

Abstract
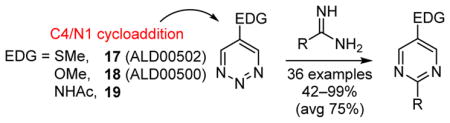
The examination of the cycloaddition reactions of 1,2,3-triazines 17–19, bearing electron-donating substituents at C5, are described. Despite the noncomplementary 1,2,3-triazine C5 substituents, amidines were found to undergo a powerful cycloaddition to provide 2,5-disubstituted pyrimidines in excellent yields (42–99%; EDG = SMe > OMe > NHAc). Even select ynamines and enamines were capable of cycloadditions with 17, but not 18 or 19, to provide trisubstituted pyridines in modest yields (37–40% and 33% respectively).
The inverse electron demand Diels–Alder reaction of electron-deficient heterocyclic azadienes is a highly effective method for the synthesis of challenging heterocyclic ring systems.1 In efforts that have expanded the range of heterocycles accessible by this powerful cycloaddition strategy, we have systematically explored the reactions of 1,2,4,5-tetrazines,2 1,2,4-triazines,3 1,3,5-triazines,4 1,3,4-oxadiazoles,5 and 1,2-diazines.6 Frequently, these methodological studies were inspired by their use as the key steps in the total synthesis of complex natural products, providing non-obvious or effective solutions to the preparation of their core structures.7–10 Additional applications include their reactions with strained olefins and alkynes as bioorthogonal bioconjugation reagents because of their extraordinarily efficient and rapid rates of cycloaddition.11 Recently, our efforts extended to 1,2,3-triazines, examining the synthesis and cycloaddition reaction scope of the parent 1,2,3-triazine and a series of 1,2,3-triazines that contain electron-withdrawing groups at sites where their electronic impact was both consonant and dissonant to that innate to the 1,2,3-triazine core (Figure 1).12 In addition to defining the scope of the 1,2,3-triazine cycloaddition reactions, these studies have also found application in the total syntheses of (−)-pyrimidoblamic acid and P-3A13 as well as the late-stage divergent total syntheses of dihydrolysergic acid, dihydrolysergol, and a series of heterocyclic derivatives14.
FIGURE 1.

Previous studies with 1,2,3-triazines.
Perhaps the most remarkable of the cycloaddition reactions of 1,2,3-triazines and the most effective of those examined to date is their reactions with amidines to provide pyrimidines. The efficiency and robust nature of these reactions led to our further study of 1,2,3-triazines that contain electron-donating groups at C5. It was expected that this modification would decrease the overall reactivity of the 1,2,3-triazines, perhaps even to the point of preventing the anticipated cycloaddition chemistry. Herein, we show that while such substituents do slow the rate of reaction of the 1,2,3-triazines, their reaction with amidines remained remarkably effective, providing the product pyrimidines in excellent yields. In selected cases, even less effective dienophiles, such as ynamines and enamines, were found to be capable of cycloaddition, albeit in more modest yields.
Three previously unreported 1,2,3-triazines were chosen to probe the effects of electron-donating substituents on the reactivity and regioselectivity of the cycloaddition reactions with amidines, ynamines, and enamines. These 1,2,3-triazines, 5-(methylthio)-1,2,3-triazine (17), 5-methoxy-1,2,3-triazine (18), and 5-(N-acetylamino)-1,2,3-triazine (19), were selected to provide a range of electronic effects on which to base an analysis (Scheme 1).
SCHEME 1.
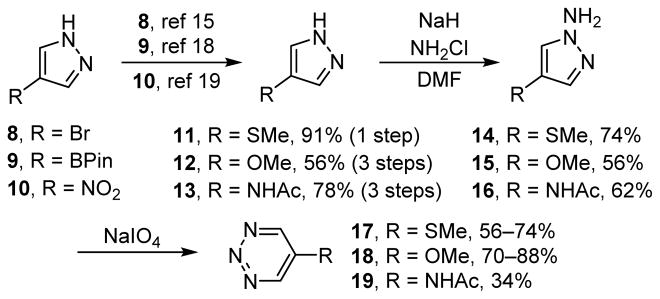
Synthesis of 1,2,3-triazines 17–19.
