

Abstract
Interleukin (IL)-4 and IL-13 were discovered approximately 30 years ago and were immediately linked to allergy and atopic diseases. Since then, new roles for IL-4 and IL-13 and their receptors in normal gestation, fetal development, neurological function and in the pathogenesis of cancer and fibrosis have been appreciated. Studying IL-4/-13 and their receptors has revealed important clues about cytokine biology and lead to the development of numerous experimental therapeutics. Here we aim to highlight new discoveries and consolidate concepts in the field of IL-4 and IL-13 structure, receptor regulation, signaling and experimental therapeutics.
Keywords: interleukin-4, interleukin-13, singling, regulation, receptor
Introduction
Interleukin (IL)-4 and IL-13 were among the first cytokines described in the early 1980s. The discovery and study of their structure, receptors and signaling laid the groundwork for much of our understanding of cytokine signaling networks, receptor complexing, immune function and powerful nature of cytokines in causing and ameliorating disease. IL-4 and IL-13 are structurally and functionally related cytokines not only involved in immune function but also in pregnancy, fetal development, mammary development and lactation, as well as in higher brain functions including memory and learning (1). Perhaps more widely known is the role of IL-4 and IL-13 in the pathogenesis of atopy, asthma, pulmonary fibrosis, and cancer.
IL-4 and IL-13 are short four α-helix bundle secreted glycoproteins with about 25% sequence similarity, encoded by adjacent genes that share several cis-acting transactivating regulatory regions. Although they share receptor subunits and signaling molecules, these two cytokines elicit overlapping but also unique biological responses. IL-4 and IL-13 are secreted by Th2-polarized T cells, granulocytes and monocytes/macrophages. In the last decade, new innate immune cell populations have been identified, including type 2 invariant natural killer T cells (iNKT2) and innate lymphoid type 2 cells (iLC2) in mice and CRTH2+ type 2 ILC in humans which secrete high levels of IL-13 but only secrete IL-4 under certain circumstances (2, 3). Every cell in the body has the potential to respond to IL-4 or IL-13, or both. In the context of the immune system, IL-4 and IL-13 trigger Th2 T cells differentiation, M2 macrophage polarization, MHCII expression, B cell and plasma cell differentiation and antibody isotype switch among others. Classically considered “anti-inflammatory” cytokines by virtue of their ability to inhibit type 1 inflammation (IFN-γ, IL-12, NO), IL-4 and IL-13 do not trigger immunological senescence, rather, they initiate potent type 2 inflammatory processes. IL-4 and IL-13 signal through cell surface receptor heterodimers composed of 3 possible subunits. These 3 subunits along with a myriad of cell signaling mediators, transcription factors and regulatory elements combine to mediate the critical physiological as well as pathophysiological manifestations of these cytokines. Herein we aim to highlight new discoveries in the field of IL-4 and IL-13 receptors and signaling as well as take a prospective look at the future of targeting this pathway to improve the human health.
Overview of IL-4/-13 Receptor Assembly and Signaling
IL-4 signals through both the type I receptor composed of the IL-4Rα and common gamma chain (γC), and the type II receptor composed of the IL-4Rα and IL-13Rα1. IL-4 binds IL-4Rα in a species-specific manner with very high affinity (KD = 20–300 pM), leading to the recruitment of either the common γC or IL-13Rα1. The γC and IL-13Rα1 subunits have lower, approximately equal affinity for the IL-4:IL-4Rα complex (KD = 500 nM). Therefore, the availability of each chain on the cell surface determines the signaling pathway activated within the responding cell (4, 5). While the IL-4Rα and IL-13Rα1 chains are widely expressed at low levels on most cell types, the γC chain is primarily expressed on hematopoietic immune cells. IL-4 binding the type I receptor complex phosphorylates JAK1/3, which in turn phosphorylates tyrosines within the IL-4Rα cytoplasmic domain. These phospho-tyrosine residues create docking sites for STAT6 or/and IRS-2 (Figure 1). Although STAT3 and other STATs can also be activated in some cell types, STAT6 and IRS-2 are acknowledged as the primary pathways involved in IL-4/-13 responsiveness. STAT6 tyrosine phosphorylation promotes phospho-STAT6 homodimerization, nuclear translocation and gene transcription. Tyrosine phosphorylation of IRS-2 leads to activation of PI3-K (6–8), AKT (9–12) and NF-κB-driven (13–15) gene transcription, activation of the cell cycle, proliferation and survival (16, 17).
Figure 1. IL-4 and IL-13 receptor structure.
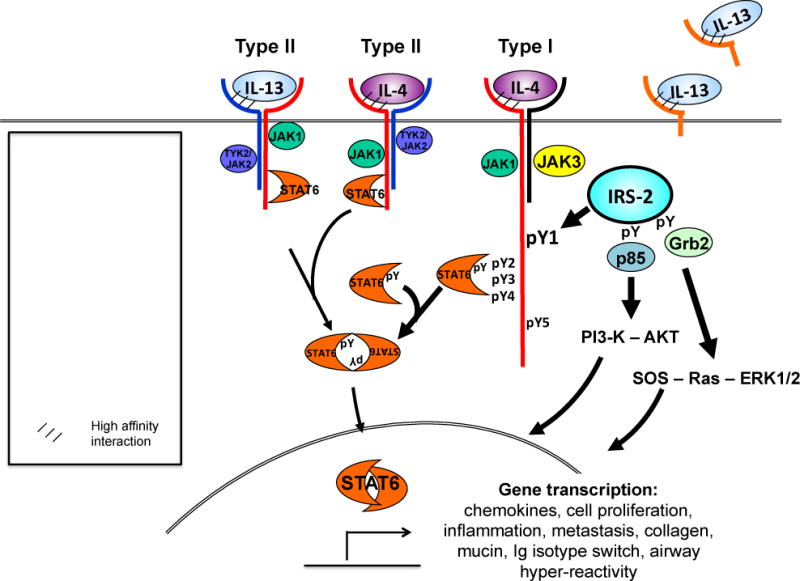
IL-4 signals through two possible receptor complexes composed of a heterodimer of the IL-4Rα (140 kDa) and γc chain (60 kDa); type I receptor or the IL-4Rα and IL-13Rα1 (65–70 kDa) chain; type II receptor. IL-4 binds IL-4Rα with high affinity, triggering dimerization with the secondary signaling chain. IL-13 binds IL-13Rα1 which complexes with the IL-4Rα, forming the type II receptor. Signaling through the type I receptor leads to activation JAKs and downstream signaling adaptor molecules STAT6 and IRS-2, whereas signaling through the type II receptor predominantly activates STAT6. IL-4 signaling also activates PI3-K and AKT. IL-13 also binds cell-surface and soluble forms of the IL-13Rα2, this so-called inhibitory subunit binds IL-13 with greater affinity than IL-13Rα1 and acts as a “cytokine sink”.
In addition to serving as a subunit of the type II IL-4 receptor, IL-13Rα1 is the ligand binding subunit for IL-13. IL-13 binds IL-13Rα1 with moderate affinity (KD = 30 nM) relative to IL-4:IL-14Rα interaction, leading to recruitment of the IL-4Rα subunit. Binding of IL-13 to the type II receptor results in activation of JAK1 or JAK2/TYK2, STAT6, STAT3 and STAT1, STAT dimerization and nuclear translocation occurs, followed by activation of gene transcription (Figure 1). Since the binding affinity of IL-4 for the IL-4Rα is markedly higher than IL-13 for the IL-13Rα1, IL-4 and IL-13 may compete for availability of the ligand binding subunits in cells expressing the type II receptor. Consequently, the concentration of IL-4 vs. IL-13 in the extracellular milieu is an important determinant of signaling through the type II receptor in non-hematopoietic cells.
IL-13 also binds the cell surface IL-13Rα2 “non-signaling” subunit. IL-13Rα2 is believed to an inhibitory subunit of the type II receptor by acting act as a “decoy” receptor for IL-13 (18). However, there is mounting evidence this may not be the case and is addressed below. In addition to cell surface-associated receptor subunits, soluble forms of the IL-4 receptor chains exist (19–21). Soluble IL-13Rα2 has been identified in mice (22, 23) but not humans (24). The discovery of soluble receptors provided important cues for cytokine neutralization therapies being investigated in pre-clinical and clinical studies (25–27).
IL-4 and IL-13 receptor expression and regulation in health and disease
IL-4 and IL-13 receptor expression in cancer
The IL-4 and IL-13 receptor subunits are expressed at low levels under homeostatic conditions and are influenced by hormones, cellular/oxidative stress, infection and inflammation (28–32). In solid tumors, such as breast cancer, high IL-4Rα expression is associated with increased cancer cell proliferation, epithelial invasion and more aggressive metastasis (33–35). Venmar et al. recently showed in human breast cancer, as well as in mouse models of metastatic breast cancer, that attenuating IL-4Rα expression reduced tumor survival as well as metastatic potential by blunting AKT, ERK and mTOR signaling (36). These findings agree with the observations of others showing that phospho-STAT6, downstream of IL-4/-13 receptor engagement, regulates pro-metastatic behaviors, such as proliferation, migration and tissue invasion (37–39). In Hodgkin/Reed-Sternberg lymphoma (HL), transformed B-cells highly expressed IL-13Rα1, secreted IL-13 and displayed over-phosphorylation of STAT6 (40). In these cells, blocking IL-4Rα and inhibiting STAT6 activation increased sensitivity to chemotherapeutics (40). In blood/bone marrow cancers such as HL, increased IL-13Rα1 (or IL-4Rα) expression may serve as a biomarker for predicting how aggressive a cancer may be as well as provide a method to monitor effectiveness of cancer therapy.
IL-4 responsiveness in the brain
Traditionally the brain is regarded as a site of immune privilege, separate from the rest of the immune system. As such, little attention has focused on investigating the role of the IL-4/-13 receptors in maintaining brain function and homeostasis. It has long been noted that T cell deficient mice have cognitive impairments and reduced adult neurogenesis, which is rescued by the adoptive transfer of Th2 polarized T cells (41–43). Aging in humans and mice is associated with decreased cognitive function that is inversely correlated with inflammation (44–46). This combined with the observations that neurological disorders such as multiple sclerosis (MS) (47, 48), Alzheimer’s disease (AD) (49, 50) and Parkinson’s disease (PD) (51) are associated with increased type 1 inflammation, suggests IL-4/-13 play an important regulatory role in the brain. Rodent models have revealed brain cells are not only sensitive to IL-4 through the expression of the IL-4 receptors, but can also produce IL-4, which is critical for optimal cognitive function and memory formation (43). Two macrophage-like cell types, astrocytes and microglial cells, not only express the IL-4Rα (but not the γc, suggesting type II receptor signaling) (52) but also produce IL-4 and possess an M2-like phenotype (53). These cells subsequently influence neuronal cells through the production of growth factors such as brain-derived neurophic factor (43), nerve growth factor (54, 55) and regulating pro-inflammatory mediators such as interferon-γ, IL-1β, IL-6 and nitric oxide (54, 56). IL-4 induced M2 skewing of microglial cells alleviates some elements of AD pathology in rodent models and promotes amyloid-β phagocytosis (57, 58). IL-4 skewing of microglia is also critical to regulate neuro-inflammation in murine experimental autoimmune encephalitis (EAE), a model of human MS (53). Adoptive transfer of M2 polarized macrophages attenuated disease symptoms (59, 60). Evidence for the importance of IL-4 in balancing autoimmune neuro-inflammation and age-associated inflammation is strong. However most of the effects of IL-4 in the brain have been restricted to astrocytes and microglia. This view was recently challenged with the finding that following CNS injury, Th2-derived IL-4 bound to the IL-4Rα on neurons. This binding not only prevented cell death but actually promoted neuronal re-growth, thus preserving cognitive function (61). This finding challenges those of other studies that suggest IL-4 in the brain solely functions to attenuate type 1 inflammation indirectly through astrocyte and glial cells. Rather it suggests IL-4 may act directly as a cyto-protective cytokine in neurons themselves. These findings are exciting and highlight several aspects of neuro-immunology that merit further investigation, particularly in the context of brain development, injury and stroke.
In contrast to evidence that the IL-4/-13 signaling axis positively influences brain function, increased expression of IL-4Rα and IL-13Rα1 has been implicated in neurodegenerative disorders notably Parkinson’s and Alzheimer’s disease. While the exact mechanisms remain unclear, the increased expression of IL-4/13 receptor subunits is believed to cause inappropriate neuronal sensitivity to IL-4/-13 (62–65). Studies in mice revealed neurons co-express the dopaminergic receptor and the IL-13Rα1, the same neurons that are lost in the progression of Parkinson’s disease (65). In this case, the IL-13Rα1 reportedly potentiated oxidative stress and cell death. It is important to note that many of these findings indicate that the IL-4/-13 receptors do not drive these disease processes alone (65). Another stressor, be it oxidative stress, a toxicant or inflammation, must also be present to drive pathology. These findings challenge our previous understanding of immune function in the brain and bring attention to a previously unappreciated role for IL-4 in maintaining homeostasis. Certainly IL-4 receptor subunits and IL-4 are necessary for normal brain development and maintaining an M2-remenicent environment, however, we are reminded that the brain exists in an exquisitely delicate balance. IL-4 signaling, while effective at attenuating type 1 inflammation, is none-the-less pro-inflammatory and aberrant receptor expression/signaling may in fact contribute to disease pathogenesis.
These new studies correlating specific IL-4/-13 receptor subunits in brain function, repair and disease in the absence of robust mechanistic studies, while very interesting, are still in their infancy. There are still many aspects of neurological IL-4 receptor expression, regulation and signaling that remain to be examined. Studying many of these aspects pose significant technical challenges and the use of animal models will be limiting. However, advances in cell isolation techniques, in vivo imaging and the development of small molecules that penetrate the blood-brain barrier suggest renewed effort into understanding immune function within the brain and central nervous system could yield important insights into neurological diseases and elucidate novel therapeutic targets. Importantly findings highlight the current lack of understanding of the mechanisms that regulate receptor expression. Moreover, it is not yet clear if these changes are the driving force behind the disease or a downstream effect of other dysregulated pathways.
