins, whereas misfolded proteins are retro-translocated across the ER membrane and degraded in the cytosol69,70. This process generally results in inefficient expression of unstable proteins that adopt a higher ratio of misfolded to native structures. However, the observed correlation between yeast surface expression level and stability is likely limited to proteins of low stability, as proteins above a certain stability threshold are generally expected to escape the ER quality control mechanism. In support of this assumption, variants of a highly thermostable three-helix bundle protein α3D with varying Tm values all above 80°C showed no correlation between yeast surface expression level and thermal stability71.
A second strategy for increasing protein stability involves application of heat stress (up to 85 °C for 10 minutes) directly to a yeast-displayed library prior to cell sorting (Figure 3B)65,72,73. This approach may be better suited for proteins with higher inherent thermal stabilities, assuming irreversible denaturation will occur under these experimental conditions. Yeast-displayed variants that resist thermal denaturation are discriminated by FACS, based on their ability to bind to a fluorescently labeled antibody or ligand specific to the native protein fold67. As an example, a library of IgG1-Fc scaffold variants was displayed on yeast and subjected to heat stress (79 °C for 10 minutes)21. Stable IgG1-Fc variants that bound to a conformationally specific antibody or a soluble Fcγ receptor after heat stress were enriched using multiple rounds of FACS, and isolated variants were analyzed for thermal stability. IgG1-Fc variants were identified with increased TM values up to 91.0 ± 0.1°C, compared to a TM of 82.6 ± 0.0°C for wild-type IgG1-Fc. Using this general strategy, a Her2/neu-binding antibody fragment variant was engineered with an increase in TM from ~70 °C (parent IgG1-Fc) to ~75 °C and an increased resistance to aggregation74. Similarly, variants of a monomeric yeast-enhanced green fluorescent protein (GFPM) were engineered with 3- to 6-fold increased resistance to thermal denaturation at 70 °C72. Notably, increased thermal stability did not confer increased yeast-surface expression levels for the GFPM variants, which further supports the addition of a heat stress step to the library screening protocol when engineering proteins with high intrinsic thermostabilities. An important consideration is that although yeast cells remain intact and can be efficiently sorted by FACS even after heat stress (e.g., 72 °C for 90 minutes or 85 °C for 10 minutes)73, their viability is compromised at temperatures above 42 °C75. Thus, after each round of heat stress and sorting, plasmid DNA from yeast cells should be isolated, amplified by PCR, and used to transform viable cells for a subsequent round of screening20,21. Additionally, this strategy involving heat stress is only applicable to proteins/domains that denature irreversibly; refolding after heat stress would prevent discrimination between variants of different thermal stabilities.
Alternatively, a third strategy exists for increasing protein stability that harnesses both the advantages of increased temperature and the quality control mechanisms of the ER to select stable variants. In this method, expression of surface-displayed protein is induced for 24 hours at temperatures up to 37 °C (compared to 20 °C or 30 °C which is typically used) to shift the equilibrium of protein structures toward the misfolded state during protein synthesis and post-translational processing in the ER, while maintaining yeast cell viability. Generally, only proteins that are efficiently folded and processed at the elevated induction temperatures will avoid the ER quality control machinery and be efficiently exported to the cell surface. The library can then be sorted as described in the first strategy (Figure 3A) to select variants with increased stabilities. For example, this strategy was applied to enhance the stability of a scTCR65, and to engineer a hepatocyte growth factor fragment with a 15 °C increase in TM and a 40-fold increase in expression yield relative to the wild-type protein fragment76. In many cases, increased thermal stability also correlates to increased recombinant expression of the soluble form of the protein relative to the wild-type protein66,67,77–80, highlighting an additional benefit of these techniques for protein engineering.
Enzyme engineering
Directed evolution is a powerful technique for enzyme engineering. However, developing a strategy for linking the genotype of an enzyme mutant to its phenotype (e.g., catalytic activity or substrate specificity) poses a difficult challenge since in most cases substrate turnover results in a product that is diffusible and not covalently bound to the surface of a phage or cell. Thus, lower-throughput microtiter or colony based screening methods (103–104 variants) have historically been used for enzyme engineering81–83. Several alternate techniques have recently been developed to address these challenges, including oil-water emulsion methods that encapsulate genetic material, the translated enzyme, and its catalyzed product into droplets that can be sorted by flow cytometry84,85.
More recently, unique strategies incorporating yeast display have allowed libraries of up to 108 enzyme variants to be screened for increased activity and substrate specificity using FACS. In this approach, yeast cells are fluorescently labeled as a result of enzymatic activity, which allows discrimination of enzyme variants based on their level of activity and/or specificity. In one example, a library of horseradish peroxidase (HRP) enzyme variants was differentially labeled based on substrate specificity86. The library was incubated with a fluorescently labeled substrate, which produced a functionalized fluorophore byproduct that covalently attached to tyrosines on the yeast cell surface. As a result, cells displaying the most active HRP variants were labeled with higher levels of fluorophore and subsequently selected by FACS. Using this strategy, an HRP variant with 8-fold altered enantioselectivity was evolved using a combination of positive and negative selection86. A similar strategy was applied to evolve HRP variants with increased selectivity toward either substrate enantiomer by up to 2 orders of magnitude87. In another example, a general strategy for evolving bond-forming enzymes was developed (Figure 4) and applied to identify a bacterial transpeptidase sortase A enzyme with a 140-fold enhancement in catalytic activity88. Yeast display has been applied to enhance the activity and/or substrate selectivity of a variety of other enzymes, including firefly luciferase89, Rhizomucor miehei lipase90, the adenylation domain of a nonribosomal peptide synthetase91, E. coli lipoic acid ligase92, and a Tobacco Etch virus protease93. In general, the success of these strategies was contingent on the enzyme substrate being labeled with an affinity handle or fluorescent probe. Thus, yeast display methodology is currently limited to engineering a subset of enzymes, as not all will be tolerant to such substrate modifications.
Figure 4.

