

Abstract
DNA interstrand cross-links (ICLs) are repaired in S phase by a complex, multistep mechanism involving translesion DNA polymerases. After replication forks collide with an ICL, the leading strand approaches to within one nucleotide of the ICL (“approach”), a nucleotide is inserted across from the unhooked lesion (“insertion”), and the leading strand is extended beyond the lesion (“extension”). How DNA polymerases bypass the ICL is incompletely understood. Here, we use repair of a site-specific ICL in Xenopus egg extracts to study the mechanism of lesion bypass. Deep sequencing of ICL repair products showed that the approach and extension steps are largely error-free. However, a short mutagenic tract is introduced in the vicinity of the lesion, with a maximum mutation frequency of ∼1%. Our data further suggest that approach is performed by a replicative polymerase, while extension involves a complex of Rev1 and DNA polymerase ζ. Rev1–pol ζ recruitment requires the Fanconi anemia core complex but not FancI–FancD2. Our results begin to illuminate how lesion bypass is integrated with chromosomal DNA replication to limit ICL repair-associated mutagenesis.
Keywords: Fanconi anemia, genome stability, interstrand cross-link repair, translesion synthesis
Introduction
DNA interstrand cross-links (ICLs) covalently link the two strands of the double helix and thereby block DNA unwinding, which is essential for DNA replication and transcription. The repair of ICLs is an intricate process involving multiple DNA repair enzymes including structure-specific endonucleases, translesion synthesis (TLS) DNA polymerases, recombinases, and the Fanconi anemia (FA) pathway (Kim & D’Andrea, 2012; Clauson et al, 2013). By monitoring replication of a plasmid containing a site-specific ICL in Xenopus egg extracts, we previously delineated a detailed mechanism of replication-coupled ICL repair (Räschle et al, 2008; Knipscheer et al, 2009; Long et al, 2011). Briefly, replication initiates at a single, randomly selected site on the plasmid and two replication forks quickly converge on the ICL and stall (Fig1A). The 3′ ends of the stalled leading strands are located 20–40 nucleotides from the lesion due to steric hindrance by the CMG helicase, which translocates ahead of DNA polymerase on the leading strand template [Fig1A, blue hexamer (Fu et al, 2011)]. After CMG unloading, which requires the BRCA1–BARD1 tumor suppressor complex (Long et al, 2014) and convergence of two replication forks on the ICL (Zhang et al, 2015), one of the leading strands advances and stalls again one nucleotide from the lesion (Fig1B). Incisions on either side of the ICL unhook the cross-link, and a nucleotide is inserted opposite the adducted base, presumably by a TLS polymerase (Fig1C). The leading strand is then extended beyond the ICL by pol ζ (Räschle et al, 2008), (Fig1D, red arrow), but the extent of this synthesis step is unknown. A replicative DNA polymerase may further extend the strand until it reaches a downstream Okazaki fragment (Fig1D, green arrow). Ligation restores one daughter DNA duplex (Fig1D), which serves as a template for repair of the other, incised daughter by homologous recombination (HR) (Fig1E). In vivo, the cross-linked adduct attached to one parental strand is probably removed by excision repair, but this reaction is inefficient in our system (Räschle et al, 2008).
Figure 1.
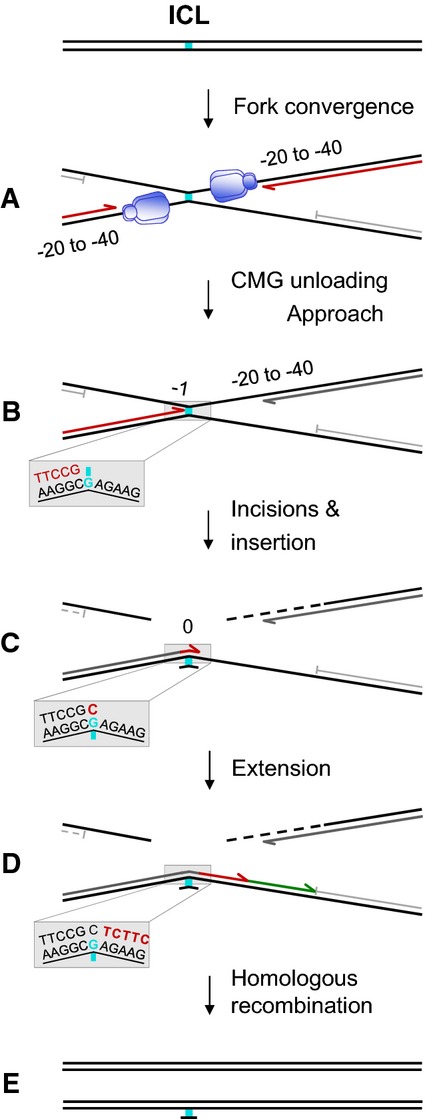
Model for ICL repair in Xenopus egg extracts
See Introduction for details on (A–E). Insets in panels (B–D) depict lesion bypass at nucleotide resolution. Blue hexamer, CMG helicase.
TLS employs specialized low-fidelity DNA polymerases to replicate across DNA lesions that cannot be copied by replicative DNA polymerases. Each vertebrate TLS polymerase is thought to bypass a particular class of lesion (Prakash et al, 2005), and lesion bypass often requires the sequential action of two different TLS polymerases. The first polymerase inserts a nucleotide across from the damaged base, often generating a mismatched and/or distorted primer terminus (Fig1C). This structure is extended by a second TLS polymerase, usually pol ζ, a B-family polymerase composed of a catalytic subunit, Rev3, and a regulatory subunit, Rev7. Pol ζ is remarkably efficient at extending abnormal primer termini (Johnson et al, 2000; Prakash & Prakash, 2002; Gan et al, 2008). We showed previously that immunodepletion of pol ζ from Xenopus egg extracts inhibits the extension step during ICL repair (Räschle et al, 2008).
Pol ζ interacts physically and genetically with Rev1, a Y-family DNA polymerase. Cells deficient in pol ζ or Rev1 show similar sensitivities to DNA damage and are hypersensitive to cross-linking agents (Kim & D’Andrea, 2012; and references therein). The interaction between Rev3, Rev7, and Rev1 is essential for resistance to cisplatin (Hara et al, 2010). As a dCMP transferase, Rev1 inserts only dCMPs opposite abasic sites and certain guanine adducts (Haracska et al, 2002; Zhang et al, 2002; Washington et al, 2004). Notably, the main function of Rev1 in TLS is independent of its catalytic activity, but requires the formation of protein–protein interactions via its BRCT domain, UBM domains, and unique C-terminus. Rev1 also interacts with pol η, pol ι, pol κ, Rev7, PCNA, and the Faap20 subunit of the FA core complex (Guo et al, 2003, 2006a,b; Ohashi et al, 2004; Ross et al, 2005). Because of its ability to bind a number of TLS polymerases, Rev1 is viewed as a scaffold that recruits TLS polymerases to DNA lesions.
TLS polymerases are inherently error-prone. Cells therefore strictly regulate their access to the primer terminus. This is achieved, at least in part, by PCNA ubiquitylation. In response to replication fork stalling, PCNA is mono-ubiquitylated by Rad6/Rad18 on lysine 164 (Kannouche et al, 2004; Watanabe et al, 2004). In addition to a PCNA-interacting motif, TLS polymerases contain ubiquitin-binding domains, which are thought to stimulate their interaction with ubiquitylated PCNA at the fork (Bienko et al, 2005). However, some TLS events in vertebrate cells are not controlled by PCNA ubiquitylation, but depend on Rev1 (Arakawa et al, 2006; Edmunds et al, 2008; Szuts et al, 2008). If Rev1 function is partly independent of PCNA ubiquitylation, the question arises how Rev1 is recruited to a stalled replication fork. The interaction with unmodified PCNA might be sufficient; however, Rev1 might also bind other proteins at the fork. A plausible candidate for such a binding partner is the FA core complex.
Fanconi anemia is caused by mutations in any one of 16 genes and is characterized by bone marrow failure, cancer predisposition, chromosomal abnormalities, and extreme cellular sensitivity to ICL-inducing agents (Kim & D’Andrea, 2012). The products of eight FA genes (FancA, FancB, FancC, FancE, FancF, FancG, FancL, and FancM) and three FA-associated proteins (Faap20, Faap24, and Faap100) form a multi-subunit “core complex” containing E3 ubiquitin ligase activity (Alpi et al, 2008; Longerich et al, 2009; Sato et al, 2012; Rajendra et al, 2014). In response to DNA damage and replication stress, the core complex mono-ubiquitylates the FancI–FancD2 heterodimer. Ubiquitylation enables localization of FancI–FancD2 to chromatin, where it promotes XPF-dependent incisions that unhook the cross-link during ICL repair (Fig1B and C) (Knipscheer et al, 2009; Klein Douwel et al, 2014). The core complex has also been implicated in regulating TLS (Kim & D’Andrea, 2012). In chicken DT-40 cells, Rev1 and Rev3 are epistatic to FancC and FancA for sensitivity to cross-linking agents. FancC-, FancA-, and FancG-deficient cells show a reduced frequency of both spontaneous and UV-induced point mutations (Papadopoulo et al, 1990a,1990b; Niedzwiedz et al, 2004; Hinz et al, 2007; Mirchandani et al, 2008; Kim et al, 2012). The core complex is also required for efficient formation of spontaneous and damage-induced Rev1 foci. Moreover, Faap20, a recently identified component of the core complex, directly interacts with Rev1, suggesting that the core complex promotes TLS by recruiting Rev1 to DNA lesions (Mirchandani et al, 2008; Kim et al, 2012). These data strongly suggest that the FA core complex helps anchor TLS polymerases to ICLs, perhaps in collaboration with ubiquitylated PCNA.
We analyzed the mechanism of TLS during repair of a site-specific cisplatin ICL in Xenopus egg extracts using a variety of approaches, including chromatin immunoprecipitation (ChIP), immunodepletion, and deep sequencing of repair products. While most of approach and extension are error-free, ICL repair generates a mutagenic tract of a few nucleotides surrounding the lesion. Our data suggest that a replicative DNA polymerase carries out the approach of the leading strand from the −20 position to the ICL. The Rev1–pol ζ complex is dispensable for insertion, but it is required for the extension step. The efficient binding of Rev1–pol ζ to ICLs requires the FA core complex but not FancI–FancD2. Our results provide a framework to understand how TLS is integrated with chromosomal DNA replication to limit mutagenesis during ICL repair.