Molecular regulation of receptor subunit expression
Several stimuli that increase IL-4/-13 receptor subunit expression have been described. However, the mechanisms that regulate expression of each subunit and thus regulate responsiveness to IL-4/-13 remain largely undefined. A newly identified mechanism that regulates expression of IL-4Rα under homeostatic conditions is the binding of Hepatocyte growth factor-regulated tyrosine kinase substrate (Hrs) to IL-4Rα. Hrs binding to the cytoplasmic tail of surface-bound IL-4Rα targets the complex to the late endosome, thereby limiting cell surface accumulation of the receptor (Figure 2A). This process is independent of ligand (cytokine) binding, ubiquitin-independent and appears to be a common regulatory mechanism for regulating homeostatic expression levels of other cytokine receptors including the IL-2Rβ (66, 67). Wei et al. reported a novel mechanism by which the IL-4Rα expression is regulated by STIP1 homology and U Box containing protein (STUB)1 in response to cytokine stimulation (68). The authors showed that STUB1 interacts with the IL-4Rα and facilitates IL-4Rα ubiquitination and proteasomal degradation (Figure 2B). Importantly, they show that this is a cytokine-inducible event and a critical negative regulatory process in the development of allergic asthma. While they do not address whether STUB1 expression itself is induced by IL-4/-13 stimulation, it provides mechanistic insight into how cells attempt to regulate IL-4 signaling through the down regulation of the high affinity IL-4Rα subunit. These findings highlight an important aspect of IL-4R regulation that has been largely overlooked. It has long been appreciated that MHC II expression is regulated by endosomal receptor internalization under steady-state as well as pathological conditions. However, this has not translated into the field of cytokine receptor regulation until this recent publication. This may reflect the technical challenges associated with low IL-4 receptor expression and the need to rely on cell culture systems for basic discovery.
Figure 2. Regulation of signaling receptors.
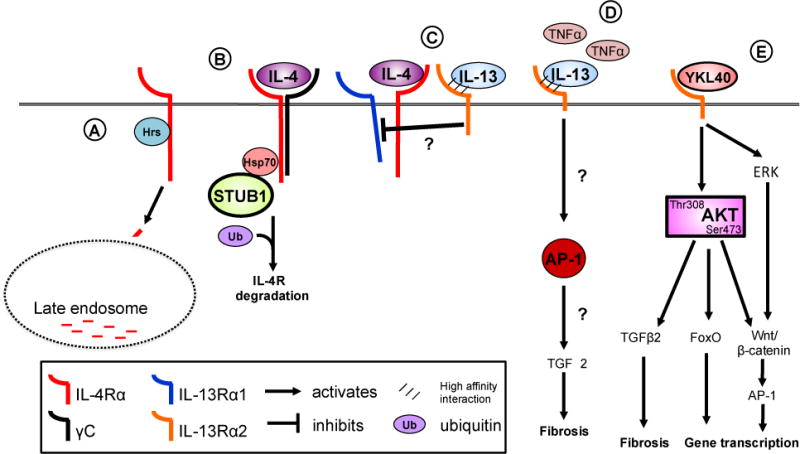
IL-4Rα expression is regulated in the steady state by Hepatocyte growth factor-regulated tyrosine substrate (Hrs) (A). Hrs binding targets the IL-4Rα to the late endosome for degradation. Downregulation of IL-4Rα occurs through STUB1 binding the cytoplasmic domain of the receptor through Hsp70 (B). Subsequent ubiquitination and degradation the IL-4Rα reduces cell surface expression and cellular responsiveness to IL-4. The IL-13Rα2 acts as a negative regulator of IL-4- but not IL-13-induced signaling through the type II IL-4 receptor (C). The IL-13Rα2 acts as a negative regulator of IL-4- but not IL-13-induced signaling through the type II IL-4 receptor although the precise mechanisms of this inhibition are not understood. IL-13Rα2 expression is induced by TNFα in fibroblasts and leads to AP-1 driven pro-fibrotic reprogramming (D). YKL40 binds binds to the IL-13Rα2 chain leading to serine 473 phosphorylation in addition to triggering ERK signaling (E). AKT phosphorylation following binding of YKL40 to IL-13Rα2 activates a myriad of signaling pathways including FoxO1/3 and the Wnt/b-catenin pathway. Hepatocyte growth factor-regulated tyrosine substrate, Hrs: STIP1 homology and U-Box containing protein 1, STUB1; heat shock protein 70, Hsp70; forkhead box protein O, FoxO; transforming growth factor-β, TGF-β; activator protein-1, AP-1.
Given these new discoveries in cancer and brain function, alongside those already described in asthma and atopy, we hope to see increased importance placed on understanding the dynamics and regulation of expression of the IL-4 receptor subunits between the cell surface and intracellular compartments. In-depth studies will provide meaningful insight to deciphering the mechanisms that regulate receptor subunit expression, localization of IL-4 receptor subunits and termination of signaling events in disease phenotypes.
IL-13Rα2 comes of age, more than just a cytokine decoy receptor
The IL-13Rα2 has greater than 35% homology to the IL-13Rα1 and binds IL-13 with extraordinarily high affinity [<10−15 M, (18)], although does not bind IL-4. Primarily regarded as a “decoy” receptor for IL-13 because of its lack of cytoplasmic tail signaling motifs and high affinity for IL-13, the pathophysiological and therapeutic potential of this receptor chain has garnered attention in recent years. Structurally, the IL-13Rα2 does not contain a signaling tail and does not associate with the other known IL-4 receptor subunits. IL-13Rα2 is expressed at the surface of the cell before it is release in its soluble form as well as within the intracellular compartment (69). In vivo analyses report conflicting anti-asthmatic/inflammatory properties (70–72) and pro-asthmatic/inflammatory properties (73–76). The IL-13Rα2 is predominantly expressed on structural cells, such as epithelial cells, but has also been identified on fibroblasts (77) and in soluble form in the circulation in mice only (24, 78–80). Recently, it was reported that TNFα alone or in synergy with IL-17 increases expression of IL-13Rα2 on normal human lung fibroblasts (81, 82). In fibroblasts, IL-13Rα2 binds IL-13 but does not initiate the typical IL-13 signaling cascades. Rather, there are reports that the IL-13Rα2 regulates IL-4 signaling. Andrews et al. reported IL-13Rα2 attenuated IL-4 and IL-13 mediated STAT6 signaling in a human airway epithelial cell line, BEAS-2B (83). They further show that the cytoplasmic domain of the IL-13Rα2 is required to attenuate IL-4-mediated signaling but not IL-13-mediated signaling, which is universally blocked by overexpression of the IL-13Rα2. These findings support previous findings that IL-13Rα2 binds IL-13 with high affinity to block STAT6 signaling while only partially attenuating IL-4-driven STAT6 signaling (82, 84). Furthermore, they suggest that the IL-13Rα2 cytoplasmic domain may inhibit IL-4 signaling by preventing dimerization with a functional secondary chain or possibly by blocking recruitment of the IL13Rα1 (Figure 2C). While these studies strongly support the idea that the IL-13Rα2 does not signal to promote IL-4-driven inflammation, compelling studies from in vivo models suggest IL-13Rα2 is more than just a decoy receptor for IL-4/-13 signaling. Fichtner-Feigl et al. reported that the IL-13Rα2 is induced by TNFα resulting in increased IL-13 signaling through the IL-13Rα2 to promote AP1-driven TGF-β production in monocytes (Figure 2D) (85). This enhanced IL-13 signaling along with downstream TGF-β production promoted lung remodeling and fibrosis. These findings were further supported in a mouse model of chronic allograft rejection that demonstrates that IL-13Rα2 expression promotes TGF-β production by graft-infiltrating cells, pro-fibrotic changes in gene expression and collagen deposition in the heart (86). Both these publications conclude that since IL-13 expression was increased and increased expression of IL-13Rα2 co-localized with fibrosis and TGF-β, IL-13 was driving the fibrotic process. While this does indeed seem plausible, recent findings published by He et al. suggest this may not be the only mechanism by which IL-13Rα2 initiates signaling. The authors report IL-13Rα2 binds Chitinase 3-like 1 (Chi3l1, YKL-40) to promote MAPK, Akt/PKB, and Wnt/β-catenin signaling leading to decreased oxidant injury and apoptosis/pyropoptosis (Figure 2E) (87). They also noted activation of anti-bacterial defenses, inhibition of the inflammasome, stimulation of TGF-β production and increased lung metastasis in mouse models. They suggest that IL-13 can complex with Chi3l1 to bind IL-13Rα2. Taken together with the two studies discussed above, there is compelling evidence that the IL-13Rα2 may indeed have yet undefined binding partners and can act alone as well as synergistically with IL-13. This possibility may help us begin to understand the apparently contradictory findings for IL-13Rα2 in animal models and humans. Closer examination of the diseases, models and cytokine milieu may elucidate the function of the IL-13Rα2. It is not yet clear how the short 17 amino acid tail of IL-13Rα2 with no apparent signaling motifs is able to stimulate such profound, diverse biological effects. Several of these responses resemble the biological functions of IL-4/-13-stimulated cell signaling, although not all of them can be attributed to IL-4/-13. Presumably these effects occur through differential association with other signaling receptors or signaling adaptor proteins, but this remains to be validated. These results are interesting nonetheless, in that they provide unique insight into the biological function of Chi3l1/YKL-40. Several studies suggest that YKL-40 is an inflammatory biomarker because it predicts severity of several diseases including asthma (88–91), cancer (92), neurodegenerative diseases (93) and glioblastoma multiforme (94–96), in spite of its largely unknown biological function. These developments finally help to reconcile the in vivo observations that IL-13Rα2 is pro-inflammatory with in vitro findings that in response to IL-13, the IL-13Rα2 has inhibitory activity. We can now appreciate that in the context of in vivo, inflammation, IL-13Rα2 is more than a simple decoy receptor and the aforementioned studies help to refine our understanding of the IL-13Rα2 in disease pathogenesis.
IL-4/-13 receptor polymorphisms in human disease
Polymorphisms in the promoters of the genes for IL4, IL13 and the IL-4 receptor subunits have previously identified. Polymorphisms that both enhance and diminish the expression or activity of the gene products have been reported in various disease states. A single nucleotide polymorphism (SNP) in the IL4 gene promoter, C590T has been shown to be associated increased IL4 transcriptional activity and positively correlated with arthritis (97, 98) multiple sclerosis (MS) (99–101) and asthma (102, 103). Two SNPs in the IL13 gene, C1055T in the promoter region and R130Q in exon 4 were reported to be associated with enhanced IL-13 biological activity and progression of pulmonary fibrosis (104), chronic obstructive pulmonary disease (COPD) (105) and asthma (106, 107). In addition, three SNPs in the IL4RA exons, I50V, S478P and Q551R, have been widely associated with hyper-IgE and atopy (102, 103, 108–110). SNPs in IL4, IL13 and the receptor subunits are reportedly associated with glioma (111, 112). These polymorphisms highlight key features of IL-4/-13 receptor signaling that regulate disease susceptibility and severity, i.e. ligand binding, signaling and its regulation. Many functional consequences of these polymorphisms remain unexamined yet may reveal new therapeutic targets to diminish unwanted inflammation and identify unappreciated regulatory pathways. The I50V SNP in the extracellular domain of the IL-4Ra has been highly correlated with many atopic diseases, allergies and asthma (106, 113). Several studies undertook a methodical examination of the functional consequences of this SNP on IL-4 ligand binding to the IL-4 receptor complexes and downstream signaling. Ford et al. demonstrated that this SNP caused not only prolonged IL-4 binding to the receptor but also prolonged STAT6 activation (113, 114). A transient increase in CIS mRNA expression, a negative regulator of cytokine signaling and member of the SOCS family, was noted in cells expressing the I50V form of the receptor in response to IL-4 stimulation (113). Enhanced STAT6 signaling further correlated with increased systemic IgE in patients with Graves Disease (114). These studies provide important insight into the effect of an SNP in the extracellular domain of the IL-4Rα chain on intracellular signaling responses and biology. They also demonstrate that under normal “healthy” conditions, IL-4 signaling induced expression of its own negative regulator highlighting that regulatory mechanisms are in place to overcome a single polymorphism. Of course, these studies remind us that many diseases do not arise from a single change but rather represent one alteration in a combination of many changes. These kinds of investigations focused on the functional consequences of SNPs provide insights into the molecular mechanisms and regulation of IL-4 signaling pathways, as well as identify altered regulatory mechanisms under disease conditions. Furthermore, studies that aim to dissect the precise mechanistic and biological consequence of the SNPs must be coordinated with large-scale correlative analyses in order to better define parameters of investigation and identify appropriate endpoints.
IL-4 and IL-13 also serve an important immunoregulatory role. In the context of T-cell-driven autoimmune inflammation such as arthritis, the IL-4Rα plays an immunosuppressive role. Two major IL-4 receptor polymorphisms, the I50V and Q551R SNPs, have been linked with the susceptibility and severity of rheumatoid arthritis (RA) (115–118). Wallis et al. and Liepe et al. provided modest evidence that disease severity and IL-17 secretion is associated with the prevalence of the V allele (115, 119). Furthermore, they showed that IL-4 had little effect on modulating IL-17 production in patients with pre-existing RA or its secretion from already Th17 polarized cells. Their data suggest that the suppressive effect of IL-4 occurs at the level of T-cell priming and may not be able to influence a pre-existing Th17-phenotype due to inhibited STAT6 activation following CD4+ T cell differentiation. It must be noted, however, that IL-4R polymorphisms are equally represented in patient populations, suggesting dampened IL-4 signaling is only one of many factors that limit Th1-/Th17-driven disease. The efficacy or usefulness of IL-4 as an immunomodulator may reside therefore in its ability to limit de novo Th1/17 differentiation and help slow disease progression by limiting epitope spread/development of T cells against new epitopes. These ideas are further discussed below in the superkine section.
Targeting IL-4 and IL-13 receptors to treat disease
IL-4 triggers cell signaling, first by binding with high affinity to the IL-4Rα subunit, which causes a conformational change, followed by association with one of two low-affinity receptor subunits either the common γC or the IL-13Rα1 chain (120). IL-4 engagement of the type I IL-4 receptor induces stronger tyrosine phosphorylation of IRS-2, whereas IL-4 or IL-13 binding the type II receptor results in weak tyrosine phosphorylation of IRS-2. This signaling pattern correlates with the degree of target gene expression and maturation/differentiation in macrophages. These differential associations likely account for the recruitment of different JAKs and STAT/IRS pathways triggered and the different biological consequences downstream. To better understand how cytokine binding to the receptor elicited differential IRS-2 signaling responses, the cytoplasmic domain of the type I receptor γC was swapped with the type II receptor IL-13Ra1 cytoplasmic domain and vice versa (9). Interestingly, the enhanced phospho-IRS-2 signaling responses could not be transferred to the type II IL-4 receptor simply by replacing the IL-13Rα1 cytoplasmic domain with that of the γC chain. The reverse “tail swap” also held true: type I IL-4 receptor responses could not be diminished by the presence of the IL-13Rα1 cytoplasmic domain in place of the γC tail. These results suggested that signaling responses are dictated, for the most part, by the interaction of the cytokines IL-4 and IL-13 with the extracellular and transmembrane domains of the receptor. These results have fortuitous implications for therapies directed at modulating IL-4/-13-mediated responses. Blocking or augmenting agents of downstream signaling can be directed at the cytokine:receptor complex outside the cell and they would not necessary need to be cell-penetrative. Taken together with the success of cytokine neutralization in animal models, these findings fostered the idea that manipulating cytokine structure may result in different biological outcomes (121). Moreover, the specificity of the cytokine:receptor binding also presents a unique opportunity to develop novel therapeutics that would effectively target a single receptor and block cytokine signaling while avoiding the complications associated with antibody neutralizing strategies.
