

Abstract
dictyBase is the model organism database for the social amoeba Dictyostelium discoideum and related species. The primary mission of dictyBase is to provide the biomedical research community with well-integrated high quality data, and tools that enable original research. Data presented at dictyBase is obtained from sequencing centers, groups performing high throughput experiments such as large-scale mutagenesis studies, and RNAseq data, as well as a growing number of manually added functional gene annotations from the published literature, including Gene Ontology, strain, and phenotype annotations. Through the Dicty Stock Center we provide the community with an impressive amount of annotated strains and plasmids. Recently dictyBase accomplished a major overhaul to adapt an outdated infrastructure to the current technological advances, thus facilitating the implementation of innovative tools and comparative genomics. It also provides new strategies for high quality annotations that enable bench researchers to benefit from the rapidly increasing volume of available data. dictyBase is highly responsive to its users needs, building a successful relationship that capitalizes on the vast efforts of the Dictyostelium research community. dictyBase has become the trusted data resource for Dictyostelium investigators, other investigators or organizations seeking information about Dictyostelium, as well as educators who use this model system.
Keywords: Dictyostelium, annotations, phenotypes, Gene Ontology, relational database, Chado
Introduction
The soil amoeba Dictyostelium discoideum is a unicellular eukaryote that obtains nutrients through phagocytosis of bacteria. When challenged by starvation, Dictyostelium has the remarkable ability to form a multicellular structure. Up to 100,000 cells signal each other by releasing cyclic AMP to form a mound that is surrounded by an extracellular matrix. After mound formation, the cells further differentiate into prestalk and prespore cells, sort out, and produce a fruiting body with approximately 80,000 spores suspended on a multicellular stalk. The life cycle is completed when the spores find an environment favorable for germination, re-entering the vegetative phase of the cycle (Gaudet et al., 2008a). A worldwide community of researchers use this protozoan model organism to study a range of biological processes, including chemotaxis, phagocytosis, signal transduction, cellular differentiation, evolution, and disease (Müller-Taubenberger et al., 2013). Dictyostelium occupies a unique position in evolution as one of the earliest branches to emerge after the plant and animal split, making Dictyostelium especially valuable for comparative genomics studies (Eichinger et al., 2005), (Schaap et al., 2006). It also serves as an inexpensive model organism for teaching the next generation of biomedical researchers.
Established in 2003, dictyBase (http://www.dictybase.org) is the central repository of genome sequence data and the most comprehensive, current, and highly curated database for Dictyostelium discoideum available online. dictyBase also provides the web infrastructure supporting the Dicty Stock Center (DSC), a repository for mutant strains and plasmid vectors (Fey et al., 2013). dictyBase integrates genomic data with published research and provides research tools to analyze and retrieve data. Nearly 13,000 genes have been identified in the D. discoideum genome assembly, which serve as the central focus of dictyBase. Each gene has an individual gene page consisting of all relevant data and information pertaining to the gene and, when available, protein sequence, function, orthologs, phenotypes, literature references, and gene ontology terms. Automated processes were initially used to assign gene function, but experienced curators refine these preliminary assignments by examining experimental results from the literature. Most recently, curators have completed a manual review and, where appropriate, modification of all D. discoideum gene models (see sections 1 and 2). dictyBase also maintains a forum for collaboration within the research community, including a mailing list, a colleague database, and shared resources through the DSC. By organizing Dictyostelium research data, providing expert annotation and assuring accessibility, dictyBase maximizes the value of research to the investigator community while also facilitating future studies.
During the past year, we accomplished a comprehensive overhaul of dictyBase to enable us to accommodate the rapidly growing amount of data and provide additional analysis tools. This creates a collaborative environment for community contributions with further integration of the DSC, all in a modern web interface.
Complete Gene Model Curation of Protein Coding Genes
In 2011, dictyBase finished a first-pass gene model curation reviewing all genomic sequence. This consisted of the manual inspection and annotation of all 13,541 predicted genes (Eichinger et al., 2005). As a result, the number of protein coding genes in the Dictyostelium AX4 genomic sequence has been revised to 12,257. Researchers use dictyBase gene models to identify and clone genes, and correct gene models are essential for accurate analysis of high throughput methods such as mass spectrometry and RNA sequencing experiments. Manual curation of the D. discoideum genome will greatly facilitate the identification and automated annotation of genes in related amoebozoa using D. discoideum as the “reference genome” for other Dictyosteliids.
Gene Curation Practice
dictyBase curators completed the manual review of all gene predictions that have at least one source of supporting data, to produce what we refer to as the “Curated Model”. Supportive data include sequences submitted to GenBank by researchers, ESTs, sequence similarity with other proteins (BLAST results), and deep RNAseq coverage. A specially designed curator tool displayed all supporting data allowing curators to approve the gene prediction in a single step when no changes were required. If evidence suggested that intron/exon boundaries and/or 5' and 3' ends needed to be changed, we used an external tool, Apollo (Lewis et al., 2002), to adjust the coordinates on the genome. The curated gene model and the supporting evidence are displayed on the relevant gene page. Fig. 1a shows a curated gene model modified from the originally predicted model, including the evidence supporting the modifications. Curated models tagged as “incomplete support” are those with no end-to-end support but the overall gene structure warranted approval. If a gene model was approved based on a regular structure (i.e. splice donors, acceptors, start and stop codons adhering to consensus) but no other evidence was available, it has been labeled as “supported by genomic context”. In some cases the data supporting the gene model was contradictory, these gene models are labeled with “conflicting evidence” (see Fig. 1b for an example evidence annotation).
