

Summary
The Cdk7 subunit of TFIIH phosphorylates RNA polymerase II (Pol II) during initiation and while recent studies show inhibition of human Cdk7 negatively influences transcription, the mechanisms involved are unclear. Using in vitro transcription with nuclear extract, we demonstrate that THZ1, a covalent Cdk7 inhibitor, causes defects in Pol II phosphorylation, co-transcriptional capping, promoter proximal pausing, and productive elongation. THZ1 does not affect initiation but blocks essentially all Pol II large subunit C-terminal domain (CTD) phosphorylation. We found that guanylylation of nascent RNAs is exquisitely length-dependent and modulated by a THZ1-sensitive factor present in nuclear extract. THZ1 impacts pausing through a capping-independent block of DSIF and NELF loading. The P-TEFb-dependent transition into productive elongation was also inhibited by THZ1, likely due to loss of DSIF. Capping and pausing were also reduced in THZ1-treated cells. Our results provide mechanistic insights into THZ1 action and how Cdk7 broadly influences transcription and capping.
Graphical abstract
Introduction
Cdk7, Cyclin H, and Mat1 comprise a kinase module in the Pol II initiation factor TFIIH (Egly and Coin, 2011) which is responsible for phosphorylation of the CTD of the large subunit of human Pol II during initiation. The CTD contains 52 repeats of a heptapeptide with the consensus sequence Y1S2P3T4S5P6S7 and Cdk7 phosphorylates Ser5 and Ser7 residues, the first of several CTD phosphorylation and dephosphorylation events during the transcription cycle (Heidemann et al., 2013). CTD phosphorylation is thought to be important in the recruitment of RNA processing factors and histone modification enzymes (Heidemann et al., 2013). In mammals, Cdk7 is also the major cyclin-dependent kinase activating kinase (CAK) and is involved in regulating the cell cycle (Fisher, 2012).
A prominent feature of transcription in metazoans is promoter proximal pausing of Pol II (Guo and Price, 2013). Pausing is induced by the DRB sensitivity inducing factor, DSIF, in conjunction with the negative elongation factor NELF (Yamaguchi et al., 2013) and these factors accumulate with Pol II downstream of the transcription start site of the majority of mammalian genes (Liu et al., 2014; Rahl et al., 2010). Gdown1 has also been implicated in stable pausing of Pol II near promoters (Cheng et al., 2012). To transition into productive elongation, the Cdk9 kinase subunit of P-TEFb must phosphorylate both DSIF, which remains associated during elongation, and NELF, which is released (Guo and Price, 2013). Cdk9 (Marshall et al., 1996) and Cdk12 (Bartkowiak et al., 2010; Bowman and Kelly, 2014) have been implicated in phosphorylation of CTD Ser2 during the transition into productive elongation and at the 3′ ends of genes.
Capping occurs co-transcriptionally and is the first of many processing steps required to generate a functional mRNA (Bentley, 2014). Initially, an RNA triphosphatase removes the terminal phosphate and then RNA guanylyltransferase mediates the 5′-5′ addition of GMP. Cap methylation occurs in a separate step and the final m7G cap influences downstream events. Guanylylation of nascent transcripts can occur as soon as the RNA emerges from Pol II (Coppola et al., 1983; Moteki and Price, 2002; Rasmussen and Lis, 1993) and human capping enzyme (HCE) is 4 to 5 orders of magnitude more efficient when the RNA substrate is associated with an elongation complex (Moteki and Price, 2002). This is aided in part by capping enzyme association with the phosphorylated CTD (Chiu et al., 2002; Cho et al., 1997; McCracken et al., 1997; Yue et al., 1997), although the site of this interaction is not conserved between yeast and mammalian capping enzymes (Ghosh et al., 2011). In fission yeast, an otherwise lethal Ser5Ala CTD mutation was rescued by fusing mouse capping enzyme to the CTD (Schwer and Shuman, 2011). In mammals, CTD phosphorylation has a 4-fold effect on capping of soluble RNA (Ho and Shuman, 1999) or nascent Pol II transcripts (Mandal et al., 2004; Moteki and Price, 2002). Capping enzymes have also been shown to interact with the Spt5 subunit of DSIF in yeast (Doamekpor et al., 2014; Doamekpor et al., 2015; Pei and Shuman, 2002; Schneider et al., 2010) and human DSIF causes a 2 to 5-fold stimulation of co-transcriptional capping (Mandal et al., 2004; Wen and Shatkin, 1999). Additionally, wild type or capping-defective HCE relieved the negative influence of NELF during transcription in vitro suggesting a competition of HCE and pausing factors for Pol II (Mandal et al., 2004). This network of interactions among the capping enzyme, the Pol II CTD, and DSIF could play a role in controlling capping and elongation in mammals.
Inhibition of Cdk7 has broad effects on transcription but the mechanisms involved remain poorly understood. Early in vitro studies gave conflicting results concerning the role of Cdk7 in transcription (Akoulitchev et al., 1995; Tirode et al., 1999). Inhibition of the Cdk7 homolog Kin28 in budding yeast reduced mRNA capping in two studies. In one, global transcription was unaffected (Kanin et al., 2007) and in the other, mRNA stability was reduced (Hong et al., 2009). Other studies showed increased Pol II promoter occupancy genome-wide (Bataille et al., 2012; Wong et al., 2014). Investigations in higher eukaryotes showed decreases in promoter-proximal pausing (Chipumuro et al., 2014; Glover-Cutter et al., 2009; Kelso et al., 2014; Kwiatkowski et al., 2014; Larochelle et al., 2012; Schwartz et al., 2003). This conflict is likely due to differences in elongation control machinery. Two recent reports described reductions in DSIF and increases in TFIIE near promoters in human cells as a result of Cdk7 inhibition (Kelso et al., 2014; Larochelle et al., 2012), but while Larochelle et al. described a concurrent loss of NELF ChIP signal, Kelso et al. found that NELF association with transcription complexes in vitro was unaffected. Curiously, Kelso et al. also showed that inhibition of Cdk7 reduced in vitro transcription but only affected 2% of expressed genes by microarray.
A newly developed covalent inhibitor of Cdk7, THZ1, negatively impacts gene expression and proliferation in cancer cell lines and animal models (Chipumuro et al., 2014; Christensen et al., 2014; Kwiatkowski et al., 2014). To elucidate the mechanism of THZ1 action we utilized an in vitro system with HeLa nuclear extract to examine how the compound affects transcription and associated events. We followed the effects of THZ1 on Pol II initiation, pausing, and productive elongation and closely examined CTD phosphorylation, capping, and elongation factor recruitment. We discovered that THZ1 causes a cascade of defects that can explain its effects in cells. This information will aid evaluation of THZ1 as a potential cancer therapy.
Results
Effects of THZ1 on transcription in vitro
To gain mechanistic insight into the effects of THZ1 we turned to an in vitro system that reconstitutes initiation, pausing, productive elongation, and coupled RNA processing (Adamson et al., 2003). We added increasing amounts of THZ1 to reactions containing HeLa nuclear extract and a CMV promoter-driven template. Preinitiation complexes were formed with a 30 minute preincubation that included the extract, template, and either THZ1 or DMSO as a control. Initiation was accomplished in a 30 second pulse with limiting α-32P-CTP. The amounts of transcripts generated were unchanged across all levels of THZ1 (Figure 1A). However, there was a dose-dependent change between 0.03 and 0.3 μM THZ1 in the pattern of transcripts that we will show later is due to a change in capping.
