

Abstract
Soluble epoxide hydrolase (sEH) has C-terminal epoxide hydrolase and N-terminal lipid phosphatase activity. Its hydrolase activity is associated with endothelial nitric oxide synthase (eNOS) dysfunction. However, little is known about the role of sEH phosphatase in regulating eNOS activity. Simvastatin, a clinical lipid-lowering drug, also has a pleiotropic effect on eNOS activation. However, whether sEH phosphatase is involved in simvastatin-activated eNOS activity remains elusive. We investigated the role of sEH phosphatase activity in simvastatin-mediated activation of eNOS in endothelial cells (ECs). Simvastain increased the phosphatase activity of sEH, which was diminished by pharmacological inhibitors of sEH phosphatase. In addition, pharmacological inhibition of sEH phosphatase or overexpressing the inactive phosphatase domain of sEH enhanced simvastatin-induced NO bioavailability, tube formation and phosphorylation of eNOS, Akt, and AMP-activated protein kinase (AMPK). In contrast, overexpressing the phosphatase domain of sEH limited the simvastatin-increased NO biosynthesis and eNOS phosphorylation at Ser1179. Simvastatin evoked epidermal growth factor receptor–c-Src–increased Tyr phosphorylation of sEH and formation of an sEH–Akt–AMPK–eNOS complex, which was abolished by the c-Src kinase inhibitor PP1 or c-Src dominant-negative mutant K298M. These findings suggest that sEH phosphatase activity negatively regulates simvastatin-activated eNOS by impeding the Akt–AMPK–eNOS signaling cascade.
Simvastatin (Zocor), an inhibitor of 3-hydroxy-3-methylglutaryl coenzyme A reductase, is a cholesterol-lowering drug that suppresses the biosynthesis of cholesterol and enhances the clearance of circulating low-density lipoprotein receptor (LDL) by upregulating the expression of hepatic LDR receptors1,2. Simvastatin is used to treat hyperlipidemia and related cardiovascular diseases2,3,4. Besides its beneficial effect on dyslipidemia, simvastatin has cholesterol-independent pleiotropic effects on the physiological function of endothelial cells (ECs) by increasing endothelial nitric oxide synthase (eNOS) activity and nitric oxide (NO) production5,6,7,8.
Endothelium-derived NO is a cricial regulator of vascular homeostasis; dysregulation of eNOS is the key event in the initiation and progression of cardiovascular diseases such as atherosclerosis and hypertension9,10. NO bioavailability is tightly controlled by eNOS activity via the complex network of kinase- and phosphatase-dependent pathways11,12,13,14,15. For instance, treatment with vascular endothelial growth factor (VEGF) increases the phosphoinositide 3-kinase (PI3K)/Akt-dependent phosphorylation of eNOS, which leads to eNOS activation and NO production in ECs11,12. By contrast, VEGF also activates protein phosphatase 2A (PP2A) to dephosphorylate eNOS and negatively regulate eNOS, thereby precisely modulating the enzymatic activity of eNOS12,14,15. However, despite extensive investigations of the molecular mechanisms of simvastatin, whether phosphatase is functionally involved in simvastatin-activated eNOS in ECs is unknown.
Soluble epoxide hydrolase (sEH) is a bifunctional enzyme with C-terminal hydrolase and N-terminal phosphatase activity16,17. sEH is expressed in several types of cells, including cardiomyocytes and ECs17. sEH hydrolase is responsible for the conversion of epoxyeicosatrienoic acids (EETs) to dihydroxyeicosatrienoic acids (DHETs). EETs are synthesized from arachidonic acid by cytochrome P450 epoxygenases and modulate cellular physiologic function including vascular relaxation, inflammatory response and fibrosis in the cardiovascular system17,18,19,20. Several lines of evidence suggest that inhibition of sEH hydrolase activity increases circulating levels of EETs and prevents the progression of hypertension and inflammatory diseases20,21,22,23,24.
In contrast to knowledge of the hyrolase activity of sEH, that of its phosphatase activity is limited. Human sEH polymorphism studies demonstrated that Arg287Gln or Lys55Arg polymorphism of sEH, encoding a mutant sEH with reduced phosphatase activity, is associated with coronary heart diseases or type 2 diabetes25,26,27,28. In addition, sEH phosphatase is involved in the cholesterol metabolism of hepatocytes, cell growth of ECs and metabolism of lysophosphatidic acids29,30,31,32. Nevertheless, whether the phosphatase activity of sEH participates in simvastatin-activated eNOS and NO production and the underlying regulatory mechanism are largely unknown.
In this study, we hypothesized that sEH phosphatase plays an important role in simvastatin-regulated eNOS activity. We investigated the change in sEH phosphatase activity with simvastatin treatment and then examined the effect of the phosphatase domain of sEH on simvastatin-induced eNOS phosphorylation and NO production as well as the Akt-AMP-activated protein kinase (AMPK) signaling pathway. We also delineated the mechanisms underlying sEH phosphatase-regulated eNOS activity in simvastatin-treated ECs. We found that sEH phosphatase is a crucial negative regulator of simvastatin-activated eNOS by activating epidermal growth factor receptor (EGFR)–c-Src signaling.