Fig. 1.

a: A gene split supported by ESTs and RNAseq expression. The light blue gene prediction (DDB0190487) spans what turned out to be two independent genes fah and cypB, reflected by the Curated Models DDB0231609 and DDB0191497, respectively (dark blue). Both curated gene structures are supported by EST TBLASTN alignments (green) and by developmental RNA expression (dark blue) supporting the exons. Note that this image has been taken using our legacy genome browser Gbrowse.
b: Evidence annotation on the gene page. A genome browser snapshot similar to what is shown in a, is depicted on each gene page. Below the snapshot, the evidence for the curated model is listed, and a green check mark indicates that the gene model is curator reviewed. In this example, the curated model (DDB0231609) of the fah gene is supported by EST and RNAseq alignments.
Special Cases
Besides curating gene predictions from the Sequencing Center (Eichinger et al., 2005) we ran a reprediction pipeline using geneid (Blanco et al., 2007) trained on approximately 3,000 Curated Models. This lead to the discovery of over 70 new genes. In addition, approximately 1,000 genes were deleted in the curation process; these were usually small, predicted genes with a non-standard structure and under 300 bp in length, spanning low complexity regions. In more than 500 cases, we found that gene prediction algorithms had either incorrectly fused two or more genes, or created multiple genes when evidence suggested a single gene, which required a “gene split” or a “gene merger”, respectively. This required either creating `new' genes, or mapping merged and thus obsolete gene IDs to the primary ID, allowing successful searches using any ID. Curators also annotated 650 pseudogenes, defined as genes that lack an open reading frame (ORF) but do have a very close protein coding homolog, and the underlying genomic sequence (usually containing one or more frame shifts) could be verified. Furthermore, curators discovered evidence for alternative splicing, adding alternative transcripts to 32 genes. A few hundred genes were inspected but not curated because the gene structure did not allow approval of the gene prediction and sequence support was unavailable. The majority of these genes turned out to be retro-transposable and transposable elements (RTEs, TEs), many of which might be non-protein coding. Finally, curators discovered several additional genes using RNA expression evidence. Please see table 1 for an exact breakdown of gene model annotation numbers.
Table 1.
Gene Model annotation numbers and statistics
| Sequencing Center Gene Predictions | 13,541 |
| Protein Coding Genes | 12,257 |
| Curated Gene Models | 11,987 |
| Curated Protein Coding Genes | 11,337 |
| Curated Pseudogenes | 650 |
| Gene Models Not Curated (but inspected) | 964 (9%)* |
| Deleted Gene Predictions | 1,026** |
| Changed Curated Models | 2,313 (19.3%)*** |
| Curated Genes, “Complete Support” | 7,206 (63.6%) |
| Curated Genes, “Incomplete Support” | 3,676 (32.4%) |
| Curated Genes, “Genomic Context” | 455 (4%) |
| Merged Gene Predictions | 432, yielding 209 curated genes |
| Split Gene Predictions | 101, yielding 208 curated genes |
| Genes with “Artificial Gaps” | 105 |
| Newly Created Genes | 99**** |
| Genes with Alternative Transcripts | 32 |
| Mean Intron Length | 129 bp |
| Genes with ≥ 1 Intron | 7,996 |
Includes 511 characterized RTE and TE genes and 453 other, uncharacterized and unsupported genes
Includes genes that were deleted during gene mergers
Includes annotated pseudogenes
From our own geneid reprediction and available RNA expression
While first-pass curation was finished a few years ago, we currently still make changes to gene structures based on researcher requests, or from newly published experimental evidence. Curators also became aware of nucleotide changes such as deletions or insertions and inaccurate stop codons in the current genome assembly, which corrupted the ORF for more than 100 genes. These genes were temporarily annotated with “artificial gaps” to create the best possible protein sequence. An important goal for the near future is to combine the knowledge we have gained with next generation sequencing (NGS) provided by Dictyostelium researchers to update the genomic sequence. This will allow us to optimize the D. discoideum reference genome annotations.
Functional Annotations
The goal of functional annotation is to provide an overview of each protein coding gene's role in Dictyostelium biology, as well as allowing the mining of annotated data in comparative and large-scale studies. Automated annotation based on sequence similarity was initially attached to as many genes as possible, but the current focus at dictyBase is manual annotation for each gene based on literature curation. The extent of annotation for any given gene depends on the amount of data available. Extensively investigated genes allow comprehensive annotation based on published experimental results including the creation of summary paragraphs while less well studied genes may have annotations predicted solely from sequence similarity to orthologous genes.
Functional annotations are displayed on each gene page, divided into separate sections representing the type of annotation: General Information, Gene Ontology (GO), Strains and their Phenotypes, and Summary Paragraphs; these are all produced during literature curation. Each week, new publications are imported into dictyBase, and curators begin the process of literature curation by matching newly published papers to their associated genes. We then extract information from the manuscript such as: categorical topics, basic annotation (gene name, gene product, etc.), GO terms, strains and phenotypes, and any additional significant research results that are included in free text descriptions and summary paragraphs. Categorical literature topics are high-level biological processes, functions and research aids attached to genes studied in the paper, e.g. chemotaxis, protein interactions, created reagents such as antibodies. These categorical topics are designed to provide a quick overview of the biological content addressed in the paper. Research papers also often provide general annotations for a gene, and curators determine if a new gene name, synonyms, or a gene product name can be extracted from a paper. dictyBase currently contains 8,545 genes that have a manually assigned gene product name. An example of a well-annotated gene can be viewed on this page: http://dictybase.org/gene/aprA.