Figure 1. Cdk7 function is not required for initiation but is required for productive elongation and influences promoter-proximal pausing.

(A) HeLa nuclear extract, template DNA, and indicated amounts of THZ1 were preincubated for 30 min and pulsed for 30 sec with limiting α-32P-CTP. Labeled transcripts were isolated and resolved by 9% Urea-PAGE.
(B) Transcription reactions as in (A) were chased for 3 min with 500 μM CTP prior to RNA isolation. RO: run-off transcripts. 6% Urea-PAGE.
(C) Transcription reactions as in (B) except THZ1 (T), Flavopiridol (F), or both (TF) were added during the 30 min preincubation (PIC) or the 30 sec pulse. 6% Urea-PAGE.
See also Figure S1.
To evaluate the effects of THZ1 on productive elongation, pulsed complexes were chased for 3 minutes by raising the CTP to 500 μM. In this assay, transcripts that quickly reach run-off are indicative of productive elongation. Run-off transcripts were reduced by the same concentrations of THZ1 that affected the pattern of transcripts during initiation (Figure 1B). These results demonstrate that while Cdk7 may be dispensable for initiation, its activity is essential for productive elongation.
Because THZ1 reduces run-off transcription, we compared its effects with those of Flavopiridol, which blocks productive elongation by inhibiting P-TEFb. We added one or both compounds to a final concentration of 1 μM at different stages of transcription. When added during the preincubation, both compounds dramatically inhibited accumulation of run-off transcripts (Figure 1C). However, the transcripts below run-off were significantly longer with THZ1. Interestingly, the combination of both compounds produced a pattern of transcripts that was nearly identical to that of THZ1 alone. These results suggest that THZ1 disrupts normal pausing. As expected for a covalent inhibitor, 1 μM THZ1 requires more than 3 minutes to covalently inhibit Cdk7 (Figure S1A). Although THZ1 is able to partially inhibit P-TEFb in a kinase assay with 30 μM ATP (Figure S1B), this inhibition should be alleviated in transcription assays with 500 μM ATP. To determine if this was actually the case, both compounds were added during the pulse (Figure 1C). As expected, Flavopiridol still inhibited the production of run-off. THZ1 had no effect by itself or in combination with Flavopiridol, indicating that the effects of THZ1 on transcription are solely due to a covalent interaction.
Inhibition of Cdk7 blocks Pol II CTD phosphorylation during initiation
We next examined CTD phosphorylation of elongation complexes generated in the presence of THZ1 or Flavopiridol using an elongation complex electrophoretic mobility shift assay. As diagrammed in Figure 2A, elongation complexes generated on an immobilized template were high salt washed to remove factors and then restriction digested to free them from the paramagnetic beads. Complexes were detected by their nascent radioactive transcripts. We have previously shown that migration of isolated elongation complexes in a native gel is sensitive to the level of CTD phosphorylation (Cheng and Price, 2008). About 90% of the control complexes had a distinct mobility and roughly 10% ran twice as fast (Figure 2A). Initiation driven by THZ1-treated extract generated almost entirely higher mobility complexes while Flavopiridol had almost no effect. Supershifts with phospho-specific antibodies demonstrated that the CTD in the main band, but not the minor band, was phosphorylated on Ser7, Ser5, and Ser2 in control and Flavopiridol treatments. However, complexes generated in the presence of THZ1 were almost entirely unphosphorylated. Antibodies to the body of the large subunit of Pol II or to the non-phosphorylated CTD shifted all complexes regardless of treatment. These results demonstrate that THZ1 blocks essentially all CTD phosphorylation during initiation in vitro. The very small amount of complexes generated in the presence of THZ1 with mobilities between the phosphorylated and unphosphorylated bands could be due to the action of other kinases or to initiation by partially phosphorylated Pol II.
Figure 2. THZ1 inhibits Pol II CTD phosphorylation by Cdk7.

(A) Migration of control, THZ1-, and Flavopiridol-treated elongation complexes before or after incubation with indicated antibodies. Elongation complexes are resolved by 4% native gel electrophoresis and visualized by their associated labeled transcripts. Final concentrations during the pulse were 1 μM for Flavopiridol and THZ1.
(B) THZ1 was added for the indicated length of time before initiation following the protocol in A except that reactions were stopped in high salt wash buffer with EDTA. Complexes were resolved before or after incubation with the indicated antibodies. Dots and labels denote specific shifts caused by 1, 2, or 3 antibodies.
(C) Migration of control and THZ1-treated elongation complexes which were stopped by direct addition of EDTA, incubated for 3 min, isolated, and reacted with mock (−) or P-TEFb kinase (K).
(D) Migration of untreated elongation complexes which were first stopped by addition of EDTA only, wash and resuspension with an EDTA-containing low salt buffer, or addition of an EDTA-containing high salt buffer. All were then incubated for indicated times prior to isolation.
(E) Migration of elongation complexes treated with indicated amounts of THZ1 which were stopped by addition of an EDTA-containing high salt buffer or EDTA only and incubated for 30 min prior to isolation.
To determine if the effect of THZ1 on CTD phosphorylation was due to covalent inhibition, reactions were performed in which THZ1 was present for the entire 30 minute preincubation, only for the last 3 minutes, or added in with the pulse mixture at zero time (Figure 2B). CTD phosphorylation was unaltered at zero time, indicating that covalent modification is required for THZ1 to have an effect. After 3 minutes, there was a mixture of both forms which is consistent with covalent inhibition of a fraction of Cdk7, a stable component of the PIC. Inhibition of all complexes was achieved with 30 minutes of preincubation but some had intermediate mobilities. The intermediate complexes seen after 3 and 30 minutes of treatment were shifted by antibodies to Ser7p, 5p and 2p. Interestingly, discrete bands formed suggesting the binding of one, two, three, or more antibodies. Because these discrete bands have the same mobilities regardless of the antibody used, the overall magnitude of any supershift is related to the number of available antibody binding sites. We therefore infer from the mobilities of supershifted control complexes in Figures 2A and 2B that the Pol II CTD is primarily phosphorylated on Ser5 with less on Ser7 and even less on Ser2.
We noticed that a fraction of the untreated control complexes had mobilities and antibody reactivities consistent with a lack of CTD phosphorylation in Figure 2A, but not 2B. The presence of these rapidly migrating complexes was traced to the way pulse reactions were stopped and the length of time before elongation complexes were isolated. The rapidly migrating fraction was recapitulated by stopping control complexes with concentrated EDTA and incubating for 3 minutes before isolation (Figure 2C). Both these and THZ1-treated complexes could be completely shifted back to the slowly migrating form by phosphorylation with P-TEFb. Incubating EDTA-stopped complexes for 3, 10, or 30 minutes led to increasing levels of the high mobility unphosphorylated form (Figure 2D). This activity remained even after washing with low salt. However, using a high salt wash buffer containing EDTA to stop reactions as in Figure 2B eliminated this activity. This behavior is best explained by the salt-sensitive association of a magnesium-independent phosphatase with the elongation complexes.
