

Abstract
Background
The transposable element (TE) content of the genomes of plant species varies from near zero in the genome of Utricularia gibba to more than 80 % in many species. It is not well understood whether this variation in genome composition results from common mechanisms or stochastic variation. The major obstacles to investigating mechanisms of TE evolution have been a lack of comparative genomic data sets and efficient computational methods for measuring differences in TE composition between species. In this study, we describe patterns of TE evolution in 14 species in the flowering plant family Asteraceae and 1 outgroup species in the Calyceraceae to investigate phylogenetic patterns of TE dynamics in this important group of plants.
Results
Our findings indicate that TE families in the Asteraceae exhibit distinct patterns of non-neutral evolution, and that there has been a directional increase in copy number of Gypsy retrotransposons since the origin of the Asteraceae. Specifically, there is marked increase in Gypsy abundance at the origin of the Asteraceae and at the base of the tribe Heliantheae. This latter shift in genome composition has had a significant impact on the diversity and abundance distribution of TEs in a lineage-specific manner.
Conclusions
We show that the TE-driven expansion of plant genomes can be facilitated by just a few TE families, and is likely accompanied by the modification and/or replacement of the TE community. Importantly, large shifts in TE composition may be correlated with major of phylogenetic transitions.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-015-1830-8) contains supplementary material, which is available to authorized users.
Background
A common feature of eukaryotic genomes is that they contain transposable elements (TEs), yet there is a remarkable amount of variation in TE content and composition between species [1, 2]. This property of eukaryotic genomes has parallels with ecological communities [3, 4], which vary in the abundance and diversity of species. While it has been shown that niche differences are an important factor in shaping species diversity [5, 6], it is generally believed that neutral processes can explain the assembly of communities over evolutionary time scales [7]. Given the ubiquitous nature of TEs and their contributions to eukaryotic genome evolution [8, 9], an important question is whether or not similar mechanisms operate to shape the genome landscape.
One possible explanation for the variation in TE content and composition between species is that random processes govern the evolution of TE communities and that chance alone determines the outcome for each TE lineage [10]. However, there is strong evidence that TEs integrate in non-random genomic locations, and TEs may show signs of positive selection [11–14]. It is important to understand the phylogenetic distribution of these patterns because TE activity may, in some cases, correlate with the diversification of their host lineages. For example, species radiations in vertebrates appear to be associated with genome repatterning and TE amplification events [15–17]. In one case, the origin of six species of Taterillus gerbils within the past 0.4 million years has been accompanied by numerous large chromosomal changes and the non-random accumulation of LINE-1 elements, with the most recently diverged species showing the greatest amount of LINE-1 accumulation [18]. Also, waves of TE amplification are associated with the radiation and subsequent speciation of four genera of salmonid fishes [19]. Similarly, massive retrotransposon amplification appears to coincide with speciation events in hybrid sunflower species [20], and non-random patterns of retrotransposon accumulation in the hybrid species’ genomes indicate a potential mechanism for chromosomal divergence between species [21]. Taken together, these results suggest that studying the properties of TE evolution may indicate the timing and nature of important evolutionary transitions. Thus, we are keenly interested in understanding the nature of TEs in the plant family Asteraceae, which harbors unparalleled species diversity in the plant kingdom [22].
The Asteraceae is the largest family of vascular plants, composed of more than 23,600 species, or 8 % of all plant species [22]. The consensus view is that the Asteraceae originated in South America within the past 40–50 million years, which is somewhat surprising given the large number of species in this family [23]. From South America, the Asteraceae spread to Central America and Africa, and the family currently has a worldwide distribution, being found on every continent except Antarctica [24]. There are 12 recognized subfamilies in the Asteraceae, though four of those subfamilies, the Mutisioideae, Carduoideae, Cichorioideae, and Asteroideae, contain 99 % of the species [24]. Within the Asteraceae, there is exceptional diversity in the ecological distribution of species. For example, there are narrow endemics, and also species such as the common sunflower (Helianthus annuus) and dandelion (Taraxacum officinale) that are found widely distributed on multiple continents. Though most species in the Asteraceae are herbaceous, there are also many shrub and tree species [24]. However, this plant family is perhaps best known for the numerous agriculturally important species such as cultivated sunflower, safflower, lettuce, and globe artichoke [25]. Given the recent evolutionary origin of this enormous plant family, as well as its global distribution, the Asteraceae represent an excellent system to study plant adaptation and speciation. However, very little is known about genome evolution and TE diversity in the Asteraceae as a whole (but see [26–29]).
In this study, we seek to understand the major features of Asteraceae genomes, and to explore the mechanistic basis of TE evolution in plants by analyzing the evolutionary history of this plant family in a lineage-specific manner. It is known that there is a major bias in genome composition towards Gypsy DNA in the common sunflower genome [28, 29], but an outstanding question is whether other Asteraceae genomes exhibit similar patterns. That is, are the genomic properties of the common sunflower unique to that lineage? More importantly, what are the mechanisms contributing to TE community structure in plants? We address these questions by generating whole-genome shotgun (WGS) sequence data from 14 species representing 5 different subfamilies in the Asteraceae, along with an outgroup, and analyzing the relative abundance of TEs in each. We use phylogenetic and linear models to investigate whether there have been lineage-specific patterns of TE evolution in the Asteraceae. We also use ecological measures of community diversity, along with simulation-based approaches, to better understand the genomic impact of TE amplification events and how changes in TE abundance influence TE diversity in the genome as a whole. Taken together, these approaches represent a novel approach to study TE properties by employing descriptive statistical approaches along with phylogenetic and ecological models to investigate the mechanisms of genome community assembly.
Results
Transposable element composition in the Asteraceae
Using WGS sequencing data, we determined that Asteraceae genomes are, on average, composed of 69.9 ± 5.3 % TEs (mean ± SD), with 53.2 ± 19.1 % of these genomes being LTR retrotransposons (LTR-RTs; Fig. 1). As expected for plant species, Class II TEs and non-LTR-RTs were lower in abundance relative to LTR-RTs, comprising just 0.60 ± 0.7 % and 0.82 ± 1.1 % of each genome, respectively. The outgroup species Nasanthus patagonicus exhibited comparable patterns of total repeat abundance (62.0 ± 0.1 %) and LTR-RT abundance (47.3 ± 3.3 %) as the Asteraceae, but contained a significantly higher abundance of Class II TEs (2.9 ± 0.1 %; P = 0.02) and a higher, though not signicantly so, abundance of non-LTR-RTs (2.0 ± 0.2 %; P = 0.20). Interestingly, in all but one species, LINE-like sequences are more prevalent (by a factor of at least 2:1) than other non-LTR-RT types. The one species that does not fit this pattern is Fulcaldea stuessyi, a member of the Barnadesioideae (the most basal subfamily of the Asteraceae), which harbors more SINE-like sequences than other non-LTR-RT types. In addition, the N. patagonicus genome contains a significantly higher abundance of endogenous retroviruses (ERVs; 1.2 ± 0.4 %; P = 0.04) than the average Asteraceae genome (0.06 ± 0.09 %), though it is likely that these sequences represent novel LTR-RTs since plant ERV sequences are more closely related to LTR-RTs than to the Retroviridae [30]. Contrasting the widespread nature of the aforementioned TE types, Penelope transposons are characterized by a sparse distribution throughout eukaryotes [31]. Consistent with this finding, Penelope transposons were found in all but two species in the Asteraceae (Fulcaldea stuessyi and Phoebanthus tenuifolius), and ERV-like sequences were absent from four species (F. stuessyi, Conoclinium coelestinum, P. tenuifolius, and H. argophyllus).