Results
Replicative DNA polymerases are enriched at a site-specific ICL
To elucidate the molecular mechanism of TLS during repair of a cisplatin ICL, we examined the binding of several replicative and translesion DNA polymerases, as well as other factors, to an ICL-containing plasmid using ChIP. In an equivalent reaction, we determined the kinetics of approach, insertion, and extension by cutting the plasmid near the ICL and monitoring the progress of the leading strand as it bypasses the lesion (Fig2A and B). Similar to what we reported previously (Räschle et al, 2008; Knipscheer et al, 2009; Long et al, 2011), by 12 min, most leading strands had arrived at the −20 to −40 position (Fig2B, lane 3), and by 40 min, leading strands had been extended to the −1 position (Fig2B, lane 6). Trace amounts of the insertion product could be detected at 40 and 65 min (Fig2B, lanes 6 and 7 and see Fig 4F), and full-length extension products reached a plateau by 65 min (Fig2B, lane 7).
Figure 2.

Correlation of TLS with ICL binding of DNA polymerases
- A Schematic representation of lesion bypass intermediates generated by the rightward fork after AflIII digestion of pICL.
- B pICL replication intermediates were digested with AflIII and separated on a denaturing gel. Nascent strands generated by the rightward fork and extension product are shown. Analogous products are generated by the leftward fork (not shown).
- C Location of primer pairs used for ChIP.
- D–J Recruitment of replication and repair proteins to the ICL and the control loci. pICL was replicated, and samples were withdrawn at the indicated times for ChIP with antibodies against (D) PCNA, (E) pol δ, (F) pol ε, (G) MCM7, (H) Rev1, (I) Rev7, or (J) FancA. Experimental replicates are shown in Supplementary Fig S1. In each graph, colored bars indicate the approximate timing of TLS events (see inset for legend).
Source data are available online for this figure.
Figure 4.
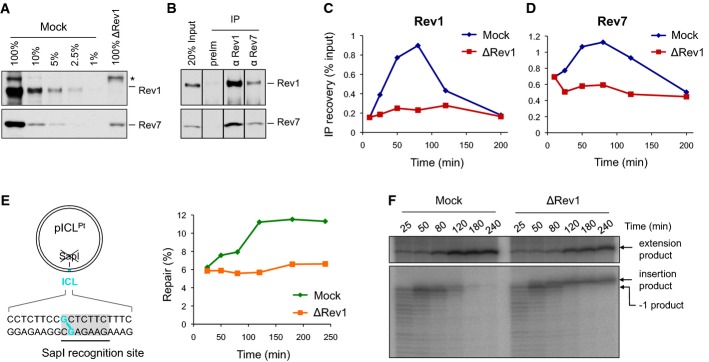
Rev1 depletion inhibits the extension step
- A Rev1-depleted NPE and a dilution series of mock-depleted NPE were analyzed by Western blotting using Rev1 and Rev7 antibodies. 100% corresponds to 0.25 μl of NPE.
- B Reciprocal co-immunoprecipitation of Rev1 and Rev7. 20% of input (0.8 μl of NPE) and proteins precipitated from 4 μl of NPE (IP) were blotted with Rev1 and Rev7. preIm, preimmune serum.
- C, D pICL was replicated in mock- or Rev1-depleted egg extracts. ChIP was performed with antibodies against Rev1 (C) and Rev7 (D) at the indicated time points. An experimental replicate of (D) is shown in Supplementary Fig S4A.
- E Left: cartoon of pICL indicating the SapI site that is blocked by the ICL. Right: DNA samples from pICL replication (performed in parallel with those shown in C and D) were cut with SapI, and repair efficiency was calculated as described (Knipscheer et al, 2012). The background level of SapI-cleavable products in the Rev1-depleted reaction is due to contamination of pICL by un-cross-linked plasmid (Knipscheer et al, 2009). The amount of SapI-cleavable products can vary between experiments due to differences in the amount of un-cross-linked plasmid and in the repair capacity of individual batches of extract.
- F Lesion bypass in mock- and Rev1-depleted extracts. DNA samples from (E) were cut with AflIII and analyzed on a denaturing gel, as described in Fig2B. A repetition of this experiment is shown in Supplementary Fig S4B.
Source data are available online for this figure.
We first used ChIP to measure the binding of PCNA, MCM7, and the replicative DNA polymerases ε and δ to the ICL locus and a control locus located 2.5 kb from the ICL (Fig2C). As shown in Fig2D–G, pol ε, pol δ, PCNA, and MCM7 bound to the ICL and control loci at early time points, concurrent with replication of the plasmid backbone (for experimental replicates, see Supplementary Fig S1A–H). Binding of PCNA and MCM7 to pCTR (an undamaged plasmid of the same sequence as pICL) closely resembled binding of these proteins to the control locus on pICL (Supplementary Fig S2A and B). By 20 min, all four proteins were largely released from the control locus, but they persisted at the ICL locus until the 65-min time point. Interestingly, like MCM7, Cdc45, and GINS (Fig2G and Fu et al, 2011), pol ε was highly enriched at the ICL locus (Fig2F). In contrast, pol δ more closely mimicked the binding of PCNA (compare Fig2D and E; see also Supplementary Fig S1). These observations are consistent with the model that a physical interaction between CMG and pol ε selects the latter as the leading strand DNA polymerase and that the high affinity of pol δ for PCNA dictates its use for lagging strand synthesis (Johansson et al, 2004; Chilkova et al, 2007; Sengupta et al, 2013; Georgescu et al, 2014). As such, they are consistent with the notion that in vertebrates, pol ε performs leading strand synthesis whereas pol δ performs lagging strand synthesis, as previously demonstrated in S. cerevisiae and S. pombe (Pursell et al, 2007; Nick McElhinny et al, 2008; Miyabe et al, 2011). In addition, they show that both replicative DNA polymerases are present near the ICL during translesion DNA synthesis.
To corroborate our ChIP results, we pulled down pICL or pCTR from egg extracts using streptavidin beads coated with biotinylated LacI protein (Supplementary Fig S3), which binds efficiently to nonspecific DNA. In this assay, binding of pol ε and PCNA to pCTR correlated with DNA replication, peaking at 5 min and dropping to background levels by 15 min (Fig3, lanes 6–9). Recovery of both proteins was abolished or greatly decreased in the absence of DNA or DNA replication (Fig3, lanes 4 and 5). In contrast, on pICL, pol ε and PCNA were retained until ∼60 min, consistent with the ChIP data (Fig3, lanes 14–20). Plasmid pull-down data with pol δ were uninterpretable due to DNA-independent sticking of the protein to beads (data not shown). Collectively, our data show that replicative DNA polymerases bind near the ICL during lesion bypass, consistent with their involvement in this process.
Figure 3.
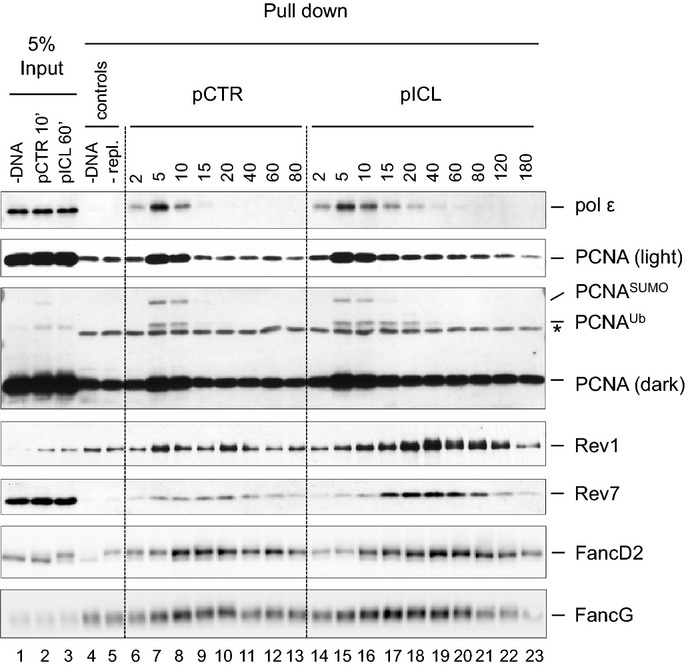
Plasmid pull-down analysis of proteins bound to pCTR and pICL
pCTR or pICL (5 ng/μl) was replicated in HSS/NPE. Plasmid DNA was pulled down by incubating aliquots of the reaction with streptavidin beads coated with biotinylated LacI. Proteins bound to the plasmids were analyzed by Western blotting with the indicated antibodies. Control reactions lacking DNA (lane 4) or containing geminin to inhibit DNA replication (lane 5) were included. Input samples (1% for PCNA and Rev1) were withdrawn at 10 min for pCTR and 60 min for pICL.Source data are available online for this figure.
Rev1 and Rev7 bind specifically to the ICL locus during lesion bypass
The main TLS polymerases genetically implicated in ICL repair are Rev1 and pol ζ (see Introduction). We previously showed that immunodepletion of pol ζ inhibits extension, but because full-length vertebrate pol ζ has not been purified, we were not able to rescue the defect. To further explore the role of pol ζ, as well as Rev1, in bypass of the cisplatin ICL, we analyzed their chromatin binding by ChIP. A small peak of Rev1 and Rev7 was detectable at the ICL and control loci at early time points (Fig2H and I; for experimental replicate, see Supplementary Fig S1J and K) which might reflect constitutive, low-level binding of these proteins to the replisome or sporadic recruitment to endogenous DNA damage. Consistent with this interpretation, binding of Rev1 was also detected during replication of undamaged pCTR plasmid (Supplementary Fig S2D). As the reaction proceeded, binding of Rev1 and Rev7 increased specifically at the ICL site, where it peaked at 40 min. At this time, −1 products, the substrates for TLS, were most abundant (Fig2B). By 65 min, most of the −1 products were fully extended, and the binding of Rev1 and Rev7 had significantly decreased. Similar results were obtained with the plasmid pull-down assay: binding of Rev1 and Rev7 to pICL was stronger and more prolonged compared to pCTR (Fig3). These results are consistent with Rev1 and pol ζ participating in TLS over a cisplatin ICL.