To determine if loss of CTD phosphorylation after THZ1 treatment was affected by this phosphatase, we added increasing amounts of THZ1 to transcription reactions and stopped with high salt to eliminate, or EDTA alone to allow phosphatase activity over a subsequent 30 minute incubation. Even in the absence of phosphatase activity, THZ1 caused loss of CTD phosphorylation on the majority of elongation complexes (Figure 2E). A small fraction was still partially phosphorylated and these were converted to the most rapidly migrating form by the phosphatase activity. Notably, 30 minutes of 0.1 μM THZ1 led to a mixture of phosphorylated and unphosphorylated complexes similar to what was found after 3 minutes of 1 μM THZ1 in Figure 2B. This non-distributive loss of phosphorylation again suggests that the Cdk7 present in TFIIH in preinitiation complexes rather than soluble CAK was responsible for CTD phosphorylation. While the phosphatase had only a small effect on CTD phosphorylation during initiation, it could be involved in later stages of transcription.
THZ1 inhibits early, efficient capping
Next, we addressed the effect of THZ1 on capping. As diagrammed in Figure 3A, complexes were pulsed for 45 seconds with limiting CTP to generate a diverse pattern of transcripts whose cap status was assessed. Capped transcripts have one less negative charge and one additional nucleoside, thus lowering their mobility in denaturing gels. To confirm cap status, transcripts were isolated, treated with the human cap methyltransferase and SAM to mature existing caps, and then captured with anti-m2,2,7G antibody beads. G16 was uncapped in both reactions because its 5′ end is still protected within Pol II (Figure 3A). In the control reaction, transcripts ending in G21 were partially capped and all larger transcripts were almost entirely capped as indicated by their mobilities and their retention by the anti-cap beads. As was seen in Figure 1A, THZ1 treatment abolished capping of G21 transcripts and reduced the capping of longer transcripts.
Figure 3. THZ1 alters mRNA capping through multiple mechanisms.

(A) Cap status determination after 30 min preincubation ± THZ1, 45 sec limiting CTP pulse, high salt wash, and optional incubation with 1 pmole HCE for 1 min. Final concentration during the pulse was 1 μM for THZ1. Below: transcript sequence with limiting CTP stops underlined. C: capped transcripts recovered with anti-m2,2,7G beads. U: uncapped transcripts found in supernatant. 9% Urea-PAGE.
(B) High salt washed elongation complexes generated as in (A) were incubated for 3 min with indicated amounts of HCE.
(C) Complexes generated as in (A) except with a low salt wash step were incubated for 3 min with indicated amounts of HCE.
(D) Complexes generated as in (A) except indicated amounts of HCE were added 20 min before initiation of transcription by pulse.
See also Figure S2.
To determine if the THZ1-dependent loss of capping of G21 and longer transcripts was due to altered CTD phosphorylation or associated factors, pulsed complexes were high salt washed and then incubated with recombinant HCE and GTP for 1 minute. G16 remained uncapped but G21 was now completely capped for both control and THZ1 complexes (Figure 3A). This suggests that the inefficient capping of G21 observed during pulse reactions is modulated by a factor that was removed by high salt wash. Longer nascent transcripts (U25, A31, G49-56) were already capped in the control elongation complexes but these transcripts, which were poorly capped during the THZ1 pulse, were still only partially capped by recombinant HCE and the longer transcripts were capped more efficiently than U25. These data demonstrate that 21 nucleotides is intrinsically the preferred length for efficient capping, but capping at this position may normally be down-modulated by a factor associated with the elongation complex. It seems likely this factor could be involved in the THZ1-dependent increase in inhibition of capping of G21.
To further examine recruitment of the capping enzyme, HCE was titrated over a range spanning the concentration of high salt washed Pol II complexes. About 30 femtomoles of elongation complexes were generated in the presence or absence of THZ1, high salt washed, and incubated for 3 minutes with 3 to 300 femtomoles of recombinant HCE. Substoichiometric addition of capping enzyme to control complexes was able to complete G21 capping (Figure 3B). Approximately 3-fold more HCE was needed to completely cap G21 transcripts associated with complexes lacking CTD phosphorylation. This is quantitatively similar to our previous observations where CTD phosphorylation had only a 4-fold effect on coupled capping activity (Moteki and Price, 2002). As seen in Figure 3A for THZ1-treated complexes, HCE efficiency was transcript length-dependent. These results support the idea that capping is most efficient at the G21 position in the absence of additional factors. They also demonstrate that CTD phosphorylation stimulates the function of the capping enzyme in the absence of other factors about 3-fold.
To assess the influence of factors associated with the elongation complex on the activity of capping enzyme, increasing amounts of HCE were added to low salt washed complexes. About one third of the G21 transcripts in control complexes were resistant to levels of HCE 1,000 times greater than that needed to completely cap high salt washed complexes (Figure 3C). These results suggest that a factor that inhibits capping of G21 was retained by a fraction of the complexes after low salt wash. THZ1 complexes behaved in a similar manner with about a third of the complexes being resistant to the highest level of HCE tested. Because THZ1 did not increase the association of the capping inhibitor, there must be additional factors or processes involved in the THZ1-mediated inhibition of capping.
To examine the efficiency of capping on complexes that were not washed at all, we titrated capping enzyme into the preinitiation complex assembly reactions. Capping of G21 during the pulse reaction for control and THZ1-treated complexes was resistant to all but the highest concentrations of HCE (Figure 3D). Based on the amount of capping enzyme needed, the efficiency of capping of G21 during the pulse was again about 3 orders of magnitude less efficient than observed with high salt washed complexes. THZ1-treated complexes were more responsive to capping at U25 and U31 than at position G21, suggesting the inhibitor functions mainly at G21. It is interesting that a fraction of the G21 transcripts are capped with the very low levels of HCE in extract, but the rest are resistant to orders of magnitude increases in HCE. This suggests that loss of the capping inhibitor at G21 may be part of an ordered exchange of factors that normally occurs during progression to the promoter proximal paused state and this transition is blocked by THZ1.
To determine if this inhibitor of capping is a required initiation factor, we supplemented a defined transcription system containing TFIIB, TBP, TFIIE, TFIIF, TFIIH, and Pol II with HCE and analyzed the transcripts produced in a pulse. Because these reactions lack DSIF and NELF, the concentration of NTPs was lowered to achieve a similar elongation rate in the absence of factor-dependent pausing. The extent of capping was dependent on the amount of capping enzyme present but unlike transcription in the complete system, THZ1 did not block capping at or near G21 (Figure S2). Evidently, the inhibitor of early capping is not present in the defined system. It did take three times more capping enzyme in THZ1 compared to control reactions to achieve similar levels of capping and this is likely due to CTD phosphorylation, as discussed above.
