

Abstract
Ruminal microbial fermentation plays an essential role in host nutrition, and as a result, the rumen microbiota have been a major focus of research examining bovine feed efficiency. Microbial communities within other sections of the gastrointestinal tract may also be important with regard to feed efficiency, since it is critical to the health and nutrition of the host. The objective of this study was to characterize the microbial communities of the colon among steers differing in feed efficiency. Individual feed intake (FI) and body weight (BW) gain were determined from animals fed the same ration, within two contemporary groups of steers. Four steers from each contemporary group within each Cartesian quadrant were sampled (n = 16/group) from the bivariate distribution of average daily BW gain and average daily FI. Bacterial 16S rRNA gene amplicons were sequenced from the colon content using next-generation sequencing technology. Within the colon content, UniFrac principal coordinate analyses did not detect any separation of microbial communities, and bacterial diversity or richness did not differ between efficiency groups. Relative abundances of microbial populations and operational taxonomic units did reveal significant differences between efficiency groups. The phylum Firmicutes accounted for up to 70% of the populations within all samples, and families Ruminococcaceae and Clostridiaceae were highly abundant. Significant population shifts in taxa were detected, including the families Ruminococcaceae, Lachnospiraceae, and Sphingomonadaceae, and the genera Butyrivibrio, Pseudobutyrivibrio, Prevotella, Faecalibacterium and Oscillospira. This study suggests the association of the colon microbial communities as a factor influencing feed efficiency at the 16S level.
Keywords: 16S rRNA, Colon, Feed efficiency, Microbiome, Operational taxonomic units
Background
Feed costs remain the largest variable cost in beef production (Arthur et al. 2005). Although it has been estimated that a 10% improvement in performance (gain) would increase profitability by only 18%, increasing the efficiency of growth of feedlot cattle by 10% could improve profitability by upwards of 43% (Fox et al. 2001). Optimization of feed efficiency in beef cattle is also important for social and environmental issues, due to increasing concerns over methane emissions of cattle, decreasing acreage for crop production (Wirsenius et al. 2010) increasing world population, and increasing divergence of traditional livestock feedstuffs for production of biofuels (Galyean et al. 2011). Genetic selection promises a lasting approach to optimizing the food produced and feed consumed per animal, but traditional selection requires costly phenotyping of individuals. A combination of traditional nutritional and management approaches, in tandem with genetic improvement of feed efficiency in beef cattle, represents a potential path to sustainably reduce feed resources required to produce beef, as well as contribute towards the environmental sustainability of beef production.
The study and improvement of feed efficiency in beef cattle has primarily focused on host-related genetic improvement technologies; however, host genetic contributions to feed efficiency in beef cattle have been difficult to identify (Saatchi et al. 2014; Sherman et al. 2010; Abo-Ismail et al. 2014). The gastrointestinal tract (GIT) contains complex and dynamic microbial communities, which have long been regarded as essential in structure, function, and overall health of the host (Fujimura et al. 2010). Among other contributing factors to beef cattle nutrition, such as host genetics, diet, and management, the microbial populations within the GIT must also be examined to comprehensively evaluate their effect on feed efficiency. Furthermore, microbial-associated feed efficiency studies have largely concentrated on ruminal associations, due to the role of the rumen in nutrient supply to the host (Kim et al. 2011; Hernandez-Sanabria et al. 2012; Jami et al. 2014; McCann et al. 2014). Myer et al. (2015) demonstrated associations between specific microbial populations and feed efficiency phenotypes in beef cattle. However, bovine ruminal microbial communities are distinct from those of the colon and feces (de Oliveira et al. 2013), therefore a full understanding of the relationship between the microbial populations along the GIT and feed efficiency, gain, and intake, requires characterization of sites distal from the rumen.
High-throughput sequencing technologies have aided researchers in the examination of microbial communities, enabling the study of the structure and function of microbial populations at great depth, and revealing significant differences within microbial communities that would not otherwise be detectable using culture-based methodologies. High-throughput molecular technologies are able to resolve complex microbial communities at finer resolutions, providing the opportunity to identify the relationships between microbial community structure and feed efficiency phenotypes.
The objective of this study was to examine the association between microbial community structure and feed efficiency phenotypes within the lower GIT, by determining the microbial community of the colon from steers differing in feed efficiency using deep 16S rRNA-based community profiling. We hypothesize that variation in the microbial populations within the colon impact or reflect variation in host feed efficiency. This study aimed to characterize the bacterial community of the cattle colon among steers differing in feed intake and growth, in order to assess the association of the microbial community profile with variation in bovine feed efficiency.
Results
Diversity of colon bacterial communities
The colon contents sampled from the 32 steers grouped into 4 feed efficiency phenotypes, produced 20,593,775 sequence reads after filtering for quality and removing apparent chimeras, for an average of 682,061 reads per sample (range 110,537–2,668,201). The average read length was 500 bp. OTUs were defined as a bin of sequence reads sharing ≥97% nucleotide sequence identity. From the cleaned sequences, a total of 323,433 OTUs were detected with an average of 10,107 ± 2,896 OTUs per individual sample. The average number of OTUs detected from each Cartesian quadrant ranged from 7,247 to 15,130 OTUs. Singletons accounted for approximately 38% of the OTUs detected within the colon content samples. The dataset reported coverage ranging from 95.82 to 99.16%, using Good’s coverage estimator as a metric for determining coverage. Bacterial diversity, as determined by Shannon diversity index, ranged from 6.84 to 8.93.
The individual samples were normalized in order to accurately compare among feed efficiency phenotype groups. The OTU table within each sample was rarefied to 100,000 sequence reads, based upon the sample rarefaction curves. The normalized samples were then used for analysis using the sample means within each quadrant. The normalized sequence reads were analyzed via alpha-diversity metrics of bacterial diversity (Shannon Index), richness (Chao-1), evenness (equitability of representation of taxa), and coverage (Good’s coverage estimator; Table 1). The number of OTUs detected within each feed efficiency group did not differ (P > 0.05), averaging 6,025 ± 1,225 OTUs per group. The Chao-1 richness metric also did not differ, estimating 10,051 ± 2,334 OTUs per group. Bacterial diversity did not indicate any differences between feed efficiency groups (P > 0.05), with a range of 7.85–8.27. Evenness, as estimated by equitability (1 = complete equitability), did not differ between groups (P > 0.05), averaging 0.59 ± 0.03 per group. Coverage was adequate, ranging from 96.53 to 97.13%, representative of the ADGlow–ADFIlow and ADGhigh–ADFIhigh groups, respectively.
Table 1.
Diversity statistics among reads from grouped samples
| Feed efficiency group | Sampling type | No. of sequences | No. of OTUsa,b | Chao1b | Shannon diversity indexb | Equitabilityb | Good’s coverage (%) |
|---|---|---|---|---|---|---|---|
| ADGhigh–ADFIchigh | Subsampled readsd | 100,000 | 5,764 ± 878 | 9,638 ± 2,004 | 7.85 ± 0.52 | 0.58 ± 0.02 | 97.13 ± 0.59 |
| ADGhigh–ADFIclow | Subsampled readsd | 100,000 | 6,098 ± 1,028 | 9,874 ± 1,853 | 8.15 ± 0.39 | 0.60 ± 0.02 | 97.02 ± 0.60 |
| ADGlow–ADFIclow | Subsampled readsd | 100,000 | 6,714 ± 1,148 | 11,538 ± 2,333 | 8.27 ± 0.49 | 0.60 ± 0.03 | 96.53 ± 0.71 |
| ADGlow–ADFIchigh | Subsampled readsd | 100,000 | 6,063 ± 1,129 | 10,052 ± 2,003 | 7.92 ± 0.74 | 0.59 ± 0.02 | 96.99 ± 0.61 |
aOTUs represents operational taxonomic units.
bWithin a column, means for the individual subsamples did not differ (P < 0.05).
c n = 8 among groups.
dMeans among the groups were compared using ANOVA and the Tukey’s test.
Principal coordinates analyses (PCoA) were conducted to determine the phylogenetic relationship between microbial community samples in the study (Fig. 1). The PCoA utilizes the phylogeny-based UniFrac method, which uses the detected OTUs to determine if the data separate into any sample clusters. This beta-diversity metric takes into account the phylogenetic divergence between the OTUs, in order to determine differences within the colonic microbial communities from each feed efficiency group (Lozupone et al. 2007). The analysis did not indicate any separation into clusters in both the weighted (quantitative) and unweighted (qualitative) UniFrac distances of the colon microbial communities (Lozupone et al. 2011).
Fig. 1.