IL-4 antagonists
Anti-cytokine therapy in the form of antibodies directed against the soluble cytokine (122–127), cytokine receptor decoys (25, 128, 129) and antibodies direct against the receptor (129, 130) have been widely studied and shown success in the clinic. Anti-cytokine therapies targeting soluble IL-4 and IL-13 have been less successful in treating human atopy/asthma, possibly because of their overlapping receptor binding properties. Several IL-4 mutants (or “muteins”) have been generated by various genetic/chemical modification strategies, including some currently in pre-clinical and clinical trials. Duppatla et al. demonstrate that chemical modifications to human IL-4 at residue 121 allowed for binding to the high affinity IL-4α chain but blocks the recruitment of either the γC or IL-13Rα, preventing IL-4R signaling in Jurkat T-cells (131). Pitrakinra (AER-001, BAY-16-9996) has been well studied in several clinical trials (129, 130). In asthmatics, pitrakinra reduced exacerbations as well as the need for rescue inhaler although these effects were predominantly observed in patients with the specific SNPs rs3024585, rs3024622, and rs4787956 (132, 133). In spite of blocking wildtype IL-4/-13 binding by up to 99%, IL-4 antagonists have not been able to block the biological activity of endogenous IL-4/-13 completely. This may be due to incomplete receptor saturation, the ability of a small number of receptors to assemble an alternative IL-4R complex configuration that leads to signaling or may reflect the very small amount of endogenous cytokine necessary to initiate a signaling response. In clinical studies, several safety aspects need to be considered including the effect of exogenous inhibitory cytokine on endogenous cytokine levels, immune function and cognitive function based on the emerging data as to the role of IL-4 in the brain. In addition, it will be important to sample a wide array of tissue in order to fully ascertain the consequences of inhibiting the IL-4/-13 signaling pathways.
IL-4 superkines
Cytokine modification can also enhance the biological properties of IL-4/-13. Junttila et al. generated modified human IL-4 that activated the IL-4R with greater affinity than endogenous cytokine and specifically activated the type I IL-4R or the type II IL-4R by increasing the affinity for the γC or the IL-13Rα1 chain respectively (4). The authors demonstrated that these superkines stimulate more robust and prolonged IL-4-induced STAT6 signaling than endogenous IL-4. In addition, a superkine with high affinity for γC only induced changes in gene transcription consistent with the type I receptor signaling, while the superkine with high affinity for the IL-13Rα1 induced a type II receptor gene profile. These findings have important implications as a therapeutic agent. These superkines would have the potential to specifically target immune cells to modulate Th1 and/or Th17-driven inflammation with out influencing structural cells or off-target effects. IL-17-producing CD4+ T cells and innate γδ T cells have been associated with extreme, corticosteroid resistant forms of asthma. To date, very few therapeutic approaches have been effective in regulating IL-17 responses. Since IL-4 is known to be a potent negative regulator of CD4+ Th1/Th17 (134) and IL-17-producing γδ, a therapeutic IL-4 superkine may be an effective way to dampen unchecked Th1/17-driven inflammation in autoimmune diseases and asthma. Since human T and B cells predominantly express γC and not IL-13Rα1, a superkine that specifically binds γC to enhance the Th2-differentiation program, without influencing non-structural cells expressing the IL-13Rα1, may be an effective way to modulate inappropriate inflammation. Such an approach may also influence the differentiation of monocytes/macrophages as well as dendritic cells. However, ex vivo experiments suggest that this may not be the case. Surprisingly, human PBMC-derived dendritic cells (DCs) exposed to super-IL-4, which preferentially recruits the γC, failed to mature, compared to DCs treated with native IL-4 (4). These observations indicate that human blood-derived DC specifically used the type II IL-4R to differentiate in response to IL-4, whereas human monocytes utilized both the γC (type I receptor) and the IL-13Rα1 (type II receptor) (4). These results suggest that a highly specific superkine may provide an important adjunct therapy specifically targeted to affect the function of relevant cell types in different disease. Ideally, combining the technologies of IL-4 antagonists and superkines could yield a highly specific therapy that could effectively target a single receptor subunit to block IL-4 signaling.
IL-4 and IL-13 signaling in health and disease
New insights into the regulation of IL-4 and IL-13 signaling
IL-4 and IL-13 take advantage of numerous signaling molecules to trigger activation and gene transcription namely 2 main pathways; the STAT6 pathway and the IRS pathway. STAT6 signaling in asthma is well characterized. In mouse models of allergic airway disease, a model of asthma, STAT6 phosphorylation is critical for Th2 differentiation (135), eosinophil migration (136, 137), mucus production and airway hyper-reactivity (138). More recently, IL-13 derived from NKT cells promotes ulcerative colitis through the activation of STAT6 (139). Rosen et al. describe a STAT6-dependent role for IL-13 signaling in the expression of claudin-2, a pore-forming protein that promotes epithelial barrier dysfunction. Furthermore, IL-13-driven STAT6 signaling promotes the production of IL-33 and thymic stromal lymphopoietin (TSLP), cytokines which promote Th2 differentiation and amplify intestinal inflammation (139).
Intrinsic factors, such as JAK/STAT inhibitors of cytokine signaling as well as extrinsic factors such as immunoregulatory cytokines, all participate to limit IL-4/-13 signaling. Intrinsic regulatory mechanisms target signal transduction molecules within the cell to limit their activation and prevent subsequent signaling. Src homology region 2 domain-containing phosphatase-1 (SHP1, also known as Tyrosine-protein phosphatase non-receptor type 6 or PTPN6), a protein tyrosine phosphatase, is a negative regulator of several signaling pathways by promoting the dephosphorylation of signaling molecules. Signaling studies revealed STAT6 is dephosphorylated by Shp1 and the IL-4Rα SH2 binding domain of the ITIM is required for efficient Stat6 phosphorylation (Figure 3A) (140). Mice deficient in Shp1 (motheaten and viable motheaten mice) develop spontaneous Th2-like disease and are highly susceptible to allergic airway disease marked by hyper-activation of Th2 cells, hyper-Ig production and lung tissue damage (141–144). In vivo studies indicate Shp1 regulates the activation and proliferation of T cell function, macrophage, epithelial cell responsiveness, mucus production and B cell development (141, 145–150). Conflicting reports have made it difficult to decipher how SHP1 deficiency regulates Th2-like disease that in vitro systems have not been able to clarify these issues. Conditional knockouts were recently generated to help address the cell-type specific role of Shp1 in regulating allergic disease. Zhou et al. reported that Shp1 regulates the development of mast cells in a murine model of anaphylaxis but reported no role for T and B cells or macrophages (151). Previous studies suggested that in T cells, Shp1 regulated TCR:MHC affinity, proliferation and Treg polarization, however, these reports were contradictory [reviewed in (152)]. To clarify the role of Shp1 in CD4+ T cells, Johnson et al. generated a conditional CD4+ T cell Shp1 knockout mouse. These mice did not develop the characteristics motheaten inflammation seen in whole body knockouts. In contrast to studies carried out in whole body knockout mice and in vitro, the authors report normal thymocyte development and normal TCR signaling in these mice, however, more activated CD4+CD44hi T cells could be detected in the periphery (153). These cells were highly responsive to IL-4 such that neutralizing IL-4 attenuated CD4+ T cell activation. Furthermore, using bone marrow chimera mice, they show that autocrine IL-4 is a potent activator of these autoreactive T cells. It is important to note that these CD4+ T cells developed an activated phenotype in the absence of a specific antigen, suggesting they are autoreactive. Phenotypic analysis revealed increased IL-4 production from these cells and enhanced serum IgE levels, supporting the idea that Shp1 negatively regulates Th2 polarization and T cell helper functions, possibly through the production of IL-4. Furthermore, they show that blocking IL-4 in vivo attenuates CD4+ T cell activation, suggesting IL-4 signaling contributes to the diffuse T cell activation in these mice (153). Although neither of these recent papers address the signaling processes reported in vivo, we can extrapolate our understanding of the function of Shp1 in regulating STAT6 phosphorylation to appreciate that these events are likely IL-4-driven. In humans, decreased SHP1 expression is linked to some lymphoma and leukemia as well as MS (152, 154, 155). Taken together, we can begin to appreciate that targeting STATs with small molecules such as FDA-approved statins [reviewed in (156)] may be an effective way to module IL-4-driven eosinophil/mast cell recruitment and differentiation, T cell activation, B cell activation and differentiation as well as hyper-Ig syndromes.
Figure 3. Regulation of signaling in response to IL-4 and IL-13.
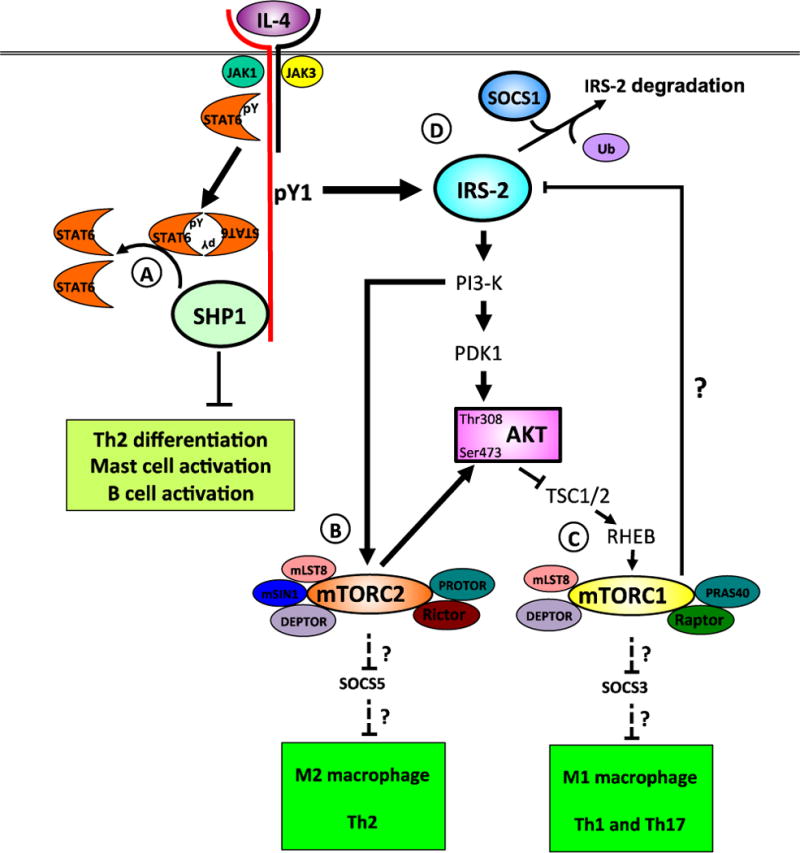
Engagement of the type I IL-4 receptor by IL-4 brings about tyrosine phosphorylation of the associated JAKs, key tyrosine residues (pY1-5) in the cytoplasmic domain in IL-4Ra, STAT6 and IRS-2. STAT6 is dephosphorylated by SHP1, a protein tyrosine phosphatase (A). De-phosphorylation of STAT6 is critical to limit autoimmune and Th2 allergic inflammation, mast cell, B cell and epithelial cell activation. IRS-2 binds the regulatory p85 subunit of PI3-K, activating the enzyme to generate PIP3. In turn, PIP3 activates PDK1 and mTORC2. The proteins comprising the two mTOR complexes 1 and 2 are shown (B). TORC2 kinase phosphorylates AKT on serine 473 and to SGK1 in CD4+ T cells, leading to Th2 polarization. AKT inactivation of the Tsc1/2 complex prevents hydrolysis of GTP bound to Rheb. GTP-bound Rheb activates mTORC1, triggering a negative feedback loop via multiple pathways on IL-4-activated IRS-2/PI3-K/AKT signaling (C). The composition of the mTORC1 complex is shown. The mTORC1 complex also regulates macrophage polarization, as Raptor-deficient macrophages have enhanced M1 and M2 responses. The mTORC1 and mTORC2 complexes also regulate by unknown mechanisms, the polarization of T cells [adapted from (170)]. An additional negative regulatory pathway triggered by IL-4 is the induction of the SOCS proteins. SOCS1 binds IRS-2 and targets it for ubiquitin-mediated degradation (D). Phosphotyrosine, pY; phosphatidylinositol-4,5-bisphosphate 3-kinase, PI3-K; phosphatidylinositol (3,4,5)-trisphosphate, PIP3; 3-phosphoinositide dependent protein kinase-1, PDK1; mTOR complex 2, mTORC2; serum and glucocorticoid-inducible kinase 1, SGK1; Ras homolog enriched in brain, RHEB; tuberous sclerosis complex 1, TSC1; regulatory-associated protein of mTOR, Raptor; mammalian lethal with Sec13 protein 8, mLST8; proline-rich Akt substrate of 40 kDa, PRAS40; DEP-domain-containing mTOR-interacting protein, DEPTOR; rapamycin-insensitive companion of TOR, RICTOR; mammalian stress-activated protein kinase-interacting protein 1, mSIN1; protein observed with RICTOR, PROTOR; suppressor of cytokine signaling 1, SOCS1.