Results
Simvastatin increases sEH phosphatase activity
We first validated the inhibitory effect of the sEH phosphatase inhibitors AFC or ebselen on sEH phosphatase activity in BAECs cells transfected with full-length sEH (WT) or the phosphatase domain of sEH (N-ter). These transfected BAECs were then treated with various concentrations of AFC (0, 12.5, 25, 50, 100 μM) or ebselen (0, 2.5, 5, 10, 20 μM) for 2 h. Our data showed that treatment with AFC or ebselen could dose-dependent inhibit the activity of sEH phosphatase activity (Fig. 1a), suggesting the two inhibitors have excellent inhibitory efficacy on phosphatase activity. However, treatment with 100 μM of AFC or 20 μM of ebselen showed cytotoxic effects to BAECs (data not shown). We therefore chose AFC at concentration 50 μM or ebselen at concentration 10 μM, respectively, for our further studies. We then evaluated the role of sEH phosphatase in regulating simvastatin-mediated effects in ECs. Time-course experiments demonstrated a significant increase in sEH phosphatase activity with 10 μM simvastatin as early as 15 min, with peak level at 30 min (Fig. 1b). Pre-treatment with AFC or ebselen totally abrogated the simvastatin-increased sEH phosphatase activity (Fig. 1c).
Figure 1. Simvastatin increases soluble epoxide hydrolase (sEH) phosphatase activity.

Phosphatase activity (PT) assay of bovine aortic endothelial cells (BAECs) (a) transfected with vector, full-length sEH (WT) or phosphatase domain of sEH (N-ter) for 48 h, then treated with or without an sEH phosphatase inhibitor, N-acetyl-S-farnesyl-L-cysteine (AFC, 0, 12.5, 25, 50, 100 μM) or ebselen (0, 2.5, 5, 10, 20 μM), for 2 h; (b) treated with vehicle or simvastatin (10 μM) for the indicated times; or (c) pretreated with AFC or ebselen (10 μM) for 2 h, then simvastatin (10 μM) for 30 min. Data are mean ± SEM from 5 independent experiments. *P < 0.05 vs. vehicle- or vector-treated cells, #P < 0.05 vs. simvastatin-treated alone cells.
sEH phosphatase negatively regulates simvastatin-induced NO production, tube formation and eNOS phosphorylation in ECs
Simvastatin can induce eNOS phosphorylation and NO production in ECs6,33. Therefore, we explored the effect of sEH phosphatase on simvastatin-mediated NO production and tube formation as well as phosphorylation of eNOS, Akt and AMPK in bovine aortic endothelial cells (BAECs). To evaluate the specific role of sEH phosphatase in these key simvastatin-induced events, BAECs were pre-treated with AFC, ebselen, or AUDA (an inhibitor of sEH hydrolase) or transfected with PTmut, inactive EH domain (EHmut) (EHmut), N-ter or C-terminal hydrolase domain (C-ter). Treatment with AFC or ebselen or transfection with PTmut further increased the simvastatin-increased NO bioavailability and tube formation (Fig. 2a,d). However, treatment with AUDA or transfection with EHmut failed to produce such effects (Fig. 2a,b). In addition, overexpression of N-ter prevented the simvastatin-induced NO production and tube formation (Fig. 2e–g). eNOS phosphorylation status is critical for its enzymatic activity in BAECs10,11. We found that treatment with AFC but not AUDA prolonged the simvastatin-mediated eNOS phosphorylation at Ser1179 (Fig. 3a).
Figure 2. Phosphatase activity of sEH negatively regulates simvastatin-induced NO production and tube formation in ECs.

(a) BAECs were pretreated with the sEH phosphatase inhibitor AFC (50 μM) or ebselen (10 μM) for 2 h or sEH hydrolase inhibitor 12-(3-adamantan-1-yl-ureido)-dodecanoic acid (AUDA, 10 μM), for 2 h and then simvastatin (10 μM) for 24 h. Level of nitrite in culture media was measured by Griess assay. (b–e) BAECs transfected with vector, Flag-tag sEH with PTmut or hydrolase mutant (EHmut); or Flag-tag N-ter or hydrolase domain of sEH (C-ter) for 48 h, then simvastatin (10 μM) for 24 h. (b,c)Western blot analysis of Flag and α-tubulin. (d,e) Level of nitrite in culture media. (f,g) Tube formation with BAECs pretreated with AFC (50 μM) or ebselen (10 μM) for 2 h or transfected with PTmut or N-ter for 48 h, then seeded on pre-coated ECL Cell Attachment Matrix with or without simvastatin (10 μM) for 24 h. Data are mean ± SEM fold branch points from 5 randomly selected microscopy views. *P < 0.05 vs. vehicle- or vector-treated cells, #P < 0.05 vs. simvastatin-treated alone cells.
Figure 3. Role of sEH phosphatase in regulating simvastatin-induced eNOS phosphorylation.

Western blot analysis of phosphorylated eNOS (p-eNOS) at Ser1179 and eNOS level in BAECs pretreated with (a) vehicle, AFC (50 μM) or AUDA (10 μM) for 2 h or (b,c) transfected with vector, Ptmut, Ehmut, N-ter or C-ter for 48 h, then simvastatin (10 μM) for 0–120 min. Data are mean ± SEM from 5 independent experiments. *P < 0.05 vs. vehicle- or vector-treated cells, #P < 0.05 vs. simvastatin-treated alone cells.
To provide further evidence that sEH phosphatase is crucial in eNOS activation, we depleted the activity of sEH phosphatase by overexpressing a full-length sEH with catalytic-inactive phosphatase. Simvastatin-mediated eNOS phosphorylation at Ser1179 was further augmented with overexpression of the inactive phosphatase domain (PTmut), whereas overexpression of the inactive EH domain (EHmut) had no effect (Fig. 3b). In contrast, overexpression of the phosphatase domain of sEH (N-ter) totally abrogated the simvastatin-increased eNOS phosphorylation at Ser1179 (Fig. 3c). sEH phosphatase may be important in regulating simvastatin-activated eNOS.