5-(Methylthio)-1,2,3-triazine (17, ALD00502) was accessed by the simultaneous deprotonation and lithium–halogen exchange of commercially available 4-bromopyrazole (8) with subsequent trap of the C-lithiate by dimethyl disulfide to provide pyrazole 11 (91%).15 Subsequent N-amination of 11 with monochloramine16 provided a mixture of N-amino pyrazole 14 and starting 11 (74%, 98% brsm, ~3.4:1) that was inseparable by chromatography. Nevertheless, when the mixture of N-amino pyrazole 14 and pyrazole 11 was treated with NaIO4 under biphasic oxidative ring expansion conditions17, 5-(methylthio)-1,2,3-triazine (17) (56–74%) was obtained in good yield and was readily separable from products derived from pyrazole 11.
Preparation of 5-methoxy-1,2,3-triazine (18, ALD00500) was achieved starting with 4-methoxypyrazole (12), which was prepared from 9 in a known 3-step sequence (56% overall).18 This pyrazole 12 was subjected to N-amination with monochloramine16 (56%) and the subsequent NaIO4 oxidative ring expansion17 to provide 18 in good yield (70–88%).
The synthesis of 5-(N-acetylamino)-1,2,3-triazine (19) began with the reduction of commercially available 4-nitropyrazole (10) (quant.) followed by bisacylation (83%) of 4-aminopyrazole and subsequent monodeacylation (94%) to provide 13.19 While it is reported that 4-aminopyrazole can be monoacylated selectively (vs N1),20 doing so was found to be challenging, frequently providing mixtures of des-, mono-, and bisacylation products in our hands. N-amination of pyrazole 13 with monochloramine16 provided the penultimate substrate 16 in good yield (62%). Oxidative ring expansion under biphasic conditions17 generates 1,2,3-triazine 19, but the solubility of the compound did not allow its extraction from the aqueous phase. As such, a new monophasic protocol, utilizing NaIO4 in acetonitrile under sonication, was developed and optimized to provide a modest yield (34%) of 5-(N-acetylamino)-1,2,3-triazine (19).
Based on previous studies12, it was expected that the reactions of 1,2,3-triazines 17–19 with free-based amidines would proceed with a C4/N1 regioselectivity to furnish symmetrical pyrimidine products. The electron-donating substituents raise the LUMOdiene relative to 1,2,3-triazine and suggest they should slow or even preclude the cycloaddition reaction (reactivity: MeS > MeO > AcNH). Initial studies with 17 revealed that reactions at 25 °C typically required 24–90 h to achieve full consumption of the limiting reagent, but that product pyrimidine was produced even under such mild reaction conditions (Figure 2, entries 1,2). Although increases in reaction time provided further yield improvements (Figure 2, entry 3), simply warming the reactions in which the amidine was the limiting reagent at 40 °C, provided acceptable reaction times and superb isolated yields (Figure 2, entry 4). Since the most valuable component of reactions is likely to be the amidine, further studies focused on a reaction stoichiometry of 1.0 equivalent of amidine and 1.5 equivalents of 1,2,3-triazine.
FIGURE 2.

Optimization of the reaction of 17 with amidines.
Finally, although not extensively examined and unlike previously disclosed examples, the inclusion of molecular sieves served neither to increase the reaction rate or yield. In fact, their inclusion was observed to decrease the isolated yield of cycloaddition products in some cases (Figure 2, entries 4,5). As a result, molecular sieves were omitted from the optimized reaction conditions.
With effective conditions in hand and with use of an extended reaction time of 24 h, the reaction of 1,2,3-triazine 17 with a wide variety of aliphatic and aryl amidines was explored (Figure 3). The cycloadditions proceeded smoothly at 40 °C to afford the 2-substituted-5-(methylthio)pyrimidines in excellent yields (generally >90%). Qualitatively, the relative reaction rates tracked with the amidine electronic character, with the electron-rich amidines (e.g. 20j) reacting most rapidly (ca. 2 h) and the electron-poor amidines (e.g. 20b and 20e) requiring the full 24 h to achieve comparable conversions. Even the sterically encumbered amidine 20k provided a superb conversion to pyrimidine 21k.
FIGURE 3.

Reaction of 1,2,3-triazine 17 with amidines.