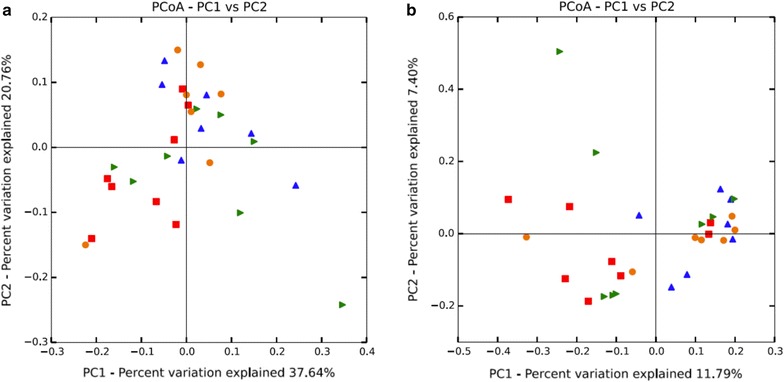
UniFrac principal coordinates analysis (PCoA) displaying correlations among the bacterial communities of the 4 feed efficiency groups. a Weighted PCoA analyzed from rarefied subsets of 100,000 sequences from each sample. b Unweighted PCoA analyzed from rarefied subsets of 100,000 sequences from each sample. n = 8, represented by differing symbols: ADGHigh–ADFIHigh orange circle, ADGHigh–ADFILow blue triangle, ADGLow–ADFILow red square, ADGLow–ADFIHigh green triangle.
Taxonomic and OTU composition
The 20,593,775 amplicon sequence reads were classified using the Greengenes 16S rRNA Gene Database (DeSantis et al. 2006) resulting in 20 phyla, 46 classes, 83 orders, 152 families, and 231 genera. The unassigned taxa accounted for approximately 1.48% of the reads. Firmicutes was the most abundant phylum within each feed efficiency group, ranging from 60 to 70% (in terms of percent of the total reads; Fig. 2a). These abundances are consistent with previous studies regarding the microbial abundances within the colon/fecal contents of cattle (de Oliveira et al. 2013; Malmuthuge and Griebel 2014). Other dominant phyla included Bacteroidetes (21–33 ± 3.4%), Spirochaetes (2.5–4.5 ± 1.3%), Tenericutes (1.2–1.9 ± 0.4%), Proteobacteria (0.3–0.5 ± 0.9%), Actinobacteria (0.23–0.33 ± 0.1%), and Fibrobacteres (0.02–0.29 ± 0.2%). No significant differences between the feed efficiency groups were observed within any of the phylum assignments. The remaining phyla accounted for <0.1% of the sequence reads, and no differences were observed between feed efficiency groups for the minor phyla abundances.
Fig. 2.
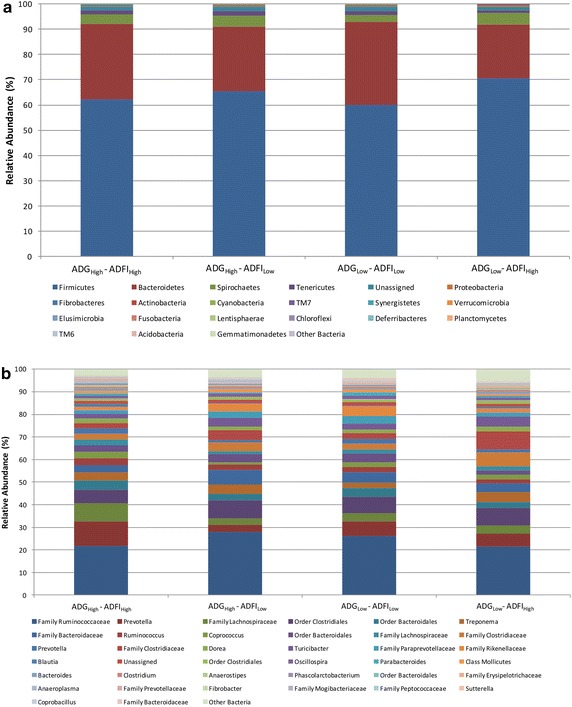
The taxonomic profiles for the relative phylum-level (a) and genus-level (b) abundance of each group classified by representation at ≥0.001% of total sequences. Taxonomic composition of the colon microbiota among the four groups was compared based on the relative abundance (reads of a taxon/total reads in a sample).
At the genus level, Prevotella (3.0–11.1 ± 2.7%), Ruminococcus (1.7–2.9 ± 0.4%), Coprococcus (1.0–2.9 ± 0.5%), Dorea (1.7–2.2 ± 0.3%), Turicibacter (1.9–4.4 ± 0.8%), Blautia (0.3–1.3 ± 0.2%), Oscillospira (1.1–1.6 ± 0.2%), and Parabacteroides (0.4–1.4 ± 0.3%) were present at greatest abundance, representing ≥1% of the total sequences (Fig. 2b). Of these genera, only Prevotella differed among the feed efficiency groups (P = 0.0259), with the ADGHigh–ADFIHigh group having the greatest abundance (Table 2). There were several taxa that were not classified to the genus level, but were present in abundances greater than 1% of the total sequences. These taxa included families Ruminococcaceae (21–28 ± 2.2%), Lachnospiraceae (2.9–7.9 ± 1.2%), Bacteroidaceae (3.3–6.4 ± 1.4%), Clostridiaceae (2.6–6.1 ± 1.1%), Paraprevotellaceae (1.6–3.4 ± 0.9%), and Rikenellaceae (1.5–4.2 ± 0.8%), as well as orders Clostridiales (6.0–7.9 ± 1.1%) and Bacteroidales (2.5–3.9 ± 0.6%). None of the aforementioned taxa at this level differed among the feed efficiency groups. Any remaining taxa were not listed and deemed non-detectable at abundances ≤ 0.001%.
Table 2.
Relative abundance of significant taxa in the four feed efficiency groups
| Classification | Percentage of sequencesa | SEM | P valueb | No. of steers with detectable taxonc | |||
|---|---|---|---|---|---|---|---|
| ADGHigh–ADFIHigh | ADGHigh–ADFILow | ADGLow–ADFILow | ADGLow–ADFIHigh | ||||
| Anaeroplasma | 0.1587 | 0.0591 | 0.0652 | 0.0854 | 0.0230 | 0.0222 | 30 |
| Cyanobacteria | 0.0187 | 0.0103 | 0.0113 | 0.0093 | 0.0024 | 0.0479 | 29 |
| Faecalibacterium | 0.1976 | 0.0276 | 0.0357 | 0.0916 | 0.0419 | 0.0361 | 23 |
| Family Barnesiellaceae | 2.49 × 10−4 | 0.0192 | 0.0216 | 9.93 × 10−4 | 0.0063 | 0.0471 | 24 |
| Family Mogibacteriaceae | 0.1013 | 0.1611 | 0.0762 | 0.2295 | 0.0358 | 0.0309 | 30 |
| Family Sphingomonadaceae | 0.0012 | 0.0011 | 2.38 × 10−4 | 1.24 × 10−4 | 3.11 × 10−4 | 0.0386 | 29 |
| Paludibacter | 1.23 × 10−4 | 4.86 × 10−5 | 0.0028 | 9.94 × 10−4 | 6.29 × 10−4 | 0.0226 | 27 |
| Prevotella | 7.9657 | 3.1272 | 3.9112 | 3.5961 | 1.1520 | 0.0259 | 30 |
| Pseudobutyrivibrio | 0.0187 | 0.0103 | 0.0113 | 0.0093 | 0.0024 | 0.0479 | 21 |
| Succinivibrio | 0.0090 | 9.76 × 10−4 | 0.0020 | 0.0037 | 0.0020 | 0.0412 | 29 |
aData is shown as LSMeans.
b P values indicate groups that differ (P < 0.05).
cAll data are defined as taxa that are present in at least 50% of the samples.
Additional taxa were identified at low relative abundances, and differences were detected between feed efficiency phenotypes. These taxa included the genera Anaeroplasma (P = 0.0222), Paludibacter (P = 0.0226), Faecalibacterium (P = 0.0361), Succinivibrio (P = 0.0412), and Pseudobutyrivibrio (P = 0.0479). Of these taxa, Anaeroplasma and Faecalibacterium were in greatest abundance within the ADGHigh–ADFIHigh group, Paludibacter was in greatest abundance within the ADGLow–ADFILow group, and Succinivibrio and Pseudobutyrivibrio were least abundant within the ADGHigh–ADFILow and ADGLow–ADFIHigh groups, respectively (Table 2). Differences between the groups were also detected within other low abundance taxa, but not classified to the genus level. These low-abundance taxa included identifications within the families Mogibacteriaceae (P = 0.0309), Sphingomonadaceae (P = 0.0386), and Barnesiellaceae (P = 0.0471), with the greatest abundance within the ADGLow–ADFIHigh group for the family Mogibacteriaceae. The families Sphingomonadaceae and Barnesiellaceae were least abundant within the ADGLow–ADFILow and ADGLow–ADFIHigh groups, respectively (Table 2). The taxa listed were observed as present in at least 50% of the samples.