Overexpression of sEH phosphatase diminishes simvastatin-induced phosphorylation of AMPK and Akt
The Akt–AMPK signaling pathway plays a crucial role in simvastatin-activated eNOS33. We analyzed whether sEH phosphatase is involved in activation of the Akt–AMPK pathway. Treatment with AFC or transfection with PTmut sustained simvastatin-induced Akt phosphorylation at Ser473 and AMPK phosphorylation at Thr172 (Fig. 4a,b). Moreover, overexpression of the phosphatase domain of sEH (N-ter) totally abolished these simvastatin-increased activities (Fig. 4a,b). Therefore, sEH phosphatase participates in simvastatin-activated Akt–AMPK signaling in ECs.
Figure 4. sEH phosphatase plays a crucial role in regulating simvastatin-induced Akt and AMPK phosphorylation.

Western blot analysis of (a) phosphorylated Akt (p-Akt) at Ser473 and Akt and (b) phosphorylated AMP-activated protein kinase (p-AMPK) at Thr172 and AMPK in BAECs pretreated with vehicle or AFC (50 μM) or transfected with vector, PTmut or N-ter for 48 h, then simvastatin (10 μM) for 0–120 min. Data are mean ± SEM from 5 independent experiments. *P < 0.05 vs. vehicle- or vector-treated cells, #P < 0.05 vs. simvastatin-treated alone cells.
Simvastatin increases the formation of an sEH–eNOS–Akt–AMPK complex
The physical interaction of eNOS with intracellular proteins such as Akt and AMPK is a key molecular mechanism in regulating eNOS activity14,34. However, whether sEH could interact directly with eNOS, Akt or AMPK remains elusive. We next investigated whether sEH phosphatase is involved in the simvastatin-induced association of eNOS, Akt and AMPK. IP assay revealed that simvastatin time-dependently increased the interaction between sEH and eNOS, Akt and AMPK (Fig. 5a–c). In addition, mammalian two-hybrid assay findings supported this notion that sEH can bind directly with eNOS, Akt and AMPK (Fig. 5d–f). sEH phosphatase might participate in simvastatin-activated Akt–AMPK–eNOS signaling by protein–protein interaction in ECs.
Figure 5. Simvastatin promotes the formation of an sEH–eNOS–Akt–AMPK complex.

BAECs were treated with simvastatin (10 μM) for 0–120 min. Cellular lysates was immunoprecipitated (IP) with anti-sEH Ab and then immunoblotted (IB) with (a) anti-eNOS, (b) anti-Akt, (c) anti-AMPK or anti-sEH Abs. Secreted human alkaline phosphatase (SEAP) activity in culture media from BAECs co-transfected with pM-sEH and (d) pVP-eNOS, (e) pVP-Akt or (f) pVP-AMPK for 48 h, then indicated concentrations of simvaststin (2.5, 5, 10 μM) for another 24 h. pM3-VP16–transfected cells were a positive control. Data are mean ± SEM from 5 independent experiments. *P < 0.05 vs. vehicle-treated group.
EGFR–c-Src signaling is required for simvastatin-increased sEH Tyr phosphorylation and formation of an eNOS–Akt–AMPK complex
EGFR–c-Src signaling was predicted to be a pathway mediating sEH Tyr phosphorylation by use of the web tools NetPhos 2.0 and NetPhosK 1.0 (http://www.cbs.dtu.dk/services/NetPhos/)35. In addition, c-Src kinase plays a key role in eNOS activation36. Therefore, we examined whether the simvastatin-induced formation of sEH–Akt–AMPK–eNOS complex requires Tyr phosphorylation of sEH by EGFR–c-Src signaling. Treatment with simvastatin time-dependently increased the phosphorylation of EGFR and c-Src (Fig. 6a,b). Pretreatment with AG1478, an inhibitor of EGFR signaling, abolished the simvastatin-increased phosphorylation of c-Src (Fig. 6c). Mammalian two-hybrid assay further revealed that simvastatin increased the interaction of EGFR and c-Src, which was prevented by treatment with AG1478, PP1 (a c-Src inhibitor) or K298M (a c-Src dominant-negative mutant) (Fig. 6d). Furthermore, simvastatin induced transient Tyr phosphorylation of sEH but not Ser or Thr phosphorylation of sEH within 7.5 to 30 min after treatment, which returned to the basal level at 60 min after treatment (Fig. 7a–c).
Figure 6. Activation of epidermal growth factor receptor (EGFR)–c-Src signaling is required for simvastatin-induced increase in sEH phosphatase activity.

BAECs were treated with simvastatin (10 μM) for 0–120 min. Cellular lysates were IB with (a) anti-phospho-epidermal growth factor receptor (p-EGFR) and EGFR and (b) anti-phospho-c-Src and c-Src. (c) Western blot analysis of anti-phosphor-c-Src and c-Src levels in BAECs pretreated with AG1478 (1 μM, a EGFR inhibitor) for 2 h, then simvastatin (10 μM) for 0–120 min. (d) Phosphatase activity (PT) assay of cells pretreated with AG1478 (1 μM) or protein phosphatase 1 (PP1; 10 μM) or transfected with K298M (a c-Src dominant-negative mutant) for 48 h, then simvastatin for 30 min. Data are mean ± SEM from 5 independent experiments. *P < 0.05 vs. vehicle- or vector-treated cells, #P < 0.05 vs. simvastatin-treated alone cells.
Figure 7. Simvastatin induces tyrosine phosphorylation of sEH.