Fig. 1.

Genomic contribution of TE superfamilies in the Asteraceae. a Phylogenetic tree of 14 Asteraceae species and one outgroup species derived from 763 nuclear loci (see Methods). Filled circles indicate nodes with >75 % bootstrap support; to the right of the tree are the subfamilies to which each species belongs; the red stars on the branches indicate the timing of whole genome duplication events based on [56]. b Barplot of the genomic composition of TE superfamilies. The x-axis indicates abundance in base pairs for each species, shown along the y-axis. Filled circles indicate the genome size for each species. Superfamilies by order and class: Copia, Gypsy, ERV, and DIRS are LTR-RTs; Helitron is in subclass II of Class II; EnSpm, MuDR, hAT, Mariner/Tc1, and Polinton are TIR Class II TEs; Crypton are unique Class II elements in the order Crypton; L1, L2, and Jockey are LINE non-LTR-RTs; Penelope TEs belong in the unique Penelope order of retrotransposons; R1 are a group of non-LTR-RTs that insert into rDNA genes. The diagonal line through each entry in the barplot legend indicates the border of each TE type in the plot, which is a solid black line
In agreement with previous studies [28, 29], we found a large bias in TE content in the genome of H. annuus, which is composed primarily of Gypsy elements (60.0 ± 3.3 %). This bias appears to be shared by all members of the subfamily Asteroideae, including all species of the genus Helianthus analyzed here (62.4 ± 2.7 %), and the most basal member of the tribe Heliantheae, P. tenuifolius (67.5 ± 5.6 %; Fig. 1). We found a significant linear increase in the genomic proportion of Gypsy LTR-RTs from the base of the Asteraceae to the most derived subfamily, the Asteroideae using a generalized least squares test (r2 = 0.996; P ≤ 2.2e-16; Fig. 2). Copia TEs exhibit an inverse pattern to that of Gypsy, with species at the base of Asteraceae containing proportionally more Copia DNA than those species in the Asteroideae (r2 = 0.915; P = 2.831e-12; Fig. 2). These phylogenetic patterns remained significant when considering only one Helianthus species (H. annuus) in the analysis, indicating that they are not due to the overrepresentation of a single genus.
Fig. 2.

Linear change in genomic composition of LTR-RTs. Shown in phylogenetic order starting with the outgroup (bottom of the y-axis) to the most derived lineages of the Asteraceae in this study (top of the y-axis) are the change in genomic proportion (shown along the x-axis) of A) Gypsy and B) Copia TEs
To further investigate the significance of the patterns, we compared the proportion of TEs at the superfamily and family levels along the phylogenetic tree to what would be expected under a Brownian motion model, and we assessed significance of these results using phylogenetically independent contrasts (PICs). We detected significant (P < 0.05) phylogenetic signal, K, for ten superfamilies of TEs (Additional file 1). Notably, Copia TEs as a whole showed significantly (P < 0.05) more phylogenetic signal (i.e., K ≥ 1) than Gypsy (i.e., K ≤ 1). At the individual TE family level, we found more LTR-RT families exhibiting significant (P < 0.05) phylogenetic signal (7 Copia families, 10 Gypsy families, 1 ERV1 family) than either non-LTR-RTs (3 L1-like families, 3 CR1 families, 1, NeSL family) or Class II TEs (1 hAT family, 2 Mariner/Tc1 family, 1 Helitron family), though the average phylogenetic signal for Class II TE families was much higher (K = 3.26 ± 0) than either LTR-RTs (K = 1.78 ± 1.13) or non-LTR-RTs (K = 3.19 ± 0.16) [see Additional files 2 and 3].
Properties of individual TE family evolution
We investigated the mechanisms of genome community assembly over large time scales by analyzing the rank abundance/dominance (RAD) for all TE families in each species in this study. We considered five ecological models and present the model that best fits the data for each species, as determined by a Bayesian Information Criterion (see Methods). Though numerous species across the Asteraceae exhibit a log-normal-like distribution of TE family abundances (6/15 species), which can be described by even abundances and few rare TE families, it is evident that the predominant pattern is for species to exhibit highly uneven TE family abundances and are thus best fit by a niche-preemption model (7/15 species; Fig. 3). For example, we found that F. stuessyi, a member of the subfamily Barnadesioideae, has a very even distribution of TE families in terms of abundance (0.33 ± 0.52 %), while members of the subfamily Asteroideae have a very uneven distribution (see Fig. 1 for subfamily description), being composed of relatively few highly abundant families and many rare families (0.92 ± 2.4 %). Six species in the Heliantheae show TE family distributions best fit by a straight line (i.e., the niche preemption model; Fig. 3). The dominance of TE families in the Heliantheae is evident when considering that the top 10 TE families in this group account for nearly 2X the genomic proportion (51.5 ± 3.14 %) as the top 10 TE families in the rest of the Asteraceae (26.8 ± 9.10 %).
Fig. 3.
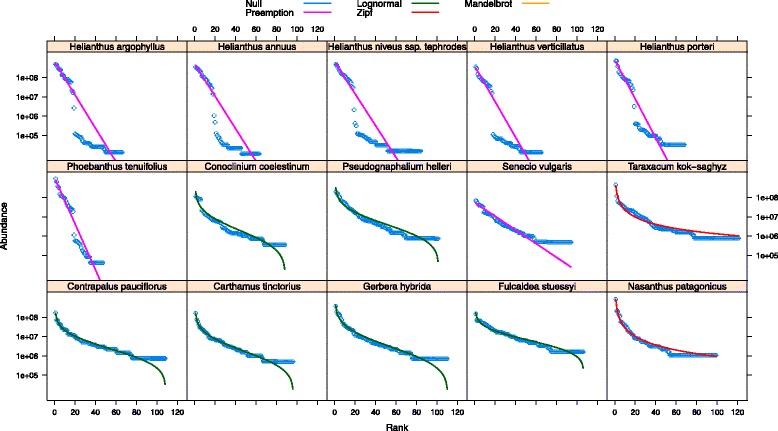
RAD plot of TE family abundance. Species are presented in phylogenetic order starting with the outgroup in the bottom right panel and the moving left to the most derived lineages of the Asteraceae in this study being displayed at the top left. The x-axis depicts the rank order of TEs by abundance, with rank 1 being given to the most abundant family, rank 2 given the second most abundant family, and so on. The y-axis depicts the log abundance of each TE family. Above the plots are the 5 ecological models used to test the fit of observed abundance. The colored line in each panel represents the best-fit model to each distribution as determined by BIC (see Methods)
While the RAD models described above demonstrate global patterns of abundance and dominance of TE families, these plots are unlabeled and do not allow investigation of specific changes in rank abundance. To infer which specific TE families have contributed the most to the rank abundance patterns observed in this study, and in the marked change in rank abundance and dominance within the Heliantheae in particular, we analyzed the rank of TE families sorted by abundance in the Asteraceae as a whole (Fig. 4) as compared to the abundance of TE families within the Heliantheae (Fig. 5). Interestingly, we found no phylogenetic patterns of rank abundance at the TE family level that are shared across the Heliantheae (Fig. 5). At the superfamily level, however, it is clear that at least the four highest-ranking TE families in the each species in the Heliantheae are members of the Gypsy superfamily.
