

Abstract
Long term survival and success of artificial tissue constructs depend greatly on vascularization. Endothelial cell (EC) differentiation and vasculature formation are dependent on spatio-temporal cues in the extracellular matrix that dynamically interact with cells, a process difficult to reproduce in artificial systems. Here we present a novel bifunctional peptide that mimics matrix-bound vascular endothelial growth factor (VEGF) and can be used to encode spatially controlled angiogenic signals in collagen scaffolds. The peptide is comprised of a collagen mimetic domain that was previously reported to bind to type I collagen by a unique hybridization mechanism, and a VEGF mimetic domain with pro-angiogenic activity. Circular dichroism and collagen binding studies confirm the triple helical structure and the collagen binding affinity of the collagen mimetic domain, and EC culture studies demonstrate the peptide’s ability to induce endothelial cell morphogenesis and network formation as a matrix-bound factor in 2D and 3D collagen scaffolds. We also show spatial modification of collagen substrates with this peptide that allows localized EC activation and network formation. These results demonstrate that the peptide can be used to present spatially directed angiogenic cues in collagen scaffolds, which may be useful for engineering organized microvasculature.
Keywords: collagen, angiogenesis, microvasculature, VEGF, tissue engineering
1. Introduction
Angiogenesis and vascularization are vital for successful tissue engineering, as newly engineered tissues require blood vessels to deliver nutrients essential for long-term survival. However, engineered tissues often lack the proper capability to form functional vasculatures.[1–2] This is because angiogenesis is a complex biological process that requires a cascade of growth factor signaling and extracellular matrix (ECM) degradation to promote proliferation, organization, and differentiation of endothelial cells (EC). Although researchers have conjugated angiogenic factors to tissue engineering scaffolds to enhance angiogenesis,[3–6] most of these studies have been conducted using simple scaffolds, such as synthetic polymers, that allow easy chemical modification without concerns for non-specific conjugation and scaffold deterioration. However, among numerous model scaffolds tested to date, only natural scaffolds, collagen and fibrin in particular, have shown the consistent ability to form thin-walled, fluid-filled endothelial cell tubes with close resemblance to natural capillaries.[7] Unlike synthetic scaffolds, scaffolds derived from natural ECM components possess unique mechano-chemical signaling capabilities that regulate vascular lumen formation and support long-term vessel stability.[8–9]
The ECM is critical to vascular biology as a scaffold for EC attachment and for supporting chemotactic migration of endothelial cells. As the primary protein component of the ECM, collagen has been identified as a regulator of angiogenesis. In concert with vascular endothelial growth factor (VEGF), type I collagen has been shown to facilitate multi-cellular reorganization, activate actin polymerization, and induce stress fiber formation in ECs, all of which are necessary for capillary morphogenesis.[10]
Despite the active role that the structural components of the ECM play in angiogenesis, development of new blood vessels is chiefly governed by VEGF, which exists in three major isoforms—VEGF121, VEGF165 and VEGF189. These three isoforms have different bioavailability dictated by differing binding affinities to the ECM.[11] The difference in binding affinity among the three VEGF isoforms is due to the presence or absence of a heparin binding domain, a sequence of fifteen basic residues encoded by exons 6 and 7 of the VEGF gene. VEGF189, an matrix-bound factor with highest ECM affinity, contains the heparin binding domain as well as additional basic residues for high heparin sulfate binding affinity. VEGF165 contains only the heparin binding domain and binds to the ECM with moderate affinity. Finally, VEGF121 contains no heparin binding domain and exists solely as a soluble factor in the ECM. Together, the three VEGF isoforms are believed to create a spatial and temporal gradient that guides angiogenesis.
There have been multiple attempts to immobilize VEGF on surfaces and scaffolds through the use of covalent conjugation chemistry (e.g. carbodiimide reaction),[12–13] or the use of a crosslinker and binding tags.[5,14–15] However, these chemical reactions are non-specific and often require organic solvents that can compromise VEGF activity and/or physico-chemical integrity of the scaffolds.[16–17] Moreover, these previous immobilization studies were primarily intended for sustained delivery of VEGF, which otherwise degrades quickly in physiological settings.[16] These methods also lack the capacity to spatially control the immobilization process, which may be necessary for directed angiogenesis.
Alternatively, other research groups have sought to emulate VEGF and cell receptor interactions using peptides and antibodies.[18–22] One of the most promising pro-angiogenic compounds comes in the form of a short peptide based on the structure of VEGF. Developed by d′Andrea et al, a 15 amino acid sequence referred to as the QK peptide mimics the α-helical receptor binding region of VEGF.[23] This peptide promoted attachment and proliferation of ECs, exhibited competitive binding to VEGF receptors, and most importantly, induced EC activation and capillary-like formation in vitro.[23–25] QK peptide is promising not only for its extensive pro-angiogenic activity, but also for its short peptide sequence that can be easily synthesized and conjugated to various peptide delivery vehicles.
Although QK peptide has shown some therapeutic promise in vivo, it was mostly used as a locally delivered soluble factor.[26–27] There have been attempts to immobilize QK to inorganic substrates[25] and self-assembling peptide scaffolds,[24] and recently, QK has been conjugated to bioactive hydrogel through poly(ethylene glycol) linkers[17] and to hydroxyapatite through a binding peptide.[28] These two recent studies have shown that the immobilized form of QK retains its biological function and is able to elicit EC response. However, these substrates lack the dynamic physico-chemical signaling capabilities of collagen, and are not optimal for supporting long-term functional vessel growth in vitro. We hoped to create a QK peptide that can bind to a collagen scaffold similar to VEGF189 binding to heparin, and evaluate this peptide’s ability to act as a matrix-bound VEGF-like signaling factor in natural collagen scaffolds which are widely used for engineering micro-vasculatures.
To achieve collagen affinity, we employed a collagen mimetic peptide (CMP) sequence which has been reported to bind to type I collagen with varying degrees of affinity.[29–32] CMPs are a widely used model system in studying the triple helical structure of natural collagen. CMP contains the Gly-X-Y sequence commonly found in natural triple helical collagen, with a majority of the X and Y positions populated respectively by hydroxyproline (Hyp) and proline (Pro). This unique sequence allows CMPs to form triple helical supermolecular structures nearly identical to those in natural collagen. Unlike natural collagen, however, these peptides form thermally reversible triple helices. Due to their small size (<3 kDa), CMPs exist in single stranded conformation when heated above their melting temperature and reform triple helical structures upon cooling. We have previously demonstrated a method to bind and modify natural collagen by addition of melted, single stranded CMPs to a pre-formed collagen matrix. [30–32] The binding took place when single stranded CMPs were allowed to fold by cooling in the presence of type I collagen. Binding of CMPs to collagen films and gels are driven by hybridization interactions believed to be mediated by strand invasion and triple helical association. The melting temperature and collagen binding affinity of CMPs can be tuned by varying the number of repeating Gly-X-Y units. CMPs with more Gly-X-Y repeating units have a higher melting temperature as well as higher collagen binding affinity.[30] The varying degrees of collagen affinity could be used to mimic the varying heparin binding affinity of natural VEGF isoforms.
Here, we present a novel bifunctional peptide, QKCMP, comprised of a VEGF mimetic QK domain and a collagen binding CMP domain that can be used to control EC morphogenesis in collagen scaffolds for tissue engineering applications. In this study, we characterize the thermal behavior of QKCMP, demonstrate its binding affinity to type I collagen, and evaluate its morphogenic effects on endothelial cells.