With 5-methoxy-1,2,3-triazine (18), the reaction rate decreased and a reoptimization of the reaction temperature was conducted. It was determined that a reaction temperature of 90 °C was effective in promoting the cycloaddition in high yields (42–97%) over a convenient timeframe (24 h) (Figure 4). Consistent with observations made with 1,2,3-triazine 17, the rate of cycloaddition of 18 tracked closely with the electronic character of the amidine and those bearing aromatic substituents generally provided higher yields of the 2-substituted-5-methoxypyrimidines than those bearing aliphatic substituents.
FIGURE 4.

Reaction of 1,2,3-triazine 18 with amidines.
The introduction of a C5 N-acetylamino substituent resulted in a further decrease in reaction rate with 19, which required reexamination of the reaction parameters. Unlike 1,2,3-triazine 18, further increasing the reaction temperature was ineffective. Instead, extended reaction times (72 h) at 90 °C were found to afford moderate to good isolated yields (43–79%) of the pyrimidine products (Figure 5).
FIGURE 5.

Reaction of 1,2,3-triazine 19 with amidines.
It is possible, even likely, that the reactions of amidines with 1,2,3-triazines represent stepwise addition-cyclization reactions proceeding through polar intermediates, although we have not yet detected such intermediates or isolated intercepted products diagnostic of their intermediacy. As a result, we conducted the reaction of 1,2,3-triazines 17 and 4 (ALD00106) with amidine 15N-20g doubly labeled with 15N. Each reaction produced the pyrimidine product in superb yields (97–98%) and each was generated with incorporation of a single 15N label (Figure 6). This is consistent with a single-step cycloaddition and, while not ruling out stepwise addition-cyclization mechanisms, the latter would have been expected to provide at least mixtures of singly and doubly 15N labeled pyrimidine products perhaps even favoring the doubly labeled product.
FIGURE 6.

Reaction of 1,2,3-triazines with 15N-labeled amidines.
We also examined the reactivity of 17–19 toward other candidate dienophiles including ynamines and enamines. Since both classes are less reactive in their cycloaddition reactions with 1,2,3-triazines than amidines, it was anticipated that their cycloadditions might be difficult to promote. An extensive survey of the reaction conditions for both ynamines and enamines was undertaken and included the exploration of the solvent, reaction time, temperature, additives, concentration, and stoichiometry. In the case of ynamines, the optimal reaction conditions identified were modestly effective in promoting the cycloaddition reaction of 1,2,3-triazine 17 (37–40%), providing the product of cycloaddition across C4/N1, whereas the 1,2,3-triazines 18 and 19 failed to react to an appreciable extent (Figure 7). Similarly, the cycloaddition reaction of selected enamines were only moderately effective in the case of 1,2,3-triazine 17 (33%) (Figure 7).
FIGURE 7.
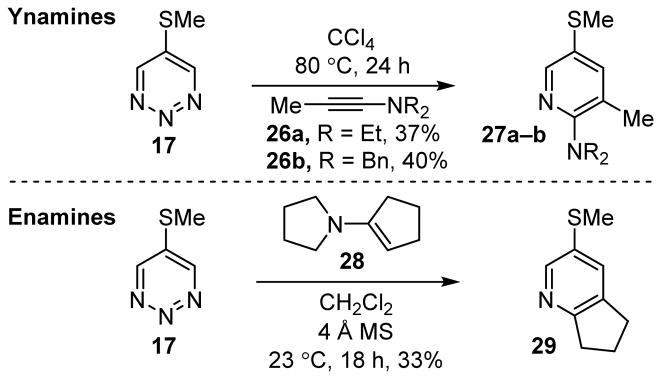
Reaction of 17 with ynamines and enamines.
A detailed study of the cycloaddition reactions of 1,2,3-triazines bearing C5 electron donating groups was conducted. Despite the unfavorable electronic effects of the substituents, 1,2,3-triazines 17–19 were found to participate in a powerful cycloaddition with a wide variety of amidines (reactivity: 17 > 18 > 19), providing the corresponding pyrimidines in excellent yields (42–99%). Remarkably, 1,2,3-triazine 17, but not 18 and 19, was even capable of reaction with a select set of ynamines and enamines, highlighting the highly reactive nature of the 1,2,3-triazine core. In addition to the intrinsic use of such 1,2,3-triazine cycloaddition reactions with amidines for the synthesis of individual substituted pyrimidines, the ability to use virtually any and all readily accessible substituted 1,2,3-triazines in the amidine cycloaddition reaction permits the late-stage divergent synthesis of a series of substituted pyrimidines from a common amidine intermediate.