Rev1 depletion inhibits the extension step during ICL repair
We next addressed the function of Rev1 in ICL repair. The dCMP transferase activity of Rev1 allows it to insert dCMPs opposite several types of DNA lesions, including a cisplatin ICL (Ho et al, 2011). Given that the ICL in pICL is formed between two guanine residues, we postulated that Rev1 performs the insertion step. To test this possibility, we immunodepleted > 99% of Rev1 from Xenopus egg extract (Fig4A, top panel). The co-depletion of 80–90% of Rev7 (Fig4A, bottom panel) is consistent with previous reports that the two proteins form a stable complex (Guo et al, 2003; Ohashi et al, 2004). Indeed, reciprocal co-IPs revealed a strong interaction between Rev1 and Rev7 that is unaffected by DNA replication or DNA damage (Fig4B and M. Budzowska & J.C. Walter, unpublished observations). Consistent with the presence of a stable Rev1–pol ζ complex, Rev1 depletion severely reduced the recruitment of Rev1 and pol ζ to chromatin (Fig4C and D; for experimental replicate, see Supplementary Fig S4A). As shown in Fig4E, Rev1 depletion reduced ICL repair to background levels, as measured by the regeneration of a SapI restriction site that overlaps with the cross-link. Strikingly, Rev1 depletion did not lead to accumulation of the −1 product, as expected if Rev1 were required for insertion; instead, we observed strong accumulation of the insertion product (Fig4F; for experimental replicate, see Supplementary Fig S4B), as seen previously after immunodepletion of Rev7 (Räschle et al, 2008). Re-addition of recombinant Xenopus Rev1 did not rescue this defect (data not shown), likely because pol ζ was co-depleted with Rev1. Our results demonstrate that Rev1 is not required for the insertion step, and they strongly suggest that a complex containing Rev1 and pol ζ performs extension.
PCNA ubiquitylation does not correlate with Rev1–pol ζ binding
PCNA ubiquitylation on lysine 164 helps recruit TLS polymerases to certain replication-blocking lesions (Lehmann et al, 2007). Testing the effect of PCNA ubiquitylation on TLS in our cell-free system would require replacement of endogenous PCNA with PCNAK164R, which cannot be ubiquitylated. However, given the high concentration of PCNA in the nuclear extract used for replication (∼30 μM; data not shown), we have not been able to functionally deplete PCNA. Instead, we addressed when PCNA ubiquitylation occurs and how it correlates with chromatin binding of TLS polymerases using the plasmid pull-down assay. PCNAUb was detected on chromatin early during replication, and it was similarly abundant on pCTR and pICL (Fig3, lanes 7–8 and 15–16). After 10 min, when replication was complete, PCNA and PCNAUb quickly decreased on pCTR (Fig3, lane 9). In contrast, PCNAUb was retained on pICL until 20 min, after which it quickly dropped to background levels (Fig3, lanes 17–19). Thus, the majority of PCNAUb disappeared from chromatin before the peak of TLS and Rev1/Rev7 binding, which occurred between 40 and 65 min (see Figs2B and 3, lane 19). These results confirm that PCNA is ubiquitylated in the course of a normal round of DNA replication (Leach & Michael, 2005). Although these results raise the possibility that ubiquitylated PCNA is not required for the retention of Rev1 and pol ζ at the site of damage during ICL repair, a more definitive experiment would be required to show this. In addition, ubiquitylated PCNA might play an important role in recruiting an insertion polymerase.
Role of the FA pathway in Rev1 and pol ζ recruitment to ICLs
To explore the role of the Fanconi anemia pathway in TLS, we first examined how the binding of FancA (a core complex component) and FancD2 to ICLs correlates with the binding of Rev1 and Rev7. FancA bound to the control and ICL sites at early time points, when the plasmid underwent replication, but then became specifically enriched at the ICL locus, where it persisted until 90 min (Fig2J). The binding of FancA overlapped with the binding of Rev1 and Rev7 (Fig2H and I). Consistent with FancA ChIP, in the plasmid pull-down assay, binding of FANCG, another core complex component, was stronger and more prolonged to pICL than to pCTR (Fig3). We analyzed binding of FancG instead of FancA because FancA bound to magnetic beads in the absence of DNA or DNA replication. FancD2 bound to both pCTR and pICL, but was retained longer on pICL (Supplementary Fig S2C, Figs5B and 3). Binding of FancD2 coincided with binding of FancG and FancA (Figs2J, 3, 5A and B), consistent with its chromatin loading being dependent on the FA core complex. These results show that the core complex and FancD2 bind to the ICL concurrently with TLS.
Figure 5.
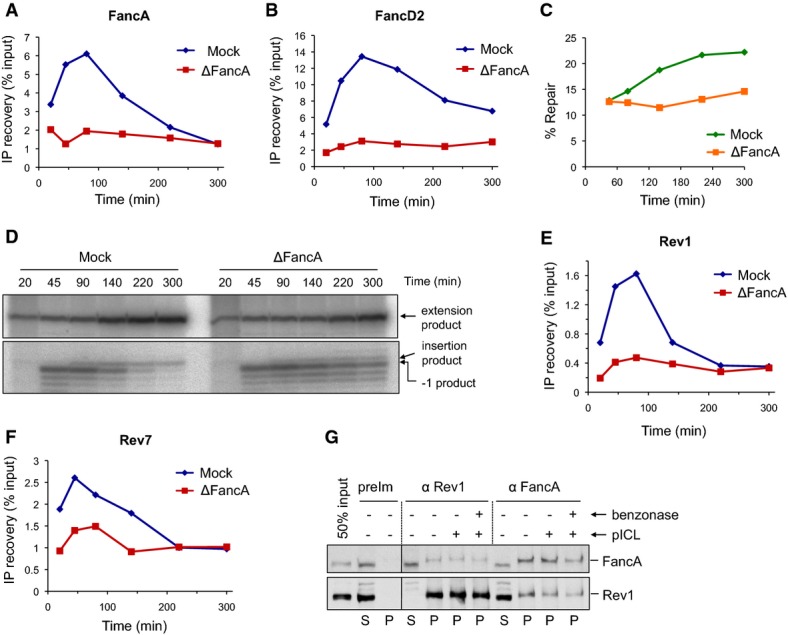
FancA depletion inhibits chromatin recruitment of TLS polymerases
- A, B pICL was replicated in mock- or FancA-depleted egg extracts, and samples were analyzed by ChIP using antibodies against FancA (A) and FancD2 (B).
- C FancA depletion inhibits ICL repair. ICL repair efficiency in the extracts used in (A) and (B) was determined as in Fig4E.
- D FancA depletion inhibits insertion. DNA samples from the reactions in (C) were cut with AflIII and resolved on a sequencing gel, as described in Fig2B.
- E, F Rev1 and Rev7 binding to the ICL locus is reduced in FancA-depleted extract. Samples from the same reactions used in (A) and (B) were analyzed by ChIP with antibodies against Rev1 (E) and Rev7 (F). An experimental replicate is shown in Supplementary Fig S5D and E.
- G Reciprocal co-immunoprecipitation of FancA and Rev1. pICL (4.2 ng/μl) was replicated in egg extract. After 40 min, 5-μl aliquots of the reaction were immunoprecipitated with the indicated antibodies. Benzonase nuclease or buffer was added during the final 15 min of the IP. 50% of input (0.3 μl of the replication reaction), supernatant (S), and precipitated proteins (P) were blotted for FancA and Rev1. preIm: preimmune serum.
Source data are available online for this figure.
To investigate the role of the core complex in the repair of pICL, we immunodepleted ∼95% of FancA from Xenopus egg extracts (Supplementary Fig S5A). As expected, this manipulation abolished FancD2 ubiquitylation (Supplementary Fig S5B) and binding of FancA and FancD2 to the ICL locus as measured by ChIP (Fig5A and B). The depletion also inhibited ICL repair as measured by regeneration of the SapI site (Fig5C). Analysis of lesion bypass revealed the accumulation of −1 products in FancA-depleted extracts, demonstrating a defect in the insertion step (Fig5D). A similar defect was caused by depletion of FancD2 (Knipscheer et al, 2009; and Fig6B) and XPF-ERCC1 (Klein Douwel et al, 2014), both of which are required for incisions. Because the core complex has not been reconstituted, we did not attempt to rescue the FancA depletion with purified proteins. However, the FancA depletion caused the expected defects in FancI–FancD2 ubiquitylation and insertion while not affecting fork convergence or activation of Chk1 phosphorylation (M. Budzowska & J.C. Walter, unpublished results), suggesting it specifically removed the core complex.
Figure 6.
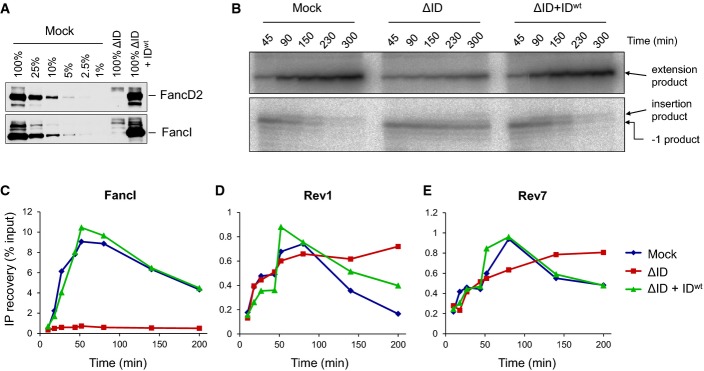
FancI–FancD2 is not essential for chromatin recruitment of Rev1 and Rev7
- A Immunodepletion of FancD2 and FancI. A dilution series of mock-depleted NPE, FancI/FancD2-depleted NPE, and FancI/FancD2-depleted NPE supplemented with recombinant wild-type xlFancD2/FancI complex were analyzed by Western blotting using FancD2 and FancI antibodies.
- B Lesion bypass in mock-depleted and FancI/FancD2-depleted extracts and in FancI–FancD2-depleted extracts supplemented with wild-type recombinant FancI–FancD2 complex was analyzed as described in Fig2B. Accumulation of the −1 product shows that the ID-depleted egg extract was functionally depleted.
- C–E Extracts depleted as in (B) were analyzed by ChIP using antibodies against FancI (C), Rev1 (D), and Rev7 (E). An experimental replicate of (E) is shown in Supplementary Fig S6B.
Source data are available online for this figure.
