

Abstract
Cleft palate, including complete or incomplete cleft palates, soft palate clefts, and submucosal cleft palates, is the most frequent congenital craniofacial anomaly in humans. Multifactorial conditions, including genetic and environmental factors, induce the formation of cleft palates. The process of palatogenesis is temporospatially regulated by transcription factors, growth factors, extracellular matrix proteins, and membranous molecules; a single ablation of these molecules can result in a cleft palate in vivo. Studies on knockout mice were reviewed in order to identify genetic errors that lead to cleft palates. In this review, we systematically describe these mutant mice and discuss the molecular mechanisms of palatogenesis.
Keywords: Tbx1, Submucosal cleft palate, Incomplete cleft palate, Palatal shelf, Palatogenesis, Knockout mice
Core tip: Cleft lip and/or palate is one of the most frequent congenital craniofacial anomalies observed. Multifactorial conditions, including genetic and environmental factors, induce the formation of cleft palates. We screened knockout mice with cleft palate phenotypes and observed approximately 180 mice with the anomaly. In order to understand the molecular regulatory mechanisms of palatogenesis and to identify genetic errors that lead to cleft palates, we aimed to review studies performed using knockout mice with cleft palates.
INTRODUCTION
Cleft lip and/or palate (CL/P) is the most frequent congenital craniofacial anomaly observed in humans, with an incidence of 1 per 700 births worldwide[1]. Furthermore, 55% of the patients with CL/P are reported to have a multiple malformation syndrome[2]. CL/P involves a multifactorial etiology, both genetic and environmental. Teratogens that cause CL/P in humans include common environmental exposures, such as alcohol, smoking, infections, dioxin, estrogen, retinoic acid, and altitude (reviewed by Murray[1]). The offspring of parents with CL/P present a higher incidence of CL/P than those without a family history[1]. Gene-environment interactions for non-syndromic CL/P have also been reported[1]. Cleft palate (CP) cases include complete CP, incomplete CP, and soft palate clefts. The mildest form of cleft palates is the soft palate cleft or bifid uvula because the initial palatal fusion occurs in the anterior region of secondary palatal shelves. Incomplete CP and soft palate clefts can manifest together with submucosal CP. This review focuses on studies performed using knockout mice with CP, aiming to clarify the molecular regulatory mechanisms of palatogenesis and to identify genetic errors underlying mammalian cleft palates.
MAMMALIAN PALATOGENESIS
The palate is formed with the primary and secondary palate. The primary palate is derived from the frontonasal prominence and becomes a small anterior part of the adult hard palate. The secondary palatal shelves extend bilaterally from the internal aspects of the maxillary prominences and will become the adult hard and soft palates. The process of palatogenesis consists of several stages: palatal shelf formation, elevation, and midline fusion of the palatal shelves (Figure 1). The secondary palatal shelves develop between embryonic day (E) 11.5 and 12.5 in the mouse embryo (Figure 1A). At E13.5, the palatal shelves grow downward on each side of the tongue (Figure 1B). As the jaws develop, the tongue descends and the palatal shelves elevate to a horizontal position above the dorsum of the tongue (E14). Continuing their growth, the bilateral palatal shelves meet at the midline and fuse between E14.5 and E15.5 (Figure 1C).
Figure 1.

Palatogenesis in mice. Hematoxylin and eosin staining of coronal sections of the head of a wild-type mouse at embryonic day (E) 12.5 (A), E13.5 (B), and E14.5 (C, D). A: Mouse palatal shelves (p) develop from the maxillary prominences; B: By E13.5, the palatal shelves grow downward on each side of the tongue (t); C and D: At E14.5, the palatal shelves face each other along the midline above the tongue and fuse, separating the oral cavity (oc) from the nasal cavity (nc). The arrow in (D) indicates the medial edge epithelial (MEE) cells that constitute the midline epithelial seam. All animal experimental procedures were reviewed and approved by the Institutional Animal Care and Use Committees of the University of Texas Southwestern Medical Center and Tokyo Medical and Dental University. mes: Mesenchyme; epi: Epithelium.
The palatal shelves are composed of the neural crest-derived mesenchyme and ectoderm-derived epithelia, which cover the palatal mesenchyme (Figure 1D). Both elevation and fusion of the secondary palatal shelves occur in the midline from anterior to posterior. The secondary palatal shelves also fuse with the primary palate, separating the oral and nasal cavities. The anterior two-thirds of the palate forms the hard palate with neural crest-derived palatal bones (Figure 2A). The posterior one-third of the palate forms the bone-free soft palate and is involved in the palatopharyngeal sealing. Disruption at any stage of the formation, elevation, growth, or fusion of the secondary palatal shelves results in CP[3].
Figure 2.