Fig. 4.
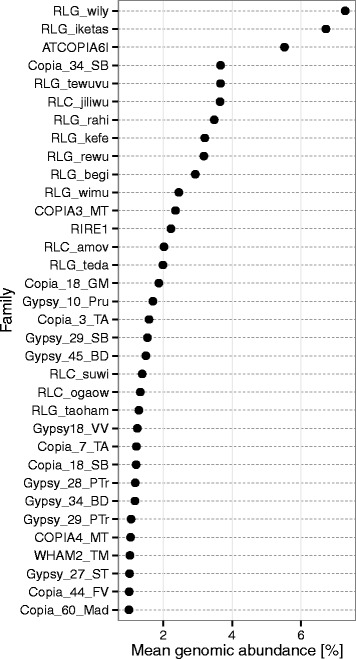
Rank abundance of TE families in the Asteraceae. The y-axis depicts the most abundant TE families in the Asteraceae, listed in decreasing rank abundance from the top the y-axis. The x-axis shows the average percent genomic abundance of each TE family in the Asteraceae
Fig. 5.

Rank abundance of TE families in the Heliantheae. Along the y-axis is the rank abundance of the top 2 % of TE families in the Heliantheae, in decreasing order. Each panel depicts the rank abundance of TE families in phylogenetic order of the tribe from the base of the plot. The x-axis shows the percent genomic abundance of each TE family
Impact of TE family abundance on TE diversity
To investigate the potential impact of changes in TE abundance on patterns of genome community diversity, we estimated the correlation of changes in TE family abundance and TE richness with genome size. As expected for plant species [1, 32, 33], the abundance of retrotransposon DNA is strongly correlated with genome size (r2 = 0.608; P = 6.06e-4; Additional file 4). These patterns were also significant when considering the non-independence of the species with a phylogenetic generalized least squares test (Copia, P = 0.0009; Gypsy, P = <0.0001; Additional file 5). However, while we did find a positive correlation with genome size and TE family size, we did not find such a correlation with genome size and TE richness (Fig. 6). To investigate the impact of genome dominance by some TE families on genome community structure, we also calculated Shannon’s diversity and evenness of TE families for each species in this study (Additional file 6), which may provide more insight into the evolution of genome community patterns than looking at TE richness alone [34]. For example, in addition to the major shift in genome composition at the base of Heliantheae, there also appears to be a reduction in Shannon’s diversity and evenness (Additional file 6). This result is further supported by a marked increase in the average TE family size in the Heliantheae, which is accompanied by a decrease in TE richness (Fig. 7).
Fig. 6.
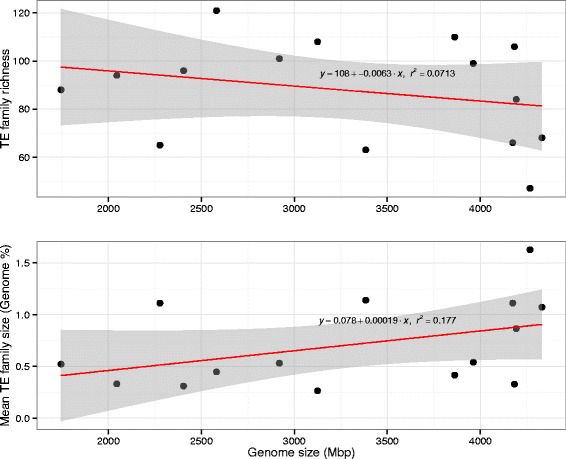
Relationship between genome size and TE family size and richness. Along the x-axis is shown the genome size of each species in mega-base pairs. a The TE richness, or total number of TE families seen, is shown along the y-axis. b The mean TE family size as a percent of the genome is depicted on the y-axis
Fig. 7.

Phylogenetic relationship between TE richness and TE family size. a The TE family richness is shown along the x-axis for each species, which are depicted in phylogenetic order from the outgroup species at the base of the y-axis to the most derived lineages in the Asteraceae at the top of the y-axis. b The mean TE family size as a percentage of the genome is shown along the x-axis. In both panels, the red vertical line indicates the mean and the horizontal dashed black line shows the base of the Heliantheae (with all species in the Heliantheae being shown above the line)
Discussion
It is well known that TEs vary in abundance and type between eukaryotic species. For example, TEs are completely absent from the genomes of some unicellular eukaryotes [36], though >50 % of the human genome is composed of TEs [35]. Similarly, the TE composition of the Saccharomyces cerevisiae genome is 4 % [37], and includes only LTR-RTs, whereas some plant genomes are >80 % TEs e.g., [29, 38–40], including hundreds of families of both Class I and Class II TEs [12]. There is also a disparity with respect to TE copy number and the occurrence of contemporary TE activity. For example, mammalian genomes contain numerous high copy number TE families though only a few recently active TE families have been discovered [41]. Conversely, there are many active TE families in the genomes of fruitfiles and pufferfish, but these families only contain a few copies [42–44]. Given the potential impact of TEs on genome structure and gene expression divergence [45–47] and the apparent variation in TE susceptibility amongst eukaryotes, an understanding of the timescales and phylogenetic patterns over which different classes of TEs are active is of great interest.
Transposable elements and genome content in the Asteraceae
Species in the Asteraceae vary tremendously in the TE composition of their genomes, especially with respect to LTR-RTs (Fig. 1). It is not surprising that the greatest magnitude of change in genome content involves LTR-RTs given that these sequences account for the largest portion of each genome. It is, however, interesting that we see such strong linear patterns of change in genome content at the LTR-RT superfamily level from the base of the Asteraceae to the crown lineages (Fig. 2). In the broad sense, these patterns fit the expectation of zero-sum change for a neutral community, which predicts that an increase in abundance in one member of a community will result in a proportional decrease in the abundance of another [7]. Though TE activity may lead to expansion of the nuclear genome [20, 38, 48], the inverse patterns of change in Gypsy and Copia abundance in the Asteraceae reflects that there are a finite number of insertion sites in the genome, and increases in copy number of one or more TE families may result in the replacement or inactivation of other TE copies.