GO annotations
GO terms are controlled vocabularies that are annotated by multiple biological databases and used by a large research community. Thus, this common platform and widespread use allows meaningful analyses between different organisms and promotes the field of comparative proteomics. dictyBase is an active member of the GO consortium (Gene Ontology Consortium, 2015), and GO annotations are a fundamental part of literature curation at dictyBase. Curators use the common annotation tool, Protein2GO (Huntley et al., 2015) provided by the European Bioinformatics Institute (EBI) to annotate GO. This common tool helps enforce the current GO consortium guidelines. We presently have 62,840 total GO terms associated with dictyBase genes, of which 25,351 have been assigned manually. A primary objective of dictyBase curation is assigning GO terms based on experimental results from published papers. We currently have 6,521, experimental GO terms manually assigned to Dictyostelium genes. GO terms have three components (process, function, and component), and defined evidence types that capture the type of experiment from which the result was obtained. GO terms are useful to the researcher because they reveal which biological processes a gene is involved in, what function the encoded protein plays and the cellular compartment to which it localizes. GO has recently implemented an “annotation extension” field, which allows adding context to the GO annotation, such as physiological conditions or developmental stages, thus providing a more comprehensive annotation.
Strain annotations
Since Dictyostelium is a haploid protozoan, experimental manipulation of genes are generated effectively with relatively small expense and time investment leading to many genetically manipulated strains. Thus, strain annotation is a major component of annotation at dictyBase. A partial list of the annotations we provide for each strain includes: strain descriptor, strain name, parental strain, genotype, genetic modification, mutagenesis method, reference, associated gene(s), strain characteristics, and strain summary. We have devised guidelines for strain descriptors and genotypes, and use controlled vocabularies for genetic modifications, mutagenesis methods, and strain characteristics. dictyBase is also tightly integrated with the Dicty Stock Center (DSC; see section below) which serves as a physical repository for Dictyostelium strains and other materials obtained from community researchers (Fey et al., 2013). In some cases, we receive strain annotations directly from the contributing researcher. This close integration improves the quality of strain annotation at dictyBase, and also highlights the value dictyBase provides, as strains must be correctly annotated in order to be of use to researchers when they are ordering strains from the DSC. dictyBase currently has 6,051 annotated strains, of which 3,127 are associated with a gene; those not linked to a gene are typically wild type strains, or chemical mutants.
Phenotype annotations
At dictyBase, phenotypes are associated with strains, and during literature curation, strains are annotated first followed by curation of their phenotype(s). Curators have developed a phenotype ontology that is pre-composed and consists of qualifiers (properties) such as “aberrant”, “delayed” or “decreased size” plus a GO process term (e.g “aberrant pseudopod formation”) or a Dicty Anatomy term (Gaudet et al., 2008b) (e.g “decreased slug size”). We also have developed both assay and environment controlled vocabularies, which further describe the experimental conditions associated with a phenotype. Phenotype and strain curation is important for functional annotation because it provides insights about a gene product's function in the model organism. Although there is some overlap between phenotypes and GO terms through the latter's use in the phenotype ontology, phenotypes broaden our knowledge with the inclusion of specific Dictyostelium anatomical features, typical assay conditions and a property, qualifying the phenotypic change. Furthermore, genetic interactions can be discerned through phenotypes of multiple mutants that can easily be generated in Dictyostelium (Linkner et al., 2012). There are currently 10,377 annotated phenotypes at dictyBase, and 1,034 genes have associated strains with phenotypes.
Summary Paragraph
As time allows, curators also write free text summaries for well-characterized genes, displayed as a summary paragraph near the bottom of the gene page. These provide an easily readable summary of the research presented in all published papers to present a comprehensive account of the reported role of that gene in Dictyostelium biology. Currently, 696 dictyBase genes have a summary paragraph. Please see table 2 for all current annotation numbers.
Table 2.
Functional annotation statistics as of March 2015
| Genes with manual gene product | 8,545 |
| Total GO annotations | 62,840 |
| Total non IEA GO annotations* | 25,351 |
| Total EXP GO annotations** | 6,521 |
| Genes with any GO annotations*** | 8,167 |
| Total annotated strains | 6,071 |
| Strains associated with genes | 3,147 |
| Total strains available at DSC | 1,970 |
| Total annotated phenotypes | 10,632 |
| Genes having strains with phenotypes | 1,044 |
| Genes with Summary | 698 |
Non-electronic (IEA) annotations contain experimental (EXP) and other manual annotations e.g., based on sequence similarity, or phylogenic ancestry.
Experimental annotations (EXP) are inferred by direct assay (IDA), protein interaction (IPI), genetic interaction (IGI), mutant phenotype (IMP), or expression pattern (IEP).
In at least one of the three aspects.
The Dicty Stock Center (DSC)
In 2009, the DSC moved from Columbia University (New York) to Northwestern University in Chicago. This has allowed the tighter integration of dictyBase and the Stock Center by streamlining the strain collection and improving curation consistency. As of March 2015, the DSC collection has grown to 1,970 strains and more than 750 plasmids. The strain collection is diverse, including natural isolates of different Dictyostelid strains (114 different species), a large collection of axenic strains including null mutants, REMI (restriction enzyme-mediated integration) mutants, labeled strains for cell biological studies, chemical mutants, tester strains for asexual genetic analysis, and bacterial strains serving as Dictyostelium food source. Other materials such as cDNA libraries and several antibodies are also available. Biological stocks can be readily ordered for a small fee via a shopping cart system and by filling out a web form.