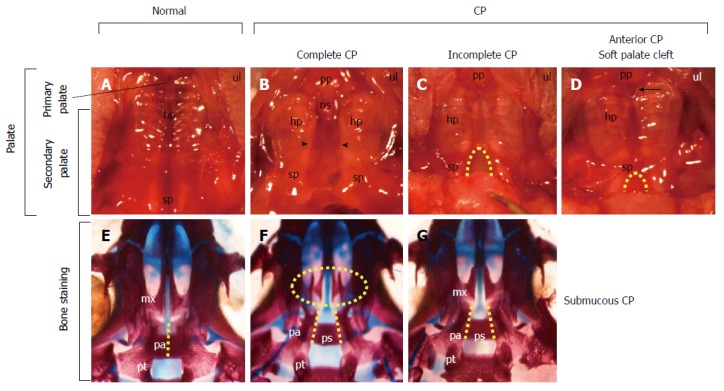
View of the palate from wild-type and Tbx1-/- mice with cleft palates. A-D: Ventral view of the maxilla of newborn wild-type (A) and Tbx1-/- mice with cleft palates (B-D). The palate consists of the primary palate (pp) and the secondary palate (sp), which consists of a hard palate (hp) and a soft palate (sp) (A). Tbx1-/- mice show complete cleft palate (CP) (arrowheads in B), incomplete CP (dashed line in C), and soft palate clefts associated with anterior CP (dashed line in D). An anterior CP (an arrow in D) is present at the junction between the primary palate and secondary palate, while the posterior palate remains fused; E-G: Ventral view of the cranial base of newborn wild-type (E) and Tbx1-/- mice (F, G) stained with alizarin red for mineralized bone and alcian blue for cartilage. Fusion of the bilateral palatal bones (pa) observed in the wild-type (dashed line in E) is absent in Tbx1-/- mice (dashed lines in F, G). The palatal shelves in the maxilla (mx) of Tbx1-/- mice with complete CP (oval dashed line in F) failed to grow toward the midline. Note the visible presphenoid bone (ps) associated with CP (F, G). Modified and used with permission from Funato et al[4]. ns: Nasal septum; pt: Pterygoid bone.
MOUSE MODELS FOR STUDYING THE MOLECULAR MECHANISMS OF PALATAL DEVELOPMENT
Major advances have been achieved regarding the molecular mechanisms that regulate palatal development using genetically engineered mice. Deletions in many genes of mice result in CP and the most frequent phenotype seen is complete CP (Figure 2B). Uniquely, Tbx1-/- mice present various phenotypes of CP[4], including complete CP (Figure 2B), incomplete CP (Figure 2C), and anterior CP (Figure 2D). Bone staining showed that some mice potentially had a submucosal CP (Figure 2G). These observations are in agreement with various CP phenotypes in humans.
In order to elucidate the molecular pathogenesis of CL/P, we conducted a literature search on PubMed (http://www.ncbi.nlm.nih.gov/pubmed) and the Mouse Genome Informatics (MGI) from the Jackson Laboratory (http://www.informatics.jax.org). The search was limited to knockout mice with CP and excluded the teratogen-induced CP (Table 1). We also investigated diseases/syndromes using the Online Mendelian Inheritance in Man (OMIM) (http://omim.org). Not all the molecules involved in cleft palates in mice are correlated to CL/P in human (Table 1). When genes in Table 1 were analyzed by biological function using BioCarta (http://www.biocarta.com) and the Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway Database (http://www.genome.jp/kegg/pathway.html), the transforming growth factor (TGF), hedgehog, Wnt, fibroblast growth factor (FGF), and mitogen-activated protein kinase (MAPK) signaling pathways were found to be critical in palatogenesis (Table 2). When genes were analyzed by molecular function using the PANTHER (Protein ANalysis THrough Evolutionary Relationships) database (http://pantherdb.org)[5], the most significantly enriched molecular function was the “transcription factor”, especially the “homeobox transcription factors” (Table 2 and Figure 3).
Table 1.