We detected significant phylogenetic signal for both Class I and Class II TEs at both the superfamily and family level (Additional files 1, 2 and 3), indicating that the genomes of related species are more similar in TE composition and abundance than expected by chance. When considering the variation in genome content between the basal and most derived lineages of the Asteraceae (Fig. 1), this result is expected. However, it seems likely that very different processes contributed to these phylogenetic patterns. For example, the phylogenetic signal seen in Penelope retrotransposons and ERVs may be a product of the sparse distribution of those sequences. The genomic composition of ERVs in N. patagonicus appears high relative to the Asteraceae, though this finding not uncommon for plant species. For example, the genomic percentage of ERVs is 2.4 % in the Amborella genome [49], twice that of N. patagonicus. Alternatively, Gypsy elements are found in all species in the Asteraceae, but there is a clear increase in the abundance of several Gypsy families at the base of Heliantheae, producing a phylogenetic pattern shared by all members of this tribe. The inverse pattern can be seen for the Copia superfamily, which also shows significant phylogenetic signal (Additional file 1), where a linear decrease in these sequences from the Barnadesioideae to the Asteroideae contributes to phylogenetic patterns across the family. The foregoing results indicate that no single evolutionary process can explain these patterns of genome evolution in the Asteraceae. Specifically, species in the basal subfamilies of the Asteraceae are strikingly different in TE composition compared with the crown subfamilies, with those species in the basal subfamilies containing a greater abundance of non-LTR-RTs and DNA transposons. Could the greater TE diversity at the base of the Asteraceae and in the outgroup species be a result of the age of those lineages, or could there be other mechanisms influencing the abundance and diversity of the genome community? While it is not currently possible for us describe the evolutionary events that produced these patterns, ongoing genome sequencing projects in the Asteraceae should enable better descriptions in future studies.
Transposable element families and genome community assembly
Although ecosystems typically vary in terms of their species abundance and diversity, most communities exhibit a very similar distribution in the relative abundance of species [7]. Specifically, most communities exhibit a log-normal-like distribution of species abundance, with few species having high abundance, many rare species with very low abundance, and numerous species lying between these extremes [7]. Interestingly, one prior study has shown that eukaryotic genomes appear to exhibit similar log-normal distributions of genetic elements, suggesting that neutral processes may best explain community assembly over evolutionary timescales, regardless of the system [50]. However, there is some doubt as to whether the log normal model is the best null hypothesis for TE abundance distributions [51]. We tested a range of neutral and niche-based abundance distribution models and asked whether Asteraceae genomes also exhibit a log-normal distribution of TE family abundances, and whether there are shared patterns of TE abundance distributions across the family. While six species in this study exhibit a log normal distribution of TE abundance, a greater number, seven species, exhibit a niche-preemption distrbution, and two species have a TE abundance distribution best fit by the Zipf model, a hierarchical distribution (Fig. 3).
Interestingly, there is a very marked break at the base of Heliantheae with all species in this tribe exhibiting numerous highly abundant TE families and many rare families. This type of distribution has been used to describe communities with poor habitat [52] and/or few species [53], or the early succession of species [54] following disturbance [55]. Typically, these patterns of uneven abundance do not fit neutral expectations [56]. While there are caveats in interpreting ecological models in a genomic context, these results, taken together with other measures of TE abundance presented here, clearly reflect a unique evolutionary history for this tribe.
What biological change facilitated the major genomic transitions in the Heliantheae? It is tempting to speculate that the whole genome duplication event at the base of the Heliantheae [57] may have provided a genomic disturbance which contributed to the biased distribution of TE family abundance in this tribe, or directed integration of Gypsy elements may have contributed to these patterns [11, 29, 58]. Clearly, more work will be required to gain a deeper understanding of the underlying processes. It is clear from this analysis, however, that whole-genome turnover and expansion events have taken place in the lineage leading to the tribe Heliantheae, which arose ca. 26–31 MYA [57, 59].
Mechanisms of change in the genome-wide level of transposable elements
Major transitions in genome content are evident in each subfamily of the Asteraceae (Fig. 1). What is the best mechanistic explanation of the patterns of TE abundance in the Asteraceae? The coexistence of species may be facilitated by niche differentiation [60], and this type of model best explains the TE abundance data we see for species in the tribe Heliantheae. However, the TE abundance and diversity for this group of species indicates a very biased composition towards Gypsy TEs (Figs. 2 and 3). The linear increase in abundance of Gypsy TEs in the Asteraceae has had at least two major influences on the genome community of TEs. First, the correlation we see with TE family size and genome size (Fig. 6) indicates an unequal contribution of TE families to the genome community. Second, it is clear that the linear pattern of increase in Gypsy is driven by only a few TE families (Fig. 3), which has lead to an increase in average family size and a decrease in overall TE richness (Fig. 7). Interestingly, we do not see different superfamilies dominating Helianthus genomes as has been observed in some species of Gossypium [61]. This may indicate that a single event at the base of Heliantheae produced the observed genomic change, and that the patterns we see in each Helianthus species are shared by phylogenetic history rather than being independent events leading to similar patterns in each species. Alternatively, Gypsyelements may have evolved features allowing them to outcompete other TEs or avoid host-silencing mechanisms. Future investigations into these questions will surely lead to a greater understanding of the processes contributing to the high levels of diversity observed within the Asteraceae, and to the processes contributing to the evolution of TE diversity across the plant kingdom as a whole.
Conclusions
The majority view of TE evolution is that these sequences evolve primarily by neutral processes and are therefore likely to generate predictable distributions of relative abundance [50]. We showed, however, that plant species may exhibit uneven distributions of TE family abundance, as exemplified by all members of the Heliantheae investigated herein. Our results indicate that these patterns may be facilitated by: 1) an unequal contribution of certain TE families over time [29, 62]; and 2) nonrandom patterns of TE accumulation across the genome, as has been shown for one species in this study, H. annuus [21, 26, 27]. Aside from species in the tribe Heliantheae, other species in the Asteraceae do exhibit TE abundance distributions that are in line with neutral expectations. This finding may indicate that the factors contributing to the relative abundance TEs vary over time. Based on these results, we believe that the relative abundance of TEs in plant genomes can be best described as a continuum of resource-based patterns (i.e., niche-preemption) to random patterns (i.e., neutral processes). Our finding of major shifts in TE composition at the base of the Asteraceae and at the base of the tribe Heliantheae provides further evidence that TE compositions contain phylogenetic signal [63], and suggests a possible role for TEs in species formation in the Asteraceae.