The examination of OTUs across all feed efficiency phenotype groups was also conducted to detect differences in abundance. Consideration was only given to OTUs detectable at abundances > 0.001% and present in at least 50% of the samples. Among the groups, 68 OTUs were identified that differed in abundance (Table 3). The most common and functionally significant OTUs identified with families Ruminococcaceae (P = 0.0029), Lachnospiraceae (P = 0.0279), and Clostridiaceae (P = 0.0460), as well as the orders Clostridiales (P = 0.0264) and Bacteroidales (P = 0.0433). At the genus level, Dorea (P = 0.0225), Butyrivibrio (P = 0.0240), Coprococcus (P = 0.0323), Prevotella (P = 0.0435), Clostridium (P = 0.0446), Oscillospira (P = 0.0456), and Pseudobutyrivibrio (P = 0.0476), as well as the species Prevotella copri (P = 0.0483) differed between groups. Coprococcus, Clostridium, Pseudobutyrivibrio, and Prevotella copri were all in greatest abundance within the ADGHigh–ADFIHigh group, while Dorea was least abundant within this group. The abundances of Butyrivibrio and Prevotella were greatest within the ADGLow–ADFIHigh group. Finally, the abundance of Oscillospira was greatest within the ADGLow–ADFILow group (Table 3).
Table 3.
Relative abundance of significant OTUs in the four feed efficiency groups
| OTU ID | Classification | Percentage of total sequencesa | SEM | P valueb | No. of steers with detectable taxonc | |||
|---|---|---|---|---|---|---|---|---|
| ADGHigh–ADFIHigh | ADGHigh–ADFILow | ADGLow–ADFILow | ADGLow–ADFIHigh | |||||
| denovo26989 | Bacteroides | 0.0163 | 0.0026 | 0.0013 | 0.0040 | 0.0034 | 0.0475 | 17 |
| denovo77346 | Blautia | 0.0028 | 0.0003 | 0.0007 | 0.0014 | 0.0006 | 0.0499 | 19 |
| denovo8562 | Butyrivibrio | 0.0003 | 0.0021 | 0.0009 | 0.0029 | 0.0004 | 0.0240 | 20 |
| denovo24149 | Butyrivibrio | 0.0003 | 0.0016 | 0.0005 | 0.0018 | 0.0004 | 0.0471 | 16 |
| denovo8243 | Clostridium | 0.0298 | 0.0018 | 0.0022 | 0.0009 | 0.0066 | 0.0446 | 23 |
| denovo8936 | Coprococcus | 0.0063 | 0.0017 | 0.0011 | 0.0010 | 0.0009 | 0.0323 | 15 |
| denovo67746 | Coprococcus | 0.0061 | 0.0014 | 0.0029 | 0.0009 | 0.0011 | 0.0441 | 19 |
| denovo33574 | Dorea | 0.0001 | 0.0010 | 0.0010 | 0.0025 | 0.0003 | 0.0225 | 18 |
| denovo12537 | Family Bacteroidaceae | 0.0001 | 0.0019 | 0.0009 | 0.0013 | 0.0004 | 0.0500 | 15 |
| denovo66919 | Family Christensenellaceae | 0.0043 | 0.0086 | 0.0030 | 0.0015 | 0.0014 | 0.0442 | 23 |
| denovo66901 | Family Christensenellaceae | 0.0021 | 0.0121 | 0.0109 | 0.0029 | 0.0024 | 0.0463 | 18 |
| denovo28680 | Family Clostridiaceae | 0.0086 | 0.0010 | 0.0016 | 0.0019 | 0.0017 | 0.0460 | 19 |
| denovo23600 | Family Lachnospiraceae | 0.0641 | 0.0126 | 0.0175 | 0.0210 | 0.0086 | 0.0279 | 29 |
| denovo25904 | Family Lachnospiraceae | 0.0018 | 0.0023 | 0.0039 | 0.0009 | 0.0006 | 0.0472 | 23 |
| denovo67326 | Family Lachnospiraceae | 0.0024 | 0.0005 | 0.0012 | 0.0005 | 0.0004 | 0.0482 | 16 |
| denovo43262 | Family Lachnospiraceae | 0.0084 | 0.0008 | 0.0025 | 0.0010 | 0.0018 | 0.0487 | 17 |
| denovo6452 | Family Lachnospiraceae | 1.5328 | 0.1368 | 0.1559 | 0.0641 | 0.3227 | 0.0495 | 30 |
| denovo37795 | Family Lachnospiraceae | 0.0021 | 0.0016 | 0.0007 | 0.0039 | 0.0007 | 0.0496 | 21 |
| denovo25335 | Family Lachnospiraceae | 0.0085 | 0.0028 | 0.0022 | 0.0028 | 0.0017 | 0.0499 | 23 |
| denovo13622 | Family Peptostreptococcaceae | 0.0001 | 0.0019 | 0.0013 | 0.0011 | 0.0004 | 0.0499 | 16 |
| denovo34577 | Family Rikenellaceae | 0.0009 | 0.0067 | 0.0384 | 0.0033 | 0.0077 | 0.0485 | 18 |
| denovo11639 | Family Rikenellaceae | 0.0155 | 0.0367 | 0.0567 | 0.0185 | 0.0104 | 0.0497 | 26 |
| denovo50904 | Family Ruminococcaceae | 0.0013 | 0.0063 | 0.0033 | 0.0005 | 0.0007 | 0.0029 | 20 |
| denovo53814 | Family Ruminococcaceae | 0.0045 | 0.0011 | 0.0013 | 0.0008 | 0.0006 | 0.0246 | 19 |
| denovo11996 | Family Ruminococcaceae | 0.0004 | 0.0004 | 0.0019 | 0.0004 | 0.0003 | 0.0387 | 15 |
| denovo43427 | Family Ruminococcaceae | 0.0044 | 0.0010 | 0.0022 | 0.0009 | 0.0007 | 0.0416 | 20 |
| denovo6911 | Family Ruminococcaceae | 0.0061 | 0.0011 | 0.0014 | 0.0018 | 0.0010 | 0.0421 | 22 |
| denovo71254 | Family Ruminococcaceae | 0.1496 | 0.5316 | 0.4293 | 0.2538 | 0.0697 | 0.0440 | 30 |
| denovo9573 | Family Ruminococcaceae | 0.0023 | 0.0076 | 0.0133 | 0.0044 | 0.0019 | 0.0445 | 21 |
| denovo19097 | Family Ruminococcaceae | 0.0024 | 0.0160 | 0.0135 | 0.0075 | 0.0028 | 0.0447 | 26 |
| denovo30835 | Family Ruminococcaceae | 0.0005 | 0.0010 | 0.0021 | 0.0003 | 0.0004 | 0.0448 | 15 |
| denovo46467 | Family Ruminococcaceae | 0.0023 | 0.0320 | 0.0111 | 0.0033 | 0.0062 | 0.0458 | 20 |
| denovo38547 | Family Ruminococcaceae | 0.0103 | 0.0078 | 0.0051 | 0.0019 | 0.0017 | 0.0459 | 27 |
| denovo359 | Family Ruminococcaceae | 0.0345 | 0.0047 | 0.0100 | 0.0124 | 0.0065 | 0.0466 | 27 |
| denovo19227 | Family Ruminococcaceae | 0.0006 | 0.0034 | 0.0029 | 0.0016 | 0.0006 | 0.0470 | 22 |
| denovo38238 | Family Ruminococcaceae | 0.3534 | 0.8847 | 0.6177 | 0.6599 | 0.1075 | 0.0476 | 30 |
| denovo25283 | Family Ruminococcaceae | 0.0228 | 0.0595 | 0.0320 | 0.0288 | 0.0078 | 0.0478 | 29 |
| denovo67138 | Family Ruminococcaceae | 0.0105 | 0.0015 | 0.0023 | 0.0030 | 0.0020 | 0.0478 | 18 |
| denovo39456 | Family Ruminococcaceae | 0.0011 | 0.0056 | 0.0016 | 0.0016 | 0.0008 | 0.0479 | 21 |
| denovo41445 | Family Ruminococcaceae | 0.0025 | 0.0125 | 0.0049 | 0.0009 | 0.0023 | 0.0479 | 23 |
| denovo16744 | Family Ruminococcaceae | 0.0019 | 0.0072 | 0.0031 | 0.0016 | 0.0012 | 0.0479 | 20 |
| denovo69598 | Family Ruminococcaceae | 0.0008 | 0.0050 | 0.0016 | 0.0013 | 0.0009 | 0.0486 | 16 |
| denovo21045 | Family Ruminococcaceae | 0.0043 | 0.0035 | 0.0012 | 0.0009 | 0.0008 | 0.0487 | 19 |
| denovo21464 | Family Ruminococcaceae | 0.0020 | 0.0013 | 0.0068 | 0.0016 | 0.0012 | 0.0492 | 17 |
| denovo59669 | Family Ruminococcaceae | 0.2908 | 0.2518 | 0.1567 | 0.0961 | 0.0449 | 0.0492 | 30 |
| denovo44398 | Family Ruminococcaceae | 0.0034 | 0.0094 | 0.0184 | 0.0051 | 0.0033 | 0.0492 | 20 |
| denovo70020 | Family Ruminococcaceae | 0.0008 | 0.0115 | 0.0065 | 0.0038 | 0.0025 | 0.0499 | 18 |
| denovo37453 | Order Bacteroidales | 0.0040 | 0.0004 | 0.0008 | 0.0010 | 0.0006 | 0.0433 | 15 |