We next asked whether FancA is required for recruitment of TLS polymerases during ICL repair. Depletion of FancA greatly reduced Rev1 binding and partially reduced Rev7 binding to the ICL locus (Fig5E and F; for experimental replicate, see Supplementary Fig S5D and E). Notably, FancA and Rev1 could be reciprocally co-immunoprecipitated independently of DNA or DNA damage (Fig5G). This interaction is likely mediated by the core complex component Faap20 (Kim et al, 2012). These data indicate that the core complex is required for recruitment of Rev1–pol ζ, possibly due to a physical interaction between the two complexes.
We next addressed whether FancI–FancD2 is required for the recruitment of TLS polymerases to the cisplatin ICL. In egg extract that was functionally depleted of FancI–FancD2 [Fig6A–C (Knipscheer et al, 2009)], recruitment of Rev1 was largely unaffected and recruitment of Rev7 was partially reduced (Fig6D and E; for experimental replicate, see Supplementary Fig S6B). However, both Rev1 and Rev7 persisted significantly longer at the ICL in the FancI–FancD2-depleted extracts, an effect that was reversed by the re-addition of recombinant FancI–FancD2 (Fig6D and E, Supplementary Fig S6B). Our results indicate that the Fanconi core complex is required for efficient Rev1–pol ζ recruitment to ICL damage, whereas FancI–FancD2 plays a less important and possibly indirect role in this process (see Discussion).
ICL repair induces a short mutagenic tract
We next investigated to what extent repair of the cisplatin ICL is error-prone. We replicated pICL, isolated DNA after 4 h when repair should be complete, PCR-amplified a 115-bp region surrounding the ICL, and deep-sequenced the amplified DNA fragments (Supplementary Fig S7A–C). In parallel, we replicated and sequenced pCTR.
Translesion DNA synthesis is expected to generate point mutations and possibly insertions/deletions (indels). During replication of pCTR, 96% of the amplified replication products were the correct length, and 4% contained indels (Supplementary Fig S9A). A total of 51% of these indels involved an 8-nucleotide deletion that removes the second of two short tandem repeats (Supplementary Fig S9B). The 8-nucleotide deletion was 26 times as common as the next most common indel and may have been produced by “slippage” of a polymerase during DNA synthesis, either in egg extract or more likely during PCR. In reactions containing pICL, 13% of the molecules contained indels, 57% of which corresponded to the 8-nucleotide deletion (Supplementary Fig S9A). It is not possible based on these data to determine precisely what fraction of deletion products arose during ICL repair in the extract and what fraction was generated during PCR amplification. However, we sometimes observe truncated extension products of pICL on sequencing gels (e.g. Fig6A in Long et al, 2014), arguing that a significant portion of the deletion products revealed by sequence analysis are genuine products of ICL repair. These repair-associated products might result from slippage of polymerases stalled at the ICL or from microhomology-mediated end joining of the incised DNA strands. Importantly, these truncated products are not cleavable with SapI and therefore do not contribute to error-free ICL repair measured by regeneration of the SapI site.
We next analyzed the frequency of nucleotide misincorporation in sequence reads of the correct length (Fig7). Because the ICL can be unhooked by incisions in the top or bottom parental strands (Fig7A), we cannot determine, for a given read, whether it derived from the leftward or rightward leading strand. As shown in Fig7A, each nucleotide corresponds to a different position relative to the ICL for the rightward or leftward leading strands. For example, the 0 position for the leftward fork (“0L”) corresponds to the −1 position for the rightward fork (“−1R”). The total misincorporation frequency at each position is therefore the sum of the misincorporation frequencies for both directions.
Figure 7.

Mutation spectrum generated during bypass of the unhooked ICL
- A schematic representation of fork convergence, incisions and lesion bypass using either of the parental strands as the template. For the nascent strand (blue arrow), the nucleotide positions relative to the position of the adduct in each parental strand are indicated (red numbers).
- pCTR and pICL replication products were recovered after 60 and 240 min, respectively. A 115-nt-long fragment surrounding the cross-link (present in pICL only) was deep-sequenced. A total of 1.7 million reads for pCTR and 2.6 million reads for pICL were obtained. For both plasmids, the misincorporation frequency in a 20-bp region surrounding the ICL is displayed. Nucleotide positions for the leftward and rightward forks are indicated, as in (A).
- Distribution of nucleotide misincorporations in (A). The height of each colored segment represents the frequency with which that nucleotide is incorporated minus the baseline frequency in pCTR. The total height of the bars represents the overall misincorporation frequency minus baseline. Nucleotide positions for the leftward and rightward forks are indicated, as in (A).
In a 78-bp region surrounding the ICL, mutations were most frequent at the 0L/−1R position, followed by the 0R/−1L position (Fig7B). Mutations at these positions occurred at a rate of 1.5 and 0.6%, respectively (Fig7B), with adenine and thymine being most frequently mis-inserted (Fig7C). The different mutation rate at these positions could reflect the influence of the sequence context on the fidelity of TLS and/or a preference for one leading strand to perform lesion bypass. Misincorporation was still well above the background on pCTR within 2–3 nucleotides of the lesion (Fig7B). Analysis of an independent pICL repair reaction yielded essentially the same results (Supplementary Fig S8B). Rev1 depletion reduced the misincorporation rate from 1.5 to 0.6% (Supplementary Fig S8C), consistent with the hypomutability of Rev1-deficient cells (Lawrence, 2004). At sites distant from the ICL, a low level of mutations was observed in all experiments that was identical between pCTR and pICL. These mutations probably resulted from sequence-dependent errors during PCR amplification and were therefore considered as background. In conclusion, summing the mutations at all positions, ICL repair is roughly 97% error-free. The low level of mutations observed is restricted to the immediate vicinity of the lesion, demonstrating that most of approach and extension are entirely error-free.
Discussion
Using Xenopus egg extracts, we have examined the mechanism of lesion bypass during replication-coupled repair of a cisplatin ICL. Advantages of our approach are that bypass intermediates can be resolved at nucleotide resolution, and the functions of specific proteins in bypass can be investigated by immunodepletion and correlated with their chromatin binding. We have also determined the frequency and location of mutations associated with ICL bypass using deep sequencing. To our knowledge, this is the first deep-sequencing analysis of a lesion bypass reaction. Using these approaches, we have shed light on key events underlying the three major steps in ICL bypass: approach, insertion, and extension.
Approach
When two replisomes converge on an ICL, the CMG helicase is evicted, which allows leading strands to approach the lesion (Fig1; Fu et al, 2011; Long et al, 2014). Three observations suggest that approach is carried out by a high-fidelity replicative DNA polymerase. First, we recently showed that approach is sensitive to aphidicolin (Long et al, 2014), which inhibits pols α, ε, δ, and ζ. Because TLS polymerases β, η, κ, ι, and λ are insensitive to aphidicolin (Nealon et al, 1996; Masutani et al, 1999; Tissier et al, 2000; Gerlach et al, 2001; Shimazaki et al, 2002) and pol ζ is not required for approach (Räschle et al, 2008), these data imply that a replicative DNA polymerase performs approach. Second, approach up to at least the −4 position involves no detectable mutagenesis (Fig7C). Third, pol ε, pol δ, and PCNA are specifically retained at the ICL locus after DNA replication of the plasmid backbone is complete. The almost identical ChIP-binding profiles of pol ε and MCM7 (Fig2F and G), together with the recently reported preferential association of pol ε with the CMG helicase (Georgescu et al, 2014), are consistent with pol ε being the leading strand polymerase. Therefore, pol ε probably occupies the stalled leading strand at the moment CMG dissociates; pol ε might then remain on the DNA long enough to carry out approach. Alternatively, it might immediately dissociate with CMG. In this case, PCNA, which should still be present on the primer, would probably recruit pol δ for approach. The fact that pol δ, like pol ε, is also retained at ICLs is consistent with this possibility. However, pol δ retention may also reflect a role in later events of ICL repair such as extension, strand displacement synthesis when the leading strand reaches a downstream lagging strand, or D-loop extension during subsequent repair of the broken sister chromatid by HR.
Insertion
The next step of lesion bypass, insertion, is presumably performed by a TLS polymerase. Deep sequencing of repair products revealed that mutations occur most frequently at the 0L/−1R and −1L/0R positions. We cannot determine the relative contributions of approach (−1) and insertion (0) to mutagenesis at these two positions. However, it seems likely that insertion of a nucleotide opposite the damaged template base is more error-prone than the approach step.
Given that the cisplatin ICL is formed between guanines and that Rev1 is a dCMP transferase, Rev1 was an attractive candidate for the primary insertion polymerase. However, in Rev1-depleted extracts, the efficiency and kinetics of insertion were completely unaffected (Fig4F), which disfavors the idea that Rev1 is the primary insertion polymerase. Our results do not, however, exclude the possibility that Rev1 normally performs a significant fraction of insertions and that in its absence another TLS polymerase carries out all the events.
Our data raise the question which polymerases other than Rev1 catalyze insertion. The best candidates are pol η and/or pol κ, whose possible involvement is consistent with biochemical and in vivo evidence (Bassett et al, 2004; Albertella et al, 2005; Chen et al, 2006; Minko et al, 2008; Shachar et al, 2009; Ho et al, 2011; Williams et al, 2012). Future work will aim to establish the identity of the insertion polymerase.
Extension
The insertion product is subsequently extended past the lesion, and our previous results showed that this event was inhibited after immunodepletion of pol ζ (Räschle et al, 2008). Here, we demonstrate that depleting Rev1 similarly inhibits extension and that Rev1 and pol ζ form a stable complex. Together with genetic experiments (see Introduction), these data provide strong evidence that a Rev1–pol ζ complex performs extension. Our sequencing analysis shows that only the first 2–3 nucleotide additions around the insertion events involve measurable misincorporations, although we cannot distinguish whether they were generated by the approach or the extension step. However, given that approach involves a replicative polymerase, it is conceivable that this step is largely error-free and that extension and insertion are mainly responsible for the elevated mutation rate in the 6-nucleotide region around the ICL. Mutagenesis at extension positions further away from the lesion could be minimized by a rapid switchback to a replicative DNA polymerase, which completes replication of the sister chromatid (Fig1). This scenario is consistent with the persistence of PCNA, pol δ, and pol ε at the ICL (Figs2 and 3). However, we cannot rule out the possibility that pol ζ synthesizes a longer product and that it is particularly error-prone near an abnormal primer template. While these issues require further investigation, our results show that a high level of point mutations arises in a narrow region surrounding a cisplatin ICL.