Molecules involved in cleft palate in mice
|
Knockout mice with cleft palates |
Humans |
||||
| Gene/category | Protein | Ref. | OMIM | Syndrome | CL/P |
| Growth factors, antagonist, and receptors | |||||
| Acvr1/Alk2 | Activin A receptor, type I | [33] | 1102576 | Fibrodysplasia ossificans progressiva | nr |
| (Wnt1-Cre-mediated ablation) | |||||
| Acvr2a | Activin A receptor, type IIA | [34] | 1102581 | nr | nr |
| Bmp4 | Bone morphogenetic protein 4 | [35] | 1112262 | Microphthalmia, syndromic 6 | r |
| Orofacial cleft 11 | |||||
| Bmp7 | Bone morphogenetic protein 7 | [36] | 1112267 | nr | nr |
| Bmpr1a/Alk3 | Bone morphogenetic protein receptor, type IA | [35] | 1601299 | Juvenile polyposis syndrome, | nr |
| (Nestin-Cre-mediated ablation) | Polyposis syndrome | ||||
| Chrd | Chordin | [37] | 1603475 | nr | nr |
| Ctgf | Connective tissue growth factor | [38] | 1121009 | nr | nr |
| Edn1 | Endothelin 1 | [39] | 2131240 | Auriculocondylar syndrome 3 | r |
| Egfr | Epidermal growth factor receptor | [17] | 1131550 | nr | nr |
| Fgf9 | Fibroblast growth factor 9 | [40] | 1600921 | uc | nr |
| Fgf10 | Fibroblast growth factor 10 | [13,41] | 1602115 | Aplasia of lacrimal and salivary glands | nr |
| LADD syndrome | |||||
| Fgf18 | Fibroblast growth factor 18 | [42,43] | 1603726 | nr | nr |
| Fgfr1 | Fibroblast growth factor receptor 1 | [44] | 1136350 | Nonsyndromic cleft lip/palate | r |
| Hartsfield syndrome | |||||
| Hypogonadotropic hypogonadism 2 | |||||
| Pfeiffer syndrome | |||||
| Fgfr2 | Fibroblast growth factor receptor 2 | [13,45] | 1176943 | Apert syndrome | r |
| (knockout) (Krt14-Cre-mediated ablation) | Crouzon syndrome | ||||
| Pfeiffer syndrome | |||||
| Saethre-Chotzen syndrome | |||||
| Fst | Follistatin | [46] | 1136470 | nr | nr |
| Gabrb3 | Gamma-aminobutyric acid A receptor, beta 3 | [47] | 1137192 | Epilepsy, childhood absence, susceptibility to, 5 | r |
| Gdf11/Bmp11 | Growth differentiation factor 11 | [48] | 1603936 | nr | nr |
| Gpr124 | G protein-coupled receptor 124 | [49] | 1606823 | nr | nr |
| Inhba | Inhibin, beta A/activin A | [50] | 1147290 | nr | nr |
| Pdgfc | Platelet-derived growth factor C | [51] | 1608452 | nr | r [52] |
| Pdgfra | Platelet-derived growth factor receptor, alpha polypeptide | [53,54] | 1173490 | Gastrointestinal stromal tumor, somatic | r |
| (knockout) (Wnt1-Cre-mediated ablation) | Hypereosinophilic syndrome, idiopathic, resistant to imatinib | ||||
| Tgfb2 | Transforming growth factor, beta 2 | [55] | 1190220 | Loeys-Dietz syndrome, type 4 | r |
| Tgfb3 | Transforming growth factor, beta 3 | [15,16,18] | 1190230 | Arrhythmogenic right ventricular dysplasia 1 | r |
| Tgfbr1/Alk5 | Transforming growth factor, beta receptor I | [56,57] | 1190181 | Loeys-Dietz syndrome, type 1 | r |
| (Wnt1-Cre-, and Nestin-Cre-mediated ablation) | |||||
| Tgfbr2 | Transforming growth factor, beta receptor II | [12,58] | 1190182 | Loeys-Dietz syndrome, type 2 | r |
| (Wnt1-Cre-, and KRT14-Cre-mediated ablation) | |||||
| Vegfa | Vascular endothelial growth factor A | [59] | 2192240 | nr | nr |
| Membrane proteins | |||||
| Ceacam1 | Carcinoembryonic antigen-related cell adhesion molecule 1 | [60] | 1109770 | nr | nr |
| Efna5 | Ephrin A5 | [61] | 1601535 | nr | nr |
| Efnb1 | Ephrin B1 | [62] | 1300035 | Craniofrontonasal dysplasia | r |
| Efnb2 | Ephrin B2 | [63] | 1600527 | nr | nr |
| Fzd2 | Frizzled class receptor 2 | [64] | 1600667 | nr | nr |
| Itga5 | Integrin alpha 5 | [65,66] | 1135620 | nr | nr |
| (knockout) (Mesp1-Cre-mediated ablation) | |||||
| Itgb1 | Integrin beta 1 | [67] | 1135630 | nr | nr |