2. Results and Discussion
2.1. Design and Characterization of QKCMP
As shown in Table 1, the target peptide designated as QKCMP consists of the VEGF mimetic QK domain and the collagen binding CMP domain. The QK domain was coupled to the N terminal side of the CMP with a three glycine spacer. We chose (ProHypGly)9 as the CMP domain because of its high collagen binding affinity.[30–31] As control samples, the QK and CMP domains were also produced separately. The peptides were prepared manually by F-moc mediated solid phase peptide synthesis and purified by high performance liquid chromatography. We used circular dichroism (CD) to investigate the structural characteristics of the QK, CMP, and QKCMP peptides (Figure 1a). As expected, CMP and QK displayed the characteristic CD spectra of triple helix[31, 33] and alpha helix[23] respectively, with CMP showing a maximum ellipticity at 225 nm and QK showing double minima at around 208 and 222 nm. The QKCMP signal corresponded to a sum of the CMP and QK CD signals, suggesting that the structures of the two domains were intact after the conjugation. Even under acidic pH conditions, QKCMP maintained the same CD spectra (Figure 1b). As previously reported, CMP’s binding affinity to natural collagen is primarily driven by its propensity to form triple helix.[30] The ability of QKCMP to retain its structure in acidic condition suggests that the peptide can be used to bind to collagen in acidic condition without concern for denaturation.
Table 1.
Amino acid sequences and molecular weights of synthetic peptides.
| Peptide | Sequence | Molecular Weight [g/mol] |
|---|---|---|
| QK | Ac*-KLTWQELYQLKYKGI | 1952 |
| CMP | (POG)9** | 2421 |
| QKCMP | Ac-KLTWQELYQLKYKGIGGG(POG)9 | 4526 |
Ac: acetyl
O: 4-hydroxyproline (Hyp)
Figure 1.
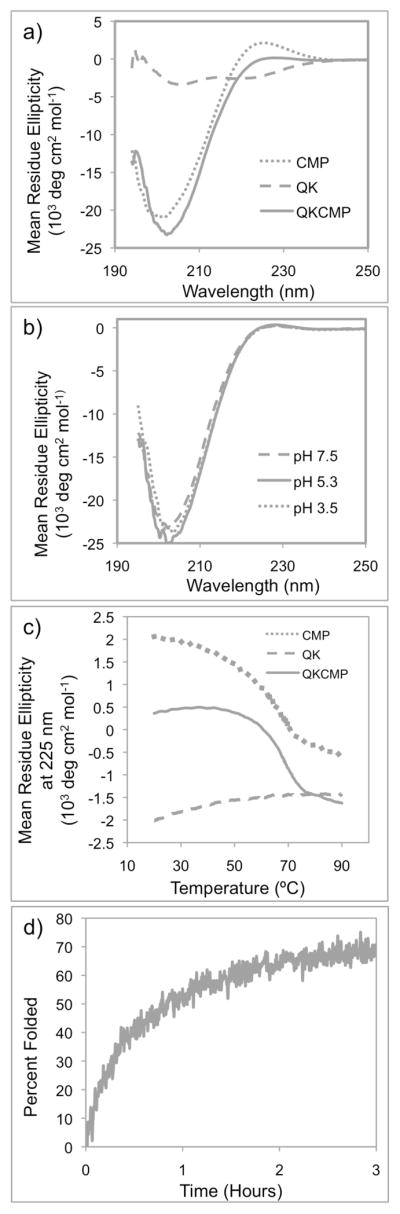
(a) Full CD spectra of CMP, QK, and QKCMP in PBS. (b) Full CD spectra of QKCMP in different pHs. (c) CD melting curves of CMP, QK, and QKCMP. (d) Triple helix refolding curve of QKCMP at 37 °C.
2.2. Thermal Folding Behavior of QKCMP
Having identified QKCMP’s triple helical structure, we investigated the thermal melting and folding behavior of the peptide by monitoring QKCMP’s triple helical CD signal as a function of temperature. Figure 1c shows the CD melting curves for CMP, QK, and QKCMP, which track their respective ellipticity at 225 nm as a function of temperature. The melting curve of CMP exhibited a sigmoidal shape indicative of a two-state, melting transition as reported earlier.[30–31] In contrast, the heating of the QK peptide did not produce a sigmoidal curve, and no melting transition was observed. This is in agreement with the previous reports that QK retains its alpha helical form even at high temperatures.[34] The peptide of interest, QKCMP, also exhibited a triple helical melting transition as demonstrated by sigmoidal melting curve in Figure 1c. By determining the minimum of the first derivatives of the sigmoidal curves,[35] the melting temperatures (Tm) of CMP and QKCMP were identified as 69 °C and 67.5 °C, respectively. The slightly lower melting temperature of QKCMP is likely due to the coulombic repulsion between the charged QK domains. The well-defined triple helical melting transition suggests that QKCMP could bind to type I collagen by the triple-helical propensity-driven hybridization mechanism previously shown for CMP and other CMP derivatives.[30–32, 36]
After establishing its melting behavior, we focused on the refolding kinetics of QKCMP. Compared to other secondary protein structures, the collagen triple helix is known to have a slower folding rate, typically on the order of minutes to hours.[33] This is because of slow cis-trans isomerization speed of Pro and Hyp, and a multi-step folding process involving chain association and registration, followed by triple helix nucleation and propagation.[37–39] Previously, we have demonstrated binding of thermally quenched CMPs (CMPs first heated to temperature above its Tm and quickly cooled to room temperature) to natural collagen by taking advantage of the slow refolding rate of CMPs, which provided a time window for single stranded CMP to hybridize with collagen before folding into homotrimeric CMP triple helices.[36] In order to study the folding rate of QKCMP, we heated QKCMP to 80 °C followed by a quick drop in temperature to 37 °C, and monitored the changes in CD ellipticity at 37 °C (Fig. 1d). As expected, QKCMP exhibited slow refolding rate with t1/2 close to 1 hr, indicating that the peptide’s folding process is similar to other CMPs, and that quenched single strands QKCMP can be used to hybridize with natural collagen.
2.3. Immobilization and Sustained Release of QKCMP in Collagen Gels and Coatings
In previous experiments,[30,31,36] collagens were modified with CMPs by adding hot or quenched CMPs to preformed collagen films and gels. Recently, we discovered high CMP immobilization when CMPs were added to the collagen solution before film and gel formation (Supporting Information, Figure S1). Rather than allowing only surface binding to take place, this new method exposes CMPs to the bulk collagens thereby maximizing the collagen loading level. The films and gels containing collagen fibers were formed by neutralization of acidic collagen solution followed by incubation at 37 °C. Figure 2a shows the SEM and TEM (inset) of type I collagen fibers prepared from a collagen solution (1.8 mg/mL) mixed in with quenched QKCMP (25 μM). The morphology of collagen fibers is nearly identically to that prepared from pure collagen solution (Fig. 2b), suggesting that the presence of QKCMP did not interfere with collagen fiber formation. Films and gels fabricated from QKCMP containing collagen solution had the physical appearance and handling characteristics similar to those made of pure collagen. It seems that CMP addition does not interfere with the collagen gelation process because the gel starts to form within a few minutes while the CMP folding and collagen hybridization may take over an hour. It is also possible that the fiber formation, which is driven by strong secondary forces such as charge-charge interactions and hydrophobic interactions, can dominate the relatively weak CMP-collagen hybridization interaction and that the CMP-collagen hybridization becomes fully active only after collagen fibers have already developed.
Figure 2.

Scanning electron and transmission electron micrographs (insets) of QKCMP modified type I collagen gel (a) and pure type I collagen gel (b). Cumulative release profiles of CF-QKCMP, CF-QK, and CF from type I collagen gels (c) and type I collagen coatings (d). Initially, 5 nmoles and 10 nmoles of each sample were added to each collagen gel and collagen coating, respectively. Day 0 and Hour 0 represent the amount of compounds remaining in the collagen matrix after removal of unbound peptide by washing with 4 °C blank buffer. Error bars represent ± SD.