Regulation of translesion synthesis
Ubiquitylation of PCNA plays a well-established role in recruiting TLS polymerases to certain DNA lesions (Lehmann et al, 2007). Our system allowed us to monitor the kinetics of PCNA ubiquitylation during synchronized replication of an undamaged plasmid, and during repair of a site-specific ICL. We found that PCNA was ubiquitylated during DNA replication, consistent with previous observations (Leach & Michael, 2005). While PCNAUb persisted on DNA after the forks collided with the ICL, its level was not higher than on undamaged DNA. Moreover, the peak of PCNAUb binding occurred early and did not coincide with the peak of TLS and Rev1/Rev7 binding. Together, these observations are in agreement with weak and delayed accumulation of ubiquitylated PCNA in human cells exposed to cross-linking agents (Niimi et al, 2008), and they support the idea that PCNA ubiquitylation is not essential for bypass of interstrand cross-links (Hicks et al, 2010). Indeed, PCNA ubiquitylation is not the sole mechanism for regulating TLS. In chicken cells exposed to DNA damaging agents, PCNA ubiquitylation is required for filling the gaps left behind the fork, while Rev1, independently of PCNA ubiquitylation, is necessary for TLS at replication forks (Edmunds et al, 2008). Moreover, the FA core complex has also been implicated in TLS: FancA deficiency results in hypomutability and abolishes damage-induced formation of Rev1 foci (Niedzwiedz et al, 2004; Mirchandani et al, 2008; Kim et al, 2012). Here, we show that the fraction of FancA bound to the ICL locus significantly increased during repair, and binding of FancA preceded binding of Rev1 and Rev7 (Fig1). Moreover, FancA depletion severely inhibited Rev1 and partially inhibited Rev7 recruitment to the ICL locus (Fig5). Together, our data are consistent with the idea that the FA core complex plays a more prominent role than PCNA ubiquitylation in recruiting these TLS polymerases to the ICL lesion.
In contrast to the core complex, whose disruption causes hypomutability, FancD2-deficient cells show increased mutagenesis (Niedzwiedz et al, 2004; Mirchandani et al, 2008; Kim et al, 2012). It has therefore been proposed that the core complex acts independently of FancD2 to promote TLS. Interestingly, we observed only a mild but reproducible reduction in TLS polymerase recruitment to the ICL locus in the absence of the FancI–FancD2 complex (Fig6). We speculate that this is an indirect effect of failure to incise the ICL. In other words, the X-shaped structure surrounding the ICL before incisions is not an efficient substrate for TLS polymerase binding.
In summary, our data help to explain how TLS is performed during cisplatin ICL repair with relatively little mutagenesis. In our model, the approach of the leading strand to the lesion is performed by a replicative DNA polymerase. Next, the FA core complex performs two independent functions to prepare the lesion for TLS: First, it ubiquitylates the FancI–FancD2 complex to promote incisions, which unhook the ICL and allow insertion by an unknown polymerase; second, it recruits the Rev1–pol ζ complex, which is required for extension past the lesion. Mutagenicity of extension is minimized, probably due to a rapid switchback to a replicative polymerase.
Materials and Methods
Xenopus egg extracts, DNA replication, and repair assays
Preparation of Xenopus egg extracts (HSS and NPE) and DNA replication reactions were performed as described previously (Walter et al, 1998). Briefly, plasmid DNA was incubated in HSS at 7.5 ng/μl for 30 min at RT, leading to the formation of pre-replication complexes (pre-RCs) on DNA. Some reactions also contained undamaged pQuant plasmid at 0.375 ng/μl, which serves as an internal control for quantification of repair efficiency. The addition of two volumes of NPE, supplemented with [α-32P]dATP, initiated DNA replication. The reaction was stopped at the indicated time points by diluting aliquots (4–6 μl) into ten volumes of stop solution A (0.5% SDS, 10 mM EDTA, 50 mM Tris pH 7.5). Samples were then supplemented with 0.13 mg/ml RNase A and incubated for 30 min at 37°C, after which proteinase K was added to 0.67 mg/ml. After overnight incubation, DNA was phenol–chloroform-extracted, precipitated with 2.5 volumes of 100% ethanol in the presence of glycogen, and dissolved in 4–6 μl of 10 mM Tris pH 7.5. A total of 1 μl of DNA was digested with HincII or HincII and SapI. DNA fragments were resolved on a 0.8% agarose gel and visualized by autoradiography. Repair efficiency was calculated as described (Knipscheer et al, 2012). Experiments in Figs4E and 5C were performed at least three times, and representative results are shown.
Antibodies and immunodepletions
Antibodies against FancD2, FancI, PCNA, Rev7, FancA, pol delta (p125), and pol epsilon (p60) were described previously (Räschle et al, 2008; Knipscheer et al, 2009; Kochaniak et al, 2009; Waga et al, 2001; Fukui et al, 2004; Sobeck et al, 2006). Rev1 antibodies were raised against residues 120–420 (N-terminal Rev1 antibody) and 841–1,230 (C-terminal Rev1 antibody) of X. laevis Rev1. Both anti-Rev1 antibodies recognized the purified antigens. Both antibodies, but not the preimmune sera, immunoprecipitated a specific band corresponding to X. laevis Rev1.
To deplete Xenopus egg extracts of Rev1, one volume of rProtein A Sepharose Fast Flow (PAS) (GE Healthcare) was bound to 1 volume of α-Rev1 N-terminal antibodies, α-Rev1 C-terminal antibodies, or preimmune serum overnight at 4°C. The beads were washed twice with 500 μl PBS, once with 500 μl ELB (10 mM Hepes–KOH at pH 7.7, 50 mM KCl, 2.5 mM MgCl2 and 250 mM sucrose), twice with 500 μl ELB + 0.5 M NaCl, and twice with ELB. Five volumes of precleared HSS or NPE was then mixed with 1 volume of the antibody-bound sepharose and incubated for 1 h at 4°C. Extracts were harvested and the depletion procedure was repeated once for HSS, and twice for NPE. For HSS, N-terminal and C-terminal α-Rev1 antibody was used in the first and second depletion round, respectively. For NPE, N-terminal antibody was used in first and second round, and C-terminal antibody was used in the last round. Depleted extracts were collected and immediately used in a replication reaction. FancA and FancD2 depletions were performed as described above, with the following changes: One volume of PAS was incubated with three and a half volumes of α-FancA antibody, preimmune serum, or FancD2–FancI antibodies (mixed at 1:1 ratio) overnight at 4°C. Five volumes (FancA depletion) or six volumes (FancD2–FancI depletion) of precleared HSS or NPE was then mixed with one volume of the antibody-bound sepharose and incubated for 20 min at RT. Extracts were harvested and the depletion procedure was repeated once.
Nascent strand analysis
Nascent strand analysis was performed as described (Räschle et al, 2008; Knipscheer et al, 2012). Briefly, DNA replication products were purified as described above and digested with AflIII. After the addition of 0.5 volumes of denaturing PAGE Gel Loading Buffer II (Life Technologies), the restriction fragments were separated on a 7% polyacrylamide sequencing gel. The gel was transferred to filter paper, dried, and visualized using a phosphorimager. Experiments in Figs4F, 5D and 6D were performed at least five times, and representative results are shown.
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) was performed as described (Long et al, 2011). Briefly, reaction samples (3–5 μl) were cross-linked with 1% formaldehyde for 10 min, followed by the addition of 0.1 volume of 1.25 M glycine. The samples were spun through Micro Bio-Spin 6 Chromatography columns (Bio-Rad), diluted with sonication buffer (20 mM Tris–HCl pH 7.5, 150 mM NaCl, 2 mM EDTA, 0.5% NP-40, 5 μg/ml aprotinin + leupeptin, and 2 mM PMSF), and sonicated to obtain DNA fragments of around 300–500 bp. Aliquots of the sonicated samples were immunoprecipitated with the indicated antibodies. Antibodies against PCNA, Rev1, Rev7, FancD2, FancI, and FancA were purified on Protein A Sepharose Fast Flow (GE Healthcare) before use, and 5 μg was used per IP. Pol δ and pol ε IPs were immunoprecipitated with 1 μl of serum per IP. Formaldehyde cross-links were reversed, and the DNA was purified by phenol–chloroform extraction and ethanol precipitation. DNA was analyzed by quantitative real-time PCR as described (Pacek et al, 2006), using the following primer pairs: ICL (5′-AGCCAGATTTTTCCTCCTCTC-3′ and 5′-CATGCATTGGTTCTGCACTT-3′) and FAR (5′-AACGCCAATAGGGACTTTCC-3′ and 5′-GGGCGTACTTGGCATATGAT-3′).
Immunoprecipitations
FancA–Rev1 interaction: For each IP reaction, 5 μl of rProtein A Sepharose Fast Flow (GE Healthcare) was incubated with 1 μl of preimmune serum, anti-FancA, or anti-Rev1 antibodies for 2 h at RT. The sepharose beads were washed two times with PBS and three times with IP buffer 1 (10 mM Hepes pH 7.7, 50 mM KCl, 2.5 mM MgCl2, 0.25% NP-40). The beads were resuspended in 50 μl of the IP buffer 1. pICL (4 ng/μl) was replicated in HSS/NPE; in control samples, 10 mM Tris pH 7.5 was used instead of pICL. At 40 min, 5-μl samples of the reaction were withdrawn and incubated with the antibodies bound to sepharose beads for 2 h at 4°C. The samples were then supplemented with 4.5 U of benzonase (Novagen) or buffer and incubated for 15 min at RT. The beads were washed four times with IP buffer 2 (10 mM Hepes pH 7.7, 50 mM KCl, 2.5 mM MgCl2, 0.5% NP-40) and resuspended in 50 μl of 2× Laemmli sample buffer.