Supplementary Material
Acknowledgments
We gratefully acknowledge the financial support of the National Institutes of Health (D.L.B., CA042056) and the award of a NSF fellowship (C.M.G.).
Footnotes
Full experimental details. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Boger DL. Tetrahedron. 1983;39:2869. [Google Scholar]; (b) Boger DL. Chem Rev. 1986;86:781. [Google Scholar]; (c) Boger DL, Weinreb SM. Hetero Diels–Alder Methodology in Organic Synthesis. Academic; San Diego: 1987. [Google Scholar]
- 2.(a) Carboni RA, Lindsey RV. J Am Chem Soc. 1959;81:4342. [Google Scholar]; (b) Sauer J, Wiest H. Angew Chem, Int Ed Engl. 1962;1:269. [Google Scholar]; (c) Boger DL, Panek JS. Tetrahedron Lett. 1983;24:4511. [Google Scholar]; (d) Boger DL, Coleman RS, Panek JS, Yohannes D. J Org Chem. 1984;49:4405. [Google Scholar]; (e) Boger DL, Panek JS, Coleman RS, Sauer J, Huber FX. J Org Chem. 1985;50:5377. [Google Scholar]; (f) Boger DL, Sakya SM. J Org Chem. 1988;53:1415. [Google Scholar]; (g) Sakya SM, Groskopf KK, Boger DL. Tetrahedron Lett. 1997;38:3805. [Google Scholar]; (h) Boger DL, Schaum RP, Garbaccio RM. J Org Chem. 1998;63:6329. doi: 10.1021/jo980795g. [DOI] [PubMed] [Google Scholar]; (i) Soenen DR, Zimpleman JM, Boger DL. J Org Chem. 2003;68:3593. doi: 10.1021/jo020713v. [DOI] [PubMed] [Google Scholar]; (j) Hamasaki A, Ducray R, Boger DL. J Org Chem. 2006;71:185. doi: 10.1021/jo051832o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Steigel A, Sauer J. Tetrahedron Lett. 1970:3357. [Google Scholar]; (b) Neunhoeffer H, Fruhauf H-W. Leibigs Ann Chem. 1972;758:120. [Google Scholar]; (c) Boger DL, Panek JS. J Org Chem. 1981;46:2179. [Google Scholar]; (d) Boger DL, Panek JS, Meier MM. J Org Chem. 1982;47:3763. [Google Scholar]; (f) Boger DL, Panek JS. Tetrahedron Lett. 1984;25:3175. [Google Scholar]
- 4.(a) Neunhoeffer H, Bachmann M. Chem Ber. 1975;108:3877. [Google Scholar]; (b) Boger DL, Schumacher J, Panek JS, Mullican MD, Patel M. J Org Chem. 1982;47:2673. [Google Scholar]; (c) Boger DL, Dang Q. Tetrahedron. 1988;44:3379. [Google Scholar]; (d) Boger DL, Dang Q. J Org Chem. 1992;57:1631. [Google Scholar]; (e) Boger DL, Kochanny MJ. J Org Chem. 1994;59:4950. [Google Scholar]
- 5.(a) Wilkie GD, Elliott GI, Blagg BSJ, Wolkenberg SE, Soenen DR, Miller MM, Pollack S, Boger DL. J Am Chem Soc. 2002;124:11292. doi: 10.1021/ja027533n. [DOI] [PubMed] [Google Scholar]; (b) Elliott GI, Fuchs JR, Blagg BSJ, Ishikawa H, Yuan Z-Q, Boger DL. J Am Chem Soc. 2006;128:10589. doi: 10.1021/ja0612549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Neunhoeffer H, Werner G. Liebigs Ann Chem. 1973:437. [Google Scholar]; (b) Boger DL, Coleman RS. J Org Chem. 1984;49:2240. [Google Scholar]; (c) Kessler SN, Wegner HA. Org Lett. 2010;12:4062. doi: 10.1021/ol101701z. [DOI] [PubMed] [Google Scholar]; (d) Sumaria CS, Tuerkmen YE, Rawal VH. Org Lett. 2014;16:3236. doi: 10.1021/ol501254h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Boger DL, Panek JS. J Am Chem Soc. 1985;107:5745. [Google Scholar]; (b) Boger