| denovo46578 | Order Bacteroidales | 0.0018 | 0.1288 | 0.0686 | 0.0301 | 0.0264 | 0.0469 | 19 |
| denovo19773 | Order Bacteroidales | 0.0126 | 0.0024 | 0.0028 | 0.0053 | 0.0022 | 0.0473 | 22 |
| denovo72787 | Order Bacteroidales | 0.0184 | 0.0018 | 0.0042 | 0.0073 | 0.0035 | 0.0475 | 16 |
| denovo8377 | Order Bacteroidales | 0.0043 | 0.0006 | 0.0013 | 0.0011 | 0.0008 | 0.0482 | 18 |
| denovo29245 | Order Bacteroidales | 0.0069 | 0.0005 | 0.0011 | 0.0029 | 0.0016 | 0.0499 | 16 |
| denovo40203 | Order Clostridiales | 0.0048 | 0.0006 | 0.0006 | 0.0008 | 0.0008 | 0.0264 | 16 |
| denovo23255 | Order Clostridiales | 0.0011 | 0.0038 | 0.0015 | 0.0054 | 0.0008 | 0.0414 | 23 |
| denovo75811 | Order Clostridiales | 0.0019 | 0.0021 | 0.0036 | 0.0068 | 0.0010 | 0.0454 | 27 |
| denovo70498 | Order Clostridiales | 0.0018 | 0.0075 | 0.0060 | 0.0020 | 0.0013 | 0.0480 | 19 |
| denovo13252 | Order Clostridiales | 0.0049 | 0.0103 | 0.0078 | 0.0149 | 0.0021 | 0.0485 | 29 |
| denovo40361 | Order Clostridiales | 0.0054 | 0.0009 | 0.0021 | 0.0015 | 0.0010 | 0.0498 | 15 |
| denovo60620 | Order Clostridiales | 0.0025 | 0.0077 | 0.0042 | 0.0015 | 0.0013 | 0.0499 | 21 |
| denovo40059 | Order Clostridiales | 0.0008 | 0.0021 | 0.0018 | 0.0004 | 0.0005 | 0.0499 | 16 |
| denovo18789 | Order Clostridiales | 0.0019 | 0.0066 | 0.0023 | 0.0071 | 0.0013 | 0.0499 | 25 |
| denovo43715 | Order Clostridiales | 0.0185 | 0.0052 | 0.0080 | 0.0110 | 0.0032 | 0.0500 | 29 |
| denovo51765 | Oscillospira | 0.0025 | 0.0046 | 0.0134 | 0.0041 | 0.0022 | 0.0456 | 25 |
| denovo26903 | Prevotella | 0.0050 | 0.0017 | 0.0027 | 0.0179 | 0.0030 | 0.0435 | 28 |
| denovo23534 | Prevotella copri | 0.0230 | 0.0011 | 0.0051 | 0.0033 | 0.0049 | 0.0483 | 17 |
| denovo60154 | Pseudobutyrivibrio | 0.0064 | 0.0007 | 0.0017 | 0.0025 | 0.0012 | 0.0476 | 21 |
| denovo45407 | Treponema | 0.0010 | 0.0237 | 0.0161 | 0.0049 | 0.0049 | 0.0477 | 21 |
aData is shown as LSMeans.
b P values indicate groups that differ (P < 0.05).
cPercentage of total sequences for steers with nondetectable OTUs were treated as 0.001%, and all data are defined as OTUs that are present in at least 50% of the samples.
Effect of gain and intake
In order to examine the microbial population associations among the contributing factors of feed efficiency, the effect of the microbial communities on ADG and ADFI were analyzed separately to determine whether the associated microbial populations differed by low vs. high ADG, low vs. high ADFI, or their interaction. The significant relative abundances of taxa and OTUs between ADG and ADFI are listed in Tables 4 and 5, respectively. No taxa were associated with gain alone (Table 4), but two taxa were determined to have either a significant effect for intake or the interaction. When examined using OTUs, a majority of the OTUs associated with either intake or the interaction (Table 5). Pertaining to significant genera, Bacteroides (P = 0.0491) and Blautia (P = 0.0497) were associated with intake, while Butyrivibrio (P = 0.0156) and Coprococcus (P = 0.0499) were significant for the interaction. Additionally, the species Prevotella copri (P = 0.0491) associated with the interaction. OTUs classified within families Ruminococcaceae, Lachnospiraceae, and Clostridiaceae were associated with intake, as well as their interaction. The genera Oscillospira (P = 0.0484) and Prevotella (P = 0.0494) were the only classifications of OTUs associated solely with gain.
Table 4.
Relative abundance of significant taxa within ADG and ADFI phenotypes
| Classification | Phenotypea | Effect | SEM | P valueb | |||
|---|---|---|---|---|---|---|---|
| ADGHigh | ADGLow | ADFIHigh | ADFILow | ||||
| Prevotella | 5.5465 | 3.7537 | 5.7809 | 3.5192 | Gain*Intake | 0.8443 | 0.0484 |
| Succinivibrio | 0.0050 | 0.0028 | 0.0064 | 0.0015 | Intake | 0.0014 | 0.0411 |
aData is shown as LSMeans.
b P values indicate groups that differ (P < 0.05).
Table 5.
Relative abundance of significant OTUs within ADG and ADFI phenotypes
| OTU ID | Classification | Phenotypea | Effect | SEM | P valueb | |||
|---|---|---|---|---|---|---|---|---|
| ADGHigh | ADGLow | ADFIHigh | ADFILow | |||||
| denovo26989 | Bacteroides | 0.0094 | 0.0027 | 0.0101 | 0.0020 | Intake | 0.0025 | 0.0491 |
| denovo77346 | Blautia | 0.0015 | 0.0011 | 0.0021 | 0.0005 | Intake | 0.0004 | 0.0497 |
| denovo8562 | Butyrivibrio | 0.0012 | 0.0019 | 0.0016 | 0.0015 | Gain*Intake | 0.0003 | 0.0156 |
| denovo8936 | Coprococcus | 0.0040 | 0.0011 | 0.0036 | 0.0014 | Gain*Intake | 0.0007 | 0.0499 |
| denovo28680 | Family Clostridiaceae | 0.0048 | 0.0017 | 0.0053 | 0.0013 | Intake | 0.0012 | 0.0486 |
| denovo43262 | Family Lachnospiraceae | 0.0046 | 0.0017 | 0.0047 | 0.0016 | Gain*Intake | 0.0013 | 0.0494 |
| denovo6452 | Family Lachnospiraceae | 0.8348 | 0.1100 | 0.7984 | 0.1464 | Gain*Intake | 0.2365 | 0.0496 |
| denovo13622 | Family Peptostreptococcaceae | 0.0010 | 0.0012 | 0.0006 | 0.0016 | Intake | 0.0003 | 0.0492 |
| denovo50904 | Family Ruminococcaceae | 0.0038 | 0.0019 | 0.0009 | 0.0048 | Intake | 0.0005 | 0.0034 |
| denovo53814 | Family Ruminococcaceae | 0.0028 | 0.0010 | 0.0026 | 0.0012 | Intake | 0.0004 | 0.0486 |
| denovo6911 | Family Ruminococcaceae | 0.0036 | 0.0016 | 0.0039 | 0.0013 | Gain*Intake | 0.0007 | 0.0492 |
| denovo69598 | Family Ruminococcaceae | 0.0029 | 0.0014 | 0.0010 | 0.0033 | Gain*Intake | 0.0006 | 0.0495 |
| denovo25283 | Family Ruminococcaceae | 0.0411 | 0.0304 | 0.0258 | 0.0458 | Intake | 0.0057 | 0.0497 |
| denovo359 | Family Ruminococcaceae | 0.0196 | 0.0112 | 0.0234 | 0.0073 | Intake | 0.0047 | 0.0499 |
| denovo8377 | Order Bacteroidales | 0.0024 | 0.0012 | 0.0027 | 0.0009 | Gain*Intake | 0.0006 | 0.0494 |
| denovo40361 | Order Clostridiales | 0.0031 | 0.0018 | 0.0034 | 0.0015 | Gain*Intake | 0.0007 | 0.0491 |
| denovo43715 | Order Clostridiales | 0.0118 | 0.0095 | 0.0148 | 0.0066 | Intake | 0.0023 | 0.0492 |
| denovo51765 | Oscillospira | 0.0035 | 0.0088 | 0.0033 | 0.0090 | Gain | 0.0016 | 0.0484 |
| denovo26903 | Prevotella | 0.0034 | 0.0103 | 0.0114 | 0.0022 | Gain | 0.0022 | 0.0494 |
| denovo23534 | Prevotella copri | 0.0121 | 0.0042 | 0.0131 | 0.0031 | Gain*Intake | 0.0036 | 0.0491 |
aData is shown as LSMeans.
b P values indicate groups that differ (P < 0.05).