Taken together, these results indicate that in the absence of additional factors, the nascent transcript is capped with high efficiency just as it emerges from the RNA exit channel at G21. However, capping at G21 is several orders of magnitude less efficient in the presence of extract which strongly suggests there is a factor that inhibits capping at this site. If capping at this “sweet spot” is missed or blocked, such as in the presence of THZ1, HCE must wait for transcripts to become longer before capping can occur and this capping is less efficient. The mechanism of THZ1 inhibition of capping at G21 likely involves an enhancement of the function of the capping inhibitor but how that is achieved in not yet clear. Loss of CTD phosphorylation may be involved in this mechanism but its small direct effect on capping cannot explain the large effect seen in the presence extract without evoking an additional unknown factor.
THZ1 effects on pausing are due to defective recruitment of DSIF and NELF, not impaired capping
While it is very clear that THZ1 affects capping, CTD phosphorylation, pausing, and productive elongation, we do not know if these effects are functionally coupled or arise from parallel pathways that may have common intermediates. To determine if loss of capping is causative or merely correlative with effects on pausing, we performed pulse/chase assays in extract with Flavopiridol to block productive elongation and examined the pattern of total, capped and uncapped transcripts. Control complexes were all paused and essentially all transcripts were capped (Figure 4A–D). Complexes treated with THZ1 generated longer transcripts overall but interestingly, half of the nascent transcripts were capped and these paused normally (Figure 4C); the uncapped transcripts were significantly longer (Figure 4D). This is consistent with capping being required for pausing. However, we discovered that 2 mM H2O2 inhibited capping without affecting pausing. This effect is best visualized after normalization to account for a slight inhibition of initiation by H2O2 (Figure 4E). We do not know how H2O2 blocks capping, but the same concentration of H2O2 did not affect CTD phosphorylation or the phosphatase activity (Figure S3A) nor did it influence the ability of HCE to cap G21 in high salt washed complexes (Figure S3B). Regardless of the mechanism of cap inhibition by H2O2, we conclude that capping is not required for pausing. However, as all capped RNAs were associated with complexes which paused normally, it is possible that capping and pausing share a Cdk7-dependent requirement.
Figure 4. THZ1-induced pause defect is independent of mRNA capping.

(A) Cap status determination of transcripts generated by a 30 min preincubation with Flavopiridol alone (Control; black) or in combination with THZ1 (red) or H2O2 (teal), 30 second limiting UTP/CTP pulse, and 3 min chase. Final concentrations during the chase were 1.3 μM for Flavopiridol and THZ1, or 2 mM for H2O2. T: total transcripts. C: capped. U: uncapped. 6% Urea-PAGE.
(B–D) Profiles from the indicated region (bracket with arrow) of total (B), capped (C), and uncapped (D) transcripts. The vertical axis height of (B) is double that of (C and D).
(E) Profiles from (B) were normalized by equalizing the areas under each curve.
See also Figure S3.
To determine if DSIF, NELF, or Gdown1 are involved in the effect of THZ1 on pausing, DSIF and NELF, NELF alone, or Gdown1 were added to control or THZ1-treated complexes after a high or low salt wash, and elongation was continued for 3 minutes. Flavopiridol was again used to block the transition into productive elongation in all reactions. Regardless of THZ1 treatment, DSIF and NELF, but not NELF alone, slowed elongation of high salt washed complexes (Figure 5) while Gdown1 had no significant effect on control or THZ1 complexes. Much of the effect of THZ1 on increasing the length of transcripts in the presence of extract (Figure 1C) was also seen after low salt wash (Figure 5A–B). Addition of DSIF and NELF or NELF alone to control complexes caused a significant increase in pausing suggesting that DSIF, but not NELF, survived the low salt wash (Figure 5A, C–D). Effects of DSIF and NELF and especially NELF alone on low salt washed THZ1 complexes were significantly blunted (Figure 5A, 5C–D). This strongly suggests that DSIF was not properly loaded and, in fact, the THZ1 complexes were highly resistant to addition of DSIF and NELF. Gdown1 had the expected negative effect on low salt washed control complexes due to the presence of GNAF (Cheng et al., 2012) and this effect was seen regardless of THZ1 (Figure 5A, E). The THZ1-dependent inhibition of DSIF loading suggests retention of a blocking factor that may or may not be the inhibitor of capping.
Figure 5. THZ1-treated complexes are resistant to pausing by DSIF and NELF.

(A) Elongation complexes were generated by preincubation with Flavopiridol alone (Control) or in combination with THZ1 and a 30 sec limiting UTP/CTP pulse. These were then isolated by high or low salt wash, incubated for 5 min with mock (−), DSIF and NELF (DN), NELF only (N), or Gdown1 (G) add-backs, and chased for 3 min. Final concentrations during the chase were 1 μM Flavopiridol and THZ1. 6% Urea-PAGE.
(B) Profiles from the indicated region (bracket with arrow) of control (black) or THZ1 (red) low salt wash mock add-backs.
(C–E) Profiles of control (left) or THZ1 (right) low salt wash mock (black) add-backs compared with DSIF/NELF (orange) (C), NELF only (pink) (D), or Gdown1 (green) (E) add-backs. (C–E) have the same vertical axis heights.
Effects of THZ1 seen in vitro also occur in cells
If THZ1 inhibits productive elongation in cells as seen in vitro (Figure 1B), it should lead to release of P-TEFb from the 7SK snRNP (Guo and Price, 2013; Liu et al., 2014). We performed glycerol gradient analysis on lysates from control and THZ1-treated HeLa cells and found that treatment with 200 nM THZ1 for 1 hour led to a dramatic increase in P-TEFb not associated with the 7SK snRNP (Figure S4A).
Nuclear run-on experiments were performed to determine if capping and pausing were also affected by THZ1 in cells. Nuclei were isolated from cells that were treated for 1 hour with DMSO, 1 μM Flavopiridol, or 1 μM THZ1 which was determined to significantly reduce CTD phosphorylation in cells (Figure S4B). Sarkosyl was used to eliminate effects of negative factors and histones during the short run-on reactions so that paused Pol II could be detected. Only α-32P-CTP was added to limit elongation to polymerases with C as their next base and after cap methylation, guanylylated transcripts were separated from uncapped transcripts by anti-m2,2,7G beads. In this experiment, the efficiency of binding to the beads was nearly quantitative as demonstrated by phosphorimaging of control pulse lanes and recovery of snRNAs with appropriate cap structures (Figure 6A). Amanitin-sensitive Pol II transcript profiles were generated by taking differences of lanes with and without α-amanitin; total Pol II profiles were made by adding capped and uncapped Pol II profiles (Figure 6B). Control nuclei had transcripts of the appropriate size (30–50 nt) for promoter proximal pausing and a large fraction of these were capped. Flavopiridol treatment resulted in increases to both the amount of pausing, as expected, and the fraction of capped transcripts. An apparent reduction of short transcripts was seen after THZ1 treatment, indicating a reduction of promoter proximal pausing, and the remaining transcripts were mostly uncapped. Similar results were obtained using different cells and a more traditional nuclear run-on protocol (Figure S5). In Figure 6, a population of uncapped nascent transcripts was observed below 25 nucleotides and these RNAs were unresponsive to calf intestinal alkaline phosphatase or high amounts of HCE (data not shown), suggesting that they are the result of 5′ degradation to the edge of the polymerase. Overall, we conclude that THZ1 leads to reduced pausing and capping both in vitro and in cells.