A general strategy for selecting bond-forming enzyme variants with increased catalytic activity from a yeast-displayed library88. A library of enzyme variants is generated and displayed as a fusion to Aga2p, and a reactive peptide handle (s6) is fused to Aga1p. Sfp phosphopantetheinyl transferase covalently links CoA-conjugated enzyme substrate A to the s6 peptide handle, where it is accessible by the displayed enzyme. Subsequently, enzyme substrate B linked to an affinity handle (shown here as biotin) is incubated with the library, resulting in A–B bond formation catalyzed by active enzyme variants. Addition of a fluorescently-labeled anti-epitope tag antibody (green star) and an affinity agent that binds to the handle on substrate B permits discrimination of enzyme variants with increased catalytic activity by FACS. Next, selected pools of yeast can be amplified for analysis or an additional round of screening, or their DNA can be extracted, subjected to mutagenesis, and used to transform new cells for further directed evolution. Application of this strategy is limited to bond-forming enzyme-substrate systems that remain functional when the substrates are tethered to the s6 peptide and an affinity handle.
Finally, many enzyme engineering examples employ site-directed saturation mutagenesis94 rather than random mutagenesis to generate protein libraries. This directed mutagenesis restricts the amino acid search space to a particular region of interest of the enzyme (typically proximal to the active site), and allows the investigation of every possible combination of mutations at selected amino acid positions. However, the maximum number of these positions one can exhaustively investigate is still limited by the throughput of the FACS instrument used to screen the library. For example, a library comprising every possible combination of amino acids at 6 or 7 sites in an enzyme (206 or 207 variants total) would require ~0.4 or 7 hours to sort at 50,000 cells per second, respectively.
Additional applications
Yeast display has been used for other purposes besides protein engineering, briefly described below, including protein epitope mapping, identification of protein-protein interactions, and generation of “armed” yeast cells for a variety of applications.
Knowledge of the critical contact sites of a protein binding pair that govern their affinity can be adventageous. Two methods for identifying these contact sites, called domain-level95 or fine96 epitope mapping, have been developed using yeast display and applied to a wide range of protein binding pairs. For domain-level epitope mapping, individual domains from one of the binding proteins are displayed on yeast and screened for binding to a soluble version of the other binding partner95. In addition, the competitive binding of two ligands has been tested to identify ligands that share overlapping binding epitopes. In contrast, fine epitope mapping has been used to identify specific amino acids at the binding interface that directly contribute to the binding affinity96. For this method, a protein library is generated using random mutagenesis, displayed on the surface of yeast, and incubated with its wild-type binding partner. Cells displaying weak-binding proteins are then selected and their encoding DNA is sequenced to identify consensus amino acid sites that substantially influence the affinity of the protein pair, and thus, are suggestive of the binding interface location. These strategies have been applied to map the binding epitopes of EGFR-specific antibodies95,96 and engineered EGFR-specific scaffold proteins97, antibodies against H1N143 and H5N198 virus hemagglutinin surface glycoprotein, gp120-binding antibodies99, and other binding pairs as reviewed elsewhere100,101.
Yeast display has also been applied to identify new protein-protein interactions. For example, an adult human testis cDNA library was displayed on yeast and screened for binding to phosphorylated peptides derived from autophosphorylation sites of EGFR and focal adhesion kinase (FAK)102. As a result, binding interactions were discovered between autophosphorylated EGFR sites and the SH2 domains of adapter protein APS and phosphoinositide 3-kinase regulatory subunit 3, as well as between autophosphorylated FAK sites and the SH2 domains of SH2B, tensin, and adapter protein APS102. Similarly, screening of a yeast-displayed human proteome library for binding to a mesothelioma-targeting single-chain variable antibody fragment (scFv) identified the specific cell surface antigen targeted by the scFv103. Yeast display has also been applied to identify interactions between proteins and small molecules. For example, a human cDNA library comprising 2 × 107 cDNA fragments from multiple human tissue samples was displayed on yeast and screened for binding to biotinylated phosphatidylinositides104. As a result, known interactions with pleckstrin homology domains, and a phosphotyrosine-binding domain were identified, and a novel interaction with a fragment of apolipoprotein H was discovered104. These and other protein-protein interactions identified using yeast display have been reviewed elsewhere100,101.
Divergent from protein engineering, yeast display technology has been used to functionalize yeast cells for a variety of biotechnology and biomedical applications, including generation of whole-cell biocatalysts, antimicrobial agents, oral vaccines, and for biosorption of various metals. In one prominent example, microbial conversion of cellulosic biomass into fuels gained substantial interest as a means of establishing a renewable energy source and an alternative to petroleum-based fuel production. Yeast display is an attractive technology for generating biofuel from cellulosic material, as it enables enzyme production, cellulose hydrolysis, and fermentation all in one step by localization of cellulolytic, amylolytic, and xylanolytic enzymes at the yeast cell surface. S. cerevisiae has been engineered using yeast display technology to convert cellulosic material into bioethanol (for reviews, see 105,106). Specifically, yeast cells were engineered to co-display endoglucanase II and β-glucosidase enzymes, and directly fermented 45 g of β-glucan per liter of media to produce 16.5 g of ethanol per liter in approximately 50 hours107. The ratio of grams of ethanol produced to grams of β-glucan utilized was 0.48 g/g (or 93.3% of the theoretical yield). Yeast co-displaying xylanase and β-xylosidase directly fermented xylan from sulfuric acid hydrolysate of wood chips108, and yeast co-displaying glucoamylase and α-amylase directly fermented raw corn starch109. Display of minicellulosomes on the surface of yeast for bioethanol production has also been achieved by simultaneously binding dockerin-tagged endoglucanase, exogluconase, and β-glucosidase enzymes to Aga2p-scaffoldin protein fusions110–113. Yeast cells have also been engineered as whole-cell biocatalysts for various other applications. For example, yeast displaying Rhizopus oryzae lipase were used as whole-cell biocatalysts to generate biodiesel from methanol and soybean oil114; yeast displaying Geotrichum sp. lipase were used to enrich docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA) from fish oil115; yeast displaying Corynebacterium diphtheria sialidase were used to transfer sialic acids for glycoprotein remodeling116, and yeast displaying glucose oxidase were used for electrochemical glucose sensing117. Yeast have also been functionalized with antimicrobial peptides118,119, pathogenic proteins for oral vaccine delivery120–122, and metal-binding proteins for bioadsporption of various metals123–129. These and other examples have recently been reviewed69.