DL, Panek JS, Duff SR, Yasuda M. J Org Chem. 1985;50:5790. [Google Scholar]; (c) Boger DL, Duff SR, Panek JS, Yasuda M. J Org Chem. 1985;50:5782. [Google Scholar]; (d) Boger DL, Coleman RS. J Am Chem Soc. 1987;109:2717. [Google Scholar]; (e) Boger DL, Coleman RS. J Am Chem Soc. 1988;110:4796. [Google Scholar]; (f) Boger DL, Zhang M. J Am Chem Soc. 1991;113:4230. [Google Scholar]; (g) Boger DL, Menezes RF, Honda T. Angew Chem Int Ed Engl. 1993;32:273. [Google Scholar]; (h) Boger DL, Honda T, Menezes RF, Colletti SL, Dang Q, Yang W. J Am Chem Soc. 1994;116:82. [Google Scholar]; (i) Boger DL, Honda T. J Am Chem Soc. 1994;116:5647. [Google Scholar]; (j) Boger DL, Colletti SL, Honda T, Menezes RF. J Am Chem Soc. 1994;116:5607. [Google Scholar]; (k) Boger DL, Honda T, Dang Q. J Am Chem Soc. 1994;116:5619. [Google Scholar]; (l) Boger DL, Hong J, Hikota M, Ishida M. J Am Chem Soc. 1999;121:2471. [Google Scholar]; (m) Boger DL, Wolkenberg SE. J Org Chem. 2000;65:9120. doi: 10.1021/jo0012546. [DOI] [PubMed] [Google Scholar]
- 8.(a) Boger DL, Patel M. J Org Chem. 1988;53:1405. [Google Scholar]; (b) Boger DL, Baldino CM. J Am Chem Soc. 1993;115:11418. [Google Scholar]; (c) Boger DL, Boyce CW, Labroli MA, Sehon CA, Jin Q. J Am Chem Soc. 1999;121:54. [Google Scholar]; (d) Boger DL, Soenen DR, Boyce CW, Hedrick MP, Jin Q. J Org Chem. 2000;65:2479. doi: 10.1021/jo9916535. [DOI] [PubMed] [Google Scholar]; (e) Boger DL, Hong J. J Am Chem Soc. 2001;123:8515. doi: 10.1021/ja011271s. [DOI] [PubMed] [Google Scholar]; (f) Hamasaki A, Zimpleman JM, Hwang I, Boger DL. J Am Chem Soc. 2005;127:10767. doi: 10.1021/ja0526416. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Oakdale JS, Boger DL. Org Lett. 2010;12:1132. doi: 10.1021/ol100146b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Boger DL, Wolkenberg SE. J Org Chem. 2002;67:7361. doi: 10.1021/jo020437k. [DOI] [PubMed] [Google Scholar]; (b) Yuan Q, Ishikawa H, Boger DL. Org Lett. 2005;7:741. doi: 10.1021/ol050017s. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ishikawa H, Elliott GI, Velcicky J, Choi Y, Boger DL. J Am Chem Soc. 2006;128:10596. doi: 10.1021/ja061256t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Ishikawa H, Colby DA, Seto S, Va P, Tam A, Kakei H, Rayl TJ, Hwang I, Boger DL. J Am Chem Soc. 2009;131:4904. doi: 10.1021/ja809842b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Ishikawa H, Colby DA, Boger DL. J Am Chem Soc. 2008;130:420. doi: 10.1021/ja078192m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Campbell EL, Zuhl AM, Liu CM, Boger DL. J Am Chem Soc. 2010;132:3009. doi: 10.1021/ja908819q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Sasaki Y, Kato D, Boger DL. J Am Chem Soc. 2010;132:13533. doi: 10.1021/ja106284s. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Lajiness JP, Jiang W, Boger DL. Org Lett. 2012;14:2078. doi: 10.1021/ol300599p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Xie J, Wolfe AL, Boger DL. Org Lett. 2013;15:868. doi: 10.1021/ol303573f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Lee K, Boger DL. J Am Chem Soc. 2014;136:3312. doi: 10.1021/ja500548e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Lee K, Boger DL. Tetrahedron. 