| (Col2a1-Cre-mediated ablation) | |||||
| Itgb8 | Integrin beta 8 | [68] | 1604160 | nr | nr |
| Jag1 | Jagged1 | [69] | 2601920 | Alagille syndrome | nr |
| (Wnt1-Cre-mediated ablation) | |||||
| Jag2 | Jagged2 | [70] | 1602570 | nr | nr |
| Kcnj2 | Potassium inwardly-rectifying channel, subfamily J, member 2 | [71] | 1600681 | Andersen syndrome | r |
| Atrial fibrillation, familial, 9 | |||||
| Short QT syndrome 3 | |||||
| Lrp6 | Low density lipoprotein receptor-related protein 6 | [72] | 1603507 | nr | nr |
| Ror2 | Receptor tyrosine kinase-like orphan receptor 2 | [73] | 1602337 | Robinow syndrome, autosomal recessive | r |
| Brachydactyly, type B1 | |||||
| Ryk | Receptor-like tyrosine kinase | [74] | 1600524 | nr | nr |
| Ryr1 | Ryanodine receptor 1, skeletal muscle | [75] | 1180901 | Central core disease | nr |
| King-Denborough syndrome | |||||
| Minicore myopathy with external ophthalmoplegia | |||||
| Sc5d/Sc5dl | Sterol-C5-desaturase (fungal ERG3, delta-5-desaturase) homolog (S. cerevisae) | [76] | 1602286 | Lathosterolosis | nr |
| Shh | Sonic hedgehog | [13,77] | 1600725 | Holoprosencephaly-3 | r |
| (KRT14-Cre-, and Sox2-Cre-mediated ablation) | Microphthalmia with coloboma 5 | ||||
| Single median maxillary central incisor | |||||
| Smo/Smoh | Smoothened, frizzled class receptor | [78] | 1601500 | Basal cell carcinoma, somatic | nr |
| (Wnt1-Cre-mediated ablation) | |||||
| Tctn2 | Tectonic family member 2 | [79] | 1613846 | Meckel syndrome 8 | r |
| Wls/Gpr177 | Wntless homolog (Drosophila) | [80] | 1611514 | nr | nr |
| (Wnt1-Cre-mediated ablation) | |||||
| Wnt5a | Wingless-type MMTV integration site family, member 5A | [81] | 1164975 | Robinow syndrome, autosomal dominant | r |
| Wnt9b | Wingless-type MMTV integration site family, member 9B | [82,83] | 1602864 | nr | nr |
| (knockout) (Foxg1-Cre-mediated ablation) | |||||
| Transcription and nucleolar factors | |||||
| Alx1 | Aristaless-like homeobox 1 | [84] | 1601527 | Frontonasal dysplasia 3 | r |
| Alx3 | Aristaless-like homeobox 3 | [85] | 1606014 | Frontonasal dysplasia 1 | r |
| Alx4 | Aristaless-like homeobox 4 | [85] | 1605420 | Frontonasal dysplasia 2 | Cleft alae nasi |
| Parietal foramina 2 | |||||
| Craniosynostosis 5 | |||||
| Anp32b | Acidic (leucine-rich) nuclear phosphoprotein 32 family, member B | [86] | nr | nr | nr |
| Arid5 | AT-rich interaction domain-containing protein 5A | [87] | 1611583 | nr | nr |
| Asxl1 | Additional sex combs like 1 | [88] | 1612990 | Bohring-Opitz syndrome | r |
| Myelodysplastic syndrome, somatic | |||||
| Barx1 | BarH-like homeobox 1 | [89] | 1603260 | nr | nr |
| Cdc42 | Cell division cycle 42 | [90] | 1116952 | nr | nr |
| (Prrx1-Cre-mediated ablation) | |||||
| Chd7 | Chromodomain helicase DNA binding protein 7 | [91,92] | 1608892 | CHARGE syndrome | r |
| (heterozygotes) (Wnt1-Cre-mediated ablation) | Hypogonadotropic hypogonadism 5 with or without anosmia | ||||
| Cited2 | CBP/p300-interacting transactivator, with Glu/Asp-rich C-terminal domain, 2 | [93] | 1602937 | Atrial septal defect 8 | nr |
| Ventricular septal defect 2 | |||||
| Crebbp/Cbp | CREB binding protein | [94] | 1600140 | Rubinstein-Taybi syndrome | nr |
| Dlx1 | Distal-less homeobox 1 | [95] | 1600029 | nr | nr |
| Dlx2 | Distal-less homeobox 2 | [95] | 1126255 | nr | nr |
| Dlx5 | Distal-less homeobox 5 | [96,97] | 1600028 | Split-hand/foot malformation 1 with sensorineural hearing loss | r |
| Dph1/Ovca1 | DPH1 homolog (S. cerevisiae) | [98] | 1603527 | nr | nr |
| Eya1 | Eyes absent 1 homolog (Drosophila) | [99] | 1601653 | Branchiootic syndrome 1 | r |