IRS-2 is also a critical mediator of IL-4 and IL-13 signaling. IL-4 induces strong phosphorylation of IRS-2 whereas IL-13 only weakly induces IL-13 phosphorylation (157). This level of phosphorylation correlates with the strength of M2 gene induction in macrophages and may account for some of the different physiological effects of these two cytokines in vivo. It has been reported that insulin signaling through IRS-2 leads to downregulation of the IRS-2 receptor (158), presumably through the induction of a negative regulator. Indeed both suppressor of cytokine signaling (SOCS)1 and SOCS3 have been shown to negatively regulate IRS-2 expression through the ubiquitination and proteasomal degradation leading to insulin insensitivity (159, 160). In the context of IL-4/-13 signaling, the role of the SOCS proteins is less well characterized. SOCS1 and SOCS3 have been implicated in macrophage polarization although studies of SOCS1 have been particularily difficult to interpret. SOCS1, but not SOCS3 is induced by IL-4 and was associated with an M2 phenotype (161). In contrast, SOCS3 was associated with an M1 phenotype, yet SOCS1 reportedly plays a role in negatively regulating M1 polarization as well (161–163). These findings have yet to define the molecular mechanisms that would account for observations in macrophage polarization and relate these observations to human disease. In line with the understanding that SOCS1 regulates M2 polarization and IRS-2 signaling, we have also found a role for SOCS1 but not SOCS3 regulation of IRS-2 in human monocytes (McCormick and Heller, manuscript in preparation). The importance of IRS expression regulation has important implications in the field of IL-4-driven cancer proliferation and metastasis. Porter et al. examined the expression of signaling adaptor proteins IRS-1 and IRS-2 in human breast cancer tissues (164). They found a correlation between high IRS-2 expression and invasive ductal carcinomas, whereas, high expression of IRS-1 correlated with poorly differentiated, less invasive cancers. Moreover and IRS-1 correlated with sensitivity to chemotherapy. Since IL-4 potently activates IRS-2 to trigger transcriptional activation, suppress apoptosis and actively promote differentiation, increased IRS-2 expression may begin to account for the exquisite sensitivity of breast cancer to IL-4/-13-driven metastasis. Furthermore, given the importance of IL-4/-13 activated tumor infiltrating macrophages with aggressive cancers, these observations suggest a possible link between macrophage IRS-2 expression and IL-4 sensitivity in tumor progression. Most importantly, the authors have identified not only a therapeutic target to dampen IL-4 signaling but also a possible prognostic marker. Investigation into strategies that modulate IL-4 signaling through the regulation of the expression of IL-4 receptor subunits or the activation of downstream STAT6 and IRS-2 signaling is warranted. Such approaches would be promising as a combinational therapy alongside standard therapies to combat aggressive tumors and metastasis.
mTOR signaling: a master regulator of IL-4-induced signal transduction
Early studies in the 1990s had uncovered that IL-4 stimulation lead to activation of PI3K through IRS-2 (165, 166) and to subsequent activation of AKT (167, 168). Other studies demonstrated a role for the IL-4/STAT6 pathway in regulating peripheral nutrient metabolism through immune cells resident in metabolic tissues such as liver, fat and skeletal muscle (168). Metabolic responses are coordinated inside the cell by the serine/threonine kinase, mTOR, acting as a sensor of glucose and amino acid availability (169). As background, mTOR is the catalytic protein that nucleates two different complexes: mTOR complex 1 (TORC1) and mTOR complex 2 (TORC2, (170)). TORC1 and TORC2 are made up of multiple proteins, with mTOR being common to both, although they localize to different regions within the cell (Figure 2C). The small GTPase, Ras homolog enriched in brain (Rheb), activates TORC1 when it binds GTP. Activation of the TORC1 kinase by Rheb is controlled by tuberous sclerosis complex 1 (TSC-1) and TSC-2. The Tsc-1/-2 complex, in turn, is phosphorylated by AKT, inhibiting its GTPase activating protein (GAP) activity and allowing GTP-bound Rheb to remain associated with TORC1. Activated TORC1 phosphorylates multiple substrates including p70 S6-kinase and 4E-BP1. The TORC1 regulates multiple cellular processes including protein synthesis, lipid biogenesis, inhibition of autophagy and other processes (169). TORC1 is sensitive to inhibition by rapamycin mediated by FKBP12. Typically, mTORC2 is not rapamycin-sensitive but long-term rapamycin treatment in some cell types inhibits this complex (171). The upstream activation of TORC2 is less well understood but thought to occur through PIP3 (172). TORC2 fully activates AKT by phosphorylation of serine 473 (173, 174). Thus, AKT is both an activator of TORC1 and a substrate of TORC2.
Although the PI3K-AKT-TOR pathway was described as activated downstream of IL-4 in T cells (175), how canonical IL-4 signaling pathways and nutrient/metabolic responses were integrated was not known. New insights into the role of the TOR complexes and TORC-activated molecules in activation and regulation of IL-4 signaling have been recently uncovered in a variety of immune cell types. The different downstream pathways and functions of mTORC1- and mTORC2-activated signaling following IL-4 stimulation has been an emerging focus of research in recent years.
The importance of mTOR-activated pathways in response to IL-4 leading to polarization of CD4+ T-cells was uncovered recently. Deletion of Rictor the mTORC2 adaptor, in CD4+ T cells leads to a deficit of Th2 polarization and IL-4 production (176, 177). However, the signaling pathways downstream of mTORC2 activation that promote Th2-polarization were not known. Heikamp et al. demonstrated that serum- and glucocorticoid-regulated kinase 1 (SGK1) is a critical regulator of Th2-cell lineage commitment downstream of mTORC2 (178). Under Th2 polarizing conditions (IL-4, α-IL-12 and α-IFN-γ), they found that SGK1 deletion in CD4+ T cells resulted in diminished Th2 cytokine production. These SGK1-deficient cells secreted much more IFN-γ, even without polarization, than wildtype CD4+ T cells under the same conditions. This lead to suppressed allergic responses in the in vivo model of allergic lung inflammation. The molecular mechanism underlying changes in T cell polarization in Rictor-deficient cells was due to the lack of SGK-mediated inhibition of the E3 ubiquitin ligase, Nedd4-2. This resulted in enhanced ubiquitin-mediated degradation of JunB, which is required for Th2-cell development.
Another important IL-4-induced mTOR-mediated response is the differentiation of macrophages into the M2 macrophage phenotype in vitro. New data have demonstrated the essential role of the TOR pathway in regulating IL-4-polarization of macrophages. Byles et al. used bone marrow macrophages from myeloid-specific Tsc1-deficient mice to highlight the role of TORC1 as a negative feedback regulator of IL-4-activated AKT signaling in macrophages (179). When Tsc1, the negative regulator of TORC1, is deleted, constitutive TORC1 activity leads to suppression of AKT signaling and deficient M2 polarization of macrophages. Validation of this study by Zhu et al. with similarly Tsc1-LyzMcre-floxed animals reinforced the finding that Tsc1 deletion in macrophages resulted in diminished AKT phosphorylation and M2 polarization following IL-4 stimulation (180). The authors further demonstrated that C/EBPβ, a critical transcription factor for the M2 fate, was also decreased in the nuclei from IL-4-stimulated macrophages derived from the Tsc1-deleted mice. Furthermore, the importance of the TORC1 complex as a regulator of M2 polarization in human macrophages was described by Mercalli et al. (181). Rapamycin treatment of human monocyte-derived macrophages polarized with IL-4 resulted in a decrease in the number of M2 macrophages and increased apoptosis. On the other hand, M1 polarization markers were enhanced. These in vitro findings were also recapitulated in monocyte-derived macrophages from patients treated with rapamycin prior to islet transplantation. The authors concluded that the mTOR pathway was essential for M2 macrophage survival although no deeper insight into the molecular mechanism in human macrophages was provided. Further investigation will be required to reconcile the apparent opposite conclusions made from mouse and human studies described above.
Taken together, these papers point to mTORC1 and 2, or TORC1 and 2-activated pathway(s), as canonical regulators of IL-4 and IL-13 signaling pathways contributing to Th2 and M2-type inflammation. Therefore, manipulation of TOR activity with rapamycin as a therapeutic in the setting of IL-4-driven Th2-mediated disease was investigated in several new publications. Mushaben et al. (182) repeated early studies by Fredricksson et al. (183) comparing the effects of rapamycin treatment during the induction phase of allergic lung inflammation versus established disease. Mushaben and colleagues described the importance of mTORC1-mediated signaling in promoting the early phases of allergic disease using the house dust mite (HDM)-induced allergic inflammation model in mice. Rapamycin had little effect on the later, effector phase of established disease. Therefore, the utility of rapamycin or mTORC1 inhibition as a therapeutic is somewhat tempered by these in vivo findings. There was no effect on AHR in the most recent study (182). In fact, AHR and airway inflammation became worse when rapamycin was given to animals with established allergic lung inflammation (183). Uncovering more details of the precise molecular underpinnings of mTORC1 or mTORC2 regulation to subvert pathogenic IL-4 signaling in allergic inflammation will aid in more effective manipulation of these pathways for therapeutic benefit.
Concluding remarks
IL-4 and IL-13 are important cytokines for cell differentiation, promoting allergic and anti-helminth inflammation, proliferation and cancer. In addition, these cytokines regulate type 1- and type 17-driven T cell inflammation, but should they not be should not be confused as anti-inflammatory cytokines. Both IL-4 and IL-13 have important functions and drive equally important and potentially pathogenic type 2 inflammation. Ubiquitous expression of the type I and II receptors on immune cells and the type II IL-4 receptor on structural cells means that IL-4 and IL-13 have the potential to affect every cell in the body. IL-4 receptor subunit expression is tightly regulated but can be modified by genetic, environmental and inflammatory stimuli. Insights into the mechanisms that regulate receptor sensitivity, density and signaling are important for identifying appropriate therapeutic targets in several diseases. In many diseases, aberrant signaling causes or certainly amplifies disease progression. Understanding the regulation of IL-4/-13 signaling is increasingly important in order to identify and develop therapies that augment the action of these pathways. The discovery of mutein cytokines have precipitated the development of antagonists as well as high affinity superkines. Superkines with modified affinity for the secondary chain could show promise in modulating activation/signaling and downstream responses in targeted cell types. These modified cytokines along with the refinement of cytotoxins could be exploited to diminish or even reverse disease severity in a variety of diseases, notably, cancer, neurological disorders, asthma and allergies.
Highlights.
-
-
IL-4 and IL-13 receptors are expressed on every cell in the body and expression is altered during disease
-
-
New mechanisms that regulate receptor expression have been identified yet highlight the lack of understanding of cytokine receptors in disease processes
-
-
IL-4/-13 and their receptors play a greater role in brain homeostasis that previously appreciated
-
-
Therapeutics that specifically target a single receptor subunit may hold promise in ameliorating treatment-refractory diseases
-
-
New insights into regulation of IL-4/-13 signaling suggest new therapeutic approaches are on the horizon
Abbreviations
- AP-1
Activator protein-1
- CNS
central nervous system
- DEPTOR
DEP-domain-containing mTORinteracting protein
- FDA
US Food and Drug Administration
- FoxO
forkhead box protein O
- Hsp70
heat shock protein 70
- Hrs
hepatocyte growth factor-regulated tyrosine substrate
- Ig
immunoglobulin
- ITIM
immunoreceptor tyrosine-based inhibition motif
- IL
interleukin
- JAK
Janus Kinase
- IRS
insulin receptor substrate
- IFN
interferon
- mLST8
mammalian lethal with Sec13 protein 8
- MHC
major histocompatibility complex
- mSIN1
mammalian stress-activated protein kinaseinteracting protein 1
- NO
nitric oxide
- PI3K
phosphatidylinositol-4,5-bisphosphate 3-kinase
- PIP3
phosphatidylinositol (3,4,5)-trisphosphate
- PDK1
3-phosphoinositide dependent protein kinase-1
- PRAS40
proline-rich Akt substrate of 40 kDa
- PROTOR
protein observed with RICTOR
- RICTOR
rapamycin-insensitive companion of TOR
- RHEB
Ras homolog enriched in brain
- TSC1
tuberous sclerosis complex 1
- Raptor
regulatory-associated protein of mTOR
- SGK1
serum and glucocorticoid-inducible kinase 1
- STAT
Signal transducer and activator of transcription
- STUB1
STIP1 homology and U-Box containing protein 1
- SOCS1
suppressor of cytokine signaling 1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gadani SP, Cronk JC, Norris GT, Kipnis J. Interleukin-4: A Cytokine to Remember. Journal of immunology (Baltimore, Md. : 1950) 2012;189:4213–4219. doi: 10.4049/jimmunol.1202246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McKenzie Andrew NJ, Spits H, Eberl G. Innate Lymphoid Cells in Inflammation and Immunity. Immunity. 2014;41:366–374. doi: 10.1016/j.immuni.2014.09.006. [DOI] [PubMed] [Google Scholar]
- 3.Hams E, Fallon PG. Innate type 2 cells and asthma. Current Opinion in Pharmacology. 2012;12:503–509. doi: 10.1016/j.coph.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 4.Junttila IS, Creusot RJ, Moraga I, Bates DL, Wong MT, Alonso MN, Suhoski MM, Lupardus P, Meier-Schellersheim M, Engleman EG, Utz PJ, Fathman CG, Paul WE, Garcia KC. Redirecting cell-type specific cytokine responses with engineered interleukin-4 superkines. Nat Chem Biol. 2012;8:990–998. doi: 10.1038/nchembio.1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heller NM, Dasgupta P, Dorsey NJ, Chapoval SP, Keegan AD. The Type I and Type II Receptor Complexes for IL-4 and IL-13 Differentially Regulate Allergic Lung Inflammation, Allergic Diseases. Pereira C, editor. Highlights in the Clinic, Mechanisms and Treatment. 2012 http://www.intechopen.com/books/allergic-diseases-highlights-in-the-clinic-mechanisms-and-treatment/the-type-i-and-type-ii-receptor-complexes-for-il-4-and-il-13-differentially-regulate-allergic-lung-i.