BAECs were treated with simvastatin (10 μM) for 0–120 min. Cellular lysates were IP with anti-sEH Ab, then IB with Ab for anti-sEH and (a) anti-Tyr phosphorylation, (b) anti-Ser phosphorylation or (c) anti-Thr phosphorylation. Data are mean ± SEM from 5 independent experiments. *P < 0.05 vs. vehicle-treated group.
We further evaluated whether c-Src is involved in simvastatin-altered Tyr phosphorylation of sEH and the formation of an sEH–Akt–AMPK–eNOS complex. Simvastatin-induced Tyr phosphorylation of sEH was abrogated by PP1 or K298M treatment (Fig. 8a,b). In addition, blocking the activation of c-Src signaling with PP1 or K298M abolished the simvastatin-induced association of sEH with eNOS (Fig. 8c,d), Akt (Fig. 8e,f) and AMPK (Fig. 8g,h). Thus, EGFR–c-Src signaling is essential for the negative regulatory mechanism of simvastatin on eNOS activation (Fig. 9).
Figure 8. c-Src signaling is essential for simvastatin-induced sEH tyrosine phosphorylation and the formation of an sEH–Akt–AMPK–eNOS complex.

BAECs were pretreated with or without PP1 (10 μM) for 2 h (a,c,e,g) or transfected with K298M for 48 h (b,d,f,h), then incubated with simvastatin (10 μM) for 15 min (a,b,e–g) or 30 min (c,d). Cellular lysates were IP with anti-sEH Ab and then IB with Ab for anti-sEH, (a,b) anti-Tyr phosphorylation, (c,d) eNOS, (e,f) Akt or (g,h) AMPK. Data are mean ± SEM from 5 independent experiments. *P < 0.05 vs. vehicle- or vector-treated group, #P < 0.05 vs. simvastatin-treated alone group.
Figure 9. Illustration of proposed mechanism of sEH phosphatase regulating simvastatin-induced eNOS activation and NO production in ECs.
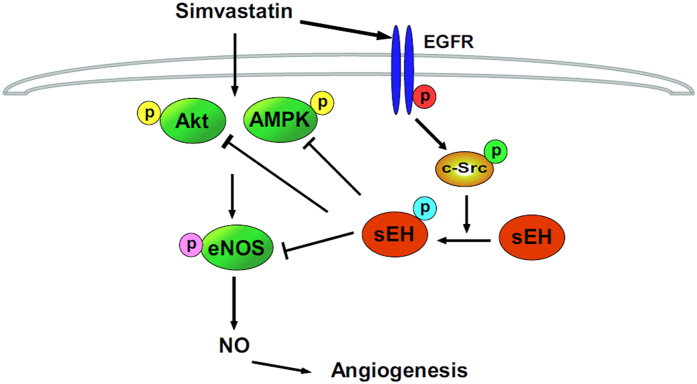
Simvastatin may activate EGFR–c-Src signaling to increase sEH Tyr phosphorylation, thus activating sEH phosphatase. This key event may negatively regulate simvastatin-activated Akt-AMPK-eNOS signaling and NO production in ECs.
Discussion
In this study, we characterized a new role for sEH phosphatase in simvastatin-mediated eNOS activation and NO production. Exposing ECs to simvastatin increased sEH phosphatase activity, which, in turn, negatively modulated the simvastatin-induced phosphorylation of Akt, AMPK and eNOS, thus leading to maintained NO production. Treatment with pharmacological inhibitors or genetic manipulation further revealed that the simvastatin-increased sEH phosphatase activity depended on activation of EGFR–c-Src signaling. Along with eNOS activation, exposing ECs to simvastatin rapidly induced Tyr phosphorylation of sEH and profoundly increased the physical interaction of sEH with Akt, AMPK and eNOS. Notably, the increased Tyr phosphorylation of sEH and sEH–Akt–AMPK–eNOS interaction was inhibited by the c-Src kinase antagonist PP1 or c-Src kinase dominant-negative mutant K298M. These results agreed with our previous findings that c-Src–sEH phosphatase signaling negatively regulates VEGF-activated eNOS37. Thus, the negative regulation of eNOS activity by sEH phosphatase may not be limited to simvastatin stimulation alone. The phosphatase activity of sEH may be pivotal in regulating eNOS activity and the physiological function of ECs.
Besides sEH phosphatase, protein serine/threonine phosphatase 1 (PP1), PP2A and PP2B are also involved in regulation of eNOS activity14. Therefore, we assessed whether PP1, PP2A or PP2B contributes to simvastatin-induced eNOS phosphorylation. Simvastatin did not affect the activity of PP1, PP2A or PP2B (data not shown). In addition, pretreatment with a PP1-, PP2A- or PP2B-specific inhibitor did not alter the status of eNOS phosphorylation elicited by simvastatin (data not shown). Thus, sEH phosphatase indeed has a unique role in regulating simvastatin-induced eNOS activation. The phosphatase activity was lower for sEH N-ter than full-length sEH in cell lysates. This result agrees with Nelson et al., who found lower phosphatase activity with purified N-ter than WT sEH in vitro38. Under physiologic conditions, sEH is a homodimer with two domains, the C-terminal epoxide hydrolase and N-terminal phosphatase activity domain16. The entire homodimer structure of sEH is important for maintaining sEH enzymatic activity32. Therefore, loss of the hydrolase domain of sEH may be attributed to decreased phosphatase activity of sEH N-ter.
sEH phosphatase is thought to have isoprenoid mono- and pyrophosphate activity and prefer hydrolysis phosphates of lipophilic compounds in cells39,40. Indeed, lysophosphatidic acids are biological substrates for sEH phosphatase31,32. Therefore, sEH phosphatase presumably is a key regulator for lipid signaling-related cellular function. However, the biological significance of sEH phosphatase activity and the detailed signaling pathways underlying its regulation of eNOS activity remain unclear. In our previous and current studies, we targeted sEH phosphatase as a regulator of eNOS activation. Inhibiting the phosphatase activity of sEH augmented VEGF-37 or simvastatin-induced eNOS phosphorylation and NO production, whereas overexpression of sEH phosphatase abolished these effects. sEH may be a crucial negative modulator of eNOS phosphorylation and NO production.