Conclusion
Yeast surface display is an effective tool for protein and cellular engineering, and has facilitated countless applications in research, biotechnology, and medicine. In contrast to other technologies such as ribosome and phage display, yeast display offers compatibility with flow cytometric analysis, enabling quantitative on-cell measurements of protein expression level, stability, affinity, and specificity without the need for soluble protein expression and purification steps. Additionally, unlike bacterial and phage display, yeast display provides a eukaryotic expression system capable of producing complex mammalian proteins containing multiple disulfide bonds. This unique combination of advantages has established yeast display as a leading technology for engineering protein stability, expression, and binding interactions, and as an emerging technology for high-throughput enzyme engineering and yeast cell engineering. Furthermore, other than requiring a FACS instrument for quantitative library screening and analysis, yeast display employs standard laboratory equipment and materials for microbial transformation and culture, and libraries can be readily generated and screened by users within a matter of weeks. As the use and applications of yeast display continue to rapidly expand, it will be exciting to see the advances this powerful technology delivers in the years to come.
Acknowledgments
Gerald M. Cherf is supported by the National Cancer Institute of the National Institutes of Health under Award Number F31CA186478, and funding from the Stanford Bioengineering Department. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- 1.He M, Taussig MJ. Antibody-ribosome-mRNA (ARM) complexes as efficient selection particles for in vitro display and evolution of antibody combining sites. Nucleic Acids Res. 1997;25:5132–4. doi: 10.1093/nar/25.24.5132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hanes J, Plückthun A. In vitro selection and evolution of functional proteins by using ribosome display. Proc Natl Acad Sci U S A. 1997;94:4937–42. doi: 10.1073/pnas.94.10.4937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roberts RW, Szostak JW. RNA-peptide fusions for the in vitro selection of peptides and proteins. Proc Natl Acad Sci U S A. 1997;94:12297–302. doi: 10.1073/pnas.94.23.12297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mattheakis LC, Bhatt RR, Dower WJ. An in vitro polysome display system for identifying ligands from very large peptide libraries. Proc Natl Acad Sci U S A. 1994;91:9022–6. doi: 10.1073/pnas.91.19.9022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mattheakis LC, Dias JM, Dower WJ. Cell-free synthesis of peptide libraries displayed on polysomes. Methods Enzymol. 1996;267:195–207. doi: 10.1016/s0076-6879(96)67013-x. [DOI] [PubMed] [Google Scholar]
- 6.Smith GP. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science. 1985;228:1315–7. doi: 10.1126/science.4001944. [DOI] [PubMed] [Google Scholar]
- 7.McCafferty J, Griffiths AD, Winter G, Chiswell DJ. Phage antibodies: filamentous phage displaying antibody variable domains. Nature. 1990;348:552–4. doi: 10.1038/348552a0. [DOI] [PubMed] [Google Scholar]
- 8.Francisco JA, Campbell R, Iverson BL, Georgiou G. Production and fluorescence-activated cell sorting of Escherichia coli expressing a functional antibody fragment on the external surface. Proc Natl Acad Sci U S A. 1993;90:10444–8. doi: 10.1073/pnas.90.22.10444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ho M, Nagata S, Pastan I. Isolation of anti-CD22 Fv with high affinity by Fv display on human cells. Proc Natl Acad Sci U S A. 2006;103:9637–42. doi: 10.1073/pnas.0603653103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beerli RR, et al. Isolation of human monoclonal antibodies by mammalian cell display. Proc Natl Acad Sci U S A. 2008;105:14336–41. doi: 10.1073/pnas.0805942105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ernst W, et al. Baculovirus surface display: construction and screening of a eukaryotic epitope library. Nucleic Acids Res. 1998;26:1718–23. doi: 10.1093/nar/26.7.1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boder ET, Wittrup KD. Yeast surface display for screening combinatorial polypeptide libraries. Nat Biotechnol. 1997;15:553–7. doi: 10.1038/nbt0697-553. [DOI] [PubMed] [Google Scholar]
- 13.Kondo A, Ueda M. Yeast cell-surface display--applications of molecular display. Appl Microbiol Biotechnol. 2004;64:28–40. doi: 10.1007/s00253-003-1492-3. [DOI] [PubMed] [Google Scholar]
- 14.Boder ET, Wittrup KD. Yeast surface display for directed evolution of protein expression, affinity, and stability. Methods Enzymol. 2000;328:430–44. doi: 10.1016/s0076-6879(00)28410-3. [DOI] [PubMed] [Google Scholar]
- 15.Kapteyn JC, Van Den Ende H, Klis FM. The contribution of cell wall proteins to the organization of the yeast cell wall. Biochim Biophys Acta. 1999;1426:373–83. doi: 10.1016/s0304-4165(98)00137-8. [DOI] [PubMed] [Google Scholar]
- 16.Roy A, Lu CF, Marykwas DL, Lipke PN, Kurjan J. The AGA1 product is involved in cell surface attachment of the Saccharomyces cerevisiae cell adhesion glycoprotein a-agglutinin. Mol Cell Biol. 1991;11:4196–206. doi: 10.1128/mcb.11.8.4196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu CF, et al. Glycosyl phosphatidylinositol-dependent cross-linking of alpha-agglutinin and beta 1,6-glucan in the Saccharomyces cerevisiae cell wall. J Cell Biol. 1995;128:333–40. doi: 10.1083/jcb.128.3.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koide S, Koide A, Lipovšek D. Target-binding proteins based on the 10th human fibronectin type III domain (10Fn3) Methods Enzymol. 2012;503:135–56. doi: 10.1016/B978-0-12-396962-0.00006-9. [DOI] [PubMed] [Google Scholar]