Methods
Taxon sampling and WGS sequencing
To investigate patterns of genome evolution across the Asteraceae, we generated paired-end Illumina Hi-Seq sequence data (100 bp in length; 400 bp insert size) for individuals from 15 taxa. The estimated genome coverage for each species ranged from 0.42x – 3.52x (Additional file 7). These species were selected to represent every major subfamily of the Asteraceae, and included an outgroup species, N. patagonicus (Additional file 7). In addition, five of the taxa were selected from the genus Helianthus in order to investigate patterns of genome evolution amongst closely related species, and to increase our understanding of the evolutionary history of the most well-studied species in the family, H. annuus, for which there have been numerous prior studies about TE properties (see [26–29]). This study was done in parallel with a previously published phylogenomic study in which the taxon sampling and library preparation methods are described [64].
Repeat identification from WGS sequences
Prior to analysis, all WGS reads were treated with PRINSEQ version 0.19.4; [65] with the parameters ‘-min_len 40 –noniupac –min_qual_mean 15 –lc_method entropy –lc_threshold 60 –trim_ns_right 10 –ns_max_p 20’ to remove low quality and short sequences. After quality filtering, we screened all chloroplast- and mitochondria-derived sequences from the WGS reads using the complete chloroplast genome sequence for cultivated sunflower line HA383 (Genbank accession number DQ383815) and a database of 10 complete plant mitochondria genome sequences obtained from Genbank, respectively. One million paired-end reads were sampled randomly from each set of screened reads and interleaved with Pairfq version 0.09; [66] prior to analysis. Repeat identification was carried out by performing an all-by-all BLAST following the methods of Staton et al. [29] with the 1 million randomly sampled paired-end reads, followed by clustering using the Louvain method [67]. Annotation of clusters was performed using blastn [68] against RepBase version 18.01; [69] and a set of full-length LTR-RTs described by Staton et al. [29]. Our repeat identification methods are implemented using the Transposome software version 0.03; [70] that we developed for this study. We performed three replicates of the above sampling and annotation procedure with Transposome for each species to minimize the statistical error in our estimates of genome composition.
To investigate the effect of varying levels of genome coverage, we simulated 10 different levels of genome coverage from the H. annuus WGS reads ranging from 0.056 to 5.1 %, with 3 replicates at each level (total of 30 read sets). The coefficient of variation in the inferred genomic composition of each TE family was measured at each level of genome coverage after analysis with Transposome to infer the appropriate level of sampling; this allowed us to maximize the level of TE diversity being captured.
Genome size estimation and prediction of changes in genome composition
In order to determine the genomic contribution of each TE family to the species in this study, and estimate the magnitude of change across the Asteraceae, we calculated genome size according to Hu et al. [71], with modifications. Using WU-BLAST with parameters “M = 1 N = -3 -Q -R 1” we mapped a reference transcriptome of 11 species from the Compositae Genome Project database (http://compgenomics.ucdavis.edu/) to 5 million WGS reads for each species, and calculated the coverage of each transcript using the formula:
where N is the total length of reads mapped and L is the transcript length. The genome size (Cval) for each species was then determined by the formula:
where P is the ploidy level, n is the total number of reads, and l is the read length. In the above formula, only alignments over 60 base pairs in length and over 70 % identity were considered. These values were chosen from a permutation test using all possible alignments from lengths 50–100 and percent identity thresholds from 50 to 100, comparing observed to expected values. The mean coverage (Covi) was trimmed to remove the top 10 % of transcripts by coverage. The estimated genome size for each species, along with the published prediction (if available), is shown in Additional files 7 and 8.
The genomic contribution of each TE superfamily was calculated from the annotation summary file generated by Transposome (Fig. 1), and was used to determine the magnitude of change in TE composition in each species. Generalized least squares tests were performed with the R programming language [72] to estimate directional change in TE content in the Asteraceae (Fig. 2). We calculated Shannon’s evenness and diversity statistics using the R package Vegan [73] to investigate the influence of genome size change on TE diversity statistics.
Phylogenetic patterns of TE family evolution
In addition to analyzing statistical patterns of repeat abundance, we also explored a mechanistic basis for TE evolution in the Asteraceae from an ecological perspective through the use of community ecology models. First, we compared RAD distributions using the R package Vegan [73] to investigate the processes leading to the inferred distribution of TE families in the Asteraceae [50]. We compared five ecological models to test whether the rank abundance distribution of TE families in each species was best fit by neutral or niche-based models (reviewed in [56]). As in previous studies (e.g., [4, 74]), we treat a TE family as analgous to a biological species, the genome as analagous to the ecological communtiy, and an individual TE is treated as an individual of a given species. The Null model fits a brokenstick model where individual TEs are randomly distributed among the observed TE families and no parameters are fitted [5]. The Lognormal and Zipf models are generalized linear models where the Lognormal model assumes the logarithm of abundances are distributed normally [73]. The Zipf model,
where a is the expected abunance of a TE family at rank r, J is the total number of individual TEs, p is the fitted proportion of the most abundant TE family, and Υ is a decay coefficient, is used to fit a particular power law distribution [73]. The Mandelbrot model is a generalization of the Zipf model and adds one nonlinear parameter to the Zipf with the remaining parameters and log-likelihood being fitted with a linear model [73]. In the Preemption model, also called the geometric series or niche preemption model, each level of TE family abundance is a sequential, constant proportion of the total number of individuals in the whole community. The preemption model rank abundance is fit by straight line in the RAD plot [75].
Second, a phylogenetic generalized least squares (pgls) test was conducted using caper [76] to test for the association of changes in TE composition with particular phylogenetic divisions within the Asteraceae and genome size. The phylogenetic tree used in the pgls analyses was generated from an alignment of 763 nuclear loci sequenced by a novel targeted enrichment method [64]. The model we tested was:
where S* is the superfamily percent genomic abundance.
To further investigate the mechanisms and timing of shifts in genome content, we calculated phylogenetic signal for each TE family by using a descriptive statistic called K, which indicates significant phylogenetic signal for a trait, in this case TE abundance, on the tree compared to a Brownian motion model, along with phylogenetic independent contrasts PICs; [77, 78]. These calculations were performed using the R package picante [79], and all statistical analyses and plotting were performed in R [72].
Data availability
All sequence data in this paper is deposited in the NCBI Short Read Archive under BioProject number PRJNA288472.
Acknowledgements
We thank Jennifer Mandel (University of Memphis) for assistance with DNA isolation and sequencing; Vicki Funk (Smithsonian Institute) for sharing plant specimens; and the Georgia Advanced Computing Research Center for cooperation and assistance with our computational analyses. Financial support was provided by the NSF Plant Genome Research Program (DBI-0820451).