Rev1–Rev7 interaction: Rev7 migrates on SDS–PAGE gels at ∼25 kDa and overlaps with the IgG light chain; therefore, cross-linking of antibodies to beads was necessary. For each IP reaction, 5 μl of rProtein A Sepharose Fast Flow (GE Healthcare) was incubated with 1 μl of preimmune serum, anti-Rev7, or anti-Rev1 N-terminal antibodies overnight at 4°C. The sepharose beads were washed three times with 50 mM sodium phosphate, pH 8. Buffer was removed, and the beads were resuspended in 10 volumes of 25 mM DMP (dimethyl pimelimidate-2HCl; Thermo Scientific) in 50 mM sodium phosphate, pH 8. The beads were rotated for 45 min at RT. The buffer was removed, and beads were washed three times with 10 volumes of TBS (50 mM Tris pH 8, 150 mM NaCl). Ten volumes of 0.1 M glycine–HCl pH 3 was then added to the beads. Beads were immediately centrifuged and washed once with PBS and three times with ELB.
For each IP, 20 μl of diluted NPE diluted [3 μl of NPE diluted with 17 μl of ELB (10 mM Hepes pH 7.7, 50 mM KCl, 2.5 mM MgCl2)] was added to 5 μl of cross-linked beads, and the suspension was incubated for 2 h at 4°C. The beads were washed three times with ELB. After the last wash, excess buffer was removed, and the beads were resuspended in 40 μl of 2× Laemmli sample buffer without β-mercaptoethanol. They were boiled for 4 min, cooled on ice, and spun down, and 35 μl of supernatant was transferred to a fresh tube. A total of 3 μl of β-mercaptoethanol was added, and the solution was boiled again for 4 min. The proteins were resolved on SDS–PAGE gels, transferred to PVDF membranes, and probed with anti-Rev1 C-terminal antibodies and anti-Rev7 antibodies.
Both IP experiments were performed at least twice, and representative results are shown.
Plasmid pull-downs
Streptavidin-coupled magnetic beads (Dynabeads M-280, Invitrogen; 6 μl per pull-down) were washed three times with wash buffer 1 (50 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA pH 8, 0.02% Tween-20). Biotinylated LacI was added to the beads (12 pmol per 6 μl beads) and incubated at RT for 40 min. The beads were washed four times with pull-down duffer 1 (10 mM Hepes pH 7.7, 50 mM KCl, 2.5 mM MgCl2, 250 mM sucrose, 0.25 mg/ml BSA, 0.02% Tween-20) and resuspended in 40 μl of the same buffer. The bead suspension was stored on ice until needed. At indicated time points, 8-μl samples of the replication reaction were withdrawn and gently mixed with LacI-streptavidin Dynabeads. The suspension was immediately placed on a rotating wheel and rotated for 30 min at 4°C. The beads were washed three times with wash buffer 2 (10 mM Hepes pH 7.7, 50 mM KCl, 2.5 mM MgCl2, 0.25 mg/ml BSA, 0.03% Tween-20). All residual buffer was removed, and the beads were resuspended in 40 μl of 2× Laemmli sample buffer). Equal volumes of the protein samples was resolved on SDS–PAGE gels, transferred onto PVDF membranes, and probed with the indicated antibodies. Pol ε, PCNA, Rev1 and Rev7 blots were developed using autoradiography films; the films were subsequently scanned. FancD2 and FancG were developed using the chemiluminescence function on Amersham Imager 600 (GE Heathcare). The experiment was performed at least three times, and representative results are shown.
Verification of polymerase stalling at the ICL adduct
Products of translesion synthesis across an unhooked ICL still contain the cross-linked adduct in the parental strand. To verify that the polymerase used for PCR amplification of the repair products will be blocked by this adduct, we performed primer extension assays. ICL repair products were isolated from the final reaction time point (240 min), purified, and precipitated as described above. Unreplicated pCTR was used as a control. The repair products and pCTR were cut with AflIII, and the digested DNA was used as a template in a one-cycle PCR. The reaction contained only one primer (F1 or R1) labeled at the 5′ end with [γ-32P]ATP. PCRs were performed using Phusion High-Fidelity DNA polymerase (NEB) and KAPA HiFi DNA Polymerase (Kapa Biosystems), according to the manufacturer’s instructions. The DNA fragments were resolved on an 8% urea-PAGE gel and visualized by autoradiography.
Sequencing of repair products
DNA samples (5 μl for pCTR, 8 μl for pICL) were taken at the final time points of replication/repair reactions (60 min for pCTR and at 240 min for pICL), and DNA was diluted with 10 volumes of stop solution A (0.5% SDS, 10 mM EDTA, 50 mM Tris pH 7.5), supplemented with 0.13 mg/ml RNase A, and incubated at 37°C for 30 min. Proteinase K was then added to 0.67 mg/ml, and samples were incubated overnight at RT. DNA was phenol–chloroform-extracted and precipitated with 100% ethanol in the presence of 0.2 mg/ml glycogen and 0.3 M sodium acetate pH 5.3. DNA was resuspended in 5 or 8 μl of 10 mM Tris pH 8. Part of the pICL DNA was digested with SapI and re-precipitated. pCTR and pICL samples were amplified with KAPA HiFi DNA polymerase (Kapa Biosystems) for 12 PCR cycles using bar-coded primers (F1: NNNN-barcode-ATGAAGATCCCTCGACCTGC; R1: NNNN-barcode-CCAATACGCAAACCGCCTC, where N represent random nucleotides to optimize sequencing, and barcode is a six-nucleotide sequence unique to a given sample [see below for complete list of primers]). The PCR samples were mixed with 5× TBE sample buffer (Invitrogen) and resolved on an 8% TBE polyacrylamide gel (Invitrogen). The gel was stained with SYBR Gold (Life Technologies), and PCR products of the appropriate size (174 nt) were excised. The gel slices were minced into smaller pieces and transferred into a 0.65-ml tube that had been pierced at the bottom with a 21-gauge needle. This tube was then placed in a 1.5-ml tube and centrifuged for 5 min at 20,000 g to break the gel slices into small pieces. Three volumes of elution buffer (10 mM Tris pH 8, 1 mM EDTA pH 8, 300 mM NaCl) was added to one volume of the gel pieces, and the mixture was agitated in Eppendorf Thermomixer R overnight at RT. The mixture was transferred to a Spin-X column (0.45 μm; Sigma) and centrifuged for 3 min at 16,000 g. The recovered supernatant was precipitated with 100% ethanol in the presence of 0.07 mg/ml glycogen and 0.3 M sodium acetate pH 5.3. DNA was resuspended in 10 mM Tris pH 8, ligated to Illumina adaptors according to the manufacturer’s instructions, and sequenced using a MiSeq sequencer (150 bp read length, paired ends). A total of 1.7 million reads for pCTR and 2.6 million reads for pICL were obtained.
Sequencing data analysis
Sequencing data were demultiplexed and exported to FASTQ files using the Cassava package (Illumina). Demultiplexed reads were analyzed using custom Python scripts (available upon request). The first read from each pair of reads was aligned against two 6-nt sequences on either end of the region amplified by PCR, and the pair was discarded if the first read did not contain an exact match to these sequences (Supplementary Table S1, rejection type 1). Reads were also discarded if the second read in the pair did not show perfect agreement to the first (Supplementary Table S1, rejection type 2). These stringent filtering criteria were chosen to minimize the contribution of sequencing errors to the observed nucleotide misincorporation rate. Once aligned, reads were classified as having the correct length or an incorrect length. Among reads of the correct length, nucleotide frequencies were tabulated at each position. Frequencies of each distinct incorrect-length sequence were tabulated, revealing that the 8-nt deletion product discussed in the Results section accounted for more than half of these reads. Files containing sequencing reads and Python scripts used to analyze the data are available upon request.
Preparation of pICLPt
pICLPt was prepared as described (Räschle et al, 2008; Enoiu et al, 2012). Briefly, two DNA oligos were cross-linked with cisplatin at a single guanine residue. The cross-linked DNA duplex was ligated into Bbs1 sites of pSVRLuc. To prepare pCTR, the unmodified duplex DNA was ligated into the same pSVRLuc backbone.
Primers used in the deep-sequencing experiment
N represents a random nucleotide. Six-nucleotide barcode sequence unique for a given sample is underlined.
pCTR:
F1: NNNNATCACGATGAAGATCCCTCGACCTGC
R1: NNNNATCACGCCAATACGCAAACCGCCTC
pICL (reaction 1):
F2: NNNNTTAGGCATGAAGATCCCTCGACCTGC
R2: NNNNTTAGGCCCAATACGCAAACCGCCTC
pICL (reaction 1) digested with SapI:
F3: NNNNACAGTGATGAAGATCCCTCGACCTGC
R3: NNNNACAGTGCCAATACGCAAACCGCCTC
pICL (reaction 2):
F4: NNNNAGTCAAATGAAGATCCCTCGACCTGC
R4: NNNNAGTCAACCAATACGCAAACCGCCTC
pCTR, Rev1 depletion:
F5: NNNNCGATGTATGAAGATCCCTCGACCTGC
R5: NNNNCGATGTCCAATACGCAAACCGCCTC
pICL, Rev1 depletion:
F6: NNNNTGACCAATGAAGATCCCTCGACCTGC
R6: NNNNTGACCACCAATACGCAAACCGCCTC
Acknowledgments
We thank Robert Fuchs, Joe Loparo, Puck Knipscheer, Markus Räschle, and members of the Walter Lab for critical reading of the manuscript. We thank Nava Gharaei for help with preparing samples for deep sequencing. We acknowledge Markus Räschle who first proposed the source of the 8-nucleotide deletion product. We thank Puck Knipscheer for expression constructs of Rev1 fragments used to generate Rev1 antibodies. We also thank Zach Herbert (DFCI Molecular Biology Core Facilities) for help with sequencing and Jieqiong Zhang for the FancI antibody. This work was supported by Human Frontiers Science Program long-term fellowship (LT000773/2010-L) and European Molecular Biology Organization long-term fellowship (ALTF 742-2009) to M.B., National Science Foundation Graduate Research Fellowship to T.G.W.G., National Science Foundation (award 1121023) to A.S., Grant-in-Aid for Scientific Research from the MEXT to S.W., and NIH grants GM62267 and HL098316 to J.C.W. J.C.W. is an investigator of the Howard Hughes Medical Institute.