- 19.Scholler N. Selection of antibody fragments by yeast display. Methods Mol Biol. 2012;907:259–80. doi: 10.1007/978-1-61779-974-7_15. [DOI] [PubMed] [Google Scholar]
- 20.Zhao Q, Zhu Z, Dimitrov DS. Yeast display of engineered antibody domains. Methods Mol Biol. 2012;899:73–84. doi: 10.1007/978-1-61779-921-1_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chao G, et al. Isolating and engineering human antibodies using yeast surface display. Nat Protoc. 2006;1:755–68. doi: 10.1038/nprot.2006.94. [DOI] [PubMed] [Google Scholar]
- 22.Feldhaus MJ, et al. Flow-cytometric isolation of human antibodies from a nonimmune Saccharomyces cerevisiae surface display library. Nat Biotechnol. 2003;21:163–70. doi: 10.1038/nbt785. [DOI] [PubMed] [Google Scholar]
- 23.Miller KD, Pefaur NB, Baird CL. Construction and screening of antigen targeted immune yeast surface display antibody libraries. Curr Protoc Cytom. 2008;Chapter 4(Unit4.7) doi: 10.1002/0471142956.cy0407s45. [DOI] [PubMed] [Google Scholar]
- 24.Wildt S, Gerngross TU. The humanization of N-glycosylation pathways in yeast. Nat Rev Microbiol. 2005;3:119–28. doi: 10.1038/nrmicro1087. [DOI] [PubMed] [Google Scholar]
- 25.De Pourcq K, De Schutter K, Callewaert N. Engineering of glycosylation in yeast and other fungi: current state and perspectives. Appl Microbiol Biotechnol. 2010;87:1617–31. doi: 10.1007/s00253-010-2721-1. [DOI] [PubMed] [Google Scholar]
- 26.Gerngross TU. Advances in the production of human therapeutic proteins in yeasts and filamentous fungi. Nat Biotechnol. 2004;22:1409–14. doi: 10.1038/nbt1028. [DOI] [PubMed] [Google Scholar]
- 27.VanAntwerp JJ, Wittrup KD. Fine affinity discrimination by yeast surface display and flow cytometry. Biotechnol Prog. 16:31–7. doi: 10.1021/bp990133s. [DOI] [PubMed] [Google Scholar]
- 28.Kieke MC, Cho BK, Boder ET, Kranz DM, Wittrup KD. Isolation of anti-T cell receptor scFv mutants by yeast surface display. Protein Eng. 1997;10:1303–10. doi: 10.1093/protein/10.11.1303. [DOI] [PubMed] [Google Scholar]
- 29.Hackel BJ, Kapila A, Wittrup KD. Picomolar affinity fibronectin domains engineered utilizing loop length diversity, recursive mutagenesis, and loop shuffling. J Mol Biol. 2008;381:1238–52. doi: 10.1016/j.jmb.2008.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boder ET, Midelfort KS, Wittrup KD. Directed evolution of antibody fragments with monovalent femtomolar antigen-binding affinity. Proc Natl Acad Sci U S A. 2000;97:10701–5. doi: 10.1073/pnas.170297297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stemmer WP. Rapid evolution of a protein in vitro by DNA shuffling. Nature. 1994;370:389–91. doi: 10.1038/370389a0. [DOI] [PubMed] [Google Scholar]
- 32.Lipovsek D, et al. Evolution of an interloop disulfide bond in high-affinity antibody mimics based on fibronectin type III domain and selected by yeast surface display: molecular convergence with single-domain camelid and shark antibodies. J Mol Biol. 2007;368:1024–41. doi: 10.1016/j.jmb.2007.02.029. [DOI] [PubMed] [Google Scholar]
- 33.Holler PD, et al. In vitro evolution of a T cell receptor with high affinity for peptide/MHC. Proc Natl Acad Sci U S A. 2000;97:5387–92. doi: 10.1073/pnas.080078297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cochran JR, Kim YS, Lippow SM, Rao B, Wittrup KD. Improved mutants from directed evolution are biased to orthologous substitutions. Protein Eng Des Sel. 2006;19:245–53. doi: 10.1093/protein/gzl006. [DOI] [PubMed] [Google Scholar]
- 35.Rao BM, Girvin AT, Ciardelli T, Lauffenburger DA, Wittrup KD. Interleukin-2 mutants with enhanced alpha-receptor subunit binding affinity. Protein Eng. 2003;16:1081–7. doi: 10.1093/protein/gzg111. [DOI] [PubMed] [Google Scholar]
- 36.Rao BM, Driver I, Lauffenburger DA, Wittrup KD. High-affinity CD25-binding IL-2 mutants potently stimulate persistent T cell growth. Biochemistry. 2005;44:10696–701. doi: 10.1021/bi050436x. [DOI] [PubMed] [Google Scholar]
- 37.Shpilman M, et al. Development and characterization of high affinity leptins and leptin antagonists. J Biol Chem. 2011;286:4429–42. doi: 10.1074/jbc.M110.196402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kariolis MS, et al. An engineered Axl ‘decoy receptor’ effectively silences the Gas6-Axl signaling axis. Nat Chem Biol. 2014;10:977–83. doi: 10.1038/nchembio.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weiskopf K, et al. Engineered SIRPα variants as immunotherapeutic adjuvants to anticancer antibodies. Science. 2013;341:88–91. doi: 10.1126/science.1238856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weiskopf K, et al. Improving macrophage responses to therapeutic antibodies by molecular engineering of SIRPα variants. Oncoimmunology. 2013;2:e25773. doi: 10.4161/onci.25773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tasumi S, et al. High-affinity lamprey VLRA and VLRB monoclonal antibodies. Proc Natl Acad Sci U S A. 2009;106:12891–6. doi: 10.1073/pnas.0904443106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Walker LM, Bowley DR, Burton DR. Efficient recovery of high-affinity antibodies from a single-chain Fab yeast display library. J Mol Biol. 2009;389:365–75. doi: 10.1016/j.jmb.2009.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shembekar N, et al. Isolation of a high affinity neutralizing monoclonal antibody against 2009 pandemic H1N1 virus that binds at the ‘Sa’ antigenic site. PLoS One. 2013;8:e55516. doi: 10.1371/journal.pone.0055516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wozniak-Knopp G, et al. Introducing antigen-binding sites in structural loops of immunoglobulin constant domains: Fc fragments with engineered HER2/neu-binding sites and antibody properties. Protein Eng Des Sel. 2010;23:289–97. doi: 10.1093/protein/gzq005. [DOI] [PubMed] [Google Scholar]