Figure 6. THZ1 inhibits proper mRNA capping and Pol II elongation in cells.

(A) Cap status determination of transcripts generated by 30 sec pulse with limiting α-32P-CTP or by nuclear run-ons performed only with α-32P-CTP in the absence or presence of 4 μg/ml α-amanitin using nuclei from HeLa cells treated 1 hr with DMSO (black), 1 μM Flavopiridol (blue), or 1 μM THZ1 (red). snRNAs with (U2, U1, U4, U5) and without (5.8S, U6) trimethyl-guanosine cap structures are indicated. 9% Urea-PAGE.
(B) Profiles from the indicated region (bracket with arrow) of Pol II transcripts generated by taking the difference between lanes with or without α-amanitin. Total profiles were generated by combining capped and uncapped lanes. All plots have the same vertical axis height.
See also Figures S4 and S5.
Discussion
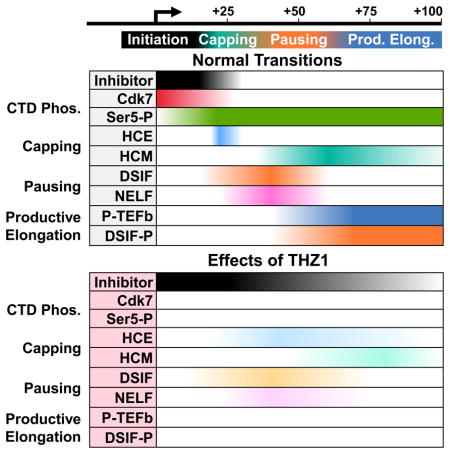
The Cdk7 inhibitor THZ1 has opened a new window through which to observe mRNA capping and the early events of Pol II elongation control. Our findings help to further define the timing of enzymatic activities and the required exchange of factors that results in capping and promoter proximal pausing (Figure 7). We propose that Cdk7 is essential for the timely execution of these transitions. Normally, CTD phosphorylation occurs swiftly and is followed by capping, pausing by DSIF and NELF, and the P-TEFb-dependent transition into productive elongation. After THZ1 treatment, CTD phosphorylation does not occur and early capping and loading of DSIF and NELF is defective. Lack of CTD phosphorylation did not affect pausing by DSIF and NELF and had only a small effect on capping using recombinant proteins on high salt washed complexes. Therefore, the major effects seen must be caused by other factors present in nuclear extract. The loss of DSIF is sufficient to inhibit productive elongation because DSIF is the functional target of P-TEFb. The sum of these disruptions in an otherwise ordered transcription system results in the reductions of capping, pausing, and productive elongation we see in cells and explains the broad impacts in promoter occupancy and gene expression noted in other recent studies (Chipumuro et al., 2014; Christensen et al., 2014; Kwiatkowski et al., 2014).
Figure 7. The role of Cdk7 in CTD phosphorylation, capping, pausing, and productive elongation.

The diagram illustrates the cascade of events dependent on Cdk7 activity (top panel) and the effects of Cdk7 inhibition by THZ1 (bottom panel). Relative CTD phosphorylation is depicted by color intensity (red > pink > light pink). An unknown factor (or factors) present in extract which blocks capping and DSIF loading is shown.
Extensive Cdk7-dependent CTD phosphorylation was observed within 30 seconds of initiation in vitro and THZ1 almost completely inhibited this phosphorylation. In addition to Ser5 and 7, we detected a lower amount of CTD phosphorylation on Ser2. This early Ser2 phosphorylation could be due to residual phosphorylation of Pol II before initiation, direct Cdk7 activity, or phosphorylation by another Ser2-specific CTD kinase. Cdk9 is unlikely to be responsible for the bulk of this phosphorylation as some Ser2 phosphorylation remained in vitro after Flavopiridol treatment (Figure 2A). While Cdk12 could be involved because it can be covalently modified by THZ1 (Kwiatkowski et al., 2014), inhibition of Cdk12 by THZ1 would require longer times and/or higher concentrations of the compound than required for its preferred target, Cdk7. Furthermore, most of the defects in CTD phosphorylation and gene expression in THZ1-treated cells was restored by expressing THZ1-resistent Cdk7 which would not rescue Cdk12 (Kwiatkowski et al., 2014). The low level of Ser2 phosphorylation of early elongation complexes observed is probably not significant since most Ser2 phosphorylation occurs near the 3′ end of genes (Rahl et al., 2010). We detected a magnesium-independent CTD phosphatase associated with elongation complexes which can dephosphorylate these complexes in an apparently processive manner. Broadly acting Fcp1 and Scp-family CTD phosphatases require magnesium to function (Kamenski et al., 2004), but Rtr1/RPAP2 (Hsu et al., 2014) and Ssu72 (Xiang et al., 2010) may not. However, Rtr1/RPAP2 and Ssu72 prefer to act on Ser5 residues while the activity we detected apparently removes phosphates from Ser7, 5 and 2 positions.
After THZ1 treatment, we observed salt-sensitive inhibitors of capping and DSIF loading. These could be initiation factors that are inappropriately retained by the transcription complex in the absence of CTD phosphorylation. One plausible candidate for the pausing inhibitor is TFIIE as its archaeal homolog TFE competes with DSIF for binding to archaeal Pol II (Grohmann et al., 2011; Martinez-Rucobo et al., 2011) and its retention increases after Cdk7 inhibition in human cells (Kelso et al., 2014; Larochelle et al., 2012). TFIIE is also a direct target for Cdk7 phosphorylation in vitro (Larochelle et al., 2012). Multiple lines of evidence suggest that Mediator could be involved. In mammals, the interaction of TFIIE and TFIIH with the preinitiation complex is stabilized by Mediator (Poss et al., 2013). A very recent structural study demonstrated that the yeast capping enzymes interact directly with the RNA exit channel and Rpb4/7 (Martinez-Rucobo et al., 2015) and this region of Pol II is normally occupied by Mediator during initiation (Plaschka et al., 2015). Importantly, the complex interaction of Mediator with Pol II is regulated in part by CTD phosphorylation (Jeronimo and Robert, 2014; Wong et al., 2014). Occam’s razor would suggest that the inhibitors of pausing and capping are the same factor. Additionally, this could explain why CTD phosphorylation has only a minor direct effect on capping of transcripts in isolated elongation complexes but a larger effect after initiation in extract with Mediator. Overall, we believe an inhibitor of capping and pausing, potentially Mediator, is present during initiation and normally requires Cdk7 activity to dissociate near G21. This departure could then enable DSIF loading which would allow NELF-mediated pausing, cap addition as transcripts emerge from the Pol II exit channel, and ultimately productive elongation.