QKCMP (labeled with carboxyfluorescein at N terminus) showed collagen binding characteristics and sustained release behavior similar to those previously reported for CMP derivatives (Fig. 2c and d).[30,36] After collagen coating or gel formation, unbound peptides were removed from the substrates by washing with excess buffer solution. The amount of peptide bound to the collagen substrates was determined using fluorescence spectrometry. In the case of the collagen gel (Fig. 2c), about 3 nmol corresponding to 60% of the QKCMPs initially applied were bound to the collagen gel after the initial wash. This correlates to roughly one QKCMP molecule for every one hundred collagen molecules. Considering the fact that the diameter of a collagen fiber is approximately 100 to 200 times of a collagen molecule, we believe that the surfaces of collagen fibers are densely decorated with QKCMP. For QK and carboxyfluorescein (CF), the level of initial binding after wash was approximate one third of the QKCMP. This background binding is likely due to the trapping effects within the pores of the collagen gel and non-specific charge-charge interactions with type I collagen. Over the next four days of 37 °C incubation, we observed a gradual release of QKCMP from the collagen gel, with 30% (1.5 nmol) remaining on day 2 and 20% (1 nmol) remaining on day 4 (Fig. 2c). We did not see significant changes in peptide release behavior in subsequent days. In contrast, the amounts of CF-labeled QK and CF in the collagen gel quickly dropped to below 10% (<0.5 nmol) on day 2. In the case of type I collagen coating, 1.8 nmol of CF-labeled QKCMP corresponding to a density of 5.53 ± 0.68 nmol/cm2 remained bound to the substrate compared to 0.6 nmol of CF and 0.6 nmol CF-tagged QK (Fig. 2d). In 24 hr of observation, we saw very little subsequent release of the peptide from the collagen coating.
The binding and release study indicates that the CMP domain of QKCMP retains collagen binding capability and can turn a soluble QK molecule into a collagen-bound factor similar to the action of the heparin binding domain of VEGF. Although there have been previous attempts to immobilize VEGF onto 2D surfaces, those studies only achieved low ligand density ranging from 3 fmol/cm2 to 6 pmol/cm2.[15,17,36,37,41] The bulkiness of the VEGF protein as well as the usage of intermediary linker molecules in these studies likely contribute to the low binding density. In comparison, immobilization of the small bioactive QK peptide onto 2D surfaces using covalently attached linkers have shown significantly higher surface density, ranging from 3 pmol/cm2 to 2.6 nmol/cm2.[17,28] Our binding studies demonstrated that QKCMP can also bind to 2D collagen substrates at a even higher density (5.53 ± 0.68 nmol/cm2), and we expected to see pronounced morphological changes in EC exposed to QKCMP immobilized on collagen scaffolds.
2.4. Endothelial Cell Morphogenesis in 2D and 3D Collagen Scaffolds
Next, we tested the ability of QKCMP to signal ECs for tubular morphogenesis in 2D and 3D environments. In nature, angiogenesis relies on a signaling cascade comprised of both soluble and insoluble growth factors. We employed QKCMP to mimic the matrix-bound VEGF isoforms by creating a peptide that has an EC signaling domain and an ECM binding domain.
We performed extensive morphogenesis studies using type I collagen coatings containing QKCMP. The collagen coatings were prepared by the same method used for the release study and the HUVECs were plated at 15,000 cells per well in a 96-well plate. As seen in Figure 3a, HUVECs plated on collagen coatings modified with QKCMP (25 and 100 μM of QKCMP in 0.1 mg/mL collagen coating solution) exhibited network morphology, while comparatively, HUVECs plated on plain collagen coating or collagen coating with QK added in the same manner (100 μM QK in 0.1 mg/mL collagen coating solution) displayed only cobble stone morphology. This indicated that QKCMP, immobilized to the collagen substrate through the CMP domain, is able to elicit a unique EC response. On the coating created with the highest QKCMP concentration (100 μM QKCMP in 0.1 mg/mL collagen coating solution), HUVECs began to show network like developments in as little as 3 hr. As the concentration of QKCMP decreased, network morphology did not develop until 6 hr in the case of intermediate QKCMP concentration (25 μM QKCMP in 0.1 mg/mL collagen coating solution) and 50 hr in the case of low QKCMP concentration (data not shown). HUVECs plated on plain collagen coating or on collagen coating with QK (100 μM QK in 0.1 mg/mL collagen coating solution) did not show any network morphological development during the 50 hr time frame. Although high QKCMP concentrations were able to quickly induce morphological changes, HUVECs stimulated under those conditions continued to migrate even after the initial network formation, and formed inactive isolated cell cluster after 24+ hr of incubation.
Figure 3.

(a) Phase contrast micrographs of HUVECs plated on type I collagen coatings modified with varying concentrations of QKCMP and QK. (b) Fluorescence micrographs of HUVECs stained with anti-PECAM-1 monoclonal antibodies (red), phalloidin (stains actin in green), and DAPI (stains nucleus in blue) plated on QKCMP modified collagen (left panel) and plain collagen coatings (right panel). Filopodia extensions are marked by arrows. Bar = 100μm.
The network morphology of HUVECs incubated on QKCMP modified collagen coating was accompanied by the presence of high concentrations of platelet-endothelial cell adhesion molecules (PECAM) dispersed throughout the cell surfaces as evidenced by immunofluorescence microscopy (Fig. 3b). PECAM is present at cell junctions of quiescent blood vessels, inhibiting EC migration and promoting stable EC-EC junctions.[42–44] However, in angiogenic ECs, PECAM is redistributed from cell junctions to cell surfaces to activate EC migration.[45–47] The diffuse presence of PECAM on the EC surface is a strong indication that the cells’ angiogenic pathways have been activated.
Furthermore, as shown in phalloidin/DAPI staining, the network morphology of HUVECs incubated on QKCMP modified collagen substrate occurred in concert with the development of filopodia extensions in multiple directions (arrows in Fig. 3b). Such filopodia extensions were not observed in the cells with cobblestone morphology (plain collagen and QK supplemented collagen). VEGF has been known to direct filopodial behavior where ECs extend thin actin processes to form stress fibers in the direction of VEGF signals for vascular expansion.[42] Model animals that only express the heparin binding form of VEGF have been shown to develop small caliber blood vessels with excessive branching which is preceded by the development of excess filopodia in multiple directions and prolonged EC migration.[42,43,48] The presence of PECAM on cell surfaces combined with multidirectional filopodia development and continued cell migration after initial network formation strongly suggest that QKCMP is giving matrix-bound VEGF-like signals to ECs in collagen substrates.
To confirm that the enhanced HUVEC morphogenesis on QKCMP modified collagen coatings was induced by EC specific stimulation, we conducted the same morphological studies with fibroblasts. Fibroblasts (human dermal neonatal fibroblasts) plated on both plain and QKCMP modified collagen coatings displayed identical fusiform morphology after 24 hr (Figure 4). The absence of morphological transformation on these fibroblasts suggests that the QKCMP’s effect on HUVEC morphology did not result from generic cell-substrate interactions (e.g. mechanical stiffness or charge interactions) but instead developed specifically from endothelial cell stimulation by the peptide’s QK domain mediated possibly by VEGF receptors.
Figure 4.

Phase contrast micrographs of human dermal neonatal fibroblasts plated on plain collagen coating (0.1 mg/mL coating solution) and QKCMP modified collagen coating (100 μM of QKCMP in 0.1 mg/mL coating solution). Bar = 100 μm.
Extracellular signal-regulated kinase 1 and 2 (ERK1/2) play a crucial role in VEGF-dependent activation of ECs. Phosphorylation of ERK1/2 is known to elicit EC angiogenesis by increasing EC cell proliferation and promoting EC migration.[23,49–51] To verify that the EC migration and morphological changes that we observed are due to ERK1/2 activation by the QK-CMP, we examined the ERK1/2 phosphorylation levels of ECs that were incubated for 1 hr on the QKCMP-modified collagen substrates (Fig. 5). In comparison to cells grown on plain collagen substrate (0.1 mg/mL collagen coating solution), cells seeded on QKCMP-modified collagen (100 μM QKCMP in 0.1 mg/mL collagen coating solution) showed a marked increase in phosphorylation level (Fig. 5b), while maintaining the same level of ERK1/2 expression (Fig. 5a). The phosphorylation level was not as high as that of the cells seeded on plain collagen supplemented with 100 ng/mL of soluble VEGF which is similar to findings by those who originally developed the QK.[23] This result indicate that the immobilized QK-CMP retains bioactivity of the QK and is able to activate the canonical pathway in VEGF signaling.