The biological role of sEH hydrolase activity and its substrate EETs in the functioning of the cardiovascular system and the development of cardiovascular diseases is well established17,18,19,20. Inhibition of sEH hydrolase activity by pharmacological antagonists or genetic deletion of sEH increases EET levels in blood, thereby alleviating the progression of angiotensin II-mediated hypertension, atherosclerosis and cardiac hypertrophy in experimental animal models24,41,42. Therefore, inhibition of sEH hydrolase by pharmacological antagonism may be a therapeutic strategy for treating cardiovascular diseases. However, sEH-knockout mice lack both phosphatase and hydrolase domains, so determining whether sEH phosphatase contributes to the beneficial effects with deletion of sEH in vivo is difficult. Indeed, several lines of evidence suggest that genetic ablation of sEH exacerbates the progression of many diseases, albeit with an unclear mechanism43,44. Further investigation delineating the exact role and molecular mechanisms of sEH phosphatase in the regulation of cardiovascular function and pathogenesis of cardiovascular diseases is warranted.
In addition to the kinase- and phosphatase-dependent regulation of eNOS activity, physical interaction of eNOS with intracellular proteins is a crucial mechanism in regulating eNOS activation with stimuli14,34. In this study, simvastatin increased the association of sEH and Akt, AMPK and eNOS in ECs and increased the Tyr phosphorylation of sEH. More importantly, c-Src kinase, a key regulator in eNOS activation by simvastatin or other agonists in ECs36,37, was the upstream signaling molecule for Tyr phosphorylation of sEH and the formation of the sEH–Akt–AMPK–eNOS complex. Our findings indicate that both kinase-dependent regulation and protein–protein interaction are involved in the simvastatin-mediated Tyr phosphorylation of sEH and interaction with kinases and eNOS as well as NO bioavailability. However, the exact site(s) of Tyr phosphorylation of sEH and the underlying molecular mechanisms in the cross-talk among c-Src, sEH Akt, AMPK and eNOS remain to be uncovered.
ECs play a crucial role in regulating vascular homeostasis, including blood cells and nutritional trafficking to tissues, coagulation and vascular tone9,10. Endothelial dysfunction is known to be a key event in the development of atherosclerosis. Statins are known to improve endothelial dysfunction by increasing eNOS activation and thus reduce cardiovascular morbidity and mortality in patients with coronary heart disease5,6,7,8. In this study, our findings suggest sEH phosphatase serves as a negative regulator in the simvastatin-induced eNOS activation. Inhibition of sEH phosphatase activity can sustain the simvastatin-induced phosphorylation of Akt, AMPK and eNOS, all of which lead to further augmented NO production. On the other hand, EETs have been reported to have protective effects on endothelial dysfunction and cardiovascular diseases18,19. Presumably, these findings imply that statin-sEH phosphatase inhibitor or sEH hydrolase-phosphatase inhibitor combination therapy might be of high therapeutic values in treating eNOS-related cardiovascular diseases.
In summary, we provide new evidence that simvastatin activates EGFR–c-Src signaling, which increases Tyr phosphorylation of sEH and sEH–Akt–AMPK–eNOS interaction. These events may work in concert to modulate simvastatin-induced eNOS activation and NO production in ECs. The molecular mechanisms we reveal may provide new information for better understanding the regulation of eNOS activation and for suggesting novel pharmacological targets for treating eNOS-related cardiovascular diseases.
Methods
Reagents
Rabbit antibody (Ab) for phosphor-eNOS at Ser1177, phosphor-Akt at Ser473, phosphor-AMPK at Thr172, phospho-epidermal growth factor receptor (EGFR) at Tyr845, phosphor-c-Src at Tyr416 and mouse Ab for phosphor-Ser and phosphor-Thr were from Cell Signaling Technology (Beverly, MA, USA). Rabbit Abs for eNOS, sEH, Akt, and AMPK; mouse Ab for EGFR; and protein A/G PLUS-Agarose were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Mouse Ab for phosphor-Tyr was from BD Biosciences (San Jose, CA, USA) and for α-tubulin, simvastatin, PP1, 4-nitrophenyl phosphate, ebselen and Griess reagent were from Sigma-Aldrich (St Louis, MO, USA). ECL Cell Attachment Matrix was from Millipore (Bedford, MA, USA). N-acetyl-S-farnesyl-L-cysteine (AFC) was from Enzo Life Sciences (Farmingdale, NY, USA). 12-(3-Adamantan-1-yl-ureido)-dodecanoic acid (AUDA) and AG1478 were from Cayman (Ann Arbor, MI, USA).
Cell culture
Bovine aortic endothelial cells (BAECs) were purchased from Cell Applications (San Diego, CA, USA) and cultured in Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum, 100 unit/ml penicillin and 100 mg/ml streptomycin (HyClone, Logan, UT, USA) in a humidified 95% air and 5% CO2 incubator at 37 °C.
Phosphatase activity assay
Treated BAECs were collected in phosphate buffered saline (PBS), which was sonicated, and supernatant was collected by brief centrifugation. The cell lysate supernatant was added to 4-nitrophenyl phosphate to 2 mM and incubated in 37 °C for 1 h. The yellow color product was detected by OD 405 nm to determine phosphatase activity.