Discussion
The function of the GIT is essential for the overall health and well-being of ruminants. In addition to many other host-microbiome interactions (Williams and Coleman 1997; McDonald et al. 2002; Chung et al. 2012), the various cellulolytic, metabolic, and fermentative functions supplied by the microbial communities throughout the bovine GIT contribute towards the overall energy and nutritional input to the host, and therefore have a significant effect on host maintenance, growth, and performance. Many microbial studies on bovine feed efficiency have focused on ruminal associations; however, the multiple sections within the GIT are specific in function and microbial community population and diversity (de Oliveira et al. 2013; Frey et al. 2010). The long large intestine and colon in cattle is the site of post-ruminal degradation of cellulose and starch, and is thought to be significant in animal digestion, particularly as it pertains to diet (Armstrong and Smithard 1979). Thus, determining host-microbe associations within the entire GIT from the sampling of digesta from one site will not accurately represent the nutritional and energy status of the host. The examination of the microbial associations with feed efficiency outside of the rumen, such as the colon, will aid in the comprehensive understanding of feed efficiency in beef cattle.
Compared to other sections within the GIT, the microbial abundance and diversity of the colon contents is far greater, even compared to that of the rumen (Myer et al. 2015; de Oliveira et al. 2013; Reti et al. 2013). To accurately capture most of the bacterial OTUs within the rectal contents of the steer, the study normalized the samples to a depth of 100,000 sequences/sample. This depth was estimated from colonic content sample rarefaction curves, as well as estimations from previous GIT studies, which have acquired adequate coverage (Jami and Mizrahi 2012). The current study was able to recover approximately 97% of all OTUs calculated at 0.03% dissimilarity, as determined by Good’s coverage estimator. Based on the coverage estimates of the current study, the colonic digesta sample rarefaction curves, and the coverage estimates from previous studies, the 100,000 sequences/sample achieved was determined to be satisfactory for colonic microbial community analyses.
Next-generation sequencing (NGS) technologies have allowed for greater sequencing depth of environmental niches and greater identification of OTUs, specifically when compared to previous studies examining the beef cattle GIT (de Oliveira et al. 2013; Reti et al. 2013). The caveat to this approach is that these technologies are limited to shorter read lengths than traditionally produced by cloning and sequencing of full-length 16S rRNA genes. The 16S rRNA gene includes multiple regions containing variable sequence interspersed with conserved regions, and determination of target variable regions is dependent upon the niche examined. In this study, the V1–V3 variable regions were selected to interrogate the colon content bacterial communities. However, the alpha-diversity analyses across the feed efficiency groups revealed no differences in the number of OTUs, richness (Chao1), diversity (Shannon Index) or evenness (equitability), despite the depth and increased detection of OTUs. The weighted and unweighted UniFrac PCoA also reflected the similarities of the bacterial communities within the colon between feed efficiency groups, as the microbial populations within the colon did not cluster by host phenotype. The weighted and unweighted UniFrac results are dependent upon and a result of the phylogenetic divergence between the OTUs. The lack of observable differences between communities at the phylogenetic level may be anticipated, where differences are usually observable in the feces of cattle fed different diets (Kim et al. 2014), but may not be detectable when examining finer nutritional changes within cattle on the same diet. This may further indicate that variation within the colon microbial comminutes as a function of differing feed efficiency arises from changes in OTU and relative taxonomic abundances, rather than the phylogenetic diversity of the community. These specific changes may have profound effects on the host. However, the observed similarities between the microbial comminutes in the colon may also partly be a result of host-specificity, which has been demonstrated in the rumen (Weimer et al. 2010).
The 16S sequences within the colon samples largely belonged to the phyla Firmicutes, Bacteroidetes, Spirochaetes, Tenericutes, Proteobacteria, Actinobacteria, and Fibrobacteres, which are present in the majority of gut-associated phylotypes in a variety of mammals (Ley et al. 2008; Shanks et al. 2011). The ubiquitous nature of these phyla within mammals suggests their critical role in the microbial ecology of the mammalian gut. There were observable differences at the phylum level, especially within the ADGHigh–ADFILow group. Shifts between the Firmicutes:Bacteroidetes ratio were evident, with an increase in the ratio within the ADGHigh–ADFILow group. Although the variable proportions of the phyla Firmicutes and Bacteroidetes were not significant, shifts in the ratio have been associated with obesity in humans (Turnbaugh et al. 2009; Ismail et al. 2010), as well as in the cattle rumen regarding energy harvesting and correlated increases of fat (Jami et al. 2014). However, any putative role of variation in these phyla in the lower GIT of beef cattle is unclear. In addition, the abundance of Firmicutes was comparable to that of the cecum and feces in similar studies, where members of the families Ruminococcaceae, Lachnospiraceae and Clostridiaceae dominated (de Oliveira et al. 2013). It is likely that these taxa also contribute to further downstream feed fermentation in the large intestine.
At the sub-phylum level, the colon content samples were dominated by orders Clostridiales and Bacteroidales, families Ruminococcaceae, Lachnospiraceae, Bacteroidaceae, and Clostridiaceae, as well as genera Prevotella, Ruminococcus, and Coprococcus. These taxa are commonly found within microbial communities across the GIT, and are also identified in the large intestine of steers (de Oliveira et al. 2013). These profiles are also similar to those identified in the fecal content of cattle (Kim et al. 2014; Jeong et al. 2011).
The colon content from the four feed efficiency groups revealed significant differences in the relative abundance of specific taxa. The putative functions of the identified organisms may provide some insight as to their potential association with regard to feed efficiency in beef cattle. Prevotella has commonly been found in cattle feces and has been associated with differences in diet (Durso et al. 2012). Its abundance was positively correlated with corn-based diets, and was nearly absent in cattle fed silage/forage (70% corn silage and 30% alfalfa haylage) (Kim et al. 2014), and may play an important role in the fecal microbial comminutes of feedlot cattle fed concentrate diets. Although found in the microbial populations of cattle feces, not much is known as to the function of the family Mogibacteriaceae in the gut. However, these populations in humans and mice have clustered with other organisms that are associated with lower body mass index (BMI) (Goodrich et al. 2014), as well as changes in the diet of non-obese diabetic (NOD) mice supplemented with cellulose, pectin, and xylan (Toivonen et al. 2014). Further study of Mogibacteriaceae is warranted in order to better determine its association with feed efficiency in cattle. The presence of Faecalibacterium in preweaned dairy calves has been associated with weight gain and the incidence of diarrhea, with greater abundance being linked with higher weight gain and less diarrhea (Oikonomou et al. 2013). The observed greater abundance in the large intestine of preweaned dairy calves may be important for maintaining proper BW and reducing enteric infections early in life. This butyrate-producing taxa is also greater in the feces of obese children than for non-obese children (Balamurugana et al. 2010), suggesting its potential function in terms of cattle health and performance. Succinivibrio species play an ecologically important role as starch digesters and ferments glucose with production of acetic and succinic acids (Bryant and Small 1956). Appropriately, these species are usually found in the rumen of cattle fed diets containing high levels of rapidly fermented carbohydrates. Finally, the butyrate-producing Pseudobutyrivibrio are core genera found to be ubiquitous in the steer GIT. Not only is its butyrate production implicated as a primary metabolic fuel for enterocytes (Scheppach et al. 1995), but the Butyrivibrio and Pseudobutyrivibrio species have gained greater interest because of their strong xylan-degrading abilities (Morgavi et al. 2013).