- 45.Rajpal A, et al. A general method for greatly improving the affinity of antibodies by using combinatorial libraries. Proc Natl Acad Sci U S A. 2005;102:8466–71. doi: 10.1073/pnas.0503543102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Boder ET, Raeeszadeh-Sarmazdeh M, Price JV. Engineering antibodies by yeast display. Arch Biochem Biophys. 2012;526:99–106. doi: 10.1016/j.abb.2012.03.009. [DOI] [PubMed] [Google Scholar]
- 47.Moore SJ, Cochran JR. Engineering knottins as novel binding agents. Methods Enzymol. 2012;503:223–51. doi: 10.1016/B978-0-12-396962-0.00009-4. [DOI] [PubMed] [Google Scholar]
- 48.Gera N, Hussain M, Rao BM. Protein selection using yeast surface display. Methods. 2013;60:15–26. doi: 10.1016/j.ymeth.2012.03.014. [DOI] [PubMed] [Google Scholar]
- 49.Silverman AP, Kariolis MS, Cochran JR. Cystine-knot peptides engineered with specificities for α(IIb)β(3) or α(IIb)β(3) and α(v)β(3) integrins are potent inhibitors of platelet aggregation. J Mol Recognit. 24:127–35. doi: 10.1002/jmr.1036. [DOI] [PubMed] [Google Scholar]
- 50.Kimura RH, Levin AM, Cochran FV, Cochran JR. Engineered cystine knot peptides that bind alphavbeta3, alphavbeta5, and alpha5beta1 integrins with low-nanomolar affinity. Proteins. 2009;77:359–69. doi: 10.1002/prot.22441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Silverman AP, Levin AM, Lahti JL, Cochran JR. Engineered cystine-knot peptides that bind alpha(v)beta(3) integrin with antibody-like affinities. J Mol Biol. 2009;385:1064–75. doi: 10.1016/j.jmb.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moore SJ, Leung CL, Norton HK, Cochran JR. Engineering agatoxin, a cystine-knot peptide from spider venom, as a molecular probe for in vivo tumor imaging. PLoS One. 2013;8:e60498. doi: 10.1371/journal.pone.0060498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kimura RH, et al. Functional mutation of multiple solvent-exposed loops in the Ecballium elaterium trypsin inhibitor-II cystine knot miniprotein. PLoS One. 2011;6:e16112. doi: 10.1371/journal.pone.0016112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Glotzbach B, et al. Combinatorial Optimization of Cystine-Knot Peptides towards High-Affinity Inhibitors of Human Matriptase-1. PLoS One. 2013;8:e76956. doi: 10.1371/journal.pone.0076956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Koide A, Bailey CW, Huang X, Koide S. The fibronectin type III domain as a scaffold for novel binding proteins. J Mol Biol. 1998;284:1141–51. doi: 10.1006/jmbi.1998.2238. [DOI] [PubMed] [Google Scholar]
- 56.Lipovsek D. Adnectins: engineered target-binding protein therapeutics. Protein Eng Des Sel. 2011;24:3–9. doi: 10.1093/protein/gzq097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bloom L, Calabro V. FN3: a new protein scaffold reaches the clinic. Drug Discov Today. 2009;14:949–55. doi: 10.1016/j.drudis.2009.06.007. [DOI] [PubMed] [Google Scholar]
- 58.Pavoor TV, Cho YK, Shusta EV. Development of GFP-based biosensors possessing the binding properties of antibodies. Proc Natl Acad Sci U S A. 2009;106:11895–900. doi: 10.1073/pnas.0902828106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lee CH, et al. Engineering of a human kringle domain into agonistic and antagonistic binding proteins functioning in vitro and in vivo. Proc Natl Acad Sci U S A. 2010;107:9567–71. doi: 10.1073/pnas.1001541107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gera N, Hussain M, Wright RC, Rao BM. Highly stable binding proteins derived from the hyperthermophilic Sso7d scaffold. J Mol Biol. 2011;409:601–16. doi: 10.1016/j.jmb.2011.04.020. [DOI] [PubMed] [Google Scholar]
- 61.Puri V, Streaker E, Prabakaran P, Zhu Z, Dimitrov DS. Highly efficient selection of epitope specific antibody through competitive yeast display library sorting. MAbs. 5:533–9. doi: 10.4161/mabs.25211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shusta EV, Kieke MC, Parke E, Kranz DM, Wittrup KD. Yeast polypeptide fusion surface display levels predict thermal stability and soluble secretion efficiency. J Mol Biol. 1999;292:949–56. doi: 10.1006/jmbi.1999.3130. [DOI] [PubMed] [Google Scholar]
- 63.Kowalski JM, Parekh RN, Wittrup KD. Secretion efficiency in Saccharomyces cerevisiae of bovine pancreatic trypsin inhibitor mutants lacking disulfide bonds is correlated with thermodynamic stability. Biochemistry. 1998;37:1264–73. doi: 10.1021/bi9722397. [DOI] [PubMed] [Google Scholar]
- 64.Kowalski JM, Parekh RN, Mao J, Wittrup KD. Protein folding stability can determine the efficiency of escape from endoplasmic reticulum quality control. J Biol Chem. 1998;273:19453–8. doi: 10.1074/jbc.273.31.19453. [DOI] [PubMed] [Google Scholar]
- 65.Shusta EV, Holler PD, Kieke MC, Kranz DM, Wittrup KD. Directed evolution of a stable scaffold for T-cell receptor engineering. Nat Biotechnol. 2000;18:754–9. doi: 10.1038/77325. [DOI] [PubMed] [Google Scholar]