Detection of nitrite production
Griess reagent was used to evaluate accumulated nitrite, the stable breakdown product of NO, in culture media. After incubation of an equal volume of Griess reagent with medium from treated cells at room temperature for 15 min, the solution was evaluated by SP-8001 UV/VIS spectrophotometry (Taipei) to detect azo dye production at OD 540 nm. Sodium nitrite was used as a standard.
Plasmid construction
Coding regions for the sEH N-terminal phosphatase domain (N-ter) and C-terminal hydrolase domain (C-ter) were amplified from mouse cDNA by PCR with the primers 5′-TTA CGC GTG CGC TGC GTG TAG CCG-3′ and 5′-GGT CTA GAC TAC CCT GTG ACC TTC TCC A-3′ for N-ter and 5′-TTA CGC GTG TCA GCC ATG GAT ATG TGA C-3′ and 5′-GGT CTA GAC TAA ATC TTG GAG GTC ACT G-3′ for the C-ter. The protocol was 2 min at 94 °C, then 15 sec at 94 °C, 30 sec at 58 °C and 2 min at 72 °C for 35 cycles. PCR products were digested with MluI and XbaI for cloning into pCMV5N-Flag vector. The sEH full-length cDNA and sEH with mutant hydrolase or phosphatase domain {WT, Ehmut, or Ptmut, respectively; kindly provided by Dr. S. Imaoka (Kwansei Gakuin University, Japan)31 were amplified by PCR (2 min at 94 °C, then 15 sec at 94 °C, 30 sec at 61 °C and 2 min at 72 °C for 35 cycles) with the primers 5′-TTA CGC GTA TGA CGC TGC GCG CGG-3′ and 5′-GGT CTA GAC TAC ATC TTT GAG ACC ACC G -3′ for the WT, EHmut or PTmut plasmid. MluI and XbaI-digested PCR products were cloned into the pCMV5 N-Flag vector. The c-Src mutant plasmid, K298M, was kindly provided by Dr. KL Guan (University of Michigan, USA).
Protein extraction and immunoblot analysis
BAECs were lyzed with SDS lysis buffer (1% Triton, 0.1% SDS, 0.2% sodium azide, 0.5% sodium deoxycholate and proteinase inhibitors [1 mM phenylmethylsulfonyl fluoride (PMSF), 10 μg/ml aprotinin, 1 μg/ml leupeptin], then centrifuged at 12,000 g for 5 min and supernatant was collected. Aliquots (50 μg) of cell lysates were separated by 8% SDS-PAGE and transblotted onto Immobilon-P membrane (Millipore, Bedford, MA, USA). After being blocked with 5% skim milk in Tween/PBS, blots were incubated with primary Abs, then with horseradish peroxidase-conjugated secondary Abs. The protein bands were detected by use of an enhanced chemiluminescence kit and quantified by use of ImageQuant 5.2 (Healthcare Bio-Sciences, Philadelphia, PA).
Immunoprecipitation (IP)
BAECs were lyzed with IP lysis buffer (50 mM Tris, pH 7.5, 5 mM EDTA, 300 mM NaCl, 1% Triton X-100, 1 mM PMSF, 10 μg/ml aprotinin, 1 μg/ml leupeptin, Tyr phosphatase cocktail I and Ser/Thr phosphatase cocktail II). After brief sonication on ice, cellular debris was removed by centrifugation at 10,000 g for 10 min. Aliquots (1000 mg) were incubated with anti-sEH Ab overnight at 4 °C, then 20 μL Protein A/G PLUS-Agarose was added for 2 h in 4 °C. Immune complexes were collected by centrifugation and washed 3 times with ice-cold PBS. The supernatant was discarded and the pellet was resuspended in SDS lysis buffer, then boiled with 1X SDS loading dye for 5 min. Protein was separated by SDS-PAGE and transferred to PVDF membranes and incubated with various Abs.
Mammalian two-hybrid assay
Full-length sEH was obtained from mouse cDNA and sub-cloned into the pM vector (Clontech, CA, USA) with MluI and XbaI cutting sites. eNOS, Akt and AMPK cDNA were obtained from human cDNA and sub-cloned into the pVP16 vector (Clontech, CA, USA) with HindIII, EcoRI and EcoRI cutting sites, respectively, and confirmed by sequencing. Secreted human alkaline phosphatase (pG5SEAP) and plasmids were co-transfected into BAECs by use of Lipofectamin 2000 for 48 h, then treated with or without simvastatin (2.5, 5, 10 μM) for an additional 24 h. Culture media was collected and underwent chemiluminescence assays by use of GreatEscAPe SEAP chemiluminescence detection kits (Clontech, CA).
Tube formation assay
Tube formation measurement was performed as described45. Briefly, ECL Cell Attachment Matrix was added to 24-well plates for overnight incubation at 37 °C. Cells were seeded onto the layer of matrix gel with indicated treatments for 4 h. Tube formation was evaluated by microscopy and quantified by counting the number of branch points in 5 random areas.
Statistical analysis
Data are shown as mean ± SEM from 5 independent experiments. Mann-Whitney test was used to compare two independent groups and Kruskal-Wallis analysis was used with Bonferroni post-hoc correction to account for multiple testing. Statistical analysis involved use of SPSS v8.0 (SPSS Inc., Chicago, IL). P < 0.05 was considered statistically significant.
Additional Information
How to cite this article: Hou, H.-H. et al. Role of phosphatase activity of soluble epoxide hydrolase in regulating simvastatin-activated endothelial nitric oxide synthase. Sci. Rep. 5, 13524; doi: 10.1038/srep13524 (2015).