The relative abundance of significant OTUs within the digesta of the terminal colon content revealed many associations previously mentioned within the taxonomic assessment, however, several additional identifications were of interest. The family Ruminococcaceae contains organisms that are known to be cellulolytic, as well as active in acetate, formate, and hydrogen production (Biddle et al. 2013). A contribution by fiber-digesting bacteria is anticipated from residual feed from the rumen and thus may also contribute to downstream feed fermentation in the large intestine. Genera within the family Rikenellaceae produce acetate, succinate, and propionate as fermentation products and are found in high abundance within the large intestine and feces of cattle (Jeong et al. 2011; Kong et al. 2010; Abe et al. 2012). The family Rikenellaceae has previously been associated with forage diets (Petri et al. 2013). This finding, along with the observation that Rikenellaceae genera clustered with other genera including Fibrobacter in diets containing fractions of bermudagrass hay rather than wheat fractions, hint that the genera within Rikenellaceae may be involved in structural carbohydrate degradation (Pitta et al. 2010). The association of Coprococcus with high grain diets was also implicated in this study (Kim et al. 2014), and its butyrate production may additionally contribute towards the enterocyte energy pool, similar to that of Butyrivibrio, Pseudobutyrivibrio, and Faecalibacterium. Oscillospira are also found in high grain diets, and cattle fed high starch diets can have high bypass starch from the rumen (Wells et al. 2009). The abundances and variability of Oscillospira in the colon between feed efficiency groups may be associated with differing levels of bypass starch (Kim et al. 2014). Lastly, Blautia spp. are common inhabitants of the GIT and feces of cattle and goats, and are ubiquitous among humans and other mammals, although at low abundance (Eren et al. 2015). Within the family Lachnospiraceae, the genus Blautia can provide energy to their host from polysaccharides that other gut microorganisms cannot degrade (Biddle et al. 2013), and thus may be integral towards the metabolic capacity of the host.
The taxa and OTUs that were identified in the study were also analyzed to determine whether their association with feed efficiency differed individually by gain (ADG) or intake (ADFI), or their interaction. At both levels of analysis, the significantly identified taxa and OTUs were associated with either ADFI or the interaction of ADG and ADFI. The gradual association with ADFI over ADG as digesta moves distally through the GIT may be expected as the metabolic function and capacity of the tissues change, from the rumen to that of the colon (Myer et al. 2015). In addition, digestible fiber that is not degraded in the rumen or escapes digestion in the rumen, partially due to high ADFI, becomes available for further processing in the lower GIT (Tan et al. 2002). Only two taxa/OTUs associated primarily with ADG alone. These included the genera Oscilliospira and Prevotella. Their association with ADG may be explained by their abundance in the rumen and potential association with bypass starch from the rumen (Wells et al. 2009). However, it is important to note that the association with ADG may also be a result of residual DNA from other sections of the GIT.
Conclusions
The taxa and OTUs identified in this study as associating with differences in feed efficiency have the potential to affect beef cattle feed efficiency based on their putative functions relating to cellulolytic and metabolic activities in the colon. No phylogenetic differences were observed between communities of differing feed efficiency phenotypes, although, significant differences were identified when examining the relative abundance of specific taxa and OTUs. This study suggests differences in the colon microbial communities at the 16S level in cattle that vary in feed efficiency. However, it is still not clear whether changes in the microbial community are contributing to differences in feed efficiency of the host or if host factors are driving changes in the microbial community.
The associations between feed efficiency and microbial populations in the bovine rumen have been reported (Myer et al. 2015), but knowledge of their role in the distal GIT is lacking. Aided by the coverage and depth of this study, specific and significant taxa and OTUs were identified and correlated with differing feed efficiency phenotypes, ADG, and ADFI. Additional study, including functional analyses based on the putative functions of the significant taxa and OTUs identified in this study, as well as a metagenomic approach may complement existing data and allow for greater resolution analyses, respectively. Many factors influence the feed efficiency of the host, and although important for the energy production and nutrient supply to the animal, the study of feed efficiency must occur in addition to and beyond the rumen in order to fully understand the processes contributing to efficient beef cattle.
Methods
Experimental design and colon sampling
Similar to Myer et al. (2015), steers selected for this study came from a population of cattle being developed to have a high percentage of the following breeds: Angus, Beefmaster, Brahman, Brangus, Braunvieh, Charolais, Chiangus, Gelbvieh, Hereford, Limousin, Maine Anjou, Red Angus, Salers, Santa Gertrudis, Shorthorn, Simmental, South Devon, and Tarentaise. Each year heifers and cows were artificially inseminated with semen from prominent industry bulls of their dominant breed. This resulted in offspring ranging from 50 to 75% of the same breed as their sire with the exception of Angus and Hereford, which ranged from 50 to 100% of the same breed as their sire. Individual feed intake was measured using an Insentec feeding system (Marknesse, The Netherlands). Steers were fed a ration (dry matter basis) of 57.35% dry-rolled corn, 30% wet distillers grain with solubles, 8% alfalfa hay, 4.25% supplement (containing 0.772 g/kg monensin), and 0.4% urea. Individual feed intake (FI) and body weight (BW) gain were measured over a 63-day period (Myer et al. 2015; Lindholm-Perry et al. 2013). Steers were selected from two contemporary groups. Group 1 (n = 148) were spring-born calves that were 371 ± 1 d of age and weighed 522 ± 4 kg at the start of the feed intake measurement. Group 2 (n = 197) were fall-born calves that were 343 ± 1 days of age and weighed 448 ± 4 kg at the start of the feed intake measurement. At the end of each feeding period, steers were ranked based on their standardized distance from the bivariate mean (average daily gain [ADG] and average daily feed intake [ADFI]) assuming a bivariate normal distribution with a calculated correlation between ADG and ADFI. Four steers with the greatest deviation within each Cartesian quadrant were sampled (n = 16/group). In the event a sire breed was over represented within a quadrant a steer with the next highest rank of a different breed was selected. The result was a 2 × 2 factorial design consisting of high and low ADFI, and high and low ADG (Myer et al. 2015). Steers were allowed ad libitum access to feed within 1 h prior to harvest. At the end of the feeding period, steers were harvested, and approximately 15 mL of colonic digesta at the rectum was sampled. The 2 feeding studies yielded 32 animals for analysis. Digesta was collected from the terminal colon adjacent to the rectum at harvest. Samples were individually stored in buffered peptone water (BPW, pH 7.0) +15% glycerol stock for processing and kept at −70°C for long-term storage post-processing.
DNA extraction, amplification and sequencing
DNA was extracted from colon samples using a repeated bead beating plus column (RBB + C) method (Yu and Morrison 2004). Briefly, 0.3 g of sample was centrifuged for 5 min at 16,000×g to pellet solids including bacterial cells, and then resuspended in 0.2 mL TE (Tris–EDTA, pH 8.0) buffer. Cell lysis was achieved by bead beating 0.15 g of the resuspended sample in ZR BashingBead Lysis Tubes (Zymo Research Corp, Santa Ana, CA, USA) using the TissueLyser II system (Qiagen, Hilden, Germany) for 3 min at 21 Hz, in the presence of 4% (w/v) sodium dodecyl sulfate (SDS), 500 mM NaCl, and 50 mM EDTA. After mechanical and chemical cell lysis, 10 M ammonium acetate (260 µL) was used to precipitate and remove the impurities and SDS, followed by equal volume isopropanol precipitation for the recovery of the nucleic acids. Supernatants were treated with 2 µL RNase (10 mg/mL) and proteinase K (QIAamp DNA Stool Mini Kit), followed by the use of QIAamp columns from the Qiagen DNA Stool Mini Kit (Qiagen, Hilden, Germany). Genomic DNA concentration was determined using a Nanodrop 1000 spectrophotometer (ThermoScientific, Wilmington, DE, USA).
Amplicon library preparation was performed by PCR amplification of the V1–V3 region of 16S rRNA gene, using modified universal primers 27F (5′-Adapter/Index/AGAGTTTGATCCTGGCTCAG) and 519R (5′ Adapter/Index/GTATTACCGCGGCTGCTG) including TruSeq® adapter sequences and indices, as well as AccuPrime™ Taq high fidelity DNA Polymerase (Life Technologies, Carlsbad, CA). Amplification consisted of 23 cycles, with an annealing temperature of 58°C. Products were purified using AmPure® bead purification (Agencourt, Beverly, MA, USA) and all libraries were quantified by the PicoGreen® dsDNA quantitation kit (Invitrogen, Carlsbad, CA, USA) and by real-time PCR on the LightCycler 480 system (Roche, Mannheim, Germany). The PCR amplicon libraries were sequenced using the 2 × 300, v3 600-cycle kit and the Illumina MiSeq® sequencing platform (Illumina, San Diego, CA, USA).
Sequence read processing and analysis
All sequences were processed using the QIIME-1.8.0 software package. Paired reads were joined using fastq-join (Aronesty 2011) and filtered for quality (≥Q25) using the Galaxy server (Blankenberg et al. 2010). Sequences that contained read lengths shorter than 400 bp were removed and adapters/index sequences were trimmed. Chimeric sequences were checked using ChimeraSlayer (Haas et al. 2011). All cleaned sequences were classified into taxa using the Greengenes 16S rRNA Gene Database (DeSantis et al. 2006). Operational taxonomic units (OTUs) were calculated using the uclust program (0.03 dissimilarity; Edgar 2010). After calculating richness for each quadrant, singletons were removed from further diversity analyses. Based on rarefaction curves, the number of OTUs was normalized via subsampling 100,000 sequences from each colon sample. A phylogenic tree was built with FastTree (Price et al. 2010) to determine alpha- and beta-diversity metrics.