Our results help explain the characteristic effects of THZ1 seen in cells and this information highlights how THZ1 might be useful in cancer treatment. The devastating effects of THZ1 on transcription we see in vitro would be partially mitigated by using lower concentrations of the compound. Of 1,151 cancer cell lines tested, THZ1 exhibited antiproliferative effects on more than half at concentrations below 200 nM (Kwiatkowski et al., 2014). Using a sub-saturating amount of the covalent inhibitor would ensure that some active Cdk7 would remain in each cell to drive sufficient transcription. An effective treatment would have to balance treatment time, the concentration of the compound, and the relative amount of compound compared to its target. -IC50s alone cannot be used as while initial non-covalent binding is concentration-dependent, covalent bond formation is slow and its success depends primarily on the off rate of the compound before covalent linkage. THZ1 appears to achieve its effect on cancer cells by reducing expression of one or a few transcription factors required for cell proliferation, such as RUNX1 in T-cell acute lymphoblastic leukemia (Kwiatkowski et al., 2014), MYCN in neuroblastoma (Chipumuro et al., 2014), and MYC family members and neuroendocrine lineage-specific factors in small cell lung cancer (Christensen et al., 2014). The common thread is that all target genes are driven by super-enhancers that normally function as amplifiers of cell-specific transcription factors in rapidly growing cells. Super-enhancers are built upon regions of the genome containing paused Pol II (Arner et al., 2015; Core et al., 2014) and we suggest that THZ1-induced pause defects lead to reduced occupancy of Pol II and the concomitant reduction in transcription factors in these enhancers. If super-enhancers amplify gene expression through the multiplicative effect of a large number of occupied sub-enhancers, impairing each sub-enhancer slightly could have a significantly larger overall effect on target gene expression. In this way, THZ1 could selectively target the super-enhancer-driven transcriptional addiction seen in many cancer cells.
Experimental Procedures
In vitro transcription
CMV templates −800 to +175 (Figure 2) or +508 (Figures 1, 3–5) and HeLa nuclear extract (HNE) described previously (Adamson et al., 2003; Cheng and Price, 2007, 2009) were incubated for 30 min in the presence of 60 mM KCl, 5 mM MgCl2, 20 mM HEPES pH 7.6, 1 mM DTT, and 0.5 U/μl SUPERase-In (Ambion AM2696) or 1 U/μl RNaseOUT (Invitrogen 10777-019). In Figure 4, 1 mM DTT was replaced with 3 mM H2O2 as indicated (2 mM final during the chase). Initiation was accomplished with a 30 or 45 sec pulse containing 60 mM KCl, 5 mM MgCl2, 20 mM HEPES pH 7.6, 0.21 μM α-32P-CTP, and 500 μM ATP/UTP/GTP (limiting CTP) or 500 μM ATP/GTP 0.1 μM UTP (limiting UTP/CTP). Elongation complexes were either stopped by addition of EDTA to 20 mM or chased for 3 min with 500 μM CTP. Before chase, immobilized complexes were optionally isolated with high salt wash (1.6 M KCl, 20 mM HEPES pH 7.6, 1 mM DTT, and 0.02% Tween20) or low salt wash (60 mM KCl, 20 mM HEPES pH 7.6, 1 mM DTT, 0.2 mg/ml BSA (New England BioLabs B9000), and 0.02% Tween20). Labeled transcripts were phenol extracted, ethanol precipitated and separated on denaturing RNA gels.
EC-EMSA
EC-EMSAs were described previously (Cheng and Price, 2008). Elongation complexes were formed as described above using a 30 sec limiting CTP (Figure 2A) or limiting UTP/CTP (Figure 2B–E) pulse. Reactions were stopped by direct addition of EDTA to a final concentration of 20 mM or after a low salt wash with EDTA or by addition of high salt wash with EDTA. Complexes were isolated by high and low salt washes and beads were detached by restriction digestion before antibody incubation. Phosphorylation of elongation complexes was accomplished during a 10 min incubation with P-TEFb (Cheng and Price, 2007) and 1 mM ATP. Elongation complexes were analyzed by native gel electrophoresis and phosphorimaging.
Factor add-backs
Recombinant human capping enzyme (Moteki and Price, 2002), recombinant DSIF (Renner et al., 2001), affinity purified NELF (Renner et al., 2001), and recombinant Gdown1 (Cheng et al., 2012) were purified as described previously. Factors were diluted in low salt wash buffer before being added back to reactions.
Transcript cap status determination
Cap status determination using anti-2,2,7-trimethylguanosine agarose beads (Calbiochem NA02A) and recombinant human cap methyltransferase (HCM) was described previously (Moteki and Price, 2002). The antibody beads were blocked with yeast RNA and BSA and extensively washed before a 2–4 hr incubation with isolated transcripts that had been subjected to methylation conditions. For Figure 6, purified anti-2,2,7-trimethylguanosine antibody was used without yeast RNA. After washing, total, bound, and unbound fractions were spiked with glycogen and phenol extracted, precipitated, and analyzed as described above.
Nuclear run-on
HeLa cells were grown in suspension. Before harvesting, DMSO optionally containing Flavopiridol or THZ1 was added to 1 μM final and cells were incubated for an additional hour. Cells were pelleted, lysed, and spun through a sucrose cushion to isolate nuclei. Pellets were resuspended in a storage buffer and frozen in aliquots at −80°. 10 6 nuclei were incubated for 3 min at 30° in an equal volume of 10 mM Tris pH 7.6, 1% sarkosyl, 300 mM KC2H3O2, 5 mM Mg(C2H3O2)2, 5 mM DTT, 0.5 U/μl SUPERase-In, 0.33 μM α-32P-CTP, and optionally 4 μg/ml α-amanitin. Reactions were stopped by addition of EDTA, extracted with Trizol LS (Ambion 10296-028), and RNA was precipitated and assayed for capping as described above.
Supplementary Material
Acknowledgments
THZ1 was generously supplied by Tinghu Zhang and Nathanael S. Gray. This research was supported by National Institutes of Health R01 GM35500-29 to DHP, National Science Foundation Grant 1121210 to DSL, American Heart Association Postdoctoral Fellowship 12POST12040106 to JG, and a University of Iowa Presidential Graduate Fellowship to KAN.