| Branchiootorenal syndrome 1, with or without cataracts | |||||
| Anterior segment anomalies with or without cataract | |||||
| Foxc2/Mfh1 | Forkhead box C2 | [100] | 1602402 | Lymphedema-distichiasis syndrome | r |
| Foxd3 | Forkhead box D3 | [101] | 1611539 | uc | nr |
| (Wnt1-Cre-mediated ablation) | |||||
| Foxe1/Titf2/Fkhl15 | Forkhead box E1 | [102] | 1602617 | Bamforth-Lazarus syndrome | r |
| Nonsyndromic orofacial clefting | |||||
| Foxf2 | Forkhead box F2 | [103] | 1603250 | nr | nr |
| Gbx2 | Gastrulation brain homeobox 2 | [104] | 1601135 | nr | nr |
| Gli2 | GLI family zinc finger 2 | [8] | 1165230 | Culler-Jones syndrome | r |
| Holoprosencephaly-9 | |||||
| Gli3 | GLI family zinc finger 3 | [105] | 1165240 | Greig cephalopolysyndactyly syndrome | r |
| Pallister-Hall syndrome | |||||
| Gsc | Goosecoid homeobox | [106] | 1138890 | Short stature, auditory canal atresia, mandibular hypoplasia, skeletal abnormalities | nr |
| Hand2/dHand | Heart and neural crest derivatives expressed 2 | [107] | 1602407 | nr | nr |
| Hic1 | Hypermethylated in cancer 1 | [108] | 1603825 | nr | nr |
| Hoxa2 | Homeobox A2 | [19] | 1604685 | Microtia with or without hearing impairment (AD) | r |
| Microtia, hearing impairment, and cleft palate (AR) | |||||
| Irf6 | Interferon regulatory factor 6 | [109,110] | 1607199 | van der Woude syndrome | r |
| Orofacial cleft 6 | |||||
| Popliteal pterygium syndrome 1 | |||||
| Jmjd6/Ptdsr | Jumonji domain containing 6 | [111] | 1604914 | nr | nr |
| Kat6a/Moz/Myst3 | K (lysine) Acetyltransferase 6A | [112] | 1601408 | nr | nr |
| Lhx7 | LIM homeobox gene 7 | [113] | nr | nr | nr |
| Lhx8 | LIM homeobox gene 8 | [11] | 1604425 | nr | r |
| Luzp1 | Leucine zipper protein 1 | [114] | 1601422 | nr | nr |
| Mef2c | MADS box transcription enhancer factor 2 | [115] | 1600662 | Chromosome 5q14.3 deletion syndrome | nr |
| (Wnt1-Cre-mediated ablation) | Mental retardation, stereotypic movements, epilepsy, and/or cerebral malformations | ||||
| Meox2 | Mesenchyme homeobox 2 | [116] | 1600535 | nr | nr |
| Mn1 | Meningioma 1 | [117] | 1156100 | Meningioma | nr |
| Mnt | Max binding protein | [118] | 1603039 | nr | nr |
| Msx1 | Msh homeobox 1 | [10,23] | 1142983 | Ectodermal dysplasia 3, Witkop type | r |
| Orofacial cleft 5 | |||||
| Tooth agenesis, selective, 1, with or without orofacial cleft | |||||
| Msx2 | Msh homeobox 2 | [119] | 1123101 | Craniosynostosis, type 2 | r |
| (missense mutation) | Parietal foramina 1 | ||||
| Parietal foramina with cleidocranial dysplasia | |||||
| Nabp2/Obfc2b/hSSB1 | Nucleic acid binding protein 2 | [120,121] | 1612104 | nr | nr |
| Osr2 | Odd-skipped related transcription factor 2 | [9] | 1611297 | nr | r |
| Pak1ip1 | PAK1 interacting protein 1 | [122] | 1607811 | nr | nr |
| Pax9 | Paired box gene 9 | [6] | 1167416 | Tooth agenesis, selective, 3 | nr |
| Pbx1 | Pre B cell leukemia homeobox 1 | [83] | 1176310 | Leukemia, acute pre-B-cell | nr |
| Pds5a | PDS5, regulator of cohesion maintenance, homolog A (S. cerevisiae) | [123] | 1613200 | nr | nr |
| Phc1/Rae28 | Polyhomeotic homolog 1 | [124] | 1602978 | uc | nr |
| Pitx1 | Paired-like homeodomain 1 | [7,125] | 1602149 | Clubfoot, congenital, with or without deficiency of long bones and/or mirror-image polydactyly | r |
| Liebenberg syndrome | |||||
| Pitx2 | Paired-like homeodomain 2 | [126] | 1601542 | Axenfeld-Rieger syndrome, type 1 | nr |
| Iridogoniodysgenesis, type 2 | |||||
| Peters anomaly | |||||
| Pnn | Pinin | [127] | 1603154 | nr | nr |
| Prdm16 | PR domain containing 16 | [128] | 1605557 | Cardiomyopathy, dilated, 1LL | nr |