Figure 5.

Effects of QKCMP modified type I collagen on the expression (a) and phosphorylation (b) of ERK1/2 in HUVECs. Western Blots experiments show that neither QKCMP nor soluble VEGF (100 ng/mL) affects the expression levels of ERK (a), while increasing the phosphorylation level of ERK (Student’s t test, p < 0.01). Error bars represent ±SD.
The network morphology and the viability of HUVECs activated by collagen bound QKCMP can be prolonged by the addition of soluble VEGF. As seen in Figure 6a, when cell culture medium was supplemented with 500 ng/mL of soluble VEGF, the network like morphology of HUVECs seeded on QKCMP modified collagen coating (100 μM QKCMP in 0.1 mg/mL collagen coating solution) was maintained up to 24 hr. This is in contrast to cells exposed to only QKCMP which form inactive cell clusters before 24 hr. Furthermore, proliferation rate assays (Fig. 6b) indicated increased EC proliferation in the presence of soluble VEGF. Although previous studies involving soluble VEGF have shown that 100 ng/mL elicits maximum cellular response,[16,53] this concentration was not able to prolong the viability of HUVECs which were also activated by our collagen bound QKCMP. This is most likely due to changes in VEGF receptor response in the presence of both soluble and matrix-bound factors, and more work is currently in progress to elucidate this phenomenon.
Figure 6.

Phase contrast micrographs of HUVECs plated on QKCMP modified type I collagen with and without 500 ng/mL soluble VEGF (a). WST-1 proliferation assay (b) shows increased cell proliferation in cells plated on QKCMP modified collagen with 100 ng/mL soluble of VEGF (Student’s t test, p < 0.05). Error bars represent ± SD. Bar = 100 μm.
The angiogenic activity of QKCMP in the presence of soluble VEGF was also observed in the 3D cell construct. We encapsulated EC spheroids[54,55] in collagen gels that contained QKCMP (25 μM) and observed tubes sprouting from EC spheroids. According to the QKCMP immobilization study (Fig. 2), we expected approximately 3 nmol of immobilized QKCMP initially and 1 nmol of QKCMP released from the collagen gel during the EC sprouting experiment. Since our 2D culture study indicated that the soluble form of QK-CMP had no morphogenic effects on ECs, we expected similarly that the QK-CMP released during the experiment would have little effect on ECs in 3D collagen gel. In Figure 7a and b, we show comparative photos of EC spheroids undergoing tubular morphogenesis in pure collagen (a) and collagen with matrix-bound QKCMP (b). Although both collagen gels induced EC sprouting possibly in response to soluble VEGF supplemented to the culture media, spheroids encapsulated in QKCMP containing collagen gels exhibited greater sprouting with more elongated tube morphology as compared to fewer and flattened sprouting morphology in the pure collagen gels. This was confirmed by examining the total number and total length of the sprouts per spheroid (Fig. 7c and d). The results suggest that the collagen bound QKCMP and soluble VEGF work synergistically in a manner similar to insoluble and soluble VEGF in native tissue to stimulate EC sprouting and tube elongation which are early indications of angiogenesis.
Figure 7.
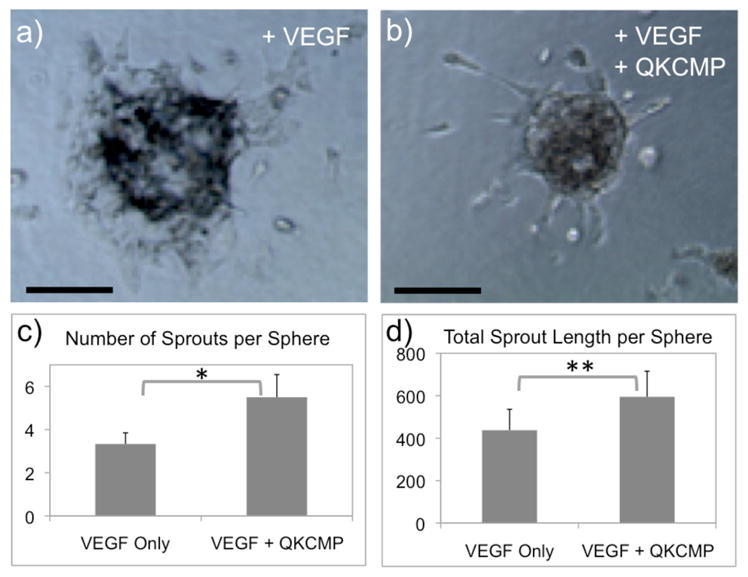
Phase contrast micrographs of 3D endothelial cell spheroids (1 day culture) encapsulated in type I collagen gel supplemented with 2.5 ng/mL of soluble VEGF (a) or 5 nmoles of QKCMP and 2.5 ng/mL of soluble VEGF (b). Quantification of sprouting from spheroids by the number of sprouts per sphere (c) and total sprout length per sphere (d). (Student’s t test: * p < 0.01, ** p < 0.05) Error bars represent ± SD. Bar = 100 μm.
Previous research demonstrated that matrix bound isoforms of VEGF promote cell migration and new vessel formation while soluble isoforms of VEGF induce EC proliferation and vessel maturation.[42,56] Model animals expressing only the matrix-bound form of VEGF suffer from hypervascularization. They develop abnormally thin blood vessels characterized by excessive branching at the expense of lumen growth and are more likely to die in utero.[42,43,56] The combinatorial expression of both matrix-binding and soluble VEGF creates a spatial gradient that guides EC migration, vascular formation, and branching development to form fully functional vascular networks. Our experiments showed the ability of collagen-bound QKCMP to activate EC morphogenesis, causing ectopic filopodia development and excessive cell migration that mirrors the beginning stages of excessive vessel branching in animals expressing only matrix-bound VEGF. The sustained network development and sprouting of HUVECs grown in 2 and 3 dimensional QKCMP-modified collagen substrates supplemented with soluble VEGF suggest that QKCMP could be used as a substitute for matrix-bound VEGF to engineer microvasculature in collagen scaffolds.
2.5. Local Activation of ECs on Collagen Substrates
One advantage of our QKCMP system in activating ECs is the ability to locally control the area of peptide binding thereby allowing spatial control of EC migration and network morphology development. We fabricated a well (48-well plate) with half of the well coated with plain collagen (0.1 mg/mL collagen coating solution) and the other half with QKCMP modified collagen (100 μM QKCMP in 0.1 mg/mL collagen coating solution). After HUVEC seeding (55,000 cells per well), the well was supplemented with 500 ng/mL of soluble VEGF. After 30 mins, there was essentially no morphological difference between the two halves of the well (Fig. 8a). The cells were homogenously dispersed within the well with a slightly rounded morphology, as the cells were just beginning to attach to the collagen substrate. The difference in morphology became apparent after 3 hr incubation, where the cells on the plain collagen coating resembled a cobblestone like morphology while the cells on the QKCMP modified collagen coating appeared elongated and starting to make cell-cell connections. The morphological differences became more apparent after 6 hr, as cells on the plain collagen retained the cobblestone morphology, while extensive network formation was seen on the QKCMP modified half of the well. In Figure 8b, phalloidin/DAPI staining shows cobble-stone like morphology on the HUVECs plated on the plain collagen half of the well. On the QKCMP modified half, the cells show a completely different morphology—the cells appear more active, as filopodia protrusions are clearly visible from a majority of the cells, indicative of cell movement and network formation.
Figure 8.

Morphology of HUVECs plated on cell culture well half coated with plain type I collagen and half coated with QKCMP modified type I collagen as visualized by phase contrast microscopy (a) and fluorescence microscopy after phalloidin and DAPI staining (b). Bar = 100 μm.