Footnotes
Author Contributions
KAN generated Figures 3–7, S1, S3B, and S5. JG generated Figures 1, 2A, and S4B and performed extensive preliminary work. MET generated Figures 2B–E and S3A. JEB generated Figure S4A. ED generated Figure S2. DHP directed the research effort. KAN and DHP produced the manuscript with extensive input from DSL and in consultation with all authors.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adamson TE, Shore SM, Price DH. Analysis of RNA polymerase II elongation in vitro. Methods Enzymol. 2003;371:264–275. doi: 10.1016/S0076-6879(03)71019-2. [DOI] [PubMed] [Google Scholar]
- Akoulitchev S, Makela TP, Weinberg RA, Reinberg D. Requirement for TFIIH kinase activity in transcription by RNA polymerase II. Nature. 1995;377:557–560. doi: 10.1038/377557a0. [DOI] [PubMed] [Google Scholar]
- Bartkowiak B, Liu P, Phatnani HP, Fuda NJ, Cooper JJ, Price DH, Adelman K, Lis JT, Greenleaf AL. CDK12 is a transcription elongation-associated CTD kinase, the metazoan ortholog of yeast Ctk1. Genes Dev. 2010;24:2303–2316. doi: 10.1101/gad.1968210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bataille AR, Jeronimo C, Jacques PE, Laramee L, Fortin ME, Forest A, Bergeron M, Hanes SD, Robert F. A universal RNA polymerase II CTD cycle is orchestrated by complex interplays between kinase, phosphatase, and isomerase enzymes along genes. Mol Cell. 2012;45:158–170. doi: 10.1016/j.molcel.2011.11.024. [DOI] [PubMed] [Google Scholar]
- Bentley DL. Coupling mRNA processing with transcription in time and space. Nat Rev Genet. 2014;15:163–175. doi: 10.1038/nrg3662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman EA, Kelly WG. RNA polymerase II transcription elongation and Pol II CTD Ser2 phosphorylation: A tail of two kinases. Nucleus. 2014;5:224–236. doi: 10.4161/nucl.29347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng B, Li T, Rahl PB, Adamson TE, Loudas NB, Guo J, Varzavand K, Cooper JJ, Hu X, Gnatt A, et al. Functional association of Gdown1 with RNA polymerase II poised on human genes. Mol Cell. 2012;45:38–50. doi: 10.1016/j.molcel.2011.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng B, Price DH. Properties of RNA polymerase II elongation complexes before and after the P-TEFb-mediated transition into productive elongation. J Biol Chem. 2007;282:21901–21912. doi: 10.1074/jbc.M702936200. [DOI] [PubMed] [Google Scholar]
- Cheng B, Price DH. Analysis of factor interactions with RNA polymerase II elongation complexes using a new electrophoretic mobility shift assay. Nucleic Acids Res. 2008;36:e135. doi: 10.1093/nar/gkn630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng B, Price DH. Isolation and functional analysis of RNA polymerase II elongation complexes. Methods. 2009;48:346–352. doi: 10.1016/j.ymeth.2009.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chipumuro E, Marco E, Christensen CL, Kwiatkowski N, Zhang T, Hatheway CM, Abraham BJ, Sharma B, Yeung C, Altabef A, et al. CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell. 2014;159:1126–1139. doi: 10.1016/j.cell.2014.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu YL, Ho CK, Saha N, Schwer B, Shuman S, Rana TM. Tat stimulates cotranscriptional capping of HIV mRNA. Mol Cell. 2002;10:585–597. doi: 10.1016/s1097-2765(02)00630-5. [DOI] [PubMed] [Google Scholar]
- Cho EJ, Takagi T, Moore CR, Buratowski S. mRNA capping enzyme is recruited to the transcription complex by phosphorylation of the RNA polymerase II carboxy-terminal domain. Genes Dev. 1997;11:3319–3326. doi: 10.1101/gad.11.24.3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen CL, Kwiatkowski N, Abraham BJ, Carretero J, Al-Shahrour F, Zhang T, Chipumuro E, Herter-Sprie GS, Akbay EA, Altabef A, et al. Targeting transcriptional addictions in small cell lung cancer with a covalent CDK7 inhibitor. Cancer Cell. 2014;26:909–922. doi: 10.1016/j.ccell.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppola JA, Field AS, Luse DS. Promoter-proximal pausing by RNA polymerase II in vitro: transcripts shorter than 20 nucleotides are not capped. Proc Natl Acad Sci U S A. 1983;80:1251–1255. doi: 10.1073/pnas.80.5.1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doamekpor SK, Sanchez AM, Schwer B, Shuman S, Lima CD. How an mRNA capping enzyme reads distinct RNA polymerase II and Spt5 CTD phosphorylation codes. Genes Dev. 2014;28:1323–1336. doi: 10.1101/gad.242768.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doamekpor SK, Schwer B, Sanchez AM, Shuman S, Lima CD. Fission yeast RNA triphosphatase reads an Spt5 CTD code. RNA. 2015;21:113–123. doi: 10.1261/rna.048181.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egly JM, Coin F. A history of TFIIH: two decades of molecular biology on a pivotal transcription/repair factor. DNA Repair (Amst) 2011;10:714–721. doi: 10.1016/j.dnarep.2011.04.021. [DOI] [PubMed] [Google Scholar]
- Fisher RP. The CDK Network: Linking Cycles of Cell Division and Gene Expression. Genes Cancer. 2012;3:731–738. doi: 10.1177/1947601912473308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A, Shuman S, Lima CD. Structural insights to how mammalian capping enzyme reads the CTD code. Mol Cell. 2011;43:299–310. doi: 10.1016/j.molcel.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover-Cutter K, Larochelle S, Erickson B, Zhang C, Shokat K, Fisher RP, Bentley DL. TFIIH-associated Cdk7 kinase functions in phosphorylation of C-terminal domain Ser7 residues, promoter-proximal pausing, and termination by RNA polymerase II. Mol Cell Biol. 2009;29:5455–5464. doi: 10.1128/MCB.00637-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grohmann D, Nagy J, Chakraborty A, Klose D, Fielden D, Ebright RH, Michaelis J, Werner F. The initiation factor TFE and the elongation factor Spt4/5 compete for the RNAP clamp during transcription initiation and elongation. Mol Cell. 2011;43:263–274. doi: 10.1016/j.molcel.2011.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J, Price DH. RNA polymerase II transcription elongation control. Chem Rev. 2013;113:8583–8603. doi: 10.1021/cr400105n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidemann M, Hintermair C, Voss K, Eick D. Dynamic phosphorylation patterns of RNA polymerase II CTD during transcription. Biochim Biophys Acta. 2013;1829:55–62. doi: 10.1016/j.bbagrm.2012.08.013. [DOI] [PubMed] [Google Scholar]
- Ho CK, Shuman S. Distinct roles for CTD Ser-2 and Ser-5 phosphorylation in the recruitment and allosteric activation of mammalian mRNA capping enzyme. Mol Cell. 1999;3:405–411. doi: 10.1016/s1097-2765(00)80468-2. [DOI] [PubMed] [Google Scholar]
- Hong SW, Hong SM, Yoo JW, Lee YC, Kim S, Lis JT, Lee DK. Phosphorylation of the RNA polymerase II C-terminal domain by TFIIH kinase is not essential for transcription of Saccharomyces cerevisiae genome. Proc Natl Acad Sci U S A. 2009;106:14276–14280. doi: 10.1073/pnas.0903642106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PL, Yang F, Smith-Kinnaman W, Yang W, Song JE, Mosley AL, Varani G. Rtr1 is a dual specificity phosphatase that dephosphorylates Tyr1 and Ser5 on the RNA polymerase II CTD. J Mol Biol. 2014;426:2970–2981. doi: 10.1016/j.jmb.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeronimo C, Robert F. Kin28 regulates the transient association of Mediator with core promoters. Nat Struct Mol Biol. 2014;21:449–455. doi: 10.1038/nsmb.2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamenski T, Heilmeier S, Meinhart A, Cramer P. Structure and mechanism of RNA polymerase II CTD phosphatases. Mol Cell. 2004;15:399–407. doi: 10.1016/j.molcel.2004.06.035. [DOI] [PubMed] [Google Scholar]