Abbreviations
- TE
Transposable element
- LTR
Long terminal repeat
- LTR-RT
Long terminal repeat retrotransposon
- ERV
Endogenous retrovirus
- LINE
Long interpersed nuclear element
- SINE
Short interspersed nuclear element
- WGS
Whole-genome shotgun, SD, Standard deviation
- PIC
Phylogenetic independent contrast
- RAD
Rank abundance dominance
- MYA
Million years ago
Additional files
Displays the phylogenetic signal for TE superfamilies in the Asteraceae. (PDF 11 kb)
Displays the phylogenetic signal for TE superfamilies. (PDF 61 kb)
Shows the TE families exhibiting significant phylogenetic signal. (PDF 63 kb)
Depicts the relationship between retrotransposon DNA and genome size. (PDF 63 kb)
Shows results from GLS and PGLS tests for the evolution of Gypsy and Copia composition. (PDF 60 kb)
Shows the genome diversity statistics for TE families. (PDF 807 kb)
Shows the raw data statistics and genome size estimates. (PDF 66 kb)
Shows published genome size estimates and genome size observations determined by the method described in this study. (PDF 54 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SES and JMB designed the study. SES performed the experiments and wrote the manuscript with feedback from JMB. All authors approved the final manuscript prior to submission.
Contributor Information
S. Evan Staton, Email: statonse@biodiversity.ubc.ca.
John M. Burke, Email: jmburke@uga.edu
References
- 1.Bennetzen JL. Transposable element contributions to plant gene and genome evolution. Plant Mol Biol. 2000;42:251–269. doi: 10.1023/A:1006344508454. [DOI] [PubMed] [Google Scholar]
- 2.Bennetzen JL, Ma J, Devos KM. Mechanisms of recent genome size variation in flowering plants. Plant Mol Biol. 2005;95:127–132. doi: 10.1093/aob/mci008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brookfield JYF. The ecology of the genome – mobile DNA elements and their hosts. Nat Rev. 2005;6:128–136. doi: 10.1038/nrg1524. [DOI] [PubMed] [Google Scholar]
- 4.Venner S, Feschotte C, Biemont C. Dynamics of transposable elements: towards a community ecology of the genome. Trends Gen. 2009;739:1–7. doi: 10.1016/j.tig.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pielou EC. Ecological diversity. New York: Wiley-Interscience; 1975. [Google Scholar]
- 6.Tokeshi M. Niche apportionment or random assortment – species abundance patters explained. J Animal Ecol. 1990;59:1129–1146. doi: 10.2307/5036. [DOI] [Google Scholar]
- 7.Hubbell SP. The unified neutral theory of biodiversity and biogeography. Princeton: Princeton University Press; 2001. [DOI] [PubMed] [Google Scholar]
- 8.Gregory TR. Evolution of the genome. San Diego: Elsevier, Inc; 2005. [Google Scholar]
- 9.Slotkin RK, Nuthikattu S, Jiang N. Plant genome diversity. Vol. 1. Vienna: Springer-Verlag Wien; 2014. The impact of transposable elements on gene and genome evolution. [Google Scholar]
- 10.Lynch M. The origins of genome architecture. Sunderland: Sinauer Associates, Inc; 2007. [Google Scholar]
- 11.Gao X, Hou Y, Ebina H, Levin HL, Voytas DF. Chromodomains direct integration of retrotransposons to heterochromatin. Genome Res. 2008;18:359–369. doi: 10.1101/gr.7146408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baucom RS, Estill JC, Chaparro C, Upshaw N, Jogi A, Deragon JM, Westerman RP, SanMiguel PJ, Bennetzen JL. Exceptional diversity, non-random distribution, and rapid evolution or retroelements in the B73 maize genome. PLoS Genet. 2009;5:1–13. doi: 10.1371/journal.pgen.1000732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baucom RS, Estill JC, Leebens-Mack J, Bennetzen JL. Natural selection on gene function drives the evolution of LTR retrotransposon families in the rice genome. Genome Res. 2009;19:243–254. doi: 10.1101/gr.083360.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nellåker C, Keane TM, Yalcin B, Wong K, Agam A, Belgard TG, Flint J, Adams DJ, Frankel WN, Ponting CP. The genomic landscape shaped by selection on transposable elements across 18 mouse strains. Genome Biol. 2012;13:R45. doi: 10.1186/gb-2012-13-6-r45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Volff JN, Korting C, Meyer A, Schartl M. Evolution and discontinuous distribution of Rex3 retrotransposons in fish. Mol Biol Evol. 2001;18:427–431. doi: 10.1093/oxfordjournals.molbev.a003819. [DOI] [PubMed] [Google Scholar]
- 16.Ray DA, Xing J, Salem AH, Batzer MA. SINEs of a nearly perfect character. Syst Biol. 2006;55:928Y935. doi: 10.1080/10635150600865419. [DOI] [PubMed] [Google Scholar]
- 17.Bohne A, Brunet F, Galiana-Arnoux D, Schultheis C, Volff J. Transposable elements as drivers of genomic and biological diversity in vertebrates. Chromosome Res. 2008;16:203–215. doi: 10.1007/s10577-007-1202-6. [DOI] [PubMed] [Google Scholar]
- 18.Dobigny G, Ozouf-Costaz C, Waters P, Bonillo C, Volobouev V. LINE-1 amplification accompanies explosive genome repatterning in Taterillus (Rodentia, Gerbillinae) Chromosome Res. 2004;12:787–793. doi: 10.1007/s10577-005-5265-y. [DOI] [PubMed] [Google Scholar]
- 19.de Boer JG, Yazawa R, Davidson WS, Koop BF. Bursts and horizontal evolution of DNA transposons in the speciation of pseudotetraploid salmonids. BMC Genomics. 2007;8:422. doi: 10.1186/1471-2164-8-422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ungerer MC, Strakosh SC, Zhen Y. Genome expansion in three hybrid sunflower species is associated with retrotransposon proliferation. Curr Biol. 2006;16:R872–R873. doi: 10.1016/j.cub.2006.09.020. [DOI] [PubMed] [Google Scholar]
- 21.Staton SE, Ungerer MC, Moore RC. The genomic organization of Ty3/gypsy-like retrotransposons in Helianthus (Asteraceae) homoploid hybrid species. Am J Bot. 2009;96:1646–1655. doi: 10.3732/ajb.0800337. [DOI] [PubMed] [Google Scholar]
- 22.Stevens PF. Angiosperm Phylogeny Website. Version 8, June 2007. [http://www.mobot.org/MOBOT/research/APweb].