- 66.Kim YS, Bhandari R, Cochran JR, Kuriyan J, Wittrup KD. Directed evolution of the epidermal growth factor receptor extracellular domain for expression in yeast. Proteins. 2006;62:1026–35. doi: 10.1002/prot.20618. [DOI] [PubMed] [Google Scholar]
- 67.Esteban O, Zhao H. Directed evolution of soluble single-chain human class II MHC molecules. J Mol Biol. 2004;340:81–95. doi: 10.1016/j.jmb.2004.04.054. [DOI] [PubMed] [Google Scholar]
- 68.Traxlmayr MW, Obinger C. Directed evolution of proteins for increased stability and expression using yeast display. Arch Biochem Biophys. 2012;526:174–80. doi: 10.1016/j.abb.2012.04.022. [DOI] [PubMed] [Google Scholar]
- 69.Ellgaard L, Helenius A. Quality control in the endoplasmic reticulum. Nat Rev Mol Cell Biol. 2003;4:181–91. doi: 10.1038/nrm1052. [DOI] [PubMed] [Google Scholar]
- 70.Sitia R, Braakman I. Quality control in the endoplasmic reticulum protein factory. Nature. 2003;426:891–4. doi: 10.1038/nature02262. [DOI] [PubMed] [Google Scholar]
- 71.Park S, et al. Limitations of yeast surface display in engineering proteins of high thermostability. Protein Eng Des Sel. 2006;19:211–7. doi: 10.1093/protein/gzl003. [DOI] [PubMed] [Google Scholar]
- 72.Pavoor TV, Wheasler JA, Kamat V, Shusta EV. An enhanced approach for engineering thermally stable proteins using yeast display. Protein Eng Des Sel. 2012;25:625–30. doi: 10.1093/protein/gzs041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Traxlmayr MW, et al. Directed evolution of stabilized IgG1-Fc scaffolds by application of strong heat shock to libraries displayed on yeast. Biochim Biophys Acta. 2012;1824:542–9. doi: 10.1016/j.bbapap.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Traxlmayr MW, et al. Directed evolution of Her2/neu-binding IgG1-Fc for improved stability and resistance to aggregation by using yeast surface display. Protein Eng Des Sel. 2013;26:255–65. doi: 10.1093/protein/gzs102. [DOI] [PubMed] [Google Scholar]
- 75.Traxlmayr MW, et al. Directed evolution of stabilized IgG1-Fc scaffolds by application of strong heat shock to libraries displayed on yeast. Biochim Biophys Acta - Proteins Proteomics. 2012;1824:542–549. doi: 10.1016/j.bbapap.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jones DS, Tsai PC, Cochran JR. Engineering hepatocyte growth factor fragments with high stability and activity as Met receptor agonists and antagonists. Proc Natl Acad Sci U S A. 2011;108:13035–40. doi: 10.1073/pnas.1102561108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Schweickhardt RL, Jiang X, Garone LM, Brondyk WH. Structure-expression relationship of tumor necrosis factor receptor mutants that increase expression. J Biol Chem. 2003;278:28961–7. doi: 10.1074/jbc.M212019200. [DOI] [PubMed] [Google Scholar]
- 78.Buonpane RA, Moza B, Sundberg EJ, Kranz DM. Characterization of T cell receptors engineered for high affinity against toxic shock syndrome toxin-1. J Mol Biol. 2005;353:308–21. doi: 10.1016/j.jmb.2005.08.041. [DOI] [PubMed] [Google Scholar]
- 79.Jones LL, et al. Engineering and characterization of a stabilized alpha1/alpha2 module of the class I major histocompatibility complex product Ld. J Biol Chem. 2006;281:25734–44. doi: 10.1074/jbc.M604343200. [DOI] [PubMed] [Google Scholar]
- 80.Weber KS, Donermeyer DL, Allen PM, Kranz DM. Class II-restricted T cell receptor engineered in vitro for higher affinity retains peptide specificity and function. Proc Natl Acad Sci U S A. 2005;102:19033–8. doi: 10.1073/pnas.0507554102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Henke E, Bornscheuer UT. Directed evolution of an esterase from Pseudomonas fluorescens. Random mutagenesis by error-prone PCR or a mutator strain and identification of mutants showing enhanced enantioselectivity by a resorufin-based fluorescence assay. Biol Chem. 380:1029–33. doi: 10.1515/BC.1999.128. [DOI] [PubMed] [Google Scholar]
- 82.Sroga GE, Dordick JS. Generation of a broad esterolytic subtilisin using combined molecular evolution and periplasmic expression. Protein Eng. 2001;14:929–37. doi: 10.1093/protein/14.11.929. [DOI] [PubMed] [Google Scholar]
- 83.Stevenson BJ, Yip SHC, Ollis DL. In vitro directed evolution of enzymes expressed by E. coli in microtiter plates. Methods Mol Biol. 2013;978:237–49. doi: 10.1007/978-1-62703-293-3_18. [DOI] [PubMed] [Google Scholar]
- 84.Tawfik DS, Griffiths AD. Man-made cell-like compartments for molecular evolution. Nat Biotechnol. 1998;16:652–6. doi: 10.1038/nbt0798-652. [DOI] [PubMed] [Google Scholar]
- 85.Griffiths AD, Tawfik DS. Directed evolution of an extremely fast phosphotriesterase by in vitro compartmentalization. EMBO J. 2003;22:24–35. doi: 10.1093/emboj/cdg014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lipovsek D, et al. Selection of horseradish peroxidase variants with enhanced enantioselectivity by yeast surface display. Chem Biol. 2007;14:1176–85. doi: 10.1016/j.chembiol.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 87.Antipov E, Cho AE, Wittrup KD, Klibanov AM. Highly L and D enantioselective variants of horseradish peroxidase discovered by an ultrahigh-throughput selection method. Proc Natl Acad Sci U S A. 2008;105:17694–9. doi: 10.1073/pnas.0809851105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chen I, Dorr BM, Liu DR. A general strategy for the evolution of bond-forming enzymes using yeast display. Proc Natl Acad Sci U S A. 2011;108:11399–404. doi: 10.1073/pnas.1101046108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fushimi T, et al. Mutant firefly luciferases with improved specific activity and dATP discrimination constructed by yeast cell surface engineering. Appl Microbiol Biotechnol. 2013;97:4003–11. doi: 10.1007/s00253-012-4467-4. [DOI] [PubMed] [Google Scholar]