- 6.Sun XJ, Wang L-M, Zhang Y, Yenush L, Myers MG, Jr, Glasheen E, Lane WS, Pierce JH, White MF. Role of IRS-2 in insulin and cytokine signalling. Nature. 1995;377:173–177. doi: 10.1038/377173a0. [DOI] [PubMed] [Google Scholar]
- 7.Dhand R, Hiles I, Panayotou G, Roche S, Fry MJ, Gout I, Totty NF, Truong O, Vicendo P, Yonezawa K. PI 3-kinase is a dual specificity enzyme: autoregulation by an intrinsic protein-serine kinase activity. The EMBO Journal. 1994;13:522–533. doi: 10.1002/j.1460-2075.1994.tb06290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ruckerl D, Jenkins SJ, Laqtom NN, Gallagher IJ, Sutherland TE, Duncan S, Buck AH, Allen JE. Induction of IL-4Ralpha-dependent microRNAs identifies PI3K/Akt signaling as essential for IL-4-driven murine macrophage proliferation in vivo. Blood. 2012;120:2307–2316. doi: 10.1182/blood-2012-02-408252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heller NM, Qi X, Gesbert F, Keegan AD. The extracellular and transmembrane domains of the gammaC and interleukin (IL)-13 receptor alpha1 chains, not their cytoplasmic domains, dictate the nature of signaling responses to IL-4 and IL-13. The Journal of biological chemistry. 2012;287:31948–31961. doi: 10.1074/jbc.M112.348896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Karlsson HR, Zierath J. Insulin signaling and glucose transport in insulin resistant human skeletal muscle. Cell Biochem Biophys. 2007;48:103–113. doi: 10.1007/s12013-007-0030-9. [DOI] [PubMed] [Google Scholar]
- 11.Landis J, Shaw LM. Insulin Receptor Substrate-2 Mediated Phosphatidylinositol-3-kinase Signaling Selectively Inhibits Glycogen Synthase Kinase-3β to Regulate Aerobic Glycolysis. Journal of Biological Chemistry. 2014;289:18603–18613. doi: 10.1074/jbc.M114.564070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Russo SJ, Bolanos CA, Theobald DE, DeCarolis NA, Renthal W, Kumar A, Winstanley CA, Renthal NE, Wiley MD, Self DW, Russell DS, Neve RL, Eisch AJ, Nestler EJ. IRS2-Akt pathway in midbrain dopamine neurons regulates behavioral and cellular responses to opiates. Nat Neurosci. 2007;10:93–99. doi: 10.1038/nn1812. [DOI] [PubMed] [Google Scholar]
- 13.Mikita T, Campbell D, Wu P, Williamson K, Schindler U. Requirements for interleukin-4-induced gene expression and functional characterization of Stat6. Molecular and Cellular Biology. 1996;16:5811–5820. doi: 10.1128/mcb.16.10.5811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shen CH, Stavnezer J. Interaction of Stat6 and NF-κB: Direct Association and Synergistic Activation of Interleukin-4-Induced Transcription. Molecular and Cellular Biology. 1998;18:3395–3404. doi: 10.1128/mcb.18.6.3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Delphin S, Stavnezer J. Characterization of an interleukin 4 (IL-4) responsive region in the immunoglobulin heavy chain germline epsilon promoter: regulation by NF-IL-4, a C/EBP family member and NF-kappa B/p50. J Exp Med. 1995;181:181–192. doi: 10.1084/jem.181.1.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hallett MA, Venmar KT, Fingleton B. Cytokine stimulation of epithelial cancer cells: the similar and divergent functions of IL-4 and IL-13. Cancer Res. 2012;72:6338–6343. doi: 10.1158/0008-5472.CAN-12-3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jenkins SJ, Ruckerl D, Thomas GD, Hewitson JP, Duncan S, Brombacher F, Maizels RM, Hume DA, Allen JE. IL-4 directly signals tissue-resident macrophages to proliferate beyond homeostatic levels controlled by CSF-1. J Exp Med. 2013;210:2477–2491. doi: 10.1084/jem.20121999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lupardus PJ, Birnbaum ME, Garcia KC. Molecular basis for shared cytokine recognition revealed in the structure of an unusually high affinity complex between IL-13 and IL-13Rα2. Structure (London, England : 1993) 2010;18:332–342. doi: 10.1016/j.str.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Silvestri T, Pulsatelli L, Dolzani P, Facchini A, Meliconi R. Elevated serum levels of soluble interleukin-4 receptor in osteoarthritis. Osteoarthritis and cartilage / OARS, Osteoarthritis Research Society. 2006;14:717–719. doi: 10.1016/j.joca.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 20.Sato TA, Widmer MB, Finkelman FD, Madani H, Jacobs CA, Grabstein KH, Maliszewski CR. Recombinant soluble murine IL-4 receptor can inhibit or enhance IgE responses in vivo. J Immunol. 1993;150:2717–2723. [PubMed] [Google Scholar]
- 21.Andrews AL, Holloway JW, Holgate ST, Davies DE. IL-4 Receptor α Is an Important Modulator of IL-4 and IL-13 Receptor Binding: Implications for the Development of Therapeutic Targets. The Journal of Immunology. 2006;176:7456–7461. doi: 10.4049/jimmunol.176.12.7456. [DOI] [PubMed] [Google Scholar]
- 22.Daines MO, Tabata Y, Walker BA, Chen W, Warrier MR, Basu S, Hershey GKK. Level of Expression of IL-13Rα2 Impacts Receptor Distribution and IL-13 Signaling. The Journal of Immunology. 2006;176:7495–7501. doi: 10.4049/jimmunol.176.12.7495. [DOI] [PubMed] [Google Scholar]
- 23.Sivaprasad U, Warrier MR, Gibson AM, Chen W, Tabata Y, Bass SA, Rothenberg ME, Khurana Hershey GK. IL-13Rα2 Has a Protective Role in a Mouse Model of Cutaneous Inflammation. The Journal of Immunology. 2010;185:6802–6808. doi: 10.4049/jimmunol.1002118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.O’Toole M, Legault H, Ramsey R, Wynn TA, Kasaian MT. A novel and sensitive ELISA reveals that the soluble form of IL-13R-α2 is not expressed in plasma of healthy or asthmatic subjects. Clinical and experimental allergy : journal of the British Society for Allergy and Clinical Immunology. 2008;38:594–601. doi: 10.1111/j.1365-2222.2007.02921.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Borish LC, Nelson HS, Corren J, Bensch G, Busse WW, Whitmore JB, Agosti JM. Efficacy of soluble IL-4 receptor for the treatment of adults with asthma. J Allergy Clin Immunol. 2001;107:963–970. doi: 10.1067/mai.2001.115624. [DOI] [PubMed] [Google Scholar]
- 26.Nestor CE, Dadfar E, Ernerudh J, Gustafsson M, Bjorkander J, Benson M, Zhang H. Sublingual immunotherapy alters expression of IL-4 and its soluble and membrane-bound receptors. Allergy. 2014;69:1564–1566. doi: 10.1111/all.12505. [DOI] [PubMed] [Google Scholar]
- 27.Benson M, Strannegard IL, Wennergren G, Strannegard O. Increase of the soluble IL-4 receptor (IL-4sR) and positive correlation between IL-4sR and IgE in nasal fluids from school children with allergic rhinitis. Allergy and asthma proceedings : the official journal of regional and state allergy societies. 2000;21:89–95. doi: 10.2500/108854100778250932. [DOI] [PubMed] [Google Scholar]
- 28.Doucet C, Giron-Michel J, Canonica GW, Azzarone B. Human lung myofibroblasts as effectors of the inflammatory process: the common receptor gamma chain is induced by Th2 cytokines, and CD40 ligand is induced by lipopolysaccharide, thrombin and TNF-alpha. European journal of immunology. 2002;32:2437–2449. doi: 10.1002/1521-4141(200209)32:9<2437::AID-IMMU2437>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 29.Yamamoto S, Kobayashi I, Tsuji K, Nishi N, Muro E, Miyazaki M, Zaitsu M, Inada S, Ichimaru T, Hamasaki Y. Upregulation of interleukin-4 receptor by interferon-gamma: enhanced interleukin-4-induced eotaxin-3 production in airway epithelium. American journal of respiratory cell and molecular biology. 2004;31:456–462. doi: 10.1165/rcmb.2004-0128OC. [DOI] [PubMed] [Google Scholar]
- 30.Bani L, David D, Fevrier M, Pialoux G, Dupont B, Sugamura K, Theze J. Interleukin-2 receptor beta and gamma chain dysregulation during the inhibition of CD4 T cell activation by human immunodeficiency virus-1 gp120. European journal of immunology. 1997;27:2188–2194. doi: 10.1002/eji.1830270911. [DOI] [PubMed] [Google Scholar]
- 31.Siegmund R, Vogelsang H, Machnik A, Herrmann D. Surface membrane antigen alteration on blood basophils in patients with Hymenoptera venom allergy under immunotherapy. J Allergy Clin Immunol. 2000;106:1190–1195. doi: 10.1067/mai.2000.110928. [DOI] [PubMed] [Google Scholar]
- 32.van der Velden VH, Naber BA, Wierenga-Wolf AF, Debets R, Savelkoul HF, Overbeek SE, Hoogsteden HC, Versnel MA. Interleukin 4 receptors on human bronchial epithelial cells. An in vivo and in vitro analysis of expression and function. Cytokine. 1998;10:803–813. doi: 10.1006/cyto.1998.0365. [DOI] [PubMed] [Google Scholar]
- 33.Koller FL, Hwang DG, Dozier EA, Fingleton B. Epithelial interleukin-4 receptor expression promotes colon tumor growth. Carcinogenesis. 2010;31:1010–1017. doi: 10.1093/carcin/bgq044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Joshi BH, Leland P, Lababidi S, Varrichio F, Puri RK. Interleukin-4 receptor alpha overexpression in human bladder cancer correlates with the pathological grade and stage of the disease. Cancer medicine. 2014;3:1615–1628. doi: 10.1002/cam4.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Burt BM, Bader A, Winter D, Rodig SJ, Bueno R, Sugarbaker DJ. Expression of Interleukin-4 Receptor Alpha in Human Pleural Mesothelioma Is Associated with Poor Survival and Promotion of Tumor Inflammation. Clinical Cancer Research. 2012;18:1568–1577. doi: 10.1158/1078-0432.CCR-11-1808. [DOI] [PubMed] [Google Scholar]
- 36.Venmar KT, Carter KJ, Hwang DG, Dozier EA, Fingleton B. IL4 Receptor ILR4α Regulates Metastatic Colonization by Mammary Tumors through Multiple Signaling Pathways. Cancer Research. 2014;74:4329–4340. doi: 10.1158/0008-5472.CAN-14-0093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lyons A, Downer EJ, Crotty S, Nolan YM, Mills KHG, Lynch MA. CD200 Ligand–Receptor Interaction Modulates Microglial Activation In Vivo and In Vitro: A Role for IL-4. The Journal of Neuroscience. 2007;27:8309–8313. doi: 10.1523/JNEUROSCI.1781-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Walker DG, Dalsing-Hernandez JE, Campbell NA, Lue L-F. Decreased expression of CD200 and CD200 receptor in Alzheimer’s disease: A potential mechanism leading to chronic inflammation. Experimental Neurology. 2009;215:5–19. doi: 10.1016/j.expneurol.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cao C, Arendash GW, Dickson A, Mamcarz MB, Lin X, Ethell DW. Aβ-specific Th2 cells provide cognitive and pathological benefits to Alzheimer’s mice without infiltrating the CNS. Neurobiology of Disease. 2009;34:63–70. doi: 10.1016/j.nbd.2008.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Natoli A, Lüpertz R, Merz C, Müller WW, Köhler R, Krammer PH, Li-Weber M. Targeting the IL-4/IL-13 signaling pathway sensitizes Hodgkin lymphoma cells to chemotherapeutic drugs. International Journal of Cancer. 2013;133:1945–1954. doi: 10.1002/ijc.28189. [DOI] [PubMed] [Google Scholar]
- 41.Ziv Y, Ron N, Butovsky O, Landa G, Sudai E, Greenberg N, Cohen H, Kipnis J, Schwartz M. Immune cells contribute to the maintenance of neurogenesis and spatial learning abilities in adulthood. Nat Neurosci. 2006;9:268–275. doi: 10.1038/nn1629. [DOI] [PubMed] [Google Scholar]
- 42.Wolf SA, Steiner B, Akpinarli A, Kammertoens T, Nassenstein C, Braun A, Blankenstein T, Kempermann G. CD4-Positive T Lymphocytes Provide a Neuroimmunological Link in the Control of Adult Hippocampal Neurogenesis. The Journal of Immunology. 2009;182:3979–3984. doi: 10.4049/jimmunol.0801218. [DOI] [PubMed] [Google Scholar]
- 43.Derecki NC, Cardani AN, Yang CH, Quinnies KM, Crihfield A, Lynch KR, Kipnis J. Regulation of learning and memory by meningeal immunity: a key role for IL-4. The Journal of Experimental Medicine. 2010;207:1067–1080. doi: 10.1084/jem.20091419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Verbitsky M, Yonan AL, Malleret G, Kandel ER, Gilliam TC, Pavlidis P. Altered Hippocampal Transcript Profile Accompanies an Age-Related Spatial Memory Deficit in Mice. Learning & Memory. 2004;11:253–260. doi: 10.1101/lm.68204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zahn JM, Poosala S, Owen AB, Ingram DK, Lustig A, Carter A, Weeraratna AT, Taub DD, Gorospe M, Mazan-Mamczarz K, Lakatta EG, Boheler KR, Xu X, Mattson MP, Falco G, Ko MSH, Schlessinger D, Firman J, Kummerfeld SK, Wood WH, III, Zonderman AB, Kim SK, Becker KG. AGEMAP: A Gene Expression Database for Aging in Mice. PLoS Genet. 2007;3:e201. doi: 10.1371/journal.pgen.0030201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lynch MA. Age-related neuroinflammatory changes negatively impact on neuronal function. Frontiers in aging neuroscience. 2010;1:6. doi: 10.3389/neuro.24.006.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Falcone M, Rajan AJ, Bloom BR, Brosnan CF. A Critical Role for IL-4 in Regulating Disease Severity in Experimental Allergic Encephalomyelitis as Demonstrated in IL-4-Deficient C57BL/6 Mice and BALB/c Mice. The Journal of Immunology. 1998;160:4822–4830. [PubMed] [Google Scholar]