- 23.Kim KJ, Choi KS, Jansen RK. Two chloroplast DNA inversions originated simultaneously during the early evolution of the sunflower family (Asteraceae) Mol Biol Evol. 2005;22:1783–1792. doi: 10.1093/molbev/msi174. [DOI] [PubMed] [Google Scholar]
- 24.Panero JL, Funk VA. The value of sampling anomalous taxa in phylogenetic studies: major clades of the Asteraceae revealed. Mol Phylogenet Evol. 2008;47:757–782. doi: 10.1016/j.ympev.2008.02.011. [DOI] [PubMed] [Google Scholar]
- 25.Funk VA. Systematics, evolution, and biogeography of the compositae. Vienna: IAPT; 2009. [Google Scholar]
- 26.Santini S, Cavallini A, Natali L, Minelli S, Maggini F, Cionini PG. Ty1/Copia- and Ty3/Gypsy-like DNA sequences in Helianthus species. Chromosoma. 2002;111:192–200. doi: 10.1007/s00412-002-0196-2. [DOI] [PubMed] [Google Scholar]
- 27.Natali L, Santini S, Giordani T, Minelli S, Maestrini P, Cionini PG, Cavallini A. Distribution of Ty3-Gypsy- and Ty1-Copia-like DNA sequences in the genus Helianthus and other Asteraceae. Genome. 2006;49:64–72. doi: 10.1139/g05-058. [DOI] [PubMed] [Google Scholar]
- 28.Cavallini A, Natali L, Zuccolo A, Giordani T, Jurman I, Ferrillo V, et al. Analysis of transposons and repeat composition of the sunflower (Helianthus annuus L.) genome. Theor Appl Genet. 2010;120:491–508. doi: 10.1007/s00122-009-1170-7. [DOI] [PubMed] [Google Scholar]
- 29.Staton SE, Hartman Bakken B, Blackman B, Chapman M, Kane N, Tang S, Ungerer M, Knapp S, Rieseberg L, Burke J. The sunflower (Helianthus annuus L.) genome reflects a recent history of biased accumulation of transposable elements. Plant J. 2012;72:142–153. doi: 10.1111/j.1365-313X.2012.05072.x. [DOI] [PubMed] [Google Scholar]
- 30.Peterson-Burch BD, Wright DA, Laten HM, Voytas DF. Retroviruses in plants? Trends Gen. 2000;16:151–152. doi: 10.1016/S0168-9525(00)01981-8. [DOI] [PubMed] [Google Scholar]
- 31.Akhipova I. Distribution and phylogeny of Penelope-like in Eukaryotes. Syst Biol. 2006;55:875–878. doi: 10.1080/10635150601077683. [DOI] [PubMed] [Google Scholar]
- 32.Bennetzen JL. Patterns in grass genome evolution. Curr Opin Plant Biol. 2007;10:176–181. doi: 10.1016/j.pbi.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 33.Devos KM. Grass genome organization and evolution. Curr Opin Plant Biol. 2010;13:139–145. doi: 10.1016/j.pbi.2009.12.005. [DOI] [PubMed] [Google Scholar]
- 34.Ma M. Species richness vs evenness: independent relationship and different responses to edaphic factors. OIKOS. 2005;111:192–198. doi: 10.1111/j.0030-1299.2005.13049.x. [DOI] [Google Scholar]
- 35.Lander E, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. Initial sequencing and analysis of the human genome. Nature. 2001;15:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 36.DeBarry JD, Kissinger JC. Jumbled genomes: missing apicomplexan synteny. Mol Biol Evol. 2011;28:2855–2871. doi: 10.1093/molbev/msr103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim JM, Vanguri S, Boeke JD, Gabriel A, Voytas DF. Transposable elements and genome organization: a comprehensive survey of retrotransposons revealed by the complete Saccharomyces cerevisiae genome sequence. Genome Res. 1998;8:464–478. doi: 10.1101/gr.8.5.464. [DOI] [PubMed] [Google Scholar]
- 38.SanMiguel P, Tikhonov A, Jin Y, Motchoulskaia N, Zakharov D, Melake-Berhan A, et al. Nested retrotransposons in the intergenic regions of the maize genome. Science. 1996;274:765–768. doi: 10.1126/science.274.5288.765. [DOI] [PubMed] [Google Scholar]
- 39.Schnable P, Ware D, Fulton RS, Stein JC, Wei F, Pastemak S, Liang C, et al. The B73 maize genome: complexity, diversity, and dynamics. Science. 2009;326:1112–1115. doi: 10.1126/science.1178534. [DOI] [PubMed] [Google Scholar]
- 40.Qin C, Yu C, Shen Y, Fang X, Chen L, Min J, Cheng J, Zhao S, Xu M, Luo Y, et al. Whole-genome sequencing of cultivated and wild peppers provides insights into Capsicum domestication and specialization. Proc Natl Acad Sci U S A. 2014;111:5135–5140. doi: 10.1073/pnas.1400975111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Furano AV, Duvernell DD, Boissinot S. L1 (LINE-1) diversity differs dramatically between mammals and fish. Trends Gen. 2004;20:9–14. doi: 10.1016/j.tig.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 42.Neafsey DE, Blumenstiel JP, Hartl DL. Different regulatory mechanisms underlie similar transposable element profiles in pufferfish and fruitfiles. Mol Biol Evol. 2014;21:2310–2318. doi: 10.1093/molbev/msh243. [DOI] [PubMed] [Google Scholar]
- 43.Eickbush TH, Furano AV. Fruit flies and humans respond differently to retrotransposons. Curr Opin Gen Dev. 2002;12:669–674. doi: 10.1016/S0959-437X(02)00359-3. [DOI] [PubMed] [Google Scholar]
- 44.Hua-Van A, Le Rouzic A, Maisonhaute C, Capy P. Abundance, distribution and dynamics of retrotransposable elements: similarities and differences. Cytogen Genome Res. 2005;110:426–440. doi: 10.1159/000084975. [DOI] [PubMed] [Google Scholar]
- 45.Xie D, Chen C, Ptaszek LM, Xiao S, Cao X, Fang F, Ng HH, Lewin HA, Cowan C, Zhong S. Rewirable gene regulatory networks in the preimplantation embryonic development of three mammalian species. Genome Res. 2010;20:804–815. doi: 10.1101/gr.100594.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Warenfors M, Pereira V, Eyre-Walker A. Transposable elements: insertion pattern and impact on gene expression evolution in Hominids. Mol Biol Evol. 2010;27:1955–1962. doi: 10.1093/molbev/msq084. [DOI] [PubMed] [Google Scholar]
- 47.Hollister JD, Smith LM, Guo Y, Ott F, Weigel D, Gaut BS. Transposable elements and small RNAs contribute to gene expression divergence between Arabidopsis thaliana and Arabidopsis lyrata. Proc Natl Acad Sci U S A. 2011;108:2322–2327. doi: 10.1073/pnas.1018222108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Piegu B, Guyot R, Picault N, Roulin A, Saniyal A, Kim H, Collura K, et al. Doubling genome size without polyploidization: dynamics of retrotransposon-mediated genome expansions in Oryza australensis, a wild relative of rice. Genome Res. 2006;16:1262–1269. doi: 10.1101/gr.5290206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Amborella genome project The Amborella genome and the evolution of flowering plants. Science. 2013;342:1241089. doi: 10.1126/science.1241089. [DOI] [PubMed] [Google Scholar]