| Left ventricular noncompaction 8 | |||||
| Prrx1/Prx1/Mhox | Paired related homeobox 1 | [129] | 1167420 | Agnathia-otocephaly complex | r |
| Ptch1/Ptc1 | Patched 1 | [130] | 1601309 | Basal cell nevus syndrome | r |
| (Wnt1-Cre-mediated ablation) | (Gorlin syndrome) | ||||
| Holoprosencephaly type 7 | |||||
| Pygo2 | Pygopus 2 | [131] | 1606903 | nr | nr |
| (CMV-Cre-mediated ablation) | |||||
| Rax | Retina and anterior neural fold homeobox | [132] | 1601881 | Microphthalmia, isolated 3 | nr |
| Recql4 | RecQ protein-like 4 | [133] | 1603780 | Baller-Gerold syndrome | r |
| RAPADILINO syndrome | |||||
| Rothmund-Thomson syndrome | |||||
| Runx2 | Runt-related transcription factor 2 | [134] | 1600211 | Cleidocranial dysplasia | r |
| Sall3 | Spalt-like transcription factor 3 | [24] | 1605079 | nr | nr |
| Satb2 | SATB homeobox 2 | [135,136] | 1608148 | Glass syndrome | r |
| Shox2 | Short stature homeobox 2 | [22] | 1602504 | nr | nr |
| Sim2 | Single-minded family bHLH transcription factor 2 | [137] | 1600892 | nr | nr |
| Smad4 (Osr2-Cre-mediated ablation) | SMAD family member 4 | [138] | 1600993 | Juvenile polyposis/hereditary hemorrhagic telangiectasia syndrome | nr |
| Myhre syndrome | |||||
| Smad7 | SMAD family member 7 | [139] | 1602932 | uc | nr |
| Snai2 | Snail family zinc finger 2 | [140] | 1602150 | Piebaldism | nr |
| Waardenburg syndrome, type 2D | |||||
| Sox5 | SRY (sex determining region Y)-box 5 | [141] | 1604975 | nr | nr |
| Sox9 (heterozygous) | SRY (sex determining region Y)-box 9 | [142,143] | 1608160 | Acampomelic campomelic dysplasia | r |
| (Wnt1-Cre-mediated ablation) | |||||
| Sox11 | SRY (sex determining region Y)-box 11 | [144] | 1600898 | Mental retardation, autosomal dominant, 27 | nr |
| Sp8 | Sp8 transcription factor | [145] | 1608306 | nr | nr |
| Tshz1 | Teashirt zinc finger family member 1 | [146] | 1614427 | Aural atresia, congenital | nr |
| Tbx1 | T-box 1 | [4,147] | 1602054 | DiGeorge syndrome | r |
| (knockout) (KRT14-Cre-mediated ablation) | Velocardiofacial syndrome | ||||
| Conotruncal anomaly face syndrome | |||||
| Tetralogy of Fallot | |||||
| Tbx2 | T-box 2 | [148] | 1600747 | nr | nr |
| Tbx22 | T-box 22 | [149] | 1300307 | Cleft palate with ankyloglossia | r |
| submucous cleft palate (SMCP) | |||||
| Tcof1 | Treacher Collins-Franceschetti syndrome 1 | [150] | 1606847 | Treacher-Collins syndrome | r |
| (heterozygous) | |||||
| Tfap2A | Transcription factor AP-2 alpha | [151] | 1107580 | Branchio-oculo-facial syndrome | r |
| (Wnt1-Cre-mediated ablation) | |||||
| Trp63/Tp63 | Transformation related protein p63 | [152] | 1603273 | Ectrodactyly, ectodermal dysplasia, and cleft lip/palate syndrome 3 | r |
| Orofacial cleft 8 | |||||
| Hay-Wells syndrome | |||||
| Limb-mammary syndrome | |||||
| Vax1 | Ventral anterior homeobox 1 | [153] | 1604294 | Microphthalmia, syndromic 11 | r |
| Whsc1 | Wolf-Hirschhorn syndrome candidate 1 | [154] | 1602952 | nr | nr |
| Zeb1 | Zinc finger E-box binding homeobox 1 | [155] | 1189909 | Corneal dystrophy | nr |
| Zfp640/Mzf6d | Zinc finger protein 640 | [87] | nr | nr | nr |
| Zic3 | Zinc finger protein of the cerebellum 3 | [156] | 1300265 | Congenital heart defects, nonsyndromic | r |
| Heterotaxy, visceral, 1 | |||||
| VACTERL association | |||||
| Cytoplasmic proteins | |||||
| Akap8/Akap95 | A kinase (PRKA) anchor protein 8 | [157] | 1604692 | nr | nr |
| Apaf1 | Apoptotic peptidase activating factor 1 | [158] | 1602233 | nr | nr |
| B9d1 | B9 protein domain 1 | [159] | 1614144 | Meckel syndrome 9 | nr |