- 48.Sofroniew MV. Astrocyte barriers to neurotoxic inflammation. Nat Rev Neurosci. 2015;16:249–263. doi: 10.1038/nrn3898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Abbas N, Bednar I, Mix E, Marie S, Paterson D, Ljungberg A, Morris C, Winblad B, Nordberg A, Zhu J. Up-regulation of the inflammatory cytokines IFN-γ and IL-12 and down-regulation of IL-4 in cerebral cortex regions of APPSWE transgenic mice. Journal of Neuroimmunology. 2002;126:50–57. doi: 10.1016/s0165-5728(02)00050-4. [DOI] [PubMed] [Google Scholar]
- 50.Wyss-Coray T, Rogers J. Inflammation in Alzheimer Disease—A Brief Review of the Basic Science and Clinical Literature. Cold Spring Harbor Perspectives in Medicine. 2012;2:a006346. doi: 10.1101/cshperspect.a006346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Perry VH. Innate Inflammation in Parkinson’s Disease. Cold Spring Harbor Perspectives in Medicine. 2012;2 doi: 10.1101/cshperspect.a009373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu H, Prayson RA, Estes ML, Drazba JA, Barnett GH, Bingaman W, Liu J, Jacobs BS, Barna BP. IN VIVO EXPRESSION OF THE INTERLEUKIN 4 RECEPTOR ALPHA BY ASTROCYTES IN EPILEPSY CEREBRAL CORTEX. Cytokine. 2000;12:1656–1661. doi: 10.1006/cyto.2000.0773. [DOI] [PubMed] [Google Scholar]
- 53.Ponomarev ED, Maresz K, Tan Y, Dittel BN. CNS-Derived Interleukin-4 Is Essential for the Regulation of Autoimmune Inflammation and Induces a State of Alternative Activation in Microglial Cells. The Journal of Neuroscience. 2007;27:10714–10721. doi: 10.1523/JNEUROSCI.1922-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brodie C, Goldreich N, Haiman T, Kazimirsky G. Functional IL-4 receptors on mouse astrocytes: IL-4 inhibits astrocyte activation and induces NGF secretion. Journal of Neuroimmunology. 1998;81:20–30. doi: 10.1016/s0165-5728(97)00154-9. [DOI] [PubMed] [Google Scholar]
- 55.Awatsuji H, Furukawa Y, Hirota M, Murakami Y, Nii S, Furukawa S, Hayashi K. Interleukin-4 and -5 as modulators of nerve growth factor synthesis/secretion in astrocytes. Journal of Neuroscience Research. 1993;34:539–545. doi: 10.1002/jnr.490340506. [DOI] [PubMed] [Google Scholar]
- 56.Hu S, Sheng WS, Peterson PK, Chao CC. Differential regulation by cytokines of human astrocyte nitric oxide production. Glia. 1995;15:491–494. doi: 10.1002/glia.440150412. [DOI] [PubMed] [Google Scholar]
- 57.Kiyota T, Okuyama S, Swan RJ, Jacobsen MT, Gendelman HE, Ikezu T. CNS expression of anti-inflammatory cytokine interleukin-4 attenuates Alzheimer’s disease-like pathogenesis in APP+PS1 bigenic mice. The FASEB Journal. 2010;24:3093–3102. doi: 10.1096/fj.10-155317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shimizu E, Kawahara K, Kajizono M, Sawada M, Nakayama H. IL-4-Induced Selective Clearance of Oligomeric β-Amyloid Peptide1–42 by Rat Primary Type 2 Microglia. The Journal of Immunology. 2008;181:6503–6513. doi: 10.4049/jimmunol.181.9.6503. [DOI] [PubMed] [Google Scholar]
- 59.Mikita J, Dubourdieu-Cassagno N, Deloire MS, Vekris A, Biran M, Raffard G, Brochet B, Canron M-H, Franconi J-M, Boiziau C, Petry KG. Altered M1/M2 activation patterns of monocytes in severe relapsing experimental rat model of multiple sclerosis. Amelioration of clinical status by M2 activated monocyte administration. Multiple Sclerosis Journal. 2011;17:2–15. doi: 10.1177/1352458510379243. [DOI] [PubMed] [Google Scholar]
- 60.Butovsky O, Landa G, Kunis G, Ziv Y, Avidan H, Greenberg N, Schwartz A, Smirnov I, Pollack A, Jung S, Schwartz M. Induction and blockage of oligodendrogenesis by differently activated microglia in an animal model of multiple sclerosis. The Journal of Clinical Investigation. 2006;116:905–915. doi: 10.1172/JCI26836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Walsh JT, Hendrix S, Boato F, Smirnov I, Zheng J, Lukens JR, Gadani S, Hechler D, Gölz G, Rosenberger K, Kammertöns T, Vogt J, Vogelaar C, Siffrin V, Radjavi A, Fernandez-Castaneda A, Gaultier A, Gold R, Kanneganti T-D, Nitsch R, Zipp F, Kipnis J. MHCII-independent CD4+ T cells protect injured CNS neurons via IL-4. The Journal of Clinical Investigation. 2015;125:0–0. doi: 10.1172/JCI76210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ribizzi G, Fiordoro S, Barocci S, Ferrari E, Megna M. Cytokine polymorphisms and Alzheimer disease: possible associations. Neurol Sci. 2010;31:321–325. doi: 10.1007/s10072-010-0221-9. [DOI] [PubMed] [Google Scholar]
- 63.Colton CA, Mott RT, Sharpe H, Xu Q, Van Nostrand WE, Vitek MP. Expression profiles for macrophage alternative activation genes in AD and in mouse models of AD. Journal of Neuroinflammation. 2006;3:27–27. doi: 10.1186/1742-2094-3-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chakrabarty P, Tianbai L, Herring A, Ceballos-Diaz C, Das P, Golde TE. Hippocampal expression of murine IL-4 results in exacerbation of amyloid deposition. Molecular Neurodegeneration. 2012;7:36–36. doi: 10.1186/1750-1326-7-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Morrison BE, Marcondes MCG, Nomura DK, Sanchez-Alavez M, Sanchez-Gonzalez A, Saar I, Kim K-S, Bartfai T, Maher P, Sugama S, Conti B. Cutting Edge: IL-13Rα1 Expression in Dopaminergic Neurons Contributes to Their Oxidative Stress–Mediated Loss following Chronic Peripheral Treatment with Lipopolysaccharide. The Journal of Immunology. 2012;189:5498–5502. doi: 10.4049/jimmunol.1102150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Amano Y, Yamashita Y, Kojima K, Yoshino K, Tanaka N, Sugamura K, Takeshita T. Hrs Recognizes a Hydrophobic Amino Acid Cluster in Cytokine Receptors during Ubiquitin-independent Endosomal Sorting. The Journal of biological chemistry. 2011;286:15458–15472. doi: 10.1074/jbc.M110.191924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Amano Y, Yoshino K, Kojima K, Takeshita T. A hydrophobic amino acid cluster inserted into the C-terminus of a recycling cell surface receptor functions as an endosomal sorting signal. Biochemical and Biophysical Research Communications. 2013;441:164–168. doi: 10.1016/j.bbrc.2013.10.019. [DOI] [PubMed] [Google Scholar]
- 68.Wei Q, Sha Y, Bhattacharya A, Fattah EA, Bonilla D, Jyothula SSSK, Pandit L, Khurana Hershey GK, Eissa NT. Regulation of IL-4 Receptor Signaling by STUB1 in Lung Inflammation. American Journal of Respiratory and Critical Care Medicine. 2013;189:16–29. doi: 10.1164/rccm.201305-0874OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Daines MO, Tabata Y, Walker BA, Chen W, Warrier MR, Basu S, Hershey GK. Level of expression of IL-13R alpha 2 impacts receptor distribution and IL-13 signaling. J Immunol. 2006;176:7495–7501. doi: 10.4049/jimmunol.176.12.7495. [DOI] [PubMed] [Google Scholar]
- 70.Wilson MS, Elnekave E, Mentink-Kane MM, Hodges MG, Pesce JT, Ramalingam TR, Thompson RW, Kamanaka M, Flavell RA, Keane-Myers A, Cheever AW, Wynn TA. IL-13Rα2 and IL-10 coordinately suppress airway inflammation, airway-hyperreactivity, and fibrosis in mice. The Journal of Clinical Investigation. 2007;117:2941–2951. doi: 10.1172/JCI31546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wood N, Whitters MJ, Jacobson BA, Witek J, Sypek JP, Kasaian M, Eppihimer MJ, Unger M, Tanaka T, Goldman SJ, Collins M, Donaldson DD, Grusby MJ. Enhanced Interleukin (IL)-13 Responses in Mice Lacking IL-13 Receptor α 2. The Journal of Experimental Medicine. 2003;197:703–709. doi: 10.1084/jem.20020906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Morimoto M, Zhao A, Sun R, Stiltz J, Madden KB, Mentink-Kane M, Ramalingam T, Wynn TA, Urban JF, Shea-Donohue T. IL-13 Receptor α2 Regulates the Immune and Functional Response to Nippostrongylus brasiliensis Infection. Journal of immunology. 2009;183:1934–1939. doi: 10.4049/jimmunol.0804299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mentink-Kane MM, Cheever AW, Thompson RW, Hari DM, Kabatereine NB, Vennervald BJ, Ouma JH, Mwatha JK, Jones FM, Donaldson DD, Grusby MJ, Dunne DW, Wynn TA. IL-13 receptor α 2 down-modulates granulomatous inflammation and prolongs host survival in schistosomiasis. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:586–590. doi: 10.1073/pnas.0305064101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sivaprasad U, Warrier MR, Gibson AM, Chen W, Tabata Y, Bass SA, Rothenberg ME, Khurana Hershey GK. IL-13Rα2 Has a Protective Role in a Mouse Model of Cutaneous Inflammation. Journal of immunology. 2010;185:6802–6808. doi: 10.4049/jimmunol.1002118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chiaramonte MG, Mentink-Kane M, Jacobson BA, Cheever AW, Whitters MJ, Goad MEP, Wong A, Collins M, Donaldson DD, Grusby MJ, Wynn TA. Regulation and Function of the Interleukin 13 Receptor α 2 During a T Helper Cell Type 2–dominant Immune Response. The Journal of Experimental Medicine. 2003;197:687–701. doi: 10.1084/jem.20020903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chen W, Sivaprasad U, Gibson AM, Ericksen MB, Cunningham CM, Bass SA, Kinker KG, Finkelman FD, Wills-Karp M, Khurana Hershey GK. IL-13 receptor α2 contributes to development of experimental allergic asthma. Journal of Allergy and Clinical Immunology. 2013;132:951–958.e956. doi: 10.1016/j.jaci.2013.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhou X, Hu H, Balzar S, Trudeau JB, Wenzel SE. MAPK Regulation of IL-4/-13 Receptors Contributes to the Synergistic Increase in CCL11/Eotaxin-1 in Response to TGF-β1 and IL-13 in Human Airway Fibroblasts. Journal of Immunology. 2012;188:6046–6054. doi: 10.4049/jimmunol.1102760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chen W, Sivaprasad U, Tabata Y, Gibson AM, Stier MT, Finkelman FD, Khurana Hershey GK. IL-13 Receptor Alpha 2 Membrane and Soluble Isoforms Differ in Human and Mouse. Journal of immunology (Baltimore, Md. : 1950) 2009;183:7870. doi: 10.4049/jimmunol.0901028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Daines MO, Masino JA, Gibson AM, Chen W, Walker BA, Warrier MR, Tabata Y, Hershey Khurana GK. Distribution of IL-13Ra2: Impact of Allergen Exposure and Level of Expression. Journal of Allergy and Clinical Immunology. 2006;117:S148. [Google Scholar]
- 80.Daines MO, Hershey GK. A novel mechanism by which interferon-gamma can regulate interleukin (IL)-13 responses. Evidence for intracellular stores of IL-13 receptor alpha -2 and their rapid mobilization by interferon-gamma. The Journal of biological chemistry. 2002;277:10387–10393. doi: 10.1074/jbc.M108109200. [DOI] [PubMed] [Google Scholar]
- 81.Badalyan V, Thompson R, Addo K, Borthwick LA, Fisher AJ, Ort T, Myers TG, Wynn TA, Ramalingam TR. TNF-α/IL-17 synergy inhibits IL-13 bioactivity via IL-13Rα2 induction. Journal of Allergy and Clinical Immunology. 2014;134:975–978.e975. doi: 10.1016/j.jaci.2014.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chandriani S, DePianto DJ, N’Diaye EN, Abbas AR, Jackman J, Bevers J, Ramirez-Carrozzi V, Pappu R, Kauder SE, Toy K, Ha C, Modrusan Z, Wu LC, Collard HR, Wolters PJ, Egen JG, Arron JR. Endogenously Expressed IL-13Rα2 Attenuates IL-13–Mediated Responses but Does Not Activate Signaling in Human Lung Fibroblasts. The Journal of Immunology. 2014;193:111–119. doi: 10.4049/jimmunol.1301761. [DOI] [PubMed] [Google Scholar]
- 83.Andrews AL, Nordgren IK, Campbell-Harding G, Holloway JW, Holgate ST, Davies DE, Tavassoli A. The association of the cytoplasmic domains of interleukin 4 receptor alpha and interleukin 13 receptor alpha 2 regulates interleukin 4 signaling. Molecular BioSystems. 2013;9:3009–3014. doi: 10.1039/c3mb70298g. [DOI] [PubMed] [Google Scholar]
- 84.Zheng T, Liu W, Oh S-Y, Zhu Z, Hu B, Homer RJ, Cohn L, Grusby MJ, Elias JA. IL-13 Receptor α2 Selectively Inhibits IL-13-Induced Responses in the Murine Lung. The Journal of Immunology. 2008;180:522–529. doi: 10.4049/jimmunol.180.1.522. [DOI] [PubMed] [Google Scholar]
- 85.Fichtner-Feigl S, Strober W, Kawakami K, Puri RK, Kitani A. IL-13 signaling through the IL-13[alpha]2 receptor is involved in induction of TGF-[beta]1 production and fibrosis. Nat Med. 2006;12:99–106. doi: 10.1038/nm1332. [DOI] [PubMed] [Google Scholar]