- Kanin EI, Kipp RT, Kung C, Slattery M, Viale A, Hahn S, Shokat KM, Ansari AZ. Chemical inhibition of the TFIIH-associated kinase Cdk7/Kin28 does not impair global mRNA synthesis. Proc Natl Acad Sci U S A. 2007;104:5812–5817. doi: 10.1073/pnas.0611505104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelso TW, Baumgart K, Eickhoff J, Albert T, Antrecht C, Lemcke S, Klebl B, Meisterernst M. Cyclin-dependent kinase 7 controls mRNA synthesis by affecting stability of preinitiation complexes, leading to altered gene expression, cell cycle progression, and survival of tumor cells. Mol Cell Biol. 2014;34:3675–3688. doi: 10.1128/MCB.00595-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski N, Zhang T, Rahl PB, Abraham BJ, Reddy J, Ficarro SB, Dastur A, Amzallag A, Ramaswamy S, Tesar B, et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature. 2014;511:616–620. doi: 10.1038/nature13393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larochelle S, Amat R, Glover-Cutter K, Sanso M, Zhang C, Allen JJ, Shokat KM, Bentley DL, Fisher RP. Cyclin-dependent kinase control of the initiation-to-elongation switch of RNA polymerase II. Nat Struct Mol Biol. 2012;19:1108–1115. doi: 10.1038/nsmb.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Xiang Y, Fujinaga K, Bartholomeeusen K, Nilson KA, Price DH, Peterlin BM. Release of positive transcription elongation factor b (P-TEFb) from 7SK small nuclear ribonucleoprotein (snRNP) activates hexamethylene bisacetamide-inducible protein (HEXIM1) transcription. J Biol Chem. 2014;289:9918–9925. doi: 10.1074/jbc.M113.539015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal SS, Chu C, Wada T, Handa H, Shatkin AJ, Reinberg D. Functional interactions of RNA-capping enzyme with factors that positively and negatively regulate promoter escape by RNA polymerase II. Proc Natl Acad Sci U S A. 2004;101:7572–7577. doi: 10.1073/pnas.0401493101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall NF, Peng J, Xie Z, Price DH. Control of RNA polymerase II elongation potential by a novel carboxyl-terminal domain kinase. J Biol Chem. 1996;271:27176–27183. doi: 10.1074/jbc.271.43.27176. [DOI] [PubMed] [Google Scholar]
- Martinez-Rucobo FW, Kohler R, van de Waterbeemd M, Heck AJ, Hemann M, Herzog F, Stark H, Cramer P. Molecular Basis of Transcription-Coupled Pre-mRNA Capping. Mol Cell. 2015 doi: 10.1016/j.molcel.2015.04.004. [DOI] [PubMed] [Google Scholar]
- Martinez-Rucobo FW, Sainsbury S, Cheung AC, Cramer P. Architecture of the RNA polymerase-Spt4/5 complex and basis of universal transcription processivity. EMBO J. 2011;30:1302–1310. doi: 10.1038/emboj.2011.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCracken S, Fong N, Rosonina E, Yankulov K, Brothers G, Siderovski D, Hessel A, Foster S, Shuman S, Bentley DL. 5′-Capping enzymes are targeted to pre-mRNA by binding to the phosphorylated carboxy-terminal domain of RNA polymerase II. Genes Dev. 1997;11:3306–3318. doi: 10.1101/gad.11.24.3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moteki S, Price D. Functional coupling of capping and transcription of mRNA. Mol Cell. 2002;10:599–609. doi: 10.1016/s1097-2765(02)00660-3. [DOI] [PubMed] [Google Scholar]
- Pei Y, Shuman S. Interactions between fission yeast mRNA capping enzymes and elongation factor Spt5. J Biol Chem. 2002;277:19639–19648. doi: 10.1074/jbc.M200015200. [DOI] [PubMed] [Google Scholar]
- Plaschka C, Lariviere L, Wenzeck L, Seizl M, Hemann M, Tegunov D, Petrotchenko EV, Borchers CH, Baumeister W, Herzog F, et al. Architecture of the RNA polymerase II-Mediator core initiation complex. Nature. 2015;518:376–380. doi: 10.1038/nature14229. [DOI] [PubMed] [Google Scholar]
- Poss ZC, Ebmeier CC, Taatjes DJ. The Mediator complex and transcription regulation. Crit Rev Biochem Mol Biol. 2013;48:575–608. doi: 10.3109/10409238.2013.840259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahl PB, Lin CY, Seila AC, Flynn RA, McCuine S, Burge CB, Sharp PA, Young RA. c-Myc regulates transcriptional pause release. Cell. 2010;141:432–445. doi: 10.1016/j.cell.2010.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen EB, Lis JT. In vivo transcriptional pausing and cap formation on three Drosophila heat shock genes. Proc Natl Acad Sci U S A. 1993;90:7923–7927. doi: 10.1073/pnas.90.17.7923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renner DB, Yamaguchi Y, Wada T, Handa H, Price DH. A highly purified RNA polymerase II elongation control system. J Biol Chem. 2001;276:42601–42609. doi: 10.1074/jbc.M104967200. [DOI] [PubMed] [Google Scholar]
- Schneider S, Pei Y, Shuman S, Schwer B. Separable functions of the fission yeast Spt5 carboxyl-terminal domain (CTD) in capping enzyme binding and transcription elongation overlap with those of the RNA polymerase II CTD. Mol Cell Biol. 2010;30:2353–2364. doi: 10.1128/MCB.00116-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz BE, Larochelle S, Suter B, Lis JT. Cdk7 is required for full activation of Drosophila heat shock genes and RNA polymerase II phosphorylation in vivo. Mol Cell Biol. 2003;23:6876–6886. doi: 10.1128/MCB.23.19.6876-6886.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwer B, Shuman S. Deciphering the RNA polymerase II CTD code in fission yeast. Mol Cell. 2011;43:311–318. doi: 10.1016/j.molcel.2011.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirode F, Busso D, Coin F, Egly JM. Reconstitution of the transcription factor TFIIH: assignment of functions for the three enzymatic subunits, XPB, XPD, and cdk7. Mol Cell. 1999;3:87–95. doi: 10.1016/s1097-2765(00)80177-x. [DOI] [PubMed] [Google Scholar]
- Wen Y, Shatkin AJ. Transcription elongation factor hSPT5 stimulates mRNA capping. Genes Dev. 1999;13:1774–1779. doi: 10.1101/gad.13.14.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong KH, Jin Y, Struhl K. TFIIH phosphorylation of the Pol II CTD stimulates mediator dissociation from the preinitiation complex and promoter escape. Mol Cell. 2014;54:601–612. doi: 10.1016/j.molcel.2014.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang K, Nagaike T, Xiang S, Kilic T, Beh MM, Manley JL, Tong L. Crystal structure of the human symplekin-Ssu72-CTD phosphopeptide complex. Nature. 2010;467:729–733. doi: 10.1038/nature09391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi Y, Shibata H, Handa H. Transcription elongation factors DSIF and NELF: promoter-proximal pausing and beyond. Biochim Biophys Acta. 2013;1829:98–104. doi: 10.1016/j.bbagrm.2012.11.007. [DOI] [PubMed] [Google Scholar]
- Yue Z, Maldonado E, Pillutla R, Cho H, Reinberg D, Shatkin AJ. Mammalian capping enzyme complements mutant Saccharomyces cerevisiae lacking mRNA guanylyltransferase and selectively binds the elongating form of RNA polymerase II. Proc Natl Acad Sci U S A. 1997;94:12898–12903. doi: 10.1073/pnas.94.24.12898. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.