Supplementary Material
Acknowledgments
The authors thank Laura Smales (BioMedEditing, Toronto, Canada) for help in language editing. This study was supported by grants from the National Science Council (101-2628-B-010-001-MY2, 102-2628-B-010-001-MY3, 103-2320-B-010-040-MY3 and 103-2811-B-010-029), VGHUST Joint Research Program, Tsou’s Foundation (VGHUST104-G7-5-2), the Department of Health of Taipei City Government (10401-62-016) and Ministry of Education, Aim for the Top University Plan, Taiwan. We thank the National Yang-Ming University-VGH Genome Research Center for technical support.
Footnotes
Author Contributions H.-H.H., Y.-J.L. and S.-H.H. performed experiments and analyzed the data. S.-K.S. and T.-S.L. designed the experiments and T.-S.L. wrote the paper.
References
- Illingworth D. R. & Bacon S. Hypolipidemic effects of HMG-CoA reductase inhibitors in patients with hypercholesterolemia. Am J. Cardiol. 60, 33G–42G (1987). [DOI] [PubMed] [Google Scholar]
- Mölgaard J., von Schenck H. & Olsson A. G. Effects of simvastatin on plasma lipid, lipoprotein and apolipoprotein concentrations in hypercholesterolaemia. Eur. Heart J. 9, 541–551 (1988). [DOI] [PubMed] [Google Scholar]
- Frey T. & De Maio A. Increased expression of CD14 in macrophages after inhibition of the cholesterol biosynthetic pathway by lovastatin. Mol. Med. 13, 592–604 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng X. et al. Statins induce the accumulation of regulatory T cells in atherosclerotic plaque. Mol. Med. 18, 598–605 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotto A. M. Jr & Farmer J. A. Pleiotropic effects of statins: do they matter? Curr. Opin. Lipidol. 12, 391–394 (2001). [DOI] [PubMed] [Google Scholar]
- Ishida K., Matsumoto T., Taguchi K., Kamata K. & Kobayashi T. Pravastatin normalizes endothelium-derived contracting factor-mediated response via suppression of Rho-kinase signalling in mesenteric artery from aged type 2 diabetic rat. Acta. Physiol (Oxf). 205, 255–265 (2012). [DOI] [PubMed] [Google Scholar]
- Jones S. P. & Lefer D. J. Cardioprotective actions of acute HMG-CoA reductase inhibition in the setting of myocardial infarction. Acta. Physiol. Scand. 173, 139–143 (2001). [DOI] [PubMed] [Google Scholar]
- Schroll S. et al. Effects of simvastatin on pulmonary fibrosis, pulmonary hypertension and exercise capacity in bleomycin-treated rats. Acta. Physiol (Oxf). 208, 191–201 (2013). [DOI] [PubMed] [Google Scholar]
- Goligorsky M. S., Noiri E., Tsukahara H., Budzikowski A. S. & Li H. A pivotal role of nitric oxide in endothelial cell dysfunction. Acta. Physiol. Scand. 168, 33–40 (2000). [DOI] [PubMed] [Google Scholar]
- Kone B. C. Protein-protein interactions controlling nitric oxide synthases. Acta Physiol Scand. 168, 27–31 (2000). [DOI] [PubMed] [Google Scholar]
- Su K. H. et al. β Common receptor integrates the erythropoietin signaling in activation of endothelial nitric oxide synthase. J. Cell Physiol. 226, 3330–3339 (2011). [DOI] [PubMed] [Google Scholar]
- Su K. H. et al. AMP-activated protein kinase mediates erythropoietin- induced activation of endothelial nitric oxide synthase. J. Cell Physiol. 227, 3053–3062 (2012). [DOI] [PubMed] [Google Scholar]
- Su K. H. et al. Valsartan regulates the interaction of angiotensin II type 1 receptor and endothelial nitric oxide synthase via Src/PI3K/Akt signaling. Cardiovasc. Res. 82, 468–475 (2009). [DOI] [PubMed] [Google Scholar]
- Ching L. C. et al. Molecular mechanisms of activation of endothelial nitric oxide synthase mediated by transient receptor potential vanilloid type 1. Cardiovasc. Res. 91, 492–501 (2011). [DOI] [PubMed] [Google Scholar]
- Ching L. C. et al. Activation of transient receptor potential vanilloid 1 decreases endothelial nitric oxide synthase phosphorylation at Thr497 by protein phosphatase 2B-dependent dephosphorylation of protein kinase C. Acta. Physiol. (Oxf). 209, 124–135 (2013). [DOI] [PubMed] [Google Scholar]
- Newman J. W., Morisseau C., Harris T. R. & Hammock B. D. The soluble epoxide hydrolase encoded by EPXH2 is a bifunctional enzyme with novel lipid phosphate phosphatase activity. Proc. Natl. Acad. Sci. USA 100, 1558–1563 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu T. et al. Dihydroxyeicosatrienoic acids are potent activators of Ca(2+)-activated K(+) channels in isolated rat coronary arterial myocytes. J Physiol. 534, 651–667 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oltman C. L., Weintraub N. L., VanRollins M. & Dellsperger K. C. Epoxyeicosatrienoic acids and dihydroxyeicosatrienoic acids are potent vasodilators in the canine coronary microcirculation. Circ Res. 83, 932–939 (1998). [DOI] [PubMed] [Google Scholar]
- Fang X. et al. Human coronary endothelial cells convert 14,15-EET to a biologically active chain-shortened epoxide. Am J Physiol Heart Circ Physiol. 283, H2306–2314 (2002). [DOI] [PubMed] [Google Scholar]
- Levick S. P., Loch D. C., Taylor S. M. & Janicki J. S. Arachidonic acid metabolism as a potential mediator of cardiac fibrosis associated with inflammation. J. Immunol. 178, 641–646 (2007). [DOI] [PubMed] [Google Scholar]