- 86.Brunner SM, Schiechl G, Kesselring R, Martin M, Balam S, Schlitt HJ, Geissler EK, Fichtner-Feigl S. IL-13 signaling via IL-13Rα(2) triggers TGF-β(1)-dependent allograft fibrosis. Transplantation Research. 2013;2:16–16. doi: 10.1186/2047-1440-2-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chuan H He, Lee Chun G, Dela Cruz Charles S, Lee C-M, Zhou Y, Ahangari F, Erica L B Ma, Stephen A Herzog, Rosenberg Y, Adel M Li, Chirag R Nour, Schmidt Parikh I, Modis Y, Cantley L, Elias Jack A. Chitinase 3-like 1 Regulates Cellular and Tissue Responses via IL-13 Receptor α2. Cell Reports. 2013;4:830–841. doi: 10.1016/j.celrep.2013.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Specjalski K, Jassem E. YKL-40 protein is a marker of asthma. The Journal of asthma : official journal of the Association for the Care of Asthma. 2011;48:767–772. doi: 10.3109/02770903.2011.611955. [DOI] [PubMed] [Google Scholar]
- 89.Konradsen JR, James A, Nordlund B, Reinius LE, Söderhäll C, Melén E, Wheelock Å, Lödrup Carlsen KC, Lidegran M, Verhoek M, Boot RG, Dahlén B, Dahlén SE, Hedlin G. The chitinase-like protein YKL-40: A possible biomarker of inflammation and airway remodeling in severe pediatric asthma. Journal of Allergy and Clinical Immunology. 2013;132:328–335.e325. doi: 10.1016/j.jaci.2013.03.003. [DOI] [PubMed] [Google Scholar]
- 90.Lee CG, Elias JA. Role of breast regression protein-39/YKL-40 in asthma and allergic responses. Allergy, Asthma & Immunology Research. 2010;2:20–27. doi: 10.4168/aair.2010.2.1.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ober C, Tan Z, Sun Y, Possick JD, Pan L, Nicolae R, Radford S, Parry RR, Heinzmann A, Deichmann KA, Lester LA, Gern JE, Lemanske RF, Nicolae DL, Elias JA, Chupp GL. Effect of Variation in CHI3L1 on Serum YKL-40 Level, Risk of Asthma, and Lung Function. The New England journal of medicine. 2008;358:1682–1691. doi: 10.1056/NEJMoa0708801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Harving ML, Christensen LH, Ringsholt M, Lausten GS, Petersen MM. YKL-40 expression in soft-tissue sarcomas and atypical lipomatous tumors: An immunohistochemical study of 49 tumors. Acta Orthopaedica. 2014;85:195–200. doi: 10.3109/17453674.2014.893496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Shao R. YKL-40 acts as an angiogenic factor to promote tumor angiogenesis. Frontiers in Physiology. 2013;4:122. doi: 10.3389/fphys.2013.00122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Iwamoto FM, Hormigo A. Unveiling YKL-40, from Serum Marker to Target Therapy in Glioblastoma. Frontiers in Oncology. 2014;4:90. doi: 10.3389/fonc.2014.00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gállego Pérez-Larraya J, Paris S, Idbaih A, Dehais C, Laigle-Donadey F, Navarro S, Capelle L, Mokhtari K, Marie Y, Sanson M, Hoang-Xuan K, Delattre J-Y, Mallet A. Diagnostic and prognostic value of preoperative combined GFAP, IGFBP-2, and YKL-40 plasma levels in patients with glioblastoma. Cancer. 2014;120:3972–3980. doi: 10.1002/cncr.28949. [DOI] [PubMed] [Google Scholar]
- 96.Preusser M. Neuro-oncology: A step towards clinical blood biomarkers of glioblastoma. Nat Rev Neurol. 2014;10:681–682. doi: 10.1038/nrneurol.2014.208. [DOI] [PubMed] [Google Scholar]
- 97.Schubert K, Bonnsdorf Hvon, Burke M, Ahlert I, Braun S, Berner R, Deichmann KA, Heinzmann A. A Comprehensive Candidate Gene Study on Bronchial Asthma and Juvenile Idiopathic Arthritis. Disease Markers. 2006;22:127–132. doi: 10.1155/2006/373620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Song GG, Bae S-C, Kim J-H, Lee YH. Interleukin-4, interleukin-4 receptor, and interleukin-18 polymorphisms and rheumatoid arthritis: a meta-analysis. Immunological Investigations. 2013;42:455–469. doi: 10.3109/08820139.2013.804084. [DOI] [PubMed] [Google Scholar]
- 99.Hulshof S, Montagne L, De Groot CJA, Van Der Valk P. Cellular localization and expression patterns of interleukin-10, interleukin-4, and their receptors in multiple sclerosis lesions. Glia. 2002;38:24–35. doi: 10.1002/glia.10050. [DOI] [PubMed] [Google Scholar]
- 100.Sholl-Franco A, Da Silva AGLS, Adão-Novaes J. Interleukin-4 as a Neuromodulatory Cytokine. Annals of the New York Academy of Sciences. 2009;1153:65–75. doi: 10.1111/j.1749-6632.2008.03962.x. [DOI] [PubMed] [Google Scholar]
- 101.Gaupp S, Cannella B, Raine CS. Amelioration of Experimental Autoimmune Encephalomyelitis in IL-4Rα(−/−) Mice Implicates Compensatory Up-Regulation of Th2-Type Cytokines. The American Journal of Pathology. 2008;173:119–129. doi: 10.2353/ajpath.2008.071156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Noguchi, Shibasaki, Arinami, Takeda, Yokouchi, Kawashima, Yanagi, Matsui, Hamaguchi Association of asthma and the interleukin-4 promoter gene in Japanese. Clinical & Experimental Allergy. 1998;28:449–453. doi: 10.1046/j.1365-2222.1998.00256.x. [DOI] [PubMed] [Google Scholar]
- 103.Noguchi E, Shibasaki M, Arinami T, Takeda K, Maki T, Miyamoto T, Kawashima T, Kobayashi K, Hamaguchi H. Evidence for Linkage Between Asthma/Atopy in Childhood and Chromosome 5q31-q33 in a Japanese Population. American Journal of Respiratory and Critical Care Medicine. 1997;156:1390–1393. doi: 10.1164/ajrccm.156.5.9702084. [DOI] [PubMed] [Google Scholar]
- 104.Ding M, Sheng H, Shen W, Zhen J, Xi L, Zeng J, Zhang Y, Wu D, Deng L. Association of the SNP rs1800925(C/T) in the interleukin-13 gene promoter with pulmonary function in Chinese Han patients with idiopathic pulmonary fibrosis. Cell Biochem Biophys. 2013;67:905–909. doi: 10.1007/s12013-013-9580-1. [DOI] [PubMed] [Google Scholar]
- 105.Beghé B, Hall IP, Parker SG, Moffatt MF, Wardlaw A, Connolly MJ, Fabbri LM, Ruse C, Sayers I. Polymorphisms in IL13 pathway genes in asthma and chronic obstructive pulmonary disease. Allergy. 2010;65:474–481. doi: 10.1111/j.1398-9995.2009.02167.x. [DOI] [PubMed] [Google Scholar]
- 106.Nie W, Zang Y, Chen J, Xiu Q. Association between interleukin-4 receptor alpha chain (IL4RA) I50V and Q551R polymorphisms and asthma risk: an update meta-analysis. PLoS One. 2013;8:e69120. doi: 10.1371/journal.pone.0069120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Graves PE, Kabesch M, Halonen M, Holberg CJ, Baldini M, Fritzsch C, Weiland SK, Erickson RP, von Mutius E, Martinez FD. A cluster of seven tightly linked polymorphisms in the IL-13 gene is associated with total serum IgE levels in three populations of white children. Journal of Allergy and Clinical Immunology. 2000;105:506–513. doi: 10.1067/mai.2000.104940. [DOI] [PubMed] [Google Scholar]
- 108.Bugawan TL, Mirel DB, Valdes AM, Panelo A, Pozzilli P, Erlich HA. Association and Interaction of the IL4R, IL4, and IL13 Loci with Type 1 Diabetes among Filipinos. The American Journal of Human Genetics. 2003;72:1505–1514. doi: 10.1086/375655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wenzel SE, Balzar S, Ampleford E, Hawkins GA, Busse WW, Calhoun WJ, Castro M, Chung KF, Erzurum S, Gaston B, Israel E, Teague WG, Curran-Everett D, Meyers DA, Bleecker ER. IL4Rα Mutations Are Associated with Asthma Exacerbations and Mast Cell/IgE Expression. American Journal of Respiratory and Critical Care Medicine. 2007;175:570–576. doi: 10.1164/rccm.200607-909OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Howard TD, Koppelman GH, Xu J, Zheng SL, Postma DS, Meyers DA, Bleecker ER. Gene-gene interaction in asthma: IL4RA and IL13 in a Dutch population with asthma. Am J Hum Genet. 2002;70:230–236. doi: 10.1086/338242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sun G, Wang X, Shi L, Yue X, Fu L, Chen C, Li Z, Pan T, Wan Z. Association between polymorphisms in interleukin-4Ralpha and interleukin-13 and glioma risk: a meta-analysis. Cancer epidemiology. 2013;37:306–310. doi: 10.1016/j.canep.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 112.Guo J, Shi L, Li M, Xu J, Yan S, Zhang C, Sun G. Association of the interleukin-4Ralpha rs1801275 and rs1805015 polymorphisms with glioma risk. Tumour biology : the journal of the International Society for Oncodevelopmental Biology and Medicine. 2014;35:573–579. doi: 10.1007/s13277-013-1080-9. [DOI] [PubMed] [Google Scholar]
- 113.Ford AQ, Heller NM, Stephenson L, Boothby MR, Keegan AD. An atopy-associated polymorphism in the ectodomain of the IL-4R(alpha) chain (V50) regulates the persistence of STAT6 phosphorylation. Journal of immunology. 2009;183:1607–1616. doi: 10.4049/jimmunol.0803266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Yabiku K, Hayashi M, Komiya I, Yamada T, Kinjo Y, Ohshiro Y, Kouki T, Takasu N. Polymorphisms of interleukin (IL)-4 receptor alpha and signal transducer and activator of transcription-6 (Stat6) are associated with increased IL-4Rα–Stat6 signalling in lymphocytes and elevated serum IgE in patients with Graves’ disease. Clinical and Experimental Immunology. 2007;148:425–431. doi: 10.1111/j.1365-2249.2007.03366.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Leipe J, Schramm MA, Prots I, Schulze-Koops H, Skapenko A. Increased Th17 Cell Frequency and Poor Clinical Outcome in Rheumatoid Arthritis Are Associated With a Genetic Variant in the IL4R Gene, rs1805010. Arthritis & Rheumatology. 2014;66:1165–1175. doi: 10.1002/art.38343. [DOI] [PubMed] [Google Scholar]
- 116.Prots I, Skapenko A, Wendler J, Mattyasovszky S, Yone CL, Spriewald B, Burkhardt H, Rau R, Kalden JR, Lipsky PE, Schulze-Koops H. Association of the IL4R single-nucleotide polymorphism I50V with rapidly erosive rheumatoid arthritis. Arthritis and rheumatism. 2006;54:1491–1500. doi: 10.1002/art.21832. [DOI] [PubMed] [Google Scholar]
- 117.Rahman P, Snelgrove T, Peddle L, Siannis F, Farewell V, Schentag C, Gladman D. A variant of the IL4 I50V single-nucleotide polymorphism is associated with erosive joint disease in psoriatic arthritis. Arthritis & Rheumatism. 2008;58:2207–2208. doi: 10.1002/art.23558. [DOI] [PubMed] [Google Scholar]
- 118.Burgos PI, Causey ZL, Tamhane A, Kelley JM, Brown EE, Hughes LB, Danila MI, Everdingen Avan, Conn DL, Jonas BL, Callahan LF, Smith EA, Brasington RD, Moreland LW, van der Heijde DM, Alarcón GS, Bridges SL. Association of IL4R single-nucleotide polymorphisms with rheumatoid nodules in African Americans with rheumatoid arthritis. Arthritis Research & Therapy. 2010;12:R75–R75. doi: 10.1186/ar2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wallis SK, Cooney LA, Endres JL, Lee MJ, Ryu J, Somers EC, Fox DA. A polymorphism in the interleukin-4 receptor affects the ability of interleukin-4 to regulate Th17 cells: a possible immunoregulatory mechanism for genetic control of the severity of rheumatoid arthritis. Arthritis Research & Therapy. 2011;13:R15–R15. doi: 10.1186/ar3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hage T, Sebald W, Reinemer P. Crystal Structure of the Interleukin-4/Receptor α Chain Complex Reveals a Mosaic Binding Interface. Cell. 1999;97:271–281. doi: 10.1016/s0092-8674(00)80736-9. [DOI] [PubMed] [Google Scholar]
- 121.LaPorte SL, Juo ZS, Vaclavikova J, Colf LA, Qi X, Heller NM, Keegan AD, Garcia KC. Molecular and structural basis of cytokine receptor pleiotropy in the Interleukin-4/13 system. Cell. 2008;132:259–272. doi: 10.1016/j.cell.2007.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Pelaia G, Vatrella A, Maselli R. The potential of biologics for the treatment of asthma. Nat Rev Drug Discov. 2012;11:958–972. doi: 10.1038/nrd3792. [DOI] [PubMed] [Google Scholar]
- 123.Hansbro PM, Scott GV, Essilfie A-T, Kim RY, Starkey MR, Nguyen DH, Allen PD, Kaiko GE, Yang M, Horvat JC, Foster PS. Th2 cytokine antagonists: potential treatments for severe asthma. Expert Opinion on Investigational Drugs. 2012;22:49–69. doi: 10.1517/13543784.2013.732997. [DOI] [PubMed] [Google Scholar]
- 124.Corren J, Busse W, Meltzer EO, Mansfield L, Bensch G, Fahrenholz J, Wenzel SE, Chon Y, Dunn M, Weng HH, Lin S-L. A Randomized, Controlled, Phase 2 Study of AMG 317, an IL-4Rα Antagonist, in Patients with Asthma. American Journal of Respiratory and Critical Care Medicine. 2010;181:788–796. doi: 10.1164/rccm.200909-1448OC. [DOI] [PubMed] [Google Scholar]
- 125.Corren J, Lemanske RF, Hanania NA, Korenblat PE, Parsey MV, Arron JR, Harris JM, Scheerens H, Wu LC, Su Z, Mosesova S, Eisner MD, Bohen SP, Matthews JG. Lebrikizumab Treatment in Adults with Asthma. New England Journal of Medicine. 2011;365:1088–1098. doi: 10.1056/NEJMoa1106469. [DOI] [PubMed] [Google Scholar]
- 126.Pfizer W. Study evaluating the effect of IMA-638 in Subjects with persistent asthma. accessed January 5, 2014. Available from: http://clinicaltrials.gov/ct2/show/NCT00425061.