As described in section 2, strain annotations from DSC-submissions and those from the literature are overlapping and integrated. When users submit materials, they are asked to complete a form that captures essential details about the strain. This significantly streamlines the strain and plasmid annotation at dictyBase, especially when stains are sent prior to publication. On the other hand, if strains are submitted after publication the form often provides further details important for the potential customer that might not have been present in the publication. The generosity of the Dictyostelium research community makes the DSC a success and many researchers and teachers profit from the center. In the past year, we shipped over 800 strains and 450 plasmids to 12 different countries. The collection of these biological materials in a central repository ensures that they will always be readily available to everyone in the research community.
dictyBase Software Overhaul
dictyBase was founded almost 12 years ago, but since its inception the field of genomics has changed significantly. For example, next generation sequencing technology has hastened the release of additional genomes and their variants, which in turn has resulted in new types of biological data requiring novel data formats and bioinformatics data management tools. These trends have affected Dictyostelid biology as well, necessitating a database overhaul to implement modern infrastructure, software tools, and web interfaces. Several major factors prompted the overhaul process.
-
i)
Our server hardware was built on a 32 bit linux operating system that limited both the amount of memory and CPU resources we could allocate. This not only degraded overall database performance but also hindered our ability to incorporate improvements requiring scaling for more CPU cores and memory.
-
ii)
dictyBase was originally built using an oracle database server (Chisholm et al., 2006). However Oracle is proprietary, and its enterprise design made it impossible to integrate many open source or reusable software modules from GMOD (general model organism database, http://gmod.org). As a result, we had to custom design our software tools to adjust to oracle requirements. However, over time the limited reusability and workflow forced us to spend more time customizing and maintaining oracle specific software than on creating new features for dictyBase users. In addition, oracle requires significant computer resources, and thus became untenable.
-
iii)
dictyBase was designed as a single genome database (D. discoideum AX4) and at the time using oracle was cost-effective and practical. We developed many web interfaces, libraries, curation and software tools that ran on the initial system. However, with the shift to next generation sequencing multiple species and stain genomes have become available, some with multiple assemblies. Representing multiple genomes, including different Dictyostelium strains, highlighted major design limitations in our initial software system: e.g. premeditated hardcoded rules assuming only a single genome. This monolithic design in which changes to the database affected software tools made even simple improvements increasingly difficult. In addition, a limited data model was found to be unsuitable for integrating new data types such as untranslated regions (UTRs), non-coding genome annotations, signaling pathways, or large-scale genome alignments, etc.
These issues led us to conclude that a complete and comprehensive overhaul of dictyBase's software infrastructure was needed. The overhaul has been designed with the clear objective to transform dictyBase into a next generation genomic resource. The specific goals of the overhaul were: (a) data migration from Oracle to PostgreSQL, (b) develop a new architecture and re-build our software stacks, and (c) upgrade our server and hardware infrastructures.
Data Migration
The primary objective was to migrate our existing data from the proprietary Oracle software to the PostgreSQL relational database (http://www.postgresql.org). PostgreSQL is a powerful, open source object-relational database system. Under development for more than 15 years, PostgreSQL has a proven architecture that has earned a strong reputation for reliability, and data integrity. It runs on all major operating systems, including Linux, UNIX, Mac OS X, and Windows. PostgreSQL provides a large repository (http://pgxn.org) of reusable software for integration and a defined framework for writing extensions. Despite being feature rich, PostgreSQL needs minimum resources and has a low administrative overhead compared to Oracle. GMOD (http://gmod.org) provides their chado schema (http://gmod.org/wiki/Introduction_to_Chado) in PostgreSQL, which is extensively supported and maintained by the community. Chado instances of PostgreSQL are used by many established model organism databases (MODs) as their primary database system, evidence of an emerging consensus of opinion regarding its utility.
We have completed our data migration and all existing data from Oracle now reside in our new PostgreSQL chado instance. The entire migration process was designed to be split between export and import. The export process produces human readable flat files in standard exportable format while the import process consumes and loads the flat files into the PostgreSQL instance. We have used the GFF3 format for gene models, assemblies, and sequence alignments, the obo format for ontologies, bibtex for literature, and the GPAD format for gene ontology annotations. Any data in a non-standard format (e.g. phenotype, genotype and strain data) were exported in tab-delimited files. This split approach allowed us to address some of our long-standing core issues. For example, in our Oracle database, the data model does not allow a logical separation of biological splice isoforms or of transcript models generated from different gene prediction pipelines. Instead, we have used the GFF3 data exports to provide a logical separation of these data. In the imported PostgreSQL database we are now able to use versioning for gene models and the sequence ontology (SO) for splice isoforms to provide a clear logical separation. This new separation also allows us to use the same database instance to host different species such as D.discoideum, D.purpureum, and P. pallidum, different D.discoideum strains (e.g. AX2 and AX4), and even two separate assemblies of D.discoideum AX4. Both data model and design are easily scalable and could host more than 100 genomes per instance of database.
New Data Architecture/Software stacks
Our goal for the new architecture and software tools was to avoid a monolithic structure and instead develop a set of reusable software components that can be combined to build up a complete infrastructure. Each component can be used independently and swapped without disturbing the functionality of others. The new components also define a set of contractual rules. As a result, any new piece of software, independently of the programming language, can implement those contracts and continue to work together. The software is separated into a few major components. The first component is a testing tool, Test-Chado (https://github.com/dictyBase/Test-Chado), which provides a programmer interface to test any tool that manipulates data within the chado database schema. The other components described below incorporate Test-Chado to test their functionality.