The morphological differences between the two halves of the well were maintained up to 24 hr. The morphologies of the HUVECs plated on the plain collagen and QKCMP modified surfaces are comparable to the results discussed in the previous sections. The patterning experiment, however, highlights the novelty of our system: for the first time, we demonstrated that the EC morphology on collagen coating within a single tissue culture well can be spatially controlled through the use of QKCMP peptides that mimic the ECM binding and angiogenic activities of matrix-bound VEGF. We were able to induce local EC network formation which holds great promise for patterning tissue scaffold for directed angiogenesis.
3. Conclusions
Recognition of the critical roles that the various VEGF isoforms play in the development of vasculature prompted us to develop a new peptide that can mimic the matrix-bound VEGF isoform (VEGF189) that is known to have high binding affinity to ECM. We have successfully designed and synthesized QKCMP, a bifunctional peptide that contains an EC activating domain (QK) and a collagen binding domain (CMP). Our studies have shown that this peptide folds into triple helical structure and can bind to type I collagen when allowed to fold in the presence of collagen, similar to previous CMP derivatives as previously reported.[30–32,36] Over 1 nmol of QKCMP was immobilized on type I collagen coatings covering the 96 well culture plate. ECs plated on the QKCMP-immobilized collagen coatings exhibited pro-angiogenic behaviors as evidenced by diffusive presence of PECAM, multiple filopodia protrusions, and cell migration which resulted in the development of cellular networks. EC sprouting studies of EC spheroids in 3D collagen gel showed elevated tubulogenic behavior for QKCMP modified collagen gel. We also demonstrated that such cell morphogenesis can be induced in pre-defined areas within wider collagen-based cell culture constructs by spatially controlling the areas of QKCMP immobilization. The ability to encode spatially defined morphogenic signals in natural scaffolds is expected to provide new pathways for engineering complex tissues for regenerative medicine. While others have immobilized VEGF and VEGF mimetic peptides to matrices,[16–17] the work presented in this article, along with the work by Lee and coworkers,[28] is one of the first attempts in designing and studying peptides that directly mimics not only the angiogenic activity but also the ECM binding affinity of matrix-bound VEGF with the intent to enhance or spatially direct microvasculature formation. In the long run, the use of this matrix-bound VEGF mimetic may allow production of organized vasculatures that are more akin to natural tissues.
4. Experimental
Materials
All amino acids and peptide synthesis reagents including N-methylpyrrolidone (NMP), 2-(1H-benzo-triazole-1-yl)-1,1,3,3-tetra-methyluronium hexafluorophosphate (HBTU) and trifluoroacetic acid (TFA) were purchased from Advanced ChemTech (Louisville, KY). Rink-type TentaGel R Ram resin was purchased from Peptides International (Louisville, KY). Carboxyfluorescein (CF), piperidine and triisopropylsilane (TIS) were purchased from Sigma-Aldrich (St. Louis, MO). Acid soluble rat tail type I collagen was obtained from BD Bioscience (San Jose, CA). All electron microscopy fixatives and staining agents were obtained from Electron Microscopy Sciences (Hatfield, PA). HUVECs and endothelial cell media were obtained from Lonza (Walkersville, MD), and VEGF (VEGF recombinant human) was obtained from Invitrogen (Carlsbad, CA). Fibroblasts (human dermal, neonatal) were purchased from American Type Culture Collection (Manassas, VA) and fibroblast media were obtained from Invitrogen. WST-1 proliferation assay was obtained from Roche Applied Science (Indianapolis, IN). For Western Blots, all primary and secondary antibodies were obtained from Cell Signaling Technologies (Beverly, MA). For immunohistochemical staining, 10% neutral buffered formalin was obtained from Sigma-Aldrich, tris-buffered saline (TBS) was purchased from Bio-Rad Laboratories (Hercules, CA), and monoclonal mouse anti-human CD31/PECAM-1 primary antibody and Cy3 conjugated anti-mouse secondary antibody were purchased from BD Bioscience and Jackson ImmunoResearch Laboratories Inc (West Grove, PA). Alexa-Fluor 488 phalloidin and 4,6-diamidino-2-phenylindole (DAPI) were purchased from Invitrogen and Roche Applied Science, respectively.
Peptide Synthesis
All peptides were synthesized by conventional solid phase peptide synthesis methods on TentaGel R Ram resin in NMP using 4 molar excess of Fmoc protected amino acids activated with HBTU. Fmoc protection groups were removed with 20% piperidine in NMP. The efficacy of the coupling and deprotection reactions was monitored with ninhydrin and chloranil tests. For non-fluorescently labeled peptides, the N-terminus of the peptide was capped with an acetyl group by treatment with acetic anhydride. After completing the synthesis of the target sequence, the peptide as well as all side-chain protection groups were cleaved by treating the resin with a cleavage cocktail (95% TFA, 2.5% deionized water and 2.5% TIS ) for 3 hr. The crude peptides were precipitated by addition to cold diethyl ether and dried in a vacuum.
The crude peptides were purified using semipreparative reverse-phase high performance liquid chromatography (HPLC) on a Varian Polaris 210 series liquid chromatograph (Agilent Technologies, Santa Clara, CA) with UV detection at 275 nm (for general peptides) or 493 nm (for CF containing peptides), using a Vydac C-4 reverse-phased column from Grace Davison Discovery Sciences (Deerfield, IL). The HPLC was run at a flow rate of 4 mL/min using a solvent gradient comprised of deionized water (0.1% TFA) and methanol (0.1% TFA). The elutions corresponding to the target peak in the chromatogram were combined and lyophilized. The purified peptides were analyzed by matrix assisted laser desorption/ionization time of flight (MALDI-ToF) mass spectrometry (Voyager DE-STR, Applied Biosystems, Foster City, CA.) The mass spectra of all peptides are shown in Figure S2.
Circular Dichroism and Thermal Melting Experiments
Prior to CD measurements, peptides (100 μM) were incubated at 4 °C overnight in one of the three buffer solutions: 1 × PBS (pH 7.5), phosphate buffer (pH 5.3), and acetic acid (pH 3.3). The concentrations of the samples were determined by UV-Vis spectrophotometry at 275 nm or 493 nm. CD spectra were measured on a JASCO 710 spectrometer with JASCO PTC-348 WI temperature controller (Easton, MD). For thermal melting curves, the molar ellipticity at 225 nm was monitored between 5 °C and 90 °C with a heating rate of 1 °C/min.
Determination of Refolding Yields of QKCMP
To measure the refolding rate, QKCMP (100 μM) preincubated at 80 °C for 30 mins was quenched to 37 °C in an ice bath and the change in molar ellipticity at 225 nm was monitored for 3 hr at 37 °C. The refolding yield was determined from the following equation:
θA = Molar ellipticity at 225 nm of quenched sample at a given time (helical content of quenched sample during refolding)
θB = Molar ellipticity at 225 nm of sample after overnight incubation at 4 °C (helical content of fully folded sample)
θC = Molar ellipticity at 225 nm of sample after 3 hr incubation at 80°C (helical content of fully unfolded sample)
Immobilization of QKCMP in Collagen Gels and Coatings
To immobilize QKCMP to collagen, CF labeled QKCMP solution (200 μL, 25 μM) was heated to 80 °C for at least 30 mins to ensure melting. The hot solution was quenched to 37 °C in an ice water bath and immediately added to acid soluble rat tail type I collagen (100 μL). The mixture solution was neutralized with dilute sodium hydroxide (100 μL), added to a 48-well cell culture plate, and incubated at 37 °C for 3 hr to allow gel formation and peptide binding. The final volume of the gel was 400 μL with 1 mg/mL collagen concentration. The collagen gel was then washed ten times, using 500 μL of 4°C 1 × PBS, to remove any unbound peptides. After the wash, each well was reloaded with 500 μL of 1 × PBS and incubated at 37 °C for the release experiment. The PBS solution was replaced every two days.