- 127.De Boever EH, Ashman C, Cahn AP, Locantore NW, Overend P, Pouliquen IJ, Serone AP, Wright TJ, Jenkins MM, Panesar IS, Thiagarajah SS, Wenzel SE. Efficacy and safety of an anti–IL-13 mAb in patients with severe asthma: A randomized trial. Journal of Allergy and Clinical Immunology. 2014;133:989–996.e984. doi: 10.1016/j.jaci.2014.01.002. [DOI] [PubMed] [Google Scholar]
- 128.Borish LC, Nelson HS, Lanz MJ, Claussen L, Whitmore JB, Agosti JM, Garrison L. Interleukin-4 receptor in moderate atopic asthma. A phase I/II randomized, placebo-controlled trial. Am J Respir Crit Care Med. 1999;160:1816–1823. doi: 10.1164/ajrccm.160.6.9808146. [DOI] [PubMed] [Google Scholar]
- 129.Maes T, Joos GF, Brusselle GG. Targeting Interleukin-4 in Asthma: Lost in Translation? American journal of respiratory cell and molecular biology. 2012;47:261–270. doi: 10.1165/rcmb.2012-0080TR. [DOI] [PubMed] [Google Scholar]
- 130.Wenzel S, Wilbraham D, Fuller R, Getz EB, Longphre M. Effect of an interleukin-4 variant on late phase asthmatic response to allergen challenge in asthmatic patients: results of two phase 2a studies. The Lancet. 2007;370:1422–1431. doi: 10.1016/S0140-6736(07)61600-6. [DOI] [PubMed] [Google Scholar]
- 131.Duppatla V, Gjorgjevikj M, Schmitz W, Hermanns HM, Schäfer CM, Kottmair M, Müller T, Sebald W. IL-4 Analogues with Site-Specific Chemical Modification at Position 121 Inhibit IL-4 and IL-13 Biological Activities. Bioconjugate Chemistry. 2013;25:52–62. doi: 10.1021/bc400307k. [DOI] [PubMed] [Google Scholar]
- 132.Slager RE, Otulana BA, Hawkins GA, Yen YP, Peters SP, Wenzel SE, Meyers DA, Bleecker ER. IL-4 receptor polymorphisms predict reduction in asthma exacerbations during response to an anti-IL-4 receptor alpha antagonist. J Allergy Clin Immunol. 2012;130:516–522.e514. doi: 10.1016/j.jaci.2012.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Burmeister Getz E, Fisher DM, Fuller R. Human pharmacokinetics/pharmacodynamics of an interleukin-4 and interleukin-13 dual antagonist in asthma. Journal of clinical pharmacology. 2009;49:1025–1036. doi: 10.1177/0091270009341183. [DOI] [PubMed] [Google Scholar]
- 134.Lazarski CA, Ford J, Katzman SD, Rosenberg AF, Fowell DJ. IL-4 Attenuates Th1-Associated Chemokine Expression and Th1 Trafficking to Inflamed Tissues and Limits Pathogen Clearance. PLoS ONE. 2013;8:e71949. doi: 10.1371/journal.pone.0071949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hoshino A, Tsuji T, Matsuzaki J, Jinushi T, Ashino S, Teramura T, Chamoto K, Tanaka Y, Asakura Y, Sakurai T, Mita Y, Takaoka A, Nakaike S, Takeshima T, Ikeda H, Nishimura T. STAT6-mediated signaling in Th2-dependent allergic asthma: critical role for the development of eosinophilia, airway hyper-responsiveness and mucus hypersecretion, distinct from its role in Th2 differentiation. Int Immunol. 2004;16:1497–1505. doi: 10.1093/intimm/dxh151. [DOI] [PubMed] [Google Scholar]
- 136.Heller NM, Gwinn WM, Donnelly RP, Constant SL, Keegan AD. IL-4 engagement of the type I IL-4 receptor complex enhances mouse eosinophil migration to eotaxin-1 in vitro. PLoS One. 2012;7:e39673. doi: 10.1371/journal.pone.0039673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Stokes K, LaMarche NM, Islam N, Wood A, Huang W, August A. Cutting Edge: STAT6 Signaling in Eosinophils Is Necessary for Development of Allergic Airway Inflammation. The Journal of Immunology. 2015;194:2477–2481. doi: 10.4049/jimmunol.1402096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kuperman D, Schofield B, Wills-Karp M, Grusby MJ. Signal Transducer and Activator of Transcription Factor 6 (Stat6)-deficient Mice Are Protected from Antigen-induced Airway Hyperresponsiveness and Mucus Production. The Journal of Experimental Medicine. 1998;187:939–948. doi: 10.1084/jem.187.6.939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Rosen MJ, Chaturvedi R, Washington MK, Kuhnhein LA, Moore PD, Coggeshall SS, McDonough EM, Weitkamp J-H, Singh AB, Coburn LA, Williams CS, Yan F, Kaer LVan, Peebles RS, Wilson KT. STAT6 Deficiency Ameliorates Severity of Oxazolone Colitis by Decreasing Expression of Claudin-2 and Th2-Inducing Cytokines. The Journal of Immunology. 2013;190:1849–1858. doi: 10.4049/jimmunol.1201373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Hanson EM, Dickensheets H, Qu C-K, Donnelly RP, Keegan AD. Regulation of the Dephosphorylation of Stat6: PARTICIPATION OF TYR-713 IN THE INTERLEUKIN-4 RECEPTOR α, THE TYROSINE PHOSPHATASE SHP-1, AND THE PROTEASOME. Journal of Biological Chemistry. 2003;278:3903–3911. doi: 10.1074/jbc.M211747200. [DOI] [PubMed] [Google Scholar]
- 141.Cho YS, Oh SY, Zhu Z. Tyrosine phosphatase SHP-1 in oxidative stress and development of allergic airway inflammation. American journal of respiratory cell and molecular biology. 2008;39:412–419. doi: 10.1165/rcmb.2007-0229OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Oh SY, Zheng T, Kim YK, Cohn L, Homer RJ, McKenzie AN, Zhu Z. A critical role of SHP-1 in regulation of type 2 inflammation in the lung. American journal of respiratory cell and molecular biology. 2009;40:568–574. doi: 10.1165/rcmb.2008-0225OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Jang MK, Kim SH, Lee KY, Kim TB, Moon KA, Park CS, Bae YJ, Zhu Z, Moon HB, Cho YS. The tyrosine phosphatase, SHP-1, is involved in bronchial mucin production during oxidative stress. Biochem Biophys Res Commun. 2010;393:137–143. doi: 10.1016/j.bbrc.2010.01.102. [DOI] [PubMed] [Google Scholar]
- 144.Zhang L, Oh SY, Wu X, Oh MH, Wu F, Schroeder JT, Takemoto CM, Zheng T, Zhu Z. SHP-1 deficient mast cells are hyperresponsive to stimulation and critical in initiating allergic inflammation in the lung. J Immunol. 2010;184:1180–1190. doi: 10.4049/jimmunol.0901972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Pao LI, Lam K-P, Henderson JM, Kutok JL, Alimzhanov M, Nitschke L, Thomas ML, Neel BG, Rajewsky K. B Cell-Specific Deletion of Protein-Tyrosine Phosphatase Shp1 Promotes B-1a Cell Development and Causes Systemic Autoimmunity. Immunity. 2007;27:35–48. doi: 10.1016/j.immuni.2007.04.016. [DOI] [PubMed] [Google Scholar]
- 146.Kamata T, Yamashita M, Kimura M, Murata K, Inami M, Shimizu C, Sugaya K, Wang CR, Taniguchi M, Nakayama T. src homology 2 domain-containing tyrosine phosphatase SHP-1 controls the development of allergic airway inflammation. J Clin Invest. 2003;111:109–119. doi: 10.1172/JCI15719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Plas DR, Johnson R, Pingel JT, Matthews RJ, Dalton M, Roy G, Chan AC, Thomas ML. Direct Regulation of ZAP-70 by SHP-1 in T Cell Antigen Receptor Signaling. Science. 1996;272:1173–1176. doi: 10.1126/science.272.5265.1173. [DOI] [PubMed] [Google Scholar]
- 148.Kim CH, Qu C-K, Hangoc G, Cooper S, Anzai N, Feng G-S, Broxmeyer HE. Abnormal Chemokine-Induced Responses of Immature and Mature Hematopoietic Cells from Motheaten Mice Implicate the Protein Tyrosine Phosphatase Shp-1 in Chemokine Responses. The Journal of Experimental Medicine. 1999;190:681–690. doi: 10.1084/jem.190.5.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Minoo P, Zadeh MM, Rottapel R, Lebrun JJ, Ali S. A novel SHP-1/Grb2– dependent mechanism of negative regulation of cytokine-receptor signaling: contribution of SHP-1 C-terminal tyrosines in cytokine signaling. 2004 doi: 10.1182/blood-2003-07-2617. [DOI] [PubMed] [Google Scholar]
- 150.Cho SH, Oh SY, Lane AP, Lee J, Oh M-H, Lee S, Zheng T, Zhu Z. Regulation of Nasal Airway Homeostasis and Inflammation in Mice by SHP-1 and Th2/Th1 Signaling Pathways. PLoS ONE. 2014;9:e103685. doi: 10.1371/journal.pone.0103685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Zhou L, Oh SY, Zhou Y, Yuan B, Wu F, Oh MH, Wang Y, Takemoto C, Rooijen NVan, Zheng T, Zhu Z. SHP-1 Regulation of Mast Cell Function in Allergic Inflammation and Anaphylaxis. PLoS ONE. 2013;8:e55763. doi: 10.1371/journal.pone.0055763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Christophi GP, Panos M, Hudson CA, Christophi RL, Gruber RC, Mersich AT, Blystone SD, Jubelt B, Massa PT. Macrophages of multiple sclerosis patients display deficient SHP-1 expression and enhanced inflammatory phenotype. Laboratory investigation; a journal of technical methods and pathology. 2009;89:742–759. doi: 10.1038/labinvest.2009.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Johnson DJ, Pao LI, Dhanji S, Murakami K, Ohashi PS, Neel BG. Shp1 regulates T cell homeostasis by limiting IL-4 signals. The Journal of Experimental Medicine. 2013;210:1419–1431. doi: 10.1084/jem.20122239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Wu C, Sun M, Liu L, Zhou GW. The function of the protein tyrosine phosphatase SHP-1 in cancer. Gene. 2003;306:1–12. doi: 10.1016/s0378-1119(03)00400-1. [DOI] [PubMed] [Google Scholar]
- 155.Christophi GP, Hudson CA, Gruber RC, Christophi CP, Mihai C, Mejico LJ, Jubelt B, Massa PT. SHP-1 deficiency and increased inflammatory gene expression in PBMCs of multiple sclerosis patients. Laboratory investigation; a journal of technical methods and pathology. 2008;88:243–255. doi: 10.1038/labinvest.3700720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Miklossy G, Hilliard TS, Turkson J. Therapeutic modulators of STAT signalling for human diseases. Nat Rev Drug Discov. 2013;12:611–629. doi: 10.1038/nrd4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Heller NM, Qi X, Junttila IS, Shirey KA, Vogel SN, Paul WE, Keegan AD. Type I IL-4Rs selectively activate IRS-2 to induce target gene expression in macrophages. Science signaling. 2008;1:ra17. doi: 10.1126/scisignal.1164795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Hirashima Y, Tsuruzoe K, Kodama S, Igata M, Toyonaga T, Ueki K, Kahn CR, Araki E. Insulin down-regulates insulin receptor substrate-2 expression through the phosphatidylinositol 3-kinase/Akt pathway. The Journal of endocrinology. 2003;179:253–266. doi: 10.1677/joe.0.1790253. [DOI] [PubMed] [Google Scholar]
- 159.Rui L, Yuan M, Frantz D, Shoelson S, White MF. SOCS-1 and SOCS-3 Block Insulin Signaling by Ubiquitin-mediated Degradation of IRS1 and IRS2. Journal of Biological Chemistry. 2002;277:42394–42398. doi: 10.1074/jbc.C200444200. [DOI] [PubMed] [Google Scholar]
- 160.Kershaw NJ, Murphy JM, Liau NPD, Varghese LN, Laktyushin A, Whitlock EL, Lucet IS, Nicola NA, Babon JJ. SOCS3 binds specific receptor–JAK complexes to control cytokine signaling by direct kinase inhibition. Nat Struct Mol Biol. 2013;20:469–476. doi: 10.1038/nsmb.2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Whyte CS, Bishop ET, Rückerl D, Gaspar-Pereira S, Barker RN, Allen JE, Rees AJ, Wilson HM. Suppressor of cytokine signaling (SOCS)1 is a key determinant of differential macrophage activation and function. Journal of Leukocyte Biology. 2011;90:845–854. doi: 10.1189/jlb.1110644. [DOI] [PubMed] [Google Scholar]
- 162.Liu Y, Stewart KN, Bishop E, Marek CJ, Kluth DC, Rees AJ, Wilson HM. Unique expression of suppressor of cytokine signaling 3 is essential for classical macrophage activation in rodents in vitro and in vivo. J Immunol. 2008;180:6270–6278. doi: 10.4049/jimmunol.180.9.6270. [DOI] [PubMed] [Google Scholar]
- 163.Qin H, Holdbrooks AT, Liu Y, Reynolds SL, Yanagisawa LL, Benveniste EN. SOCS3 Deficiency Promotes M1 Macrophage Polarization and Inflammation. The Journal of Immunology. 2012;189:3439–3448. doi: 10.4049/jimmunol.1201168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Porter HA, Perry A, Kingsley C, Tran NL, Keegan AD. IRS1 is highly expressed in localized breast tumors and regulates the sensitivity of breast cancer cells to chemotherapy, while IRS2 is highly expressed in invasive breast tumors. Cancer letters. 2013;338:239–248. doi: 10.1016/j.canlet.2013.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Wang LM, Keegan AD, Paul WE, Heidaran MA, Gutkind JS, Pierce JH. IL-4 activates a distinct signal transduction cascade from IL-3 in factor-dependent myeloid cells. EMBO J. 1992;11:4899–4908. doi: 10.1002/j.1460-2075.1992.tb05596.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Izuhara K, Harada N. Interleukin-4 (IL-4) induces protein tyrosine phosphorylation of the IL-4 receptor and association of phosphatidylinositol 3-kinase to the IL-4 receptor in a mouse T cell line, HT2. The Journal of biological chemistry. 1993;268:13097–13102. [PubMed] [Google Scholar]
- 167.Cerezo A, Martinez AC, Lanzarot D, Fischer S, Franke TF, Rebollo A. Role of Akt and c-Jun N-terminal kinase 2 in apoptosis induced by interleukin-4 deprivation. Molecular biology of the cell. 1998;9:3107–3118. doi: 10.1091/mbc.9.11.3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Scheid MP, Duronio V. Dissociation of cytokine-induced phosphorylation of Bad and activation of PKB/akt: involvement of MEK upstream of Bad phosphorylation. Proc Natl Acad Sci U S A. 1998;95:7439–7444. doi: 10.1073/pnas.95.13.7439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Laplante M, Sabatini DM. mTOR signaling at a glance. Journal of Cell Science. 2009;122:3589–3594. doi: 10.1242/jcs.051011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Saleiro D, Platanias LC. Intersection of mTOR and STAT signaling in immunity. Trends in immunology. 2015;36:21–29. doi: 10.1016/j.it.2014.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Molecular cell. 2006;22:159–168. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 172.Gan X, Wang J, Su B, Wu D. Evidence for direct activation of mTORC2 kinase activity by phosphatidylinositol 3,4,5-trisphosphate. The Journal of biological chemistry. 2011;286:10998–11002. doi: 10.1074/jbc.M110.195016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and Regulation of Akt/PKB by the Rictor-mTOR Complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 174.Hart JR, Vogt PK. Phosphorylation of AKT: a mutational analysis. Oncotarget. 2011;2:467–476. doi: 10.18632/oncotarget.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Stephenson LM, Park DS, Mora AL, Goenka S, Boothby M. Sequence motifs in IL-4R alpha mediating cell-cycle progression of primary lymphocytes. J Immunol. 2005;175:5178–5185. doi: 10.4049/jimmunol.175.8.5178. [DOI] [PubMed] [Google Scholar]
- 176.Lee K, Gudapati P, Dragovic S, Spencer C, Joyce S, Killeen N, Magnuson MA, Boothby M. Mammalian target of rapamycin protein complex 2 regulates differentiation of Th1 and Th2 cell subsets via distinct signaling pathways. Immunity. 2010;32:743–753. doi: 10.1016/j.immuni.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Delgoffe GM, Pollizzi KN, Waickman AT, Heikamp E, Meyers DJ, Horton MR, Xiao B, Worley PF, Powell JD. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat Immunol. 2011;12:295–303. doi: 10.1038/ni.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Heikamp EB, Patel CH, Collins S, Waickman A, Oh M-H, Sun I-H, Illei P, Sharma A, Naray-Fejes-Toth A, Fejes-Toth G, Misra-Sen J, Horton MR, Powell JD. The AGC kinase SGK1 regulates TH1 and TH2 differentiation downstream of the mTORC2 complex. Nat Immunol. 2014;15:457–464. doi: 10.1038/ni.2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Byles V, Covarrubias AJ, Ben-Sahra I, Lamming DW, Sabatini DM, Manning BD, Horng T. The TSC-mTOR pathway regulates macrophage polarization. Nat Commun. 2013;4 doi: 10.1038/ncomms3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Zhu L, Yang T, Li L, Sun L, Hou Y, Hu X, Zhang L, Tian H, Zhao Q, Peng J, Zhang H, Wang R, Yang Z, Zhang L, Zhao Y. TSC1 controls macrophage polarization to prevent inflammatory disease. Nat Commun. 2014;5 doi: 10.1038/ncomms5696. [DOI] [PubMed] [Google Scholar]
- 181.Mercalli A, Calavita I, Dugnani E, Citro A, Cantarelli E, Nano R, Melzi R, Maffi P, Secchi A, Sordi V, Piemonti L. Rapamycin unbalances the polarization of human macrophages to M1. Immunology. 2013;140:179–190. doi: 10.1111/imm.12126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Mushaben EM, Hershey GK, Pauciulo MW, Nichols WC, Cras TDLe. Chronic Allergic Inflammation Causes Vascular Remodeling and Pulmonary Hypertension in Bmpr2 Hypomorph and Wild-Type Mice. PLoS ONE. 2012;7:e32468. doi: 10.1371/journal.pone.0032468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Fredriksson K, Fielhaber JA, Lam JK, Yao X, Meyer KS, Keeran KJ, Zywicke GJ, Qu X, Yu Z-X, Moss J, Kristof AS, Levine SJ. Paradoxical Effects of Rapamycin on Experimental House Dust Mite-Induced Asthma. PLoS ONE. 2012;7:e33984. doi: 10.1371/journal.pone.0033984. [DOI] [PMC free article] [PubMed] [Google Scholar]