The second component handles database management and bulk loading of large datasets. The first tool in this group is Chado-Sqitch (https://github.com/dictyBase/Chado-Sqitch), which manages the chado database through version control. It allows us to track every change in the database through a version upgrade or to rollback to any previous version. This permits the seamless addition of any custom dictyBase modification. The second tool is the Modware-Loader (https://github.com/dictyBase/Modware-Loader), responsible for handling the majority of the data migration process. It also includes a general purpose GFF3 loader, in addition to loaders for GPAD, ontology, ontology path, and bibtex. These loaders are reusable, which allows us to bulk load various kinds of bioinformatics datasets, extending well beyond our import process.
The third component is data access software that uses the RESTful principle (http://en.wikipedia.org/wiki/Representational_state_transfer) to expose web services through the HTTP interface. The data access software is built in two pieces: data access specification and the default implementation. The data access specifications (https://github.com/dictyBase/dictyrestspec) describe a set of HTTP resources and the prescribed way to access data using a standard HTTP client including any web browser. The specifications also define the type of data format (json/xml/text/csv etc.) and data structures that are available for each resource. The default software implementation (https://github.com/dictyBase/dictyrest) runs using a standard HTTP server, and connects with the chado PostgreSQL instance of dictyBase. All of our web interfaces then use this server to retrieve and store data from our chado instance.
Infrastructure upgrade
The objective is to upgrade our server hardware with more CPU cores, memory and hard drive space, running a 64 bit baseline centos 6.5 linux server. The software components and PostgreSQL are hosted in separate physical machines. With the 64 bit architecture, each software component will run in its own virtual container utilizing docker (http://www.docker.com/whatisdocker) and will be allowed to address more memory and CPU cores as required by demand. The server with software components generally starts with 4–6 CPU cores, 4–8 GB of RAM and ~40–60 GB of hard drive space. We have started the PostgreSQL database server with 8 CPU cores and 8 GB of RAM configuration. We are monitoring the servers, particularly the database resource usage, and will change the configuration based on demand.
New Web Design
dictyBase has been redesigned with the goal of improving navigation, facilitating the integration of new tools, and modernizing the user interface. The overall structure was built using HTML5 and CSS3 under the open source front-end frameworks of AngularJS and Bootstrap. AngularJS (https://angularjs.org/) embraces and extends HTML, CSS, and Javascript to frame a powerful infrastructure for developing dynamic web apps. Bootstrap provides components for responsive web design, increasing the accessibility of dictyBase to devices ranging from standard Windows and OSX machines to those using IOS and Android systems. Thus, Bootstrap's fluid grid system enables the development of scaffolds that easily adapts to a wide range of screen sizes.
The emphasis of the new web design is to improve the usability of dictyBase. The primary target audience of dictyBase is the dicty research community. Some elements of the new layout are directly inspired by their feedback. However, we also seek to reach scientists familiar with GMOD databases, and any user, such as teachers and students, interested in learning about this model organism.
The new homepage is currently under active development and a design prototype is shown in Fig. 2.The structure of the page is divided into header, main body, and footer. Although the background of both header and footer extends to the size of the browser, the content area will be constrained to a maximum and minimum of 1100 and 320 pixels, respectively, which facilitates the control of the layout on every screen size. The new footer offers a broad map view of the structure of the entire site, facilitating navigation. The header maintains the previous basic structure consisting of the dicty logo, horizontal navigation bar, quick links, and search box. The `guided search box', a frequently used design element, adopts a larger size. Following intuitive conventions, the `quick links' will be available from the top of the search box.
Fig. 2.

The prototype of new dictyBase front page. The AngularJS infrastructure allows displaying content in blocks that allows efficient updates. Some information, such as `Latest Publications', DSC `New Materials', and `Recent Annotations' are updated automatically. At the time of writing, the front page has been designed and is under active development.
The main body of the frontpage has seven main blocks of content: images, papers, conferences, news, quick links, the DSC, and `annotations'. A carousel of images seeks to reinforce the identity of the site by displaying pictures of social amoebae submitted by the community of researchers. `News' and `Meetings' are blocks of information that will be updated frequently with issues concerning the Dicty community and their research interests; `Papers' will be updated automatically on a weekly schedule when new Dictyostelium publications are imported into dictyBase. `Quick links' will provide fast access to popular tools and sections and will be available under the format of widgets, which will be updated, e.g., when new tools become available. Similarly, access to the DSC will be available through the front page, automatically highlighting new available items. Finally, a block `Recent Annotations' will provide our users with links to newly curated publications and the latest comprehensively annotated genes.
Automated Data Processing at dictyBase
As a widely used online resource, the database content of dictyBase is always kept up to date to provide users with the latest information. The update system involves automated execution of software pipelines that run a sequential series of software processes to complete the entire task. These pipelines share a common pattern of harvesting the latest data from the resource followed by updating the content of dictybase and then optionally disseminating the data in a prescribed format. We describe below two pipelines that we have created to automate the workflow of updating ontologies and ontology-based annotations. The annotation refresh is periodic whereas the ontology update is on demand.