To determine the amount of peptides bound to the collagen gels, the fluorescence intensity of the gels (after melting) was measured with a Gemini EM Fluorescence Microplate Reader (Molecular Devices, Sunnyvale, CA). For each time point in the binding release study, excess PBS was first removed from the collagen gel. The remaining collagen gels were melted at 80 °C for 45 mins. The solutions were then cooled to 4 °C and their fluorescence intensities were measured at 533 nm emission and 489 nm excitation wavelengths. The fluorescence readings were then converted to a concentration of CF labeled peptides using a standard curve generated for each type of peptide studied.
To measure QKCMP binding to collagen coating, a 1 mM stock solution of CF-labeled peptide solution was created in 1 × PBS. The solution was heated at 80 °C for at least 30 mins to enable triple helical melting. Separately, a collagen solution was created by mixing rat tail type I collagen with 1 × PBS to a final collagen concentration of 0.1 mg/mL. The heated CF labeled peptide solution was then added directly to the collagen coating solution, creating a final peptide concentration of 100 μM. No quenching was required since the heated peptides were quickly cooled during the mixing step due to the large volume of collagen coating solution (10 to 40 fold dilution). The mixture solution (100 μL) was added to the wells of a 96-well plate and incubated for 1 hr. After the incubation, to ensure that the unbound peptides were removed from the collagen-coated well, all excess solutions were removed from the cell culture well and saved for fluorescence readings. The wells were then replenished with 100 μL of fresh 1 × PBS. Subsequently, the culture plate was incubated at 37 °C and each well was replaced with 100 μL fresh 1 × PBS at each noted time point for the peptide release study. The PBS buffer removed during the study was saved for fluorescence readings. The amounts of CF-labeled peptides remaining in the collagen coating were determined from the fluorescence intensities of the released peptides in the PBS solutions which were converted to a concentration using a custom calibration curve.
Electron Microscopy
QKCMP modified collagen gel (400 μL with 1.8 mg/mL collagen concentration) was created as described above. Plain collagen gel was created by the same method, but 1 × PBS was used instead of the QKCMP solution. The gels were first fixed for 1 hr with 100 mM sodium cacodylate solution containing 3% paraformaldehyde, 1.5% glutaraldehyde and 2.5% sucrose, and stained with 1% osmium tetroxide solution for 30 min. For scanning electron microscopy, the samples were dehydrated with ethanol and dried with a supercritical point dryer (Tousimis 795, Rockville, MD). The samples were then sputter coated with a 4 nm thick platinum layer (Anatach Sputter Coater, Union City, CA) and observed under high vacuum in FEI Quanta ESEM 200 (Hillsboro, OR). For transmission electron microscopy, the samples were additionally stained with Kellenberger’s uranyl acetate overnight before ethanol dehydration. The dehydrated samples were then embedded in Embed 812 resin and sectioned into 80–100 nm thick slices. The sample slices were placed on a TEM grid and observed with FEI Tecnai 12 (Hillsboro, OR).
Cell Culture
HUVECs were cultured in a complete endothelial cell growth medium (EGM-2) and human dermal neonatal fibroblasts were cultured in 1:1 Dulbecco’s Modified Eagle Medium/Ham’s F-12 Nutrient Mixture (DMEM/F-12 with GlutaMAX, Invitrogen) supplemented with 10% FBS. Both cell types were grown at 37 °C with a fully humidified atmosphere and 5% CO2. The growth media were changed every other day. The cells were harvested with trypsin-EDTA treatment at 70–80% confluency, and only passage 2–7 cells were used in the 2D and 3D tubulogenesis experiments. All phase contrast images were taken with EVOS phase contrast microscopy (Advanced Microscopy Group, Botwell, WA).
Endothelial Cells Cultured on 2D Collagen Coating
A collagen solution was prepared by mixing rat tail type I collagen with 1 × PBS to a final collagen concentration of 0.1 mg/mL. For plain collagen coating, 100 μL of the solution was added at room temperature to the wells of a 96-well tissue culture plate and incubated at 37 °C for 1 hr. For peptide-modified collagen coating, a stock solution of the peptide (1 mM) was first heated at 80 °C for at least 30 mins to enable melting. This heated peptide solution was then added directly to the collagen coating solution at 1:10 and 1:40 dilutions (by volume), creating a final peptide concentration of 100 μM and 25 μM, respectively. No quenching was required since the heated peptides were quickly cooled during the mixing step due to the large volume of the collagen coating solution. The mixture solution (100 μL) was added to the wells of a 96-well plate and incubated for 1 hr. After the incubation, all excess solutions were removed from the cell culture well. Subsequently, 15,000 HUVECs in 100 μL of endothelial basal media (EBM) supplemented with 2% fetal bovine serum (FBS) were seeded in each well (96 well plate). For VEGF supplemented experiments, the 100 μL of growth media were further supplemented with 500 ng/mL of VEGF.
ERK Assay
Cells were first serum starved in reduced serum media (EBM supplemented with 0.25% FBS) for 4 hours. For plain collagen coating, 1.5 mL of the collagen solution (0.1 mg/mL) was added at room temperature to the wells of a 6-well tissue culture plate and incubated at 37 °C for 1 hr. For peptide-modified collagen coating, a stock solution of the QKCMP peptide (1 mM) was first heated at 80 °C for at least 30 min to enable melting. This heated peptide solution was then added directly to the collagen coating solution at 1:10 dilution (by volume), creating a final peptide concentration of 100 μM. The QKCMP-collagen solutions (1.5 mL) were then added to wells of the same 6-well tissue culture plate. The plate was incubated at 37 °C for 1 hr, and the remaining coating solution was removed. Each well was then seeded with the serum starved HUVECs (485,000 cells/well) in 1.5 mL of EBM media supplemented with 2% FBS. As a positive control, cells seeded on one of the plain collagen coated wells were supplemented with 100 ng/mL of soluble VEGF. The cells were first cultured for 1 hr at 37 °C and then lysed with lysis buffer (25 mM Tris-HCL, 0.5% Triton X-100, 200 mM NaCl, 2 mM EDTA, phosphotase and protease inhibitors) (Roche Applied Science). The cell lysate was spun at 15,000 g for 15 min at 4 °C, and the supernatant was collected. Protein concentration within the supernatant was determined using the Pierce BCA assay kit (Thermo Fisher Scientific, Rockford, IL). The lysate (17 μg) was loaded into a NuPAGE Novex 10% Bis-Tris gel (Invitrogen) for protein separation by electrophoresis. The separated proteins were then transferred onto a nitrocellulose membrane using the iBlot Dry Blotting System (Invitrogen). The membrane was blocked with 3% milk for 1 hr at room temperature, and then stained overnight at 4 °C with specific antibodies for total ERK1/2, and Phospho-p44/42 ERK1/2. Afterwards, the membranes were stained with anti-rabbit horseradish peroxidase conjugated secondary antibody for 1 hr at room temperature. Amersham ECL detection reagent (GE Healthcare Life Sciences, UK) was used to reveal the bands on films, and the bands were quantified using Amersham ImageQuant TL (GE Healthcare Life Science). Data and analysis were taken from 3 independent trials.
Endothelial Cell Sprouting in 3D Collagen Gels
Endothelial cell spheroids for 3D tubulogenesis study were constructed as previously described [46,47]. Briefly, methylcellulose was dissolved in 30 mL of EGM-2 at a concentration of 24 mg/mL. HUVECS were suspended in the methycellulose/EGM-2 solution and incubated overnight in round bottom 96 well plates (500 cells per well) to allow spheroid formation. Spheroids were harvested with a multichannel pipette and dispersed in neutralized type I collagen solution (1.8 mg/mL) or QKCMP modified collagen solutions (25 μM QKCMP in 1.8 mg/mL collagen) prepared as previously described. The spheroid-collagen solution was delivered to the wells of 48 well plates (200 μL) and incubated at 37 °C for gel formation and spheroid encapsulation. After the gel formation, 500 μL of media (EBM supplemented with 2% FBS and soluble growth factors at noted concentrations) were added to each well followed by incubation at 37 °C. The spheroids were imaged after 24 hr, and at least four spheroids were studied under each condition.