Statistical analysis
All analyses were conducted using SAS 9.4 (SAS Inst. Inc., Cary, NC, USA). The mean abundances (n = 8) of data metrics and each taxon were compared among the feed efficiency groups using a model of contemporary group and Cartesian quadrant [high ADG, high ADFI (ADGHigh–ADFIHigh); high ADG, low ADFI (ADGHigh–ADFILow); low ADG, low ADFI (ADGLow–ADFILow); low ADG, high ADFI (ADGLow–ADFIHigh)] as fixed effects. Significant differences were determined at P < 0.05 with the Benjamini–Hochberg method used for multiple-testing corrections (Benjamini and Hochberg 1995). Multiple-testing corrections were made for the number of phyla, the number of OTU groups, and other classified taxa groups. Linear contrasts were then applied to significant quadrants to separate whether microbial populations varied by low vs. high ADG, low vs. high ADFI, or their interaction (P < 0.05). Principal coordinates analysis (PCoA) was performed using weighted and unweighted UniFrac analyses (Lozupone and Knight 2005).
Authors’ contributions
PRM carried out all sample manipulations, nucleic acid extraction, amplification, and preparation for sequencing; participated in all statistical analyses; generated all figures; participated in design of the study; and drafted the manuscript. JEW participated in design of the study and contributed to conclusions. TPLS oversaw all sequence data generation and participated in design of the study. LAK oversaw and participated in all statistical analyses and participated in design of the study. HCF aided in the development of the model; participated in design of the study; was instrumental in animal selection; and aided in sample harvesting. All authors read and approved the final manuscript.
Acknowledgements
We thank Bob Lee, Sue Hauver, and Renee Godtel for technical assistance. This project is partially supported by Agriculture and Food Research Initiative Competitive Grant No. 2011-68004-30214 from the USDA National Institute of Food and Agriculture.
Mention of a trade name, proprietary product, or specific equipment does not constitute a guarantee or warranty by the USDA and does not imply approval to the exclusion of other products that may be suitable.
Sequence data is available from the NCBI Sequence Read Archive (SRA Accession SRP047292). Additional descriptive information is associated with NCBI BioProject PRJNA261425.
USDA is an equal opportunity provider and employer.
Compliance with ethical guidelines
Competing interests The authors declare that they have no competing interests.
Ethical approval This experiment was approved by the U.S. Meat Animal Research Center Animal Care and Use Committee.
Abbreviations
- ADFI
average daily feed intake
- ADG
average daily gain
- BW
body weight
- FI
feed intake
- GIT
gastrointestinal tract
- OTU
operational taxonomic unit
Contributor Information
Phillip R Myer, Email: pmyer@utk.edu.
James E Wells, Email: jim.wells@ars.usda.gov.
Timothy P L Smith, Email: tim.smith@ars.usda.gov.
Larry A Kuehn, Email: larry.kuehn@ars.usda.gov.
Harvey C Freetly, Email: Harvey.Freetly@ars.usda.gov.
References
- Abe K, Ueki A, Ohtaki Y, Kaku N, Watanabe K, Ueki K. Anaerocella delicata gen. nov., sp. nov., a strictly anaerobic bacterium in the phylum Bacteroidetes isolated from a methanogenic reactor of cattle farms. J Gen Appl Microbiol. 2012;58:405–412. doi: 10.2323/jgam.58.405. [DOI] [PubMed] [Google Scholar]
- Abo-Ismail MK, Vander Voort G, Squires JJ, Swanson KC, Mandell IB, Liao X, et al. Single nucleotide polymorphisms for feed efficiency and performance in crossbred beef cattle. BMC Genet. 2014;15:14. doi: 10.1186/1471-2156-15-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong DG, Smithard RR. The fate of carbohydrates in the small and large intestines of the ruminant. Proc Nutr Soc. 1979;38:283–294. doi: 10.1079/PNS19790050. [DOI] [PubMed] [Google Scholar]
- Aronesty E (2011) ea-utils: command-line tools for processing biological sequencing data. http://code.google.com/p/ea-utils
- Arthur PF, Herd RM, Wilkins JF, Archer JA. Maternal productivity for Angus cows divergently selected for post-weaning residual feed intake. Aust J Exp Agric. 2005;45:985–993. doi: 10.1071/EA05052. [DOI] [Google Scholar]
- Balamurugana R, Georgea G, Kabeerdossa J, Hepsibaa J, Chandragunasekarana A, Ramakrishnaa BS. Quantitative differences in intestinal Faecalibacterium prausnitzii in obese Indian children. Br J Nutr. 2010;103:335–338. doi: 10.1017/S0007114509992182. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B. 1995;57:289–300. [Google Scholar]
- Biddle A, Stewart L, Blanchard J, Leschine S. Untangling the genetic basis of fibrolytic specialization by Lachnospiraceae and Ruminococcaceae in diverse gut communities. Diversity. 2013;5:627–640. doi: 10.3390/d5030627. [DOI] [Google Scholar]
- Blankenberg D, Gordon A, Von Kuster G, Coraor N, Taylor J, Nekrutenko A. Galaxy Team. Manipulation of FASTQ data with galaxy. Bioinformatics. 2010;26:1783–1785. doi: 10.1093/bioinformatics/btq281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant MP, Small N. Characteristics of two new genera of anaerobic curved rods isolated from the rumen of cattle. J Bacteriol. 1956;72:22. doi: 10.1128/jb.72.1.22-26.1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung H, Pamp SJ, Hill JA, Surana NJ, Edelman SM. Gut immune maturation depends on colonization with a host-specific microbiota. Cell. 2012;149:1578–1593. doi: 10.1016/j.cell.2012.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Oliveira MN, Jewell KA, Freitas FS, Benjamin LA, Tótola MR, Borges AC, et al. Characterizing the microbiota across the gastrointestinal tract of a Brazilian Nelore steer. Vet Microbiol. 2013;164:307–314. doi: 10.1016/j.vetmic.2013.02.013. [DOI] [PubMed] [Google Scholar]
- DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol. 2006;72:5069–5072. doi: 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durso LM, Wells JE, Harhay GP, Rice WC, Kuehn L, Bono JL, et al. Comparison of bacterial communities in faeces of beef cattle fed diets containing corn and wet distillers’ grain with solubles. Lett Appl Microbiol. 2012;55:109–114. doi: 10.1111/j.1472-765X.2012.03265.x. [DOI] [PubMed] [Google Scholar]
- Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- Eren AM, Sogin ML, Morrison HG, Vineis JH, Fisher JC, Newton RJ, et al. A single genus in the gut microbiome reflects host preference and specificity. ISME J. 2015;9:90–100. doi: 10.1038/ismej.2014.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox DG, Tedeschi LO, Guiroy PJ (2001) Determining feed intake and feed efficiency of individual cattle fed in groups. In: Proc Beef Impr Fed 33rd Ann Res Symp Annu Meet, vol 33, pp 80–98
- Frey J, Pell A, Berthiaume R, Lapierre H, Lee S, Ha J, et al. Comparative studies of microbial populations in the rumen, duodenum, ileum and faeces of lactating dairy cows. J Appl Microbiol. 2010;108:1982–1993. doi: 10.1111/j.1365-2672.2009.04602.x. [DOI] [PubMed] [Google Scholar]
- Fujimura KE, Slusher NA, Cabana MD, Lynch SV. Role of the gut microbiota in defining human health. Expert Rev Anti Infect Ther. 2010;8:435–454. doi: 10.1586/eri.10.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galyean ML, Ponce C, Schutz J. The future of beef production in North America. Anim Front. 2011;1:29–36. doi: 10.2527/af.2011-0013. [DOI] [Google Scholar]
- Goodrich JK, Waters JL, Poole AC, Sutter JL, Koren O, Blekhman R, et al. Human genetics shape the gut microbiome. Cell. 2014;159:789–799. doi: 10.1016/j.cell.2014.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas BJ, Gevers D, Earl AM, Feldgarden M, Ward DV, Giannoukos G, et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 2011;21:494–504. doi: 10.1101/gr.112730.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Sanabria E, Goonewardene LA, Wang Z, Durunna ON, Moore SS, Guan LL. Impact of feed efficiency and diet on adaptive variations in the bacterial community in the rumen fluid of cattle. Appl Environ Microbiol. 2012;78:1203–1214. doi: 10.1128/AEM.05114-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ismail NA, Ragab SH, ElBaky AA, Shoeib AR, Alhosary Y, Fekry D. Frequency of Firmicutes and Bacteroidetes in gut microbiota in obese and normal weight Egyptian children and adults. Arch Med Sci. 2010;7:501–507. doi: 10.5114/aoms.2011.23418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jami E, Mizrahi I. Composition and similarity of bovine rumen microbiota across individual animals. PLoS One. 2012;7:e33306. doi: 10.1371/journal.pone.0033306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jami E, White BA, Mizrahi I. Potential role of the bovine rumen microbiome in modulating milk composition and feed efficiency. PLoS One. 2014;9:e85423. doi: 10.1371/journal.pone.0085423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong JY, Park HD, Lee KH, Weon HY, Ka JO. Microbial community analysis and identification of alternative host-specific fecal indicators in fecal and river water samples using pyrosequencing. J Microbiol. 2011;49:585–594. doi: 10.1007/s12275-011-0530-6. [DOI] [PubMed] [Google Scholar]