GO Annotation update
dictyBase curators use Protein2GO from the EBI to annotate GO terms (Huntley et al., 2015) and by default those annotations reside in EBI's server and are available from QuickGO (http://www.ebi.ac.uk/QuickGO/Gannotation). To receive and process these annotations, we have created a custom pipeline that harvests the latest annotations from the EBI, updates dictyBase and then submits them to the GO consortium. The pipeline is composed of several components. The first component downloads and updates the ontology at dictyBase to keep it in sync with the ontology version at protein2GO. The ontology is downloaded from the NCBO Bioportal (http://bioportal.bioontology.org/), using their RESTful programmer interface (http://data.bioontology.org/documentation). The next step downloads the dictyBase GO annotations from EBI's quickgo webservice (http://www.ebi.ac.uk/QuickGO/WebServices.html), and then loads them into dictyBase. The loading component does some extra processing before uploading the retrieved annotations. This is required as the Protein2GO tool creates protein (sequence) centric annotations based on the UniprotKB protein database. As a result Protein2GO is unable to handle genes that produce identical proteins. In D.discoideum this concerns both genes located on a duplicated stretch on chromosome 2 and 17 actin genes that produce identical proteins. Therefore the loading component of the pipeline adds the missing annotations back to these genes so they are available at dictyBase. The pipeline then exports the complete annotations in GAF format to the GO consortium. As Protein2GO also does not handle or update alternate names and description fields, and since curators update those entries on a regular basis, the export component takes care of incorporating the latest values of those fields in the final GAF file. The final component sends the GAF file to the GO consortium server and finishes the end-to-end pipeline of annotation import, upload and export processes.
Ontology update through Github
In addition to GO annotations, dictyBase curators develop custom ontologies for data curation that cover various aspects of Dictyostelid biology, for example we have ontologies for phenotypes, assays and environmental conditions, and strain characteristics (see also strain and phenotype annotation sections above). Curators also add new ontologies as required when new data become available from researchers. These ontologies are typically edited using the OBO-Edit software (Day-Richter et al., 2007) and first saved locally. In order to update these ontologies in the database, we have created a workflow that leverages github (https://github.com) to streamline the curation process. GitHub is an online Git repository hosting service, which offers a distributed revision control for managing source code. Curators then upload the updated ontology in one of our github repositories (https://github.com/dictyBase/migration-data/tree/master/ontologies) using a graphical desktop application (https://mac.github.com). To update the ontologies in dictyBase, we are using github's developer service, specifically the webhook (https://developer.github.com/webhooks) system. Webhook allows setting up software integration with any part of the github system including the online repositories. We have installed and configured a webhook for our ontology repository that connects to our webhook server at dictyBase through a secured (https) interface. When a curator publishes an update in the ontology repository, github sends an immediate notification to the dictyBase server. The notification includes detailed information, such as the ontology name, the download location, the term changes, the time of update, and the curator who published the update. The server verifies the notification and if successful passes it along to an ontology-loading component, which downloads the updated ontology and updates it in our database. The pipeline is triggered on github notification, meaning it runs only when an ontology update is published by our curator.
New Tool
The updated database also allows us to add new tools to dictyBase, as many of our older tools are outdated or impossible to update. The JBrowse tool described below is publicly available from the dictyBase website.
JBrowse: a Genome Browser
We implemented a new Genome Browser, JBrowse (Skinner et al., 2009):, to replace Gbrowse. JBrowse is a fast, embeddable genome browser built completely with JavaScript and HTML5, which allows flexible data loading. It is especially suitable to represent deep coverage sequencing. Fast and easily scalable it allows users to effectively browse the ever-growing amount of data associated with the Dictyostelium genome.
At the time of writing, we have released the first version of this tool. The new JBrowse tool can be accessed from the Tools menu (Tools > New JBrowse) on our home page or can be reached directly from this link http://dictybase.org/tools/jbrowse (see also Fig. 3). This browser supports two major ways of retrieving data, one is directly from genomic flat files and the other is through a RESTful backend that can connect to a relational chado database. For this first release, the genome annotation data of JBrowse is served through our GFF3 annotation files. However, we are continuing its development and for the next version will switch to a RESTful chado backend.
Fig. 3.
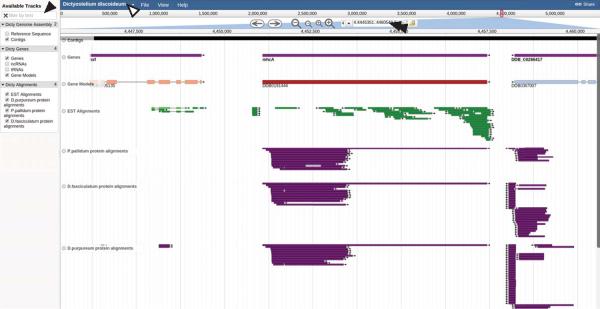
JBrowse, the modern genome browser at dictyBase. The illustration displays a segment of the D. discoideum genome and shows various menus, visual controls and a track pane of JBrowse. The left pane shows the tracks control menu (black single arrow head) and across the top are navigational and zooming controls, followed by the search box (double arrow head). The drop down on top left of the blue bar allows switching to genomes of other species (white arrow head). From top to bottom, the track pane shows the contig (black), gene (purple), canonical gene models (red and blue, for Watson and Crick orientation, respectively), EST (green) and TBLASTN alignments tracks of other species (purple; P. pallidum, D. fasciculatum, and D. pupureum).
Planned Improvements
The tools mentioned below are under development and will be made public in the near future. Here, we briefly describe the rationale of selecting those tools and how they will be integrated in our new environment.