- 90.Han S, Zhang J, Han Z, Zheng S, Lin Y. Combination of site-directed mutagenesis and yeast surface display enhances Rhizomucor miehei lipase esterification activity in organic solvent. Biotechnol Lett. 2011;33:2431–8. doi: 10.1007/s10529-011-0705-6. [DOI] [PubMed] [Google Scholar]
- 91.Zhang K, et al. Engineering the substrate specificity of the DhbE adenylation domain by yeast cell surface display. Chem Biol. 2013;20:92–101. doi: 10.1016/j.chembiol.2012.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.White KA, Zegelbone PM. Directed Evolution of a Probe Ligase with Activity in the Secretory Pathway and Application to Imaging Intercellular Protein-Protein Interactions. Biochemistry. 2013 doi: 10.1021/bi400268m. [DOI] [PubMed] [Google Scholar]
- 93.Yi L, et al. Engineering of TEV protease variants by yeast ER sequestration screening (YESS) of combinatorial libraries. Proc Natl Acad Sci U S A. 2013;110:7229–34. doi: 10.1073/pnas.1215994110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Steffens DL, Williams JGK. Efficient site-directed saturation mutagenesis using degenerate oligonucleotides. J Biomol Tech. 2007;18:147–9. [PMC free article] [PubMed] [Google Scholar]
- 95.Cochran JR, Kim YS, Olsen MJ, Bhandari R, Wittrup KD. Domain-level antibody epitope mapping through yeast surface display of epidermal growth factor receptor fragments. J Immunol Methods. 2004;287:147–58. doi: 10.1016/j.jim.2004.01.024. [DOI] [PubMed] [Google Scholar]
- 96.Chao G, Cochran JR, Wittrup KD. Fine epitope mapping of anti-epidermal growth factor receptor antibodies through random mutagenesis and yeast surface display. J Mol Biol. 2004;342:539–50. doi: 10.1016/j.jmb.2004.07.053. [DOI] [PubMed] [Google Scholar]
- 97.Boersma YL, Chao G, Steiner D, Wittrup KD, Plückthun A. Bispecific designed ankyrin repeat proteins (DARPins) targeting epidermal growth factor receptor inhibit A431 cell proliferation and receptor recycling. J Biol Chem. 2011;286:41273–85. doi: 10.1074/jbc.M111.293266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Han T, et al. Fine epitope mapping of monoclonal antibodies against hemagglutinin of a highly pathogenic H5N1 influenza virus using yeast surface display. Biochem Biophys Res Commun. 2011;409:253–9. doi: 10.1016/j.bbrc.2011.04.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mata-Fink J, et al. Rapid conformational epitope mapping of anti-gp120 antibodies with a designed mutant panel displayed on yeast. J Mol Biol. 2013;425:444–56. doi: 10.1016/j.jmb.2012.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Pepper LR, Cho YK, Boder ET, Shusta EV. A decade of yeast surface display technology: where are we now? Comb Chem High Throughput Screen. 2008;11:127–34. doi: 10.2174/138620708783744516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gai SA, Wittrup KD. Yeast surface display for protein engineering and characterization. Curr Opin Struct Biol. 2007;17:467–73. doi: 10.1016/j.sbi.2007.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bidlingmaier S, Liu B. Construction and application of a yeast surface-displayed human cDNA library to identify post-translational modification-dependent protein-protein interactions. Mol Cell Proteomics. 2006;5:533–40. doi: 10.1074/mcp.M500309-MCP200. [DOI] [PubMed] [Google Scholar]
- 103.Bidlingmaier S, et al. Identification of MCAM/CD146 as the target antigen of a human monoclonal antibody that recognizes both epithelioid and sarcomatoid types of mesothelioma. Cancer Res. 2009;69:1570–7. doi: 10.1158/0008-5472.CAN-08-1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bidlingmaier S, Liu B. Interrogating yeast surface-displayed human proteome to identify small molecule-binding proteins. Mol Cell Proteomics. 2007;6:2012–20. doi: 10.1074/mcp.M700223-MCP200. [DOI] [PubMed] [Google Scholar]
- 105.Kondo A, Tanaka T, Hasunuma T, Ogino C. Applications of yeast cell-surface display in bio-refinery. Recent Pat Biotechnol. 2010;4:226–34. doi: 10.2174/187220810793611509. [DOI] [PubMed] [Google Scholar]
- 106.Tanaka T, Yamada R, Ogino C, Kondo A. Recent developments in yeast cell surface display toward extended applications in biotechnology. Appl Microbiol Biotechnol. 2012;95:577–91. doi: 10.1007/s00253-012-4175-0. [DOI] [PubMed] [Google Scholar]
- 107.Fujita Y, et al. Direct and efficient production of ethanol from cellulosic material with a yeast strain displaying cellulolytic enzymes. Appl Environ Microbiol. 2002;68:5136–41. doi: 10.1128/AEM.68.10.5136-5141.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Katahira S, Mizuike A, Fukuda H, Kondo A. Ethanol fermentation from lignocellulosic hydrolysate by a recombinant xylose- and cellooligosaccharide-assimilating yeast strain. Appl Microbiol Biotechnol. 2006;72:1136–43. doi: 10.1007/s00253-006-0402-x. [DOI] [PubMed] [Google Scholar]
- 109.Shigechi H, et al. Direct production of ethanol from raw corn starch via fermentation by use of a novel surface-engineered yeast strain codisplaying glucoamylase and alpha-amylase. Appl Environ Microbiol. 2004;70:5037–40. doi: 10.1128/AEM.70.8.5037-5040.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Tsai SL, DaSilva NA, Chen W. Functional display of complex cellulosomes on the yeast surface via adaptive assembly. ACS Synth Biol. 2013;2:14–21. doi: 10.1021/sb300047u. [DOI] [PubMed] [Google Scholar]