Author contributions
TGWG analyzed the sequencing data. AS and SW supplied nonrenewable antibody reagents. MB and JCW designed and analyzed experiments; MB performed experiments; and MB and JCW prepared the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary Figures, Supplementary Table S1
Source Data for Supplementary Figure S3
Source Data for Supplementary Figure S4
Source Data for Supplementary Figure S5
Source Data for Supplementary Figure S7
Review Process File
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
References
- Albertella MR, Green CM, Lehmann AR, O’Connor MJ. A role for polymerase eta in the cellular tolerance to cisplatin-induced damage. Cancer Res. 2005;65:9799–9806. doi: 10.1158/0008-5472.CAN-05-1095. [DOI] [PubMed] [Google Scholar]
- Alpi AF, Pace PE, Babu MM, Patel KJ. Mechanistic insight into site-restricted monoubiquitination of FANCD2 by Ube2t, FANCL, and FANCI. Mol Cell. 2008;32:767–777. doi: 10.1016/j.molcel.2008.12.003. [DOI] [PubMed] [Google Scholar]
- Arakawa H, Moldovan G-L, Saribasak H, Saribasak NN, Jentsch S, Buerstedde J-M. A role for PCNA ubiquitination in immunoglobulin hypermutation. PLoS Biol. 2006;4:e366. doi: 10.1371/journal.pbio.0040366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett E, King NM, Bryant MF, Hector S, Pendyala L, Chaney SG, Cordeiro-Stone M. The role of DNA polymerase eta in translesion synthesis past platinum-DNA adducts in human fibroblasts. Cancer Res. 2004;64:6469–6475. doi: 10.1158/0008-5472.CAN-04-1328. [DOI] [PubMed] [Google Scholar]
- Bienko M, Green CM, Crosetto N, Rudolf F, Zapart G, Coull B, Kannouche P, Wider G, Peter M, Lehmann AR, Hofmann K, Dikic I. Ubiquitin-binding domains in Y-family polymerases regulate translesion synthesis. Science. 2005;310:1821–1824. doi: 10.1126/science.1120615. [DOI] [PubMed] [Google Scholar]
- Chen Y-W, Cleaver JE, Hanaoka F, Chang C-F, Chou K-M. A novel role of DNA polymerase eta in modulating cellular sensitivity to chemotherapeutic agents. Mol Cancer Res. 2006;4:257–265. doi: 10.1158/1541-7786.MCR-05-0118. [DOI] [PubMed] [Google Scholar]
- Chilkova O, Stenlund P, Isoz I, Stith CM, Grabowski P, Lundström E-B, Burgers PM, Johansson E. The eukaryotic leading and lagging strand DNA polymerases are loaded onto primer-ends via separate mechanisms but have comparable processivity in the presence of PCNA. Nucleic Acids Res. 2007;35:6588–6597. doi: 10.1093/nar/gkm741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clauson C, Schärer OD, Niedernhofer L. Advances in understanding the complex mechanisms of DNA interstrand cross-link repair. Cold Spring Harb Perspect Med. 2013;3:a012732. doi: 10.1101/cshperspect.a012732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmunds CE, Simpson LJ, Sale JE. PCNA ubiquitination and REV1 define temporally distinct mechanisms for controlling translesion synthesis in the avian cell line DT40. Mol Cell. 2008;30:519–529. doi: 10.1016/j.molcel.2008.03.024. [DOI] [PubMed] [Google Scholar]
- Enoiu M, Ho TV, Long DT, Walter JC, Schärer OD. Construction of plasmids containing site-specific DNA interstrand cross-links for biochemical and cell biological studies. Methods Mol Biol. 2012;920:203–219. doi: 10.1007/978-1-61779-998-3_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu YV, Yardimci H, Long DT, Ho TV, Guainazzi A, Bermudez VP, Hurwitz J, van Oijen A, Schärer OD, Walter JC. Selective bypass of a lagging strand roadblock by the eukaryotic replicative DNA helicase. Cell. 2011;146:931–941. doi: 10.1016/j.cell.2011.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukui T, Yamauchi K, Muroya T, Akiyama M, Maki H, Sugino A, Waga S. Distinct roles of DNA polymerases delta and epsilon at the replication fork in Xenopus egg extracts. Genes Cells. 2004;9:179–191. doi: 10.1111/j.1356-9597.2004.00716.x. [DOI] [PubMed] [Google Scholar]
- Gan GN, Wittschieben JP, Wittschieben BO, Wood RD. DNA polymerase zeta (pol zeta) in higher eukaryotes. Cell Res. 2008;18:174–183. doi: 10.1038/cr.2007.117. [DOI] [PubMed] [Google Scholar]
- Georgescu RE, Langston L, Yao NY, Yurieva O, Zhang D, Finkelstein J, Agarwal T, O’Donnell ME. Mechanism of asymmetric polymerase assembly at the eukaryotic replication fork. Nat Struct Mol Biol. 2014;21:664–670. doi: 10.1038/nsmb.2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlach VL, Feaver WJ, Fischhaber PL, Friedberg EC. Purification and characterization of pol kappa, a DNA polymerase encoded by the human DINB1 gene. J Biol Chem. 2001;276:92–98. doi: 10.1074/jbc.M004413200. [DOI] [PubMed] [Google Scholar]
- Guo C, Fischhaber PL, Luk-Paszyc MJ, Masuda Y, Zhou J, Kamiya K, Kisker C, Friedberg EC. Mouse Rev1 protein interacts with multiple DNA polymerases involved in translesion DNA synthesis. EMBO J. 2003;22:6621–6630. doi: 10.1093/emboj/cdg626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C, Sonoda E, Tang T-S, Parker JL, Bielen AB, Takeda S, Ulrich HD, Friedberg EC. REV1 protein interacts with PCNA: significance of the REV1 BRCT domain in vitro and in vivo. Mol Cell. 2006a;23:265–271. doi: 10.1016/j.molcel.2006.05.038. [DOI] [PubMed] [Google Scholar]
- Guo C, Tang T-S, Bienko M, Parker JL, Bielen AB, Sonoda E, Takeda S, Ulrich HD, Dikic I, Friedberg EC. Ubiquitin-binding motifs in REV1 protein are required for its role in the tolerance of DNA damage. Mol Cell Biol. 2006b;26:8892–8900. doi: 10.1128/MCB.01118-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara K, Hashimoto H, Murakumo Y, Kobayashi S, Kogame T, Unzai S, Akashi S, Takeda S, Shimizu T, Sato M. Crystal structure of human REV7 in complex with a human REV3 fragment and structural implication of the interaction between DNA polymerase zeta and REV1. J Biol Chem. 2010;285:12299–12307. doi: 10.1074/jbc.M109.092403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haracska L, Prakash S, Prakash L. Yeast Rev1 protein is a G template-specific DNA polymerase. J Biol Chem. 2002;277:15546–15551. doi: 10.1074/jbc.M112146200. [DOI] [PubMed] [Google Scholar]
- Hicks JK, Chute CL, Paulsen MT, Ragland RL, Howlett NG, Gueranger Q, Glover TW, Canman CE. Differential roles for DNA polymerases eta, zeta, and REV1 in lesion bypass of intrastrand versus interstrand DNA cross-links. Mol Cell Biol. 2010;30:1217–1230. doi: 10.1128/MCB.00993-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinz JM, Nham PB, Urbin SS, Jones IM, Thompson LH. Disparate contributions of the Fanconi anemia pathway and homologous recombination in preventing spontaneous mutagenesis. Nucleic Acids Res. 2007;35:3733–3740. doi: 10.1093/nar/gkm315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho TV, Guainazzi A, Derkunt SB, Enoiu M, Schärer OD. Structure-dependent bypass of DNA interstrand crosslinks by translesion synthesis polymerases. Nucleic Acids Res. 2011;39:7455–7464. doi: 10.1093/nar/gkr448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson E, Garg P, Burgers PMJ. The Pol32 subunit of DNA polymerase delta contains separable domains for processive replication and proliferating cell nuclear antigen (PCNA) binding. J Biol Chem. 2004;279:1907–1915. doi: 10.1074/jbc.M310362200. [DOI] [PubMed] [Google Scholar]
- Johnson RE, Washington MT, Haracska L, Prakash S, Prakash L. Eukaryotic polymerases iota and zeta act sequentially to bypass DNA lesions. Nature. 2000;406:1015–1019. doi: 10.1038/35023030. [DOI] [PubMed] [Google Scholar]
- Kannouche PL, Wing J, Lehmann AR. Interaction of human DNA polymerase eta with monoubiquitinated PCNA: a possible mechanism for the polymerase switch in response to DNA damage. Mol Cell. 2004;14:491–500. doi: 10.1016/s1097-2765(04)00259-x. [DOI] [PubMed] [Google Scholar]
- Kim H, D’Andrea AD. Regulation of DNA cross-link repair by the Fanconi anemia/BRCA pathway. Genes Dev. 2012;26:1393–1408. doi: 10.1101/gad.195248.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Yang K, Dejsuphong D, D’Andrea AD. Regulation of Rev1 by the Fanconi anemia core complex. Nat Struct Mol Biol. 2012;19:164–170. doi: 10.1038/nsmb.2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein Douwel D, Boonen RACM, Long DT, Szypowska AA, Räschle M, Walter JC, Knipscheer P. XPF-ERCC1 acts in Unhooking DNA interstrand crosslinks in cooperation with FANCD2 and FANCP/SLX4. Mol Cell. 2014;54:460–471. doi: 10.1016/j.molcel.2014.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knipscheer P, Räschle M, Smogorzewska A, Enoiu M, Ho TV, Schärer OD, Elledge SJ, Walter JC. The Fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science. 2009;326:1698–1701. doi: 10.1126/science.1182372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knipscheer P, Räschle M, Schärer OD, Walter JC. Replication-coupled DNA interstrand cross-link repair in Xenopus egg extracts. Methods Mol Biol. 2012;920:221–243. doi: 10.1007/978-1-61779-998-3_16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochaniak AB, Habuchi S, Loparo JJ, Chang DJ, Cimprich KA, Walter JC, van Oijen AM. Proliferating cell nuclear antigen uses two distinct modes to move along DNA. J Biol Chem. 2009;284:17700–17710. doi: 10.1074/jbc.M109.008706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence CW. Cellular functions of DNA polymerase zeta and Rev1 protein. Adv Protein Chem. 2004;69:167–203. doi: 10.1016/S0065-3233(04)69006-1. [DOI] [PubMed] [Google Scholar]