| Cask | Calcium/calmodulin-dependent serine protein kinase | [160] | 1300172 | FG syndrome 4 | r |
| Mental retardation and microcephaly with pontine and cerebellar hypoplasia | |||||
| Cdkn1c/p57kip2 | Cyclin-dependent kinase inhibitor 1C | [161,162] | 1600856 | Beckwith-Wiedemann syndrome | r |
| IMAGe syndrome | |||||
| Chuk/Ikk1/Tcf16 | Conserved helix-loop-helix ubiquitous kinase | [163] | 1600664 | Cocoon syndrome | nr |
| Crk | v-crk sarcoma virus CT10 oncogene homolog | [164] | 1164762 | nr | nr |
| Ctnnb1 | Catenin (cadherin-associated protein), beta 1, | [165,166] | 1116806 | Mental retardation, autosomal dominant 19 | nr |
| (KRT14-Cre-mediated ablation) | |||||
| Cyp26B1 | Cytochrome P450, family 26, subfamily b, polypeptide 1 | [167] | 1605207 | Craniosynostosis with radiohumeral fusions and other skeletal and craniofacial anomalies | nr |
| Cyp51 | Cytochrome P450, family 51 | [168] | 1601637 | nr | nr |
| Dhcr7 | 7-dehydrocholesterol reductase | [169,170] | 1602858 | Smith-Lemli-Opitz syndrome | r |
| Dhrs3 | Dehydrogenase/reductase (SDR family) member 3 | [171,172] | 1612830 | nr | nr |
| Dicer1 | Dicer 1, ribonuclease type III | [29] | 1606241 | Rhabdomyosarcoma, embryonal, 2 | nr |
| (Pax2-Cre-mediated ablation) | Goiter, multinodular 1 | ||||
| Pleuropulmonary blastoma | |||||
| Dlg1/Dlgh/Sap97 | Discs large 1 | [173] | 1601014 | nr | nr |
| Fuz | Fuzzy planar cell polarity protein | [174] | 1610622 | Neural tube defects | nr |
| Gab1 | Growth factor receptor bound protein 2-associated protein 1 | [175] | 1604439 | nr | nr |
| Gad1/Gad67 | Glutamate decarboxylase 1 | [176,177] | 1605363 | Cerebral palsy, spastic quadriplegic, 1 | r |
| Glce | Glucuronyl C5-epimerase | [178] | 1612134 | nr | nr |
| Glg1 | Golgi apparatus protein 1 | [179] | 1600753 | nr | nr |
| Grb2 | Growth factor receptor bound protein 2 | [180] | 1108355 | nr | nr |
| Gsk3b | Glycogen synthase kinase 3 beta | [181] | 1605004 | nr | nr |
| Hs2st1 | Heparan sulfate 2-O-sulfotransferase 1 | [182] | 1604844 | nr | nr |
| Hspb11/Ift25 | Heat shock protein family B (small), member 11 | [183] | 1604844 | nr | nr |
| Ilk | Integrin linked kinase | [184] | 1602366 | nr | nr |
| (Col2a1-Cre-mediated ablation) | |||||
| Impad1/Jaws | Inositol monophosphatase domain containing 1 | [185] | 1614010 | Chondrodysplasia with joint dislocations, GRAPP type | r |
| Inpp5e | Inositol polyphosphate-5-phosphatase E | [186] | 1613037 | Joubert syndrome 1 | nr |
| Mental retardation, truncal obesity, retinal dystrophy, and micropenis | |||||
| Kif3a | Kinesin family member 3A | [187] | 1604683 | nr | nr |
| (Wnt1-Cre-mediated ablation) | |||||
| Map3k7/Tak1 | Mitogen-activated protein kinase kinase kinase 7 | [188,189] | 1602614 | nr | nr |
| (Wnt1-Cre-mediated ablation) | |||||
| Nprl3 | Nitrogen permease regulator-like 3 | [190] | 1600928 | nr | nr |
| Ofd1 | Oral-facial-digital syndrome 1 gene homolog (human) | [191] | 1300170 | Joubert syndrome 10 | r |
| (CAG-Cre-mediated ablation) | Orofaciodigital syndrome I | ||||
| Simpson-Golabi-Behmel syndrome, type 2 | |||||
| Pdss2 | Prenyl (solanesyl) diphosphate synthase, subunit 2 | [192] | 1610564 | Coenzyme Q10 deficiency, primary, 3 | nr |
| (Pax2-Cre-mediated ablation) | |||||
| Piga | Phosphatidylinositol glycan anchor biosynthesis, class A | [193] | 1311770 | Multiple congenital anomalies-hypotonia-seizures syndrome 2 Paroxysmal nocturnal hemoglobinuria, somatic | nr |
| (EIIa-Cre-mediated ablation) | |||||
| Pkdcc/Vlk | Protein kinase domain containing, cytoplasmic | [194,195] | 1614150 | nr | nr |