Patterned Collagen Substrate for Local EC Activation
A collagen solution (110 μL, 0.1 mg/mL) was added to individual wells of a 48-well plate with the plate tilted at a 45° angle so that only half the surface area of each well was covered with the collagen solution. The plate was then incubated at 37 °C in the tilted position for 1 hr. After removing the remaining collagen solution, the plate was tilted in the opposite direction, followed by addition of QKCMP-collagen mixture solution (110 μL, 100 μM QKCMP in 0.1 mg/mL collagen) previously prepared by adding 80 °C QKCMP (11 μL, 1 mM) to type I collagen solution (99 μL, 0.1 mg/mL). The tilted plate was then incubated at 37 °C for 1 hr, and the remaining coating solution was removed. The well was then seeded with HUVECs (55,000 cells/well) in 500 μL of EBM media supplemented with 2% FBS and VEGF (500 ng/mL).
Immunohistochemical Staining
The cells were fixed with 200 μL of 10% neutral buffered formalin for 30 min. After fixing, the cells were permeabilized and blocked with 200 μL of TBS supplemented with 5% donkey serum and 0.25% Triton X-100. Primary mouse anti-human PECAM-1 antibodies (0.5 mg/mL) were added at 1:50 dilution in TBS and the mixture was incubated at 4 °C overnight. Subsequently, Cy3 conjugated anti-mouse secondary antibodies (0.5 mg/mL) were added at 1:250 dilution in TBS and the mixture was further incubated at room temperature for 90 mins. Finally, the cells were stained with a 200 μL of PBS solution containing phalloidin (0.165 μM) and DAPI (1 μg/mL) for 30 mins. The fluorescent cell images were captured on Nikon Eclipse TE2000-E (Nikon Instruments, Melville, NY).
Supplementary Material
Figure S1. Cumulative release profiles of QKCMP from type I collagen gels, where quenched QKCMP was added in solution before gel formation (binding in solution) and to preformed gel (binding to preformed gel). Day 0 and hour 0 represent the amount of compounds remaining in the collagen matrix after removal of unbound peptide by washing with 4 °C blank buffer. Error bars represent ±SD.
Figure S2. MALDI-ToF mass spectroscopy of target peptides after purification.
Acknowledgments
The authors thank Kalina Hristova and Edwin Li for help with circular dichroism, Christopher Chen, Xingyu Liu, Jacob Koskimaki and Colette Shen for help with cell studies, J. Michael McCaffery and Erin Pryce for help with electron microscopy, and Fenghao Chen, Lijuan He and Sarvenaz Sarabipour for help with Western Blots. This work was supported by grants from NSF (DMR-0645411) and NIAMS/NIH (R01AR060484) to S.M.Y., by NSF IGERT fellowship to T.R.C., and HHMI Graduate Training Program (NBMed) and NDSEG (32 CFR 168a) fellowships to P.J.S.
Footnotes
Supporting Information is available online from Wiley InterScience or from the author.
References
- 1.Ennet AB, Mooney DJ. Expert Opin Biol Ther. 2002;2:805. doi: 10.1517/14712598.2.8.805. [DOI] [PubMed] [Google Scholar]
- 2.Soker S, Machado M, Atala A. World J Urol. 2000;18:10. doi: 10.1007/pl00007070. [DOI] [PubMed] [Google Scholar]
- 3.Phelps EA, Landázuri N, Thule PM, Taylor WR, García AJ. Proc Natl Acad Sci U S A. 2010;107:3323. doi: 10.1073/pnas.0905447107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Silva EA, Mooney DJ. J Thromb Haemost. 2007;5:590. doi: 10.1111/j.1538-7836.2007.02386.x. [DOI] [PubMed] [Google Scholar]
- 5.Zisch AH, Schenk U, Schense JC, Sakiyama-Elbert SE, Hubbell J. J Controlled Release. 2001;72:101. doi: 10.1016/s0168-3659(01)00266-8. [DOI] [PubMed] [Google Scholar]
- 6.Zisch AH, Lutolf MP, Ehrbar M, Raeber GP, Rizzi SC, Davies N, Schmökel H, Bezuidenhout D, Djonov V, Zilla P, Hubbell JA. FASEB J. 2003;17:2260. doi: 10.1096/fj.02-1041fje. [DOI] [PubMed] [Google Scholar]
- 7.Francis ME, Uriel S, Brey EM. Tissue Eng Pt B-Rev. 2008;14:19. doi: 10.1089/teb.2007.0115. [DOI] [PubMed] [Google Scholar]
- 8.Davis GE, Stratman AN, Sacharidou A, Koh W. Int Rev Cel Mol Bio. 2011;288:101. doi: 10.1016/B978-0-12-386041-5.00003-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rhodes JM, Simon M. J Cell Mol Med. 2007;11:176. doi: 10.1111/j.1582-4934.2007.00031.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davis GE, Sanger DR. Circ Res. 2005;97:1093. doi: 10.1161/01.RES.0000191547.64391.e3. [DOI] [PubMed] [Google Scholar]
- 11.Ng Y. In: VEGF in Development. Ruhrberg C, editor. Landes Bioscience; Austin: 2008. pp. 1–13. [Google Scholar]
- 12.Shen YH, Shoichet MS, Radisic M. Acta Biomater. 2008;4:477. doi: 10.1016/j.actbio.2007.12.011. [DOI] [PubMed] [Google Scholar]
- 13.Chiu LLY, Radisic M. Biomaterials. 2010;31:226. doi: 10.1016/j.biomaterials.2009.09.039. [DOI] [PubMed] [Google Scholar]
- 14.Koch S, Yao C, Grieb G, Prével, Noah EM, Steffens GCM. J Mater Sci: Mater Med. 2006;17:735. doi: 10.1007/s10856-006-9684-x. [DOI] [PubMed] [Google Scholar]
- 15.Backer MV, Patel V, Jehning BT, Claffey KP, Backer JM. Biomaterials. 2006;27:5452. doi: 10.1016/j.biomaterials.2006.06.025. [DOI] [PubMed] [Google Scholar]
- 16.Tayalia P, Mooney DJ. Adv Mater. 2009;21:3269. doi: 10.1002/adma.200900241. [DOI] [PubMed] [Google Scholar]
- 17.Leslie-Barbick JE, Saik JE, Gould DJ, Dickinson ME, West JL. Biomaterials. 2011;32:5782. doi: 10.1016/j.biomaterials.2011.04.060. [DOI] [PubMed] [Google Scholar]
- 18.Scheidegger P, Weiglhofer W, Suarez S, Console S, Waltenberger J, Pepper MS, Jaussi R, Ballmer-Hofer K. Biochem J. 2001;353:569. doi: 10.1042/0264-6021:3530569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cooke SP, Boxer GM, Lawrence L, Pedley RB, Spencer DIR, Begent RHJ, Chester KA. Cancer Res. 2001;61:3653. [PubMed] [Google Scholar]
- 20.Prewett M, Hubber J, Li Y, Santiago A, O’Connor W, King K, Overholser J, Hooper A, Pytowski B, Witte L, Bohlen P, Hicklin DJ. Cancer Res. 1999;59:5209. [PubMed] [Google Scholar]
- 21.Jia H, Jezequel S, Löhr M, Shaikh S, Davis D, Soker S, Selwood D, Zachary I. Biochem Biophys Res Commun. 2001;283:164. doi: 10.1006/bbrc.2001.4761. [DOI] [PubMed] [Google Scholar]