2015;71:3741. doi: 10.1016/j.tet.2014.07.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Boger DL, Cassidy KC, Nakahara S. J Am Chem Soc. 1993;115:10733. [Google Scholar]; (b) Boger DL, Hüter O, Mbiya K, Zhang M. J Am Chem Soc. 1995;117:11839. [Google Scholar]; (c) Boger DL, Hong J. J Am Chem Soc. 1998;120:1218. [Google Scholar]; (d) Boger DL, Ichikawa S, Jiang H. J Am Chem Soc. 2000;122:12169. [Google Scholar]; (e) Blagg BSJ, Boger DL. Tetrahedron. 2002;58:6343. [Google Scholar]; (f) Schnermann MJ, Boger DL. J Am Chem Soc. 2005;127:15704. doi: 10.1021/ja055041f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Blackman ML, Royzen M, Fox JM. J Am Chem Soc. 2008;130:13518. doi: 10.1021/ja8053805. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Devaraj NK, Weissleder R, Hilderbrand SA. Bioconjugate Chem. 2008;19:2297. doi: 10.1021/bc8004446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Anderson ED, Boger DL. Org Lett. 2011;13:2492. doi: 10.1021/ol2007428. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Anderson ED, Boger DL. J Am Chem Soc. 2011;133:12285. doi: 10.1021/ja204856a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Anderson ED, Duerfeldt AS, Zhu K, Glinkerman CM, Boger DL. Org Lett. 2014;16:5084. doi: 10.1021/ol502436n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Duerfeldt AS, Boger DL. J Am Chem Soc. 2014;136:2119. doi: 10.1021/ja412298c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee K, Poudel Y, Glinkerman CM, Boger DL. Tetrahedron. 2015 doi: 10.1016/j.tet.2015.05.093. http://dx.doi.org/10.1016/j.tet.2015.05.093. [DOI] [PMC free article] [PubMed]
- 15.Ahn K, Boehm M, Cabral S, Carpino PA, Futatsugi K, Hepworth D, Dung DW, Orr S, Wang J. Diacylglycerol Acyltransferase 2 Inhibitors. 2013/150416 A1. WO. 2013 Oct 10;
- 16.Hynes J, Doubleday WW, Kyckman AJ, Godfrey JD, Grosso JA, Kiau S, Leftheris K. J Org Chem. 2004;69:1368. doi: 10.1021/jo035587p. [DOI] [PubMed] [Google Scholar]
- 17.(a) Itoh T, Ohsawa A, Okada M, Kaihoh T, Igeta H. Chem Pharm Bull. 1985;33:3050. [Google Scholar]; (b) Itoh T, Okada M, Nagata K, Yamaguchi K, Ohsawa A. Chem Pharm Bull. 1990;38:2108. [Google Scholar]
- 18.Oslob J, McDowell R, Johnson R, Yang H, Evanchik M, Haria C, Cai H, Hu L. Heterocyclic Modulators of Lipid Synthesis. 2014/008197 A1. WO. 2014 Jan 9;
- 19.(a) Harrison R, Ramsden N, Major J, Morel A, Convery L, Sunose M, Lynch R, Adrego R, Jones A. Heterocyclyl Pyrimidine Analogues as TYK2 Inhibitors. 2013/174895 A1. WO. 2013 Nov 28;; (b) Giles D, Parnell E, Renwick J. J Chem Soc C. 1966:1179. doi: 10.1039/j39660001179. [DOI] [PubMed] [Google Scholar]; (c) Lu Y, Kraatz H. Inorg Chim Acta. 2004;357:159. [Google Scholar]
- 20.Linnanen T, Wohlfahrt G, Nanduri S, Ujjin-Amatada R, Rajagopalan S, Mukherjee S. Protein Kinase Inhibitors. 2013/053983 A1. WO. 2013 Apr 18;
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.