- 50.Serra F, Becher V, Dopazo H. Neutral theory predicts the relative abundance and diversity of genetic elements in a broad array of eukaryotic genomes. PLoS ONE. 2013;8:6. doi: 10.1371/journal.pone.0063915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Linquist S, Cotenie K, Elliott TA, Saylor B, Kremer SC, Gregory TR. Applying ecological models to communities of genetic elements: the case of Neutral Theory. Mol Ecol. 2015;24:3232–3242. doi: 10.1111/mec.13219. [DOI] [PubMed] [Google Scholar]
- 52.Keeley JE, Fotheringham CJ. Species–area relationships in Mediterranean climate plant communities. J Biogeogr. 2003;30:1629–1657. doi: 10.1046/j.1365-2699.2003.00950.x. [DOI] [Google Scholar]
- 53.Whittaker RH. Dominance and diversity in land plant communties. Science. 1965;147:250–260. doi: 10.1126/science.147.3655.250. [DOI] [PubMed] [Google Scholar]
- 54.Whittaker RH. Evolution and measurement of species diversity. Taxon. 1972;21:213–251. doi: 10.2307/1218190. [DOI] [Google Scholar]
- 55.Nummelin M. Log-normal distribution of species abundances is not a universal indicator of rain forest disturbance. J Appl Ecol. 1998;35:454–457. doi: 10.1046/j.1365-2664.1998.00309.x. [DOI] [Google Scholar]
- 56.McGill BJ, Etienne RS, Gray JS, Alonso D, Anderson MJ, Benecha HK, Dornelas M, Enquist BJ, Green JL, He F, Hurlbert AH, Magurran AE, Marquet PA, Maurer BA, Ostling A, Soykan CU, Ugland KI, White EP. Species abundance distributions: moving beyond single prediction theories to integration within an ecological framework. Ecol Lett. 2007;10:995–1015. doi: 10.1111/j.1461-0248.2007.01094.x. [DOI] [PubMed] [Google Scholar]
- 57.Barker MS, Kane NC, Matvienko M, Kozik A, Michelmore RW, Knapp SJ, Rieseberg LH. Multiple paleopolypoidizations during the evolution of the Compositae reveal parallel patterns of duplicate gene retention after millions of years. Mol Biol Evol. 2008;25:2445–2455. doi: 10.1093/molbev/msn187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Peterson-Burch BD, Nettleton D, Voytas DF. Genomic neighborhoods for Arbidopsis retrotransposons: a role for targeted integration in the distribution of the Metaviridae. Genome Biol. 2004;5:R78. doi: 10.1186/gb-2004-5-10-r78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chapman MA, Leebens-Mack JH, Burke JM. Positive selection and expression divergence following gene duplication in the sunflower CYCLOIDEA gene family. Mol Biol Evol. 2008;25:1260–1273. doi: 10.1093/molbev/msn001. [DOI] [PubMed] [Google Scholar]
- 60.Hutchinson GE. Homage to Santa Rosalia, or why are there so many kinds of animals? Am Nat. 1959;93:145–159. doi: 10.1086/282070. [DOI] [Google Scholar]
- 61.Hawkins JS, Kim H, Nason JD, Wing RA, Wendel JF. Differential lineage-specific amplification of transposable elements is responsible for genome size variation in Gossypium. Genome Res. 2006;16:1252–1261. doi: 10.1101/gr.5282906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Buti M, Giordani T, Cattonaro F, Cossu RM, Pistelli L, Vukich M, Morgante M, Cavallini A, Natali L. Temporal dynamics in the evolution of the sunflower genome as revealed by sequencing and annotation of three large genomic regions. Theor Appl Genet. 2011;5:779–791. doi: 10.1007/s00122-011-1626-4. [DOI] [PubMed] [Google Scholar]
- 63.Dodsworth S, Chase MW, Kelly LJ, Leitch IJ, Macas J, Novák P, Piednoel M, Weiss-Schneeweiss H, Leitch AR. Genomic repeat abundances contain phylogenetic signal. Syst Biol. 2015;64:112–126. doi: 10.1093/sysbio/syu080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mandel J, Dikow RB, Funk VA, Masalia R, Staton SE, Kozik A, Michelmore RW, Rieseberg LH, Burke JM. A target enrichment method for gathering phylogenetic information from hundreds of loci: an example from the Compositae. App Plant Sci. 2014;2:130085. doi: 10.3732/apps.1300085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schmieder R, Edwards R. Quality control and preprocessing of metagenomic datasets. Bioinformatics. 2011;27:863–864. doi: 10.1093/bioinformatics/btr026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Staton SE. Pairfq: sync paired-end FASTA/Q files and keep singleton reads. [https://github.com/sestaton/Pairfq].
- 67.Blondel VD, Guillaume J, Lambiotte R, Lefebvre E. Fast unfolding of communities in large networks. J Stat Mech. 2008;2008:P10008. doi: 10.1088/1742-5468/2008/10/P10008. [DOI] [Google Scholar]
- 68.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. Repbase update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 2005;110:462–467. doi: 10.1159/000084979. [DOI] [PubMed] [Google Scholar]
- 70.Staton SE, Burke JM. Transposome: annotation of transposable element families from unassembled sequence reads. Bioinformatics. 2015;31:1827–1829. doi: 10.1093/bioinformatics/btv059. [DOI] [PubMed] [Google Scholar]
- 71.Hu H, Bandyopadhyay PK, Olivera BM, Yandell M. Characterization of the Conus bullatus genome and its venom-duct transcriptome. BMC Genomics. 2011;12:60. doi: 10.1186/1471-2164-12-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.R Core Team. R: A language and environment for statistical computing. [http://www.R-project.org].
- 73.Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O’Hara RB, et al. vegan: Community Ecology Package. R package version 2.0-7. [http://CRAN.R-project.org/package=vegan].
- 74.Le Rouzic A, Dupas S, Capy P. Genome ecosystem and transposable element species. Gene. 2007;390:214–220. doi: 10.1016/j.gene.2006.09.023. [DOI] [PubMed] [Google Scholar]
- 75.Motomura I. A statistical treatment of associations. Jpn J Zool. 1932;44:379–383. [Google Scholar]
- 76.Orme D, Freckleton R, Thomas G, Petzoldt T, Fritz S, Isaac L, et al. caper: Comparative Analyses of Phylogenetics and Evolution in R. R package version 0.5. [http://CRAN.R-project.org/package=caper].
- 77.Blomberg SP, Garland T, Jr, Ives AR. Testing for phylogenetic signal in comparative data: behavioral traits are more labile. Evolution. 2003;57:717–745. doi: 10.1111/j.0014-3820.2003.tb00285.x. [DOI] [PubMed] [Google Scholar]
- 78.Felsenstein J. Phylogenies and the comparative method. Am Nat. 1985;125:1–15. doi: 10.1086/284325. [DOI] [Google Scholar]
- 79.Kembel SW, Cowan PD, Helmus MR, Cornwell WK, Morlon H, Ackerly DD, Blomberg SP, Webb SP. Picante: R tools for integrating phylogenies and ecology. Bioinformatics. 2010;26:1463–1464. doi: 10.1093/bioinformatics/btq166. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All sequence data in this paper is deposited in the NCBI Short Read Archive under BioProject number PRJNA288472.