DictyMine, advanced search tool
dictyBase is regularly used by advanced users who would favor custom queries to generate their own customized report. To enable complex searches, in the near future we plan to implement DictyMine, an instance of InterMine (Smith et al., 2012). InterMine consists of two major parts, (a) the warehouse in which a biological database is created from a range of heterogeneous data sources, and (b) the user interface, which includes a flexible query builder to generate customized reports. Reports can be exported in many common formats. InterMine is also strongly integrated with the chado database. All our data types such as genomes, proteomes, literature, strains, genotypes, phenotypes, GO annotations, signaling pathways and post-translational modifications will be integrated into DictyMine.
dictyPPI
Protein-protein interactions (PPIs) are essential for most biological processes (Arkin and Wells, 2004). dictyBase curators annotate PPIs specific for Dictyostelium using the GO term “protein binding” and its children, with the identity of the binding partner in the “with/from” column, while complexes are stored using the various GO “protein complex” terms of the “cellular component” ontology. This information, together with annotations from IntAct (Kerrien et al., 2012), will be captured in Chado as “feature relationships”, connected to the Proteomics Standards Initiative-Molecular Interaction (PSI-MI) ontology. Server side code will send data in graph format compatible with Cytoscape, and the cytoscape.js toolkit (http://cytoscape.github.io/cytoscape.js), an open source JavaScript graph theory library, will be used for visualization. Annotated protein-protein interactions will be available from the protein tab on the gene page.
dictyPTMs
Posttranslational modifications (PTMs) are the chemical modification of proteins after their translation. PTMs are broadly used to regulate protein activity (Jensen, 2004), (Eisenhaber and Eisenhaber, 2010),(Feasley et al., 2013). This mechanism of regulation might increase the molecular variants of cellular proteins by two to three orders of magnitude. Therefore, identifying and understanding PTMs is critical in the study of cell biology, signal transduction, and development. Some general features of PTMs can be studied across species but some are specific to groups of organisms. To facilitate the analysis of the patterns associated with Dictyostelium, dictyBase will be collecting and providing access to PTMs experimentally characterized for Dictyostelid species. dictyBase will store data from a quantitative phosphoproteomics project in Dictyostelium, which resulted in the identification of 3,500 phosphorylated proteins (Dr. John Nichols, personal communication). Curators will also capture PTM data during literature curation. dictyPTMs allows the selection of groups of proteins according to PTMs. Individually, for each protein, PTMs will be visualized with our implementation of the Feature Viewer from BioJS (http://www.ebi.ac.uk/Tools/biojs/).
Conclusion and Outlook
The model organism database for Dictyostelium and related organisms, dictyBase, has been serving the research community as a central resource for over 12 years. The growing information through manual annotations and addition of large-scale datasets, the tight integration of the Dicty Stock Center, and the strong scientific and technical user support have made this an indispensable resource for scientists, teachers, and students world wide.
The ongoing complete update of dictyBase enables us to represent new data much more effectively. In the near future we plan to add protein modification data, protein-protein interactions and an advanced search tool (dictyMine), as well as additional genomes of other Dictyostelid species. These are the first planned additions in our new software environment. The dictyBase update will also allow us to create a guided search tool providing an intuitive interface for simple searches. To make our curators more effective, we plan to use our new HTML5 web interface for direct editing of gene pages and other informative HTML pages, eliminating the need of separate tools for, e.g., phenotype and gene product annotations. Furthermore, direct editing provides an opportunity for inclined users to actively contribute community annotations. For the DSC we intend to create a better customer experience by creating a personal acount with views of the ordering process and shopping history. In addition, we will install Apollo on JBrowse for all genomes we host. This allows both curators and motivated users to edit gene models in those species not yet manually annotated. In sum, the modern database will facilitate the expansion, maintenance, and manual curation of dictyBase and the Dicty Stock Center.
Acknowledgments
Funding: dictyBase and the Dicty Stock Center is supported by grants from the US National Institutes of Health: GM064426, GM087371 and HG0022.
References
- Arkin MR, Wells JA. Small-molecule inhibitors of protein-protein interactions: progressing towards the dream. Nat Rev Drug Discov. 2004;3:301–317. doi: 10.1038/nrd1343. [DOI] [PubMed] [Google Scholar]
- Blanco E, Parra G, Guigó R. Using geneid to identify genes. Curr Protoc Bioinformatics. 2007;Chapter 4(Unit4.3) doi: 10.1002/0471250953.bi0403s18. [DOI] [PubMed] [Google Scholar]