- Kim M, Morrison M, Yu Z. Status of the phylogenetic diversity census of ruminal microbiomes. FEMS Microbiol Ecol. 2011;76:49–63. doi: 10.1111/j.1574-6941.2010.01029.x. [DOI] [PubMed] [Google Scholar]
- Kim M, Kim J, Kuehn LA, Bono JL, Berry ED, Kalchayanand N, et al. Investigation of bacterial diversity in the feces of cattle fed different diets. J Anim Sci. 2014;92:683–694. doi: 10.2527/jas.2013-6841. [DOI] [PubMed] [Google Scholar]
- Kong Y, Teather R, Forster R. Composition, spatial distribution, and diversity of the bacterial communities in the rumen of cows fed different forages. FEMS Microbiol Ecol. 2010;74:612–622. doi: 10.1111/j.1574-6941.2010.00977.x. [DOI] [PubMed] [Google Scholar]
- Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, Bircher JS, et al. Evolution of mammals and their gut microbes. Science. 2008;320:1647–1651. doi: 10.1126/science.1155725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindholm-Perry AK, Kuehn LA, Oliver WT, Sexten AK, Miles JR, Rempel LA, et al. Adipose and muscle tissue gene expression of two genes (NCAPG and LCORL) located in a chromosomal region associated with cattle feed intake and gain. PLoS One. 2013;8:e80882. doi: 10.1371/journal.pone.0080882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone CA, Knight R. Unifrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol. 2005;71:8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone CA, Hamady M, Kelley ST, Knight R. Quantitative and qualitative β diversity measures lead to different insights into factors that structure microbial communities. Appl Environ Microbiol. 2007;73:1576–1585. doi: 10.1128/AEM.01996-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone CA, Lladser ME, Knights D, Stombaugh J, Knight R. UniFrac: an effective distance metric for microbial community comparison. ISME J. 2011;5:169–172. doi: 10.1038/ismej.2010.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malmuthuge N, Griebel PJ. Taxonomic identification of commensal bacteria associated with the mucosa and digesta throughout the gastrointestinal tracts of preweaned calves. Appl Environ Microbiol. 2014;80:2021–2028. doi: 10.1128/AEM.03864-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCann JC, Wiley LM, Forbes TD, Rouquette FM, Jr, Tedeschi LO. Relationship between the rumen microbiome and residual feed intake efficiency of Brahman bulls stocked on bermudagrass pastures. PLoS One. 2014;9:e91864. doi: 10.1371/journal.pone.0091864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald P, Edwards RA, Greenhalgh JFD, Morgan CA. Animal nutrition. 6. Harlow: Longman; 2002. [Google Scholar]
- Morgavi DP, Kelly WJ, Janssen PH, Attwood GT. Rumen microbial (meta) genomics and its application to ruminant production. Animal. 2013;7(s1):184–201. doi: 10.1017/S1751731112000419. [DOI] [PubMed] [Google Scholar]
- Myer PR, Smith TPL, Wells JE, Kuehn LA, Freetly HC. Rumen microbiome from steers differing in feed efficiency. PLoS One. 2015;10:e0129174. doi: 10.1371/journal.pone.0129174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oikonomou G, Teixeira AGV, Foditsch C, Bicalho ML, Machado VS, Bicalho RC. Fecal microbial diversity in pre-weaned dairy calves as described by pyrosequencing of metagenomic 16S rDNA. Associations of Faecalibacterium species with health and growth. PLoS One. 2013;8:e63157. doi: 10.1371/journal.pone.0063157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petri RM, Schwaiger T, Penner GB, Beauchemin KA, Forster RJ, McKinnon JJ, et al. Characterization of the core rumen microbiome in cattle during transition from forage to concentrate as well as during and after an acidotic challenge. PLoS One. 2013;8:e83424. doi: 10.1371/journal.pone.0083424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitta DW, Pinchak WE, Dowd SE, Osterstock J, Gontcharova V, Youn E, et al. Rumen bacterial diversity dynamics associated with changing from bermudagrass hay to grazed winter wheat diets. Microb Ecol. 2010;59:511–522. doi: 10.1007/s00248-009-9609-6. [DOI] [PubMed] [Google Scholar]
- Price MN, Dehal PS, Arkin AP. FastTree 2—approximately maximum-likihood trees for large alignments. PLoS One. 2010;5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reti KL, Thomas MC, Yanke LJ, Selinger LB, Inglis GD. Effect of antimicrobial growth promoter administration on the intestinal microbiota of beef cattle. Gut Pathog. 2013;5:8. doi: 10.1186/1757-4749-5-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saatchi M, Beever JE, Decker JE, Faulkner DB, Freetly HC, Hansen SL, et al. QTLs associated with dry matter intake, metabolic mid-test weight, growth and feed efficiency have little overlap across 4 beef cattle studies. BMC Genom. 2014;15:1004. doi: 10.1186/1471-2164-15-1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheppach W, Bartram P, Richter A. Management of diversion colitis, pouchitis and distal ulcerative colitis. In: Cummings JH, Rombeau JL, Sakata T, editors. Physiological and clinical aspects of short-chain fatty acids. Great Britain: Cambridge University Press; 1995. pp. 353–360. [Google Scholar]
- Shanks OC, Kelty CA, Archibeque S, Jenkins M, Newton RJ, McLellan SL, et al. Community structures of fecal bacteria in cattle from different animal feeding operations. Appl Environ Microbiol. 2011;77:2992–3001. doi: 10.1128/AEM.02988-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman EL, Nkrumah JD, Moore SS. Whole genome single nucleotide polymorphism associations with feed intake and feed efficiency in beef cattle. J Anim Sci. 2010;88:16–22. doi: 10.2527/jas.2008-1759. [DOI] [PubMed] [Google Scholar]
- Tan ZL, Lu DX, Hu M, Niu WY, Han CY, Ren XP, et al. Effect of dietary structural to nonstructural carbohydrate ratio on rumen degradability and digestibility of fiber fractions of wheat straw in sheep. Asian Australas J Anim Sci. 2002;15:1591–1598. doi: 10.5713/ajas.2002.1591. [DOI] [Google Scholar]
- Toivonen RK, Emani R, Munukka E, Rintala A, Laiho A, Pietilä S, et al. Fermentable fibres condition colon microbiota and promote diabetogenesis in NOD mice. Diabetologia. 2014;57:2183–2192. doi: 10.1007/s00125-014-3325-6. [DOI] [PubMed] [Google Scholar]
- Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weimer PJ, Stevenson DM, Mantovani HC, Man SLC. Host specificity of the ruminal bacterial community in the dairy cow following near-total exchange of ruminal contents. J Dairy Sci. 2010;93:5902–5912. doi: 10.3168/jds.2010-3500. [DOI] [PubMed] [Google Scholar]
- Wells JE, Shackelford SD, Berry ED, Kalchayanand N, Guerini MN, Varel VH, et al. Prevalence and level of Escherichia coli O157:H7 in feces and on hides of feedlot steers fed diets with or without wet distillers grains with solubles. J Food Prot. 2009;72:1624–1633. doi: 10.4315/0362-028x-72.8.1624. [DOI] [PubMed] [Google Scholar]
- Williams AG, Coleman GS. The rumen protozoa. In: Hobson PN, Stewart CS, editors. The rumen microbial ecosystem. 2. New York: Elsevier Applied Science; 1997. pp. 73–120. [Google Scholar]
- Wirsenius S, Azar C, Berndes G. How much land is needed for global food production under scenarios of dietary changes and livestock productivity increases in 2030? Agric Syst. 2010;103:621–638. doi: 10.1016/j.agsy.2010.07.005. [DOI] [Google Scholar]
- Yu Z, Morrison M. Improved extraction of PCR-quality community DNA from digesta and fecal samples. Biotechniques. 2004;36:808–813. doi: 10.2144/04365ST04. [DOI] [PubMed] [Google Scholar]