- 22.El-Mousawi M, Tchistiakova L, Yurchenko L, Pietrzynski G, Moreno M, Stanimirovic D, Ahmad D, Alakhov V. J Biol Chem. 2003;278:46681. doi: 10.1074/jbc.M308681200. [DOI] [PubMed] [Google Scholar]
- 23.D’Andrea LD, Iaccarino G, Fattorusso R, Sorriento D, Carannante C, Capasso D, Trimarco B, Pedone C. Proc Natl Acad Sci U S A. 2005;102:14215. doi: 10.1073/pnas.0505047102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang X, Horii A, Zhang S. Soft Matter. 2008;4:2388. [Google Scholar]
- 25.Zhang K, Sugawara A, Tirrell DA. ChemBioChem. 2009;10:2617. doi: 10.1002/cbic.200900542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dudar GK, D’Andrea LD, Di Stasi R, Pedone C, Wallace JL. Am J Physiol Gastrointest Liver Physiol. 2008;295:374. doi: 10.1152/ajpgi.90325.2008. [DOI] [PubMed] [Google Scholar]
- 27.Santulli G, Ciccarelli M, Palumbo G, Campnile A, Galasso G, Ziaco B, Altobelli GG, Cimini V, Piscione F, D’Andrea LD, Pedone C, Trimarco B, Iaccarino G. J Transl Med. 2009;7:41. doi: 10.1186/1479-5876-7-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee JS, Johnson AJW, Murphy WL. Adv Mater. 2010;22:5494. doi: 10.1002/adma.201002970. [DOI] [PubMed] [Google Scholar]
- 29.Heidemann ER, Harrap BS, Schiele HD. J Biol Chem. 1973;12:2958. doi: 10.1021/bi00740a002. [DOI] [PubMed] [Google Scholar]
- 30.Wang AY, Foss CA, Leong S, Mo X, Pomper MG, Yu SM. Biomacromolecules. 2008;9:1755. doi: 10.1021/bm701378k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang AY, Mo X, Chen CS, Yu SM. J Am Chem Soc. 2005;127:4130. doi: 10.1021/ja0431915. [DOI] [PubMed] [Google Scholar]
- 32.Mo X, An Y, Yun C, Yu SM. Angew Chem Int Ed. 2006;45:2267. doi: 10.1002/anie.200504529. [DOI] [PubMed] [Google Scholar]
- 33.Ackerman MS, Bhate M, Shenoy N, Beck K, Ramshaw JAM, Brodsky B. J Biol Chem. 1999;274:7668. doi: 10.1074/jbc.274.12.7668. [DOI] [PubMed] [Google Scholar]
- 34.Diana D, Ziaco B, Colombo G, Scarabelli G, Romanelli A, Pedone C, Fattorusso R, D’Andrea LD. Chem-Eur J. 2008;14:4164. doi: 10.1002/chem.200800180. [DOI] [PubMed] [Google Scholar]
- 35.Gauba V, Hartgerink JD. J Am Chem Soc. 2007;129:2683. doi: 10.1021/ja0683640. [DOI] [PubMed] [Google Scholar]
- 36.Wang AY, Leong S, Liang Y, Huang RCC, Chen CS, Yu SM. Biomacromolecules. 2008;9:2929. doi: 10.1021/bm800727z. [DOI] [PubMed] [Google Scholar]
- 37.Engel J, Prockop DJ. Annu Rev Biophys Biophys Chem. 1991;20:137. doi: 10.1146/annurev.bb.20.060191.001033. [DOI] [PubMed] [Google Scholar]
- 38.McLaughlin SH, Bulleid NJ. Matrix Biol. 1997;16:369. doi: 10.1016/s0945-053x(98)90010-5. [DOI] [PubMed] [Google Scholar]
- 39.Bulleid NJ. Semin Cell Dev Biol. 1996;7:667. [Google Scholar]
- 40.Anderson SM, Chen TT, Iruela-Arispe ML, Segura T. Biomaterials. 2009;30:4618. doi: 10.1016/j.biomaterials.2009.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Müller S, Koenig G, Charpiot A, Debry C, Voegel J, Lavalle P, Vautier D. Adv Funct Mater. 2008;18:1767. [Google Scholar]
- 42.Ruhrberg C, Gerhardt H, Golding M, Watson R, Ioannidou S, Fujisawa H, Betsholtz C, Shima DT. Genes Dev. 2002;16:2684. doi: 10.1101/gad.242002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stalmans I, Ng Y, Rohan R, Fruttiger M, Bouché A, Ÿuce A, Fujisawa H, Hermans B, Shani M, Jansen S, Hicklin D, Anderson DJ, Gardiner T, Hammes H, Moons L, Dewerchin M, Collen D, Carmeliet P, D’Amore PA. J Clin Invest. 2002;109:327. doi: 10.1172/JCI14362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gerhardt H, Golding M, Fruttiger M, Ruhrberg C, Lundkvist A, Abramsson A, Jeltsch M, Mitchell C, Alitalo K, Shima D, Betsholtz C. J Cell Biol. 2003;161:1163. doi: 10.1083/jcb.200302047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cao G, O’Brien CD, Zhou Z, Sanders SM, Greenbaum JN, Makrigiannakis A, DeLisser HM. Am J Physiol Cell Physiol. 2002:C1181. doi: 10.1152/ajpcell.00524.2001. [DOI] [PubMed] [Google Scholar]
- 46.DeLisser HM, Christofidou-Solomidou M, Strieter RM, Burdick MD, Robinson CS, Wexler RS, Kerr JS, Garlanda C, Merwin JR, Madri JA, Albelda SM. Am J Pathol. 1997;151:671. [PMC free article] [PubMed] [Google Scholar]
- 47.Romer LH, McLean NV, Yan H, Daise M, Sun J, DeLisser HM. J Immunol. 1995;154:6582. [PubMed] [Google Scholar]
- 48.Küsters B, de Waal RMW, Wesseling P, Verrijp K, Maass C, Heerschap A, Barentsz JO, Sweep F, Ruiter DJ, Leenders WPJ. Cancer Res. 2003;63:5408. [PubMed] [Google Scholar]
- 49.Berra E, Milanini J, Richard DE, Le Gall M, Viñals F, Gothié E, Roux D, Pagés G, Pouysségur J. Biochem Pharmacol. 2000;60:1171. doi: 10.1016/s0006-2952(00)00423-8. [DOI] [PubMed] [Google Scholar]
- 50.Eliceiri B, Klemke R, Strömblad S, Cheresh D. J Cell Biol. 1998;140:1255. doi: 10.1083/jcb.140.5.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rush S, Khan G, Bamisaiye A, Bidwell P, Leaver HA, Rizzo MT. Exp Cell Res. 2007;313:121. doi: 10.1016/j.yexcr.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 52.Chen TT, Luque A, Lee S, Anderson SM, Segura T, Iruela-Arispe ML. J Cell Biol. 2010;188:595. doi: 10.1083/jcb.200906044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen RR, Silva EA, Yuen WW, Brock AA, Fischbach C, Lin AS, Guldberg RE, Mooney DJ. FASEB J. 2007;21:3896. doi: 10.1096/fj.06-7873com. [DOI] [PubMed] [Google Scholar]
- 54.Korff T, Augustin HG. J Cell Sci. 1999;112:3249. doi: 10.1242/jcs.112.19.3249. [DOI] [PubMed] [Google Scholar]
- 55.Korff T, Kimmina S, Martiny-Baron G, Augustin HG. FASEB J. 2001;15:452. doi: 10.1096/fj.00-0139com. [DOI] [PubMed] [Google Scholar]
- 56.Ruhrberg C. BioEssays. 2003;25:1052. doi: 10.1002/bies.10351. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Cumulative release profiles of QKCMP from type I collagen gels, where quenched QKCMP was added in solution before gel formation (binding in solution) and to preformed gel (binding to preformed gel). Day 0 and hour 0 represent the amount of compounds remaining in the collagen matrix after removal of unbound peptide by washing with 4 °C blank buffer. Error bars represent ±SD.
Figure S2. MALDI-ToF mass spectroscopy of target peptides after purification.