- Chisholm R, Gaudet P, Just E, Pilcher K, Fey P, Merchant S, Kibbe W. dictyBase, the model organism database for Dictyostelium discoideum. Nucleic Acids Res. 2006;34:D423–D427. doi: 10.1093/nar/gkj090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day-Richter J, Harris MA, Haendel M, Gene Ontology OBO-Edit Working Group. Lewis S. OBO-Edit--an ontology editor for biologists. Bioinformatics. 2007;23:2198–2200. doi: 10.1093/bioinformatics/btm112. [DOI] [PubMed] [Google Scholar]
- Eichinger L, Pachebat JA, Glöckner G, Rajandream MA, Sucgang R, Berriman M, Song J, Olsen R, Szafranski K, Xu Q, Tunggal B, Kummerfeld S, Madera M, Konfortov BA, Rivero F, Bankier AT, Lehmann R, Hamlin N, Davies R, Gaudet P, Fey P, Pilcher K, Chen G, Saunders D, Sodergren E, Davis P, Kerhornou A, Nie X, Hall N, Anjard C, Hemphill L, Bason N, Farbrother P, Desany B, Just E, Morio T, Rost R, Churcher C, Cooper J, Haydock S, van Driessche N, Cronin A, Goodhead I, Muzny D, Mourier T, Pain A, Lu M, Harper D, Lindsay R, Hauser H, James K, Quiles M, Madan Babu M, Saito T, Buchrieser C, Wardroper A, Felder M, Thangavelu M, Johnson D, Knights A, Loulseged H, Mungall K, Oliver K, Price C, Quail MA, Urushihara H, Hernandez J, Rabbinowitsch E, Steffen D, Sanders M, Ma J, Kohara Y, Sharp S, Simmonds M, Spiegler S, Tivey A, Sugano S, White B, Walker D, Woodward J, Winckler T, Tanaka Y, Shaulsky G, Schleicher M, Weinstock G, Rosenthal A, Cox EC, Chisholm RL, Gibbs R, Loomis WF, Platzer M, Kay RR, Williams J, Dear PH, Noegel AA, Barrell B, Kuspa A. The genome of the social amoeba Dictyostelium discoideum. Nature. 2005;435:43–57. doi: 10.1038/nature03481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenhaber B, Eisenhaber F. Prediction of posttranslational modification of proteins from their amino acid sequence. Methods Mol. Biol. 2010;609:365–384. doi: 10.1007/978-1-60327-241-4_21. [DOI] [PubMed] [Google Scholar]
- Feasley CL, Hykollari A, Paschinger K, Wilson IBH, West CM. N-glycomic and N-glycoproteomic studies in the social amoebae. Methods Mol. Biol. 2013;983:205–229. doi: 10.1007/978-1-62703-302-2_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fey P, Dodson RJ, Basu S, Chisholm RL. One stop shop for everything Dictyostelium: dictyBase and the Dicty Stock Center in 2012. In: Eichinger L, Rivero F, editors. Methods Mol. Biol. Springer Protocol. Vol. 983. 2013. pp. 59–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudet P, Fey P, Chisholm R. Cold Spring Harb Protoc 2008. 2008a. Multicellular development of dictyostelium. pdb.prot5100. [DOI] [PubMed] [Google Scholar]
- Gaudet P, Williams JG, Fey P, Chisholm RL. An anatomy ontology to represent biological knowledge in Dictyostelium discoideum. BMC Genomics. 2008b;9:130. doi: 10.1186/1471-2164-9-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gene Ontology Consortium Gene Ontology Consortium: going forward. Nucleic Acids Res. 2015;43:D1049–D1056. doi: 10.1093/nar/gku1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntley RP, Sawford T, Mutowo-Meullenet P, Shypitsyna A, Bonilla C, Martin MJ, O'Donovan C. The GOA database: gene Ontology annotation updates for 2015. Nucleic Acids Res. 2015;43:D1057–D1063. doi: 10.1093/nar/gku1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen ON. Modification-specific proteomics: characterization of post-translational modifications by mass spectrometry. Curr Opin Chem Biol. 2004;8:33–41. doi: 10.1016/j.cbpa.2003.12.009. [DOI] [PubMed] [Google Scholar]
- Kerrien S, Aranda B, Breuza L, Bridge A, Broackes-Carter F, Chen C, Duesbury M, Dumousseau M, Feuermann M, Hinz U, Jandrasits C, Jimenez RC, Khadake J, Mahadevan U, Masson P, Pedruzzi I, Pfeiffenberger E, Porras P, Raghunath A, Roechert B, Orchard S, Hermjakob H. The IntAct molecular interaction database in 2012. Nucleic Acids Res. 2012;40:D841–D846. doi: 10.1093/nar/gkr1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis SE, Searle SMJ, Harris N, Gibson M, Lyer V, Richter J, Wiel C, Bayraktaroglir L, Birney E, Crosby MA, Kaminker JS, Matthews BB, Prochnik SE, Smithy CD, Tupy JL, Rubin GM, Misra S, Mungall CJ, Clamp ME. Apollo: a sequence annotation editor. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-12-research0082. RESEARCH0082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linkner J, Nordholz B, Junemann A, Winterhoff M, Faix J. Highly effective removal of floxed Blasticidin S resistance cassettes from Dictyostelium discoideum mutants by extrachromosomal expression of Cre. Eur. J. Cell Biol. 2012;91:156–160. doi: 10.1016/j.ejcb.2011.11.001. [DOI] [PubMed] [Google Scholar]
- Müller-Taubenberger A, Kortholt A, Eichinger L. Simple system--substantial share: the use of Dictyostelium in cell biology and molecular medicine. Eur. J. Cell Biol. 2013;92:45–53. doi: 10.1016/j.ejcb.2012.10.003. [DOI] [PubMed] [Google Scholar]
- Schaap P, Winckler T, Nelson M, Alvarez-Curto E, Elgie B, Hagiwara H, Cavender J, Milano-Curto A, Rozen DE, Dingermann T, Mutzel R, Baldauf SL. Molecular phylogeny and evolution of morphology in the social amoebas. Science (New York, N.Y.) 2006;314:661–663. doi: 10.1126/science.1130670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner ME, Uzilov AV, Stein LD, Mungall CJ, Holmes IH. JBrowse: a next-generation genome browser. Genome Res. 2009;19:1630–1638. doi: 10.1101/gr.094607.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RN, Aleksic J, Butano D, Carr A, Contrino S, Hu F, Lyne M, Lyne R, Kalderimis A, Rutherford K, Stepan R, Sullivan J, Wakeling M, Watkins X, Micklem G. InterMine: a flexible data warehouse system for the integration and analysis of heterogeneous biological data. Bioinformatics. 2012;28:3163–3165. doi: 10.1093/bioinformatics/bts577. [DOI] [PMC free article] [PubMed] [Google Scholar]