- 111.Kim S, Baek SH, Lee K, Hahn JS. Cellulosic ethanol production using a yeast consortium displaying a minicellulosome and β-glucosidase. Microb Cell Fact. 2013;12:14. doi: 10.1186/1475-2859-12-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Tsai SL, Oh J, Singh S, Chen R, Chen W. Functional assembly of minicellulosomes on the Saccharomyces cerevisiae cell surface for cellulose hydrolysis and ethanol production. Appl Environ Microbiol. 2009;75:6087–93. doi: 10.1128/AEM.01538-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Tsai SL, Goyal G, Chen W. Surface display of a functional minicellulosome by intracellular complementation using a synthetic yeast consortium and its application to cellulose hydrolysis and ethanol production. Appl Environ Microbiol. 2010;76:7514–20. doi: 10.1128/AEM.01777-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Matsumoto T, Fukuda H, Ueda M, Tanaka A, Kondo A. Construction of yeast strains with high cell surface lipase activity by using novel display systems based on the Flo1p flocculation functional domain. Appl Environ Microbiol. 2002;68:4517–22. doi: 10.1128/AEM.68.9.4517-4522.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Pan XX, et al. Efficient display of active Geotrichum sp lipase on Pichia pastoris cell wall and its application as a whole-cell biocatalyst to enrich EPA and DHA in fish oil. J Agric Food Chem. 2012;60:9673–9. doi: 10.1021/jf301827y. [DOI] [PubMed] [Google Scholar]
- 116.Kim S, Oh DB, Kwon O, Kang HA. Construction of an in vitro trans-sialylation system: surface display of Corynebacterium diphtheriae sialidase on Saccharomyces cerevisiae. Appl Microbiol Biotechnol. 2010;88:893–903. doi: 10.1007/s00253-010-2812-z. [DOI] [PubMed] [Google Scholar]
- 117.Wang H, et al. Yeast surface displaying glucose oxidase as whole-cell biocatalyst: construction, characterization, and its electrochemical glucose sensing application. Anal Chem. 2013;85:6107–12. doi: 10.1021/ac400979r. [DOI] [PubMed] [Google Scholar]
- 118.Ren R, et al. Display of adenoregulin with a novel Pichia pastoris cell surface display system. Mol Biotechnol. 2007;35:103–8. doi: 10.1007/BF02686102. [DOI] [PubMed] [Google Scholar]
- 119.Jo JH, Im EM, Kim SH, Lee HH. Surface display of human lactoferrin using a glycosylphosphatidylinositol-anchored protein of Saccharomyces cerevisiae in Pichia pastoris. Biotechnol Lett. 2011;33:1113–20. doi: 10.1007/s10529-011-0536-5. [DOI] [PubMed] [Google Scholar]
- 120.Shibasaki S, et al. An oral vaccine against candidiasis generated by a yeast molecular display system. Pathog Dis. 2013;69:262–8. doi: 10.1111/2049-632X.12068. [DOI] [PubMed] [Google Scholar]
- 121.Tamaru Y, et al. Application of the arming system for the expression of the 380R antigen from red sea bream iridovirus (RSIV) on the surface of yeast cells: a first step for the development of an oral vaccine. Biotechnol Prog. 22:949–53. doi: 10.1021/bp060130x. [DOI] [PubMed] [Google Scholar]
- 122.Wasilenko JL, Sarmento L, Spatz S, Pantin-Jackwood M. Cell surface display of highly pathogenic avian influenza virus hemagglutinin on the surface of Pichia pastoris cells using alpha-agglutinin for production of oral vaccines. Biotechnol Prog. 26:542–7. doi: 10.1002/btpr.343. [DOI] [PubMed] [Google Scholar]
- 123.Kotrba P, Ruml T. Surface display of metal fixation motifs of bacterial P1-type ATPases specifically promotes biosorption of Pb(2+) by Saccharomyces cerevisiae. Appl Environ Microbiol. 2010;76:2615–22. doi: 10.1128/AEM.01463-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kuroda K, Shibasaki S, Ueda M, Tanaka A. Cell surface-engineered yeast displaying a histidine oligopeptide (hexa-His) has enhanced adsorption of and tolerance to heavy metal ions. Appl Microbiol Biotechnol. 2001;57:697–701. doi: 10.1007/s002530100813. [DOI] [PubMed] [Google Scholar]
- 125.Kuroda K, Ueda M. Bioadsorption of cadmium ion by cell surface-engineered yeasts displaying metallothionein and hexa-His. Appl Microbiol Biotechnol. 2003;63:182–6. doi: 10.1007/s00253-003-1399-z. [DOI] [PubMed] [Google Scholar]
- 126.Kuroda K, Ueda M, Shibasaki S, Tanaka A. Cell surface-engineered yeast with ability to bind, and self-aggregate in response to, copper ion. Appl Microbiol Biotechnol. 2002;59:259–64. doi: 10.1007/s00253-002-1014-8. [DOI] [PubMed] [Google Scholar]
- 127.Kuroda K, Nishitani T, Ueda M. Specific adsorption of tungstate by cell surface display of the newly designed ModE mutant. Appl Microbiol Biotechnol. 2012;96:153–9. doi: 10.1007/s00253-012-4069-1. [DOI] [PubMed] [Google Scholar]
- 128.Kuroda K, Ueda M. Effective display of metallothionein tandem repeats on the bioadsorption of cadmium ion. Appl Microbiol Biotechnol. 2006;70:458–63. doi: 10.1007/s00253-005-0093-8. [DOI] [PubMed] [Google Scholar]
- 129.Nishitani T, Shimada M, Kuroda K, Ueda M. Molecular design of yeast cell surface for adsorption and recovery of molybdenum, one of rare metals. Appl Microbiol Biotechnol. 2010;86:641–8. doi: 10.1007/s00253-009-2304-1. [DOI] [PubMed] [Google Scholar]
- 130.Midelfort KS, et al. Substantial energetic improvement with minimal structural perturbation in a high affinity mutant antibody. J Mol Biol. 2004;343:685–701. doi: 10.1016/j.jmb.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 131.Pettersen EF, et al. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–12. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]