- Kim J., Imig J. D., Yang J., Hammock B. D. & Padanilam B. J. Inhibition of soluble epoxide hydrolase prevents renal interstitial fibrosis and inflammation. Am J. Physiol. Renal Physiol. 307, F971–F980 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. et al. Pharmacological inhibition of soluble epoxide hydrolase prevents renal interstitial fibrogenesis in obstructive nephropathy. Am J. Physiol. Renal Physiol. 308, F131–F139 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang C. W. et al. Genetic deletion of soluble epoxide hydrolase attenuates inflammation and fibrosis in experimental obstructive nephropathy. Mediators Inflamm. 2015, 693260 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ai D. et al. Soluble epoxide hydrolase plays an essential role in angiotensin II-induced cardiac hypertrophy. Proc. Natl. Acad. Sci. USA 106, 564–569 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przybyla-Zawislak B. D. et al. Polymorphisms in human soluble epoxide hydrolase. Mol. Pharmacol. 64, 482–490 (2003). [DOI] [PubMed] [Google Scholar]
- Srivastava P. K., Sharma V. K., Kalonia D. S. & Grant D. F. Polymorphisms in human soluble epoxide hydrolase: effects on enzyme activity, enzyme stability, and quaternary structure. Arch. Biochem. Biophys. 427, 164–169 (2004). [DOI] [PubMed] [Google Scholar]
- Fornage M. et al. Polymorphism of the soluble epoxide hydrolase is associated with coronary artery calcification in African-American subjects: The Coronary Artery Risk Development in Young Adults (CARDIA) study. Circulation 109, 335–339 (2004). [DOI] [PubMed] [Google Scholar]
- Ohtoshi K. et al. Association of soluble epoxide hydrolase gene polymorphism with insulin resistance in type 2 diabetic patients. Biochem. Biophys. Res. Commun. 331, 347–350 (2005). [DOI] [PubMed] [Google Scholar]
- EnayetAllah A. E. et al. Opposite regulation of cholesterol levels by the phosphatase and hydrolase domains of soluble epoxide hydrolase. J. Biol. Chem. 283, 36592–36598 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oguro A., Sakamoto K., Suzuki S. & Imaoka S. Contribution of hydrolase and phosphatase domains in soluble epoxide hydrolase to vascular endothelial growth factor expression and cell growth. Biol. Pharm. Bull. 32, 1962–1967 (2009). [DOI] [PubMed] [Google Scholar]
- Morisseau C. et al. Role of soluble epoxide hydrolase phosphatase activity in the metabolism of lysophosphatidic acids. Biochem. Biophys. Res. Commun. 419, 796–800 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oguro A. & Imaoka S. Lysophosphatidic acids are new substrates for the phosphatase domain of soluble epoxide hydrolase. J. Lipid Res. 53, 505–512 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su K. H. et al. The essential role of transient receptor potential vanilloid 1 in simvastatin-induced activation of endothelial nitric oxide synthase and angiogenesis. Acta. Physiol (Oxf). 212, 191–204 (2014). [DOI] [PubMed] [Google Scholar]
- Sun W. et al. Statins activate AMP-activated protein kinase in vitro and in vivo. Circulation 114, 2655–2662 (2006). [DOI] [PubMed] [Google Scholar]
- Liu T. & Altman R. B. Prediction of calcium-binding sites by combining loop-modeling with machine learning. BMC Struct. Biol. 9, 72 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su K. H. et al. Valsartan regulates the interaction of angiotensin II type 1 receptor and endothelial nitric oxide synthase via Src/PI3K/Akt signaling. Cardiovasc Res. 82, 468–475 (2009). [DOI] [PubMed] [Google Scholar]
- Hou H. H. et al. N-terminal domain of soluble epoxide hydrolase negatively regulates the VEGF-mediated activation of endothelial nitric oxide synthase. Cardiovasc. Res. 93, 120–129 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson J. W., Subrahmanyan R. M., Summers S. A., Xiao X. & Alkayed N. J. Soluble epoxide hydrolase dimerization is required for hydrolase activity. J. Biol. Chem. 288, 7697–7703 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enayetallah A. E. & Grant D. F. Effects of human soluble epoxide hydrolase polymorphisms on isoprenoid phosphate hydrolysis. Biochem. Biophys. Res. Commun. 341, 254–260 (2006). [DOI] [PubMed] [Google Scholar]
- Hahn S. et al. Complementary screening techniques yielded fragments that inhibit the phosphatase activity of soluble epoxide hydrolase. Chem Med Chem. 6, 2146–2149 (2011). [DOI] [PubMed] [Google Scholar]
- Ulu A. et al. An omega-3 epoxide of docosahexaenoic acid lowers blood pressure in angiotensin-II-dependent hypertension. J. Cardiovasc. Pharmacol. 64, 87–99 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris T. R. & Hammock B. D. Soluble epoxide hydrolase: gene structure, expression and deletion. Gene 526, 61–74 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L. et al. Opposite effects of gene deficiency and pharmacological inhibition of soluble epoxide hydrolase on cardiac fibrosis. PLoS One 9, e94092 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchens M. P. et al. Soluble epoxide hydrolase gene deletion reduces survival after cardiac arrest and cardiopulmonary resuscitation. Resuscitation 76, 89–94 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ching L. C. et al. Implication of AMP-activated protein kinase in transient receptor potential vanilloid type 1-mediated activation of endothelial nitric oxide synthase. Mol. Med. 18, 805–815 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.