- Leach CA, Michael WM. Ubiquitin/SUMO modification of PCNA promotes replication fork progression in Xenopus laevis egg extracts. J Cell Biol. 2005;171:947–954. doi: 10.1083/jcb.200508100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann AR, Niimi A, Ogi T, Brown S, Sabbioneda S, Wing JF, Kannouche PL, Green CM. Translesion synthesis: Y-family polymerases and the polymerase switch. DNA Repair. 2007;6:891–899. doi: 10.1016/j.dnarep.2007.02.003. [DOI] [PubMed] [Google Scholar]
- Long DT, Räschle M, Joukov V, Walter JC. Mechanism of RAD51-dependent DNA interstrand cross-link repair. Science. 2011;333:84–87. doi: 10.1126/science.1204258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long DT, Joukov V, Budzowska M, Walter JC. BRCA1 promotes unloading of the CMG Helicase from a stalled DNA replication fork. Mol Cell. 2014;56:174–185. doi: 10.1016/j.molcel.2014.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longerich S, San Filippo J, Liu D, Sung P. FANCI binds branched DNA and is monoubiquitinated by UBE2T-FANCL. J Biol Chem. 2009;284:23182–23186. doi: 10.1074/jbc.C109.038075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masutani C, Araki M, Yamada A, Kusumoto R, Nogimori T, Maekawa T, Iwai S, Hanaoka F. Xeroderma pigmentosum variant (XP-V) correcting protein from HeLa cells has a thymine dimer bypass DNA polymerase activity. EMBO J. 1999;18:3491–3501. doi: 10.1093/emboj/18.12.3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minko IG, Harbut MB, Kozekov ID, Kozekova A, Jakobs PM, Olson SB, Moses RE, Harris TM, Rizzo CJ, Lloyd RS. Role for DNA polymerase kappa in the processing of N2-N2-guanine interstrand cross-links. J Biol Chem. 2008;283:17075–17082. doi: 10.1074/jbc.M801238200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirchandani KD, McCaffrey RM, D’Andrea AD. The Fanconi anemia core complex is required for efficient point mutagenesis and Rev1 foci assembly. DNA Repair. 2008;7:902–911. doi: 10.1016/j.dnarep.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyabe I, Kunkel TA, Carr AM. The major roles of DNA polymerases epsilon and delta at the eukaryotic replication fork are evolutionarily conserved. PLoS Genet. 2011;7:e1002407. doi: 10.1371/journal.pgen.1002407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nealon K, Nicholl ID, Kenny MK. Characterization of the DNA polymerase requirement of human base excision repair. Nucleic Acids Res. 1996;24:3763–3770. doi: 10.1093/nar/24.19.3763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nick McElhinny SA, Gordenin DA, Stith CM, Burgers PMJ, Kunkel TA. Division of labor at the eukaryotic replication fork. Mol Cell. 2008;30:137–144. doi: 10.1016/j.molcel.2008.02.022. ) . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niedzwiedz W, Mosedale G, Johnson M, Ong CY, Pace P, Patel KJ. The Fanconi anaemia gene FANCC promotes homologous recombination and error-prone DNA repair. Mol Cell. 2004;15:607–620. doi: 10.1016/j.molcel.2004.08.009. [DOI] [PubMed] [Google Scholar]
- Niimi A, Brown S, Sabbioneda S, Kannouche PL, Scott A, Yasui A, Green CM, Lehmann AR. Regulation of proliferating cell nuclear antigen ubiquitination in mammalian cells. Proc Natl Acad Sci USA. 2008;105:16125–16130. doi: 10.1073/pnas.0802727105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi E, Murakumo Y, Kanjo N, Akagi J-I, Masutani C, Hanaoka F, Ohmori H. Interaction of hREV1 with three human Y-family DNA polymerases. Genes Cells. 2004;9:523–531. doi: 10.1111/j.1356-9597.2004.00747.x. [DOI] [PubMed] [Google Scholar]
- Pacek M, Tutter AV, Kubota Y, Takisawa H, Walter JC. Localization of MCM2-7, Cdc45, and GINS to the site of DNA unwinding during eukaryotic DNA replication. Mol Cell. 2006;21:581–587. doi: 10.1016/j.molcel.2006.01.030. [DOI] [PubMed] [Google Scholar]
- Papadopoulo D, Guillouf C, Mohrenweiser H, Moustacchi E. Hypomutability in Fanconi anemia cells is associated with increased deletion frequency at the HPRT locus. Proc Natl Acad Sci USA. 1990a;87:8383–8387. doi: 10.1073/pnas.87.21.8383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulo D, Porfirio B, Moustacchi E. Mutagenic response of Fanconi’s anemia cells from a defined complementation group after treatment with photoactivated bifunctional psoralens. Cancer Res. 1990b;50:3289–3294. [PubMed] [Google Scholar]
- Prakash S, Prakash L. Translesion DNA synthesis in eukaryotes: a one- or two-polymerase affair. Genes Dev. 2002;16:1872–1883. doi: 10.1101/gad.1009802. [DOI] [PubMed] [Google Scholar]
- Prakash S, Johnson RE, Prakash L. Eukaryotic translesion synthesis DNA polymerases: specificity of structure and function. Annu Rev Biochem. 2005;74:317–353. doi: 10.1146/annurev.biochem.74.082803.133250. [DOI] [PubMed] [Google Scholar]
- Pursell ZF, Isoz I, Lundström E-B, Johansson E, Kunkel TA. Yeast DNA polymerase epsilon participates in leading-strand DNA replication. Science. 2007;317:127–130. doi: 10.1126/science.1144067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendra E, Oestergaard VH, Langevin F, Wang M, Dornan GL, Patel KJ, Passmore LA. The genetic and biochemical basis of FANCD2 monoubiquitination. Mol Cell. 2014;54:858–869. doi: 10.1016/j.molcel.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Räschle M, Knipscheer P, Enoiu M, Angelov T, Sun J, Griffith JD, Ellenberger TE, Schärer OD, Walter JC. Mechanism of replication-coupled DNA interstrand crosslink repair. Cell. 2008;134:969–980. doi: 10.1016/j.cell.2008.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross A-L, Simpson LJ, Sale JE. Vertebrate DNA damage tolerance requires the C-terminus but not BRCT or transferase domains of REV1. Nucleic Acids Res. 2005;33:1280–1289. doi: 10.1093/nar/gki279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato K, Toda K, Ishiai M, Takata M, Kurumizaka H. DNA robustly stimulates FANCD2 monoubiquitylation in the complex with FANCI. Nucleic Acids Res. 2012;40:4553–4561. doi: 10.1093/nar/gks053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta S, van Deursen F, de Piccoli G, Labib K. Dpb2 integrates the leading-strand DNA polymerase into the eukaryotic replisome. Curr Biol. 2013;23:543–552. doi: 10.1016/j.cub.2013.02.011. [DOI] [PubMed] [Google Scholar]
- Shachar S, Ziv O, Avkin S, Adar S, Wittschieben J, Reissner T, Chaney S, Friedberg EC, Wang Z, Carell T, Geacintov N, Livneh Z. Two-polymerase mechanisms dictate error-free and error-prone translesion DNA synthesis in mammals. EMBO J. 2009;28:383–393. doi: 10.1038/emboj.2008.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimazaki N, Yoshida K, Kobayashi T, Toji S, Tamai K, Koiwai O. Over-expression of human DNA polymerase lambda in E. coli and characterization of the recombinant enzyme. Genes Cells. 2002;7:639–651. doi: 10.1046/j.1365-2443.2002.00547.x. [DOI] [PubMed] [Google Scholar]
- Sobeck A, Stone S, Costanzo V, de Graaf B, Reuter T, de Winter J, Wallisch M, Akkari Y, Olson S, Wang W, Joenje H, Christian JL, Lupardus PJ, Cimprich KA, Gautier J, Hoatlin ME. Fanconi anemia proteins are required to prevent accumulation of replication-associated DNA double-strand breaks. Mol Cell Biol. 2006;26:425–437. doi: 10.1128/MCB.26.2.425-437.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szuts D, Marcus AP, Himoto M, Iwai S, Sale JE. REV1 restrains DNA polymerase zeta to ensure frame fidelity during translesion synthesis of UV photoproducts in vivo. Nucleic Acids Res. 2008;36:6767–6780. doi: 10.1093/nar/gkn651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tissier A, McDonald JP, Frank EG, Woodgate R. poliota, a remarkably error-prone human DNA polymerase. Genes Dev. 2000;14:1642–1650. [PMC free article] [PubMed] [Google Scholar]
- Waga S, Masuda T, Takisawa H, Sugino A. DNA polymerase epsilon is required for coordinated and efficient chromosomal DNA replication in Xenopus egg extracts. Proc Natl Acad Sci USA. 2001;98:4978–4983. doi: 10.1073/pnas.081088798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter J, Sun L, Newport J. Regulated chromosomal DNA replication in the absence of a nucleus. Mol Cell. 1998;1:519–529. doi: 10.1016/s1097-2765(00)80052-0. [DOI] [PubMed] [Google Scholar]
- Washington MT, Minko IG, Johnson RE, Haracska L, Harris TM, Lloyd RS, Prakash S, Prakash L. Efficient and error-free replication past a minor-groove N2-guanine adduct by the sequential action of yeast Rev1 and DNA polymerase zeta. Mol Cell Biol. 2004;24:6900–6906. doi: 10.1128/MCB.24.16.6900-6906.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K, Tateishi S, Kawasuji M, Tsurimoto T, Inoue H, Yamaizumi M. Rad18 guides poleta to replication stalling sites through physical interaction and PCNA monoubiquitination. EMBO J. 2004;23:3886–3896. doi: 10.1038/sj.emboj.7600383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams HL, Gottesman ME, Gautier J. Replication-independent repair of DNA interstrand crosslinks. Mol Cell. 2012;47:140–147. doi: 10.1016/j.molcel.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Wu X, Rechkoblit O, Geacintov NE, Taylor J-S, Wang Z. Response of human REV1 to different DNA damage: preferential dCMP insertion opposite the lesion. Nucleic Acids Res. 2002;30:1630–1638. doi: 10.1093/nar/30.7.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Dewar JM, Budzowska M, Motnenko A, Cohn MA, Walter JC. DNA interstrand cross-link repair requires replication-fork convergence. Nat Struct Mol Biol. 2015;22:242–247. doi: 10.1038/nsmb.2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures, Supplementary Table S1
Source Data for Supplementary Figure S3
Source Data for Supplementary Figure S4
Source Data for Supplementary Figure S5
Source Data for Supplementary Figure S7
Review Process File
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6