| (Sox2-Cre-mediated ablation) | |||||
| Prickle1 | Prickle homolog 1 | [196] | 1608500 | Epilepsy, progressive myoclonic 1B | nr |
| Rad23b | RAD23b homolog (S. cerevisiae) | [197] | 1600062 | nr | nr |
| Rspo2 | R-spondin 2 homolog (Xenopus laevis) | [198,199] | 1610575 | nr | nr |
| Schip1 | Schwannomin interacting protein 1 | [87] | nr | nr | nr |
| Sdccag8 | Serologically defined colon cancer antigen 8 | [200] | 1613524 | Bardet-Biedl syndrome 16 | nr |
| Senior-Loken syndrome 7 | |||||
| Slc32a1/Viaat | Solute carrier family 32, member 1 | [201,202] | nr | nr | nr |
| Spry1 | Sprouty homolog 1 | [203] | 1602465 | nr | nr |
| (Wnt1-Cre-mediated ablation) | |||||
| Spry2 | Sprouty homolog 2 | [204] | 1602466 | nr | nr |
| Sumo1 | SMT3 suppressor of mif two 3 homolog 1 (yeast) | [205] | 1601912 | Orofacial cleft 10 | r |
| (heterozygous) | |||||
| Ugdh | UDP-glucose dehydrogenase | [206] | 1603370 | nr | nr |
| (Wnt1-Cre-mediated ablation) | |||||
| Wdpcp | WD repeat containing planar cell polarity effector | [207] | 1613580 | uc | nr |
| Extracellular proteins | |||||
| Col2a1 | Collagen, type II, alpha 1 | [208] | 2120140 | Achondrogenesis, type II | r |
| Stickler syndrome, type I | |||||
| Kniest dysplasia | |||||
| Hspg2 | Heparan sulfate proteoglycan 2, perlecan | [209,210] | 1142461 | Dyssegmental dysplasia | nr |
| Schwartz-Jampel syndrome, type 1 | |||||
| Serpinh1/Hsp47 | Serpine peptidase inhibitor, clade H, member 1 | [211] | 1600943 | Osteogenesis imperfecta, type X | nr |
| (Col2a1-Cre-mediated ablation) | |||||
| Smoc1 | SPARC related modular calcium binding 1 | [212] | 1608488 | Microphthalmia with limb anomalies | r |
Before an OMIM entry number indicates a gene;
Before an OMIM entry number indicates that the entry includes a description of a gene of known sequence and a phenotype. OMIM: Online Mendelian Inheritance in Man (http://omim.org); CL/P: Cleft lip and/or palate; r: Reported; nr: Not reported; uc: Unclarified.
Table 2.
Classification of genes associated with cleft palate in mice
| Genes | |
| Signaling pathway | |
| TGF-beta signaling pathway | Acvr1/Alk2, Acvr2a, Bmp41, Bmp7, Bmpr1a/Alk3, Cdc42, Chrd, Crebbp/Cbp, Cited2, Foxc2/Mfh11, Foxd3, Foxe1/Titf2/Fkhl151, Foxf2, Fst, Inhba, Gdf11/Bmp11, Map3k7/Tak1, Pitx2, Smad4, Smad7, Tgfb21, Tgfb31, Tgfbr1/Alk51, Tgfbr21 |
| Hedgehog signaling pathway | Bmp41, Bmp7, Crebbp/Cbp, Gli21, Gli31, Gsk3b, Ptch1/Ptc11, Shh1, Smo/Smoh, Wnt5a1, Wnt9b |
| Wnt signaling pathway | Acvr1/Alk2, Ctnnb1, Crebbp/Cbp, Edn11, Fzd2, Gsk3b, Lrp6, Map3k7/Tak1, Prickle1, Smad4, Smo/Smoh, Wnt5a1, Wnt9b |
| FGF signaling pathway | Fgf10, Fgf18, Fgf9, Fgfr11, Fgfr21, Grb2, Spry1, Spry2 |
| MAPK signaling pathway | Cdc42, Chuk/Ikk1/Tcf16, Egfr, Fgf10, Fgf18, Fgf9, Fgfr11, Fgfr21, Grb2, Map3k7/Tak1, Pdgfra1, Tgfb21, Tgfb31, Tgfbr1/Alk51, Tgfbr21, Crk, Itgb1 |
| Cytokine-cytokine receptor interaction | Acvr1/Alk2, Acvr2a, Bmp7, Bmpr1a/Alk3, Egfr, Inhba, Pdgfra1, Pdgfc1, Tgfb21, Tgfb31, Tgfbr1/Alk51, Tgfbr21, Vegfa |
| CBL mediated ligand- induced downregulation of EGF receptors | Egfr, Grb2, Pdgfra1 |
| Sprouty regulation of tyrosine kinase signals | Egfr, Grb2, Spry2, Spry1 |
| NFkB activation | Crebbp/Cbp, Chuk/Ikk1/Tcf16, Map3k7/Tak1, Smad4, Tgfbr1/Alk51, Tgfbr21 |
| Adherens junction | Crebbp/Cbp, Ctnnb1, Cdc42, Egfr, Fgfr11, Map3k7/ Tak1, Smad4, Snai2, Tgfbr1/Alk51, Tgfbr21 |
| Focal adhesion | Ctnnb1, Cdc42, Col2a11, Crk, Egfr, Gsk3b, Grb2, Itga5, Itgb1, Itgb8, Ilk, Pdgfra1, Pdgfc1, Vegfa |
| Steroid biosynthesis | Cyp51, Dhcr71, Sc5d/Sc5dl |
| Cell cycle | Crebbp/Cbp, Cdkn1c/p57kip21, Gsk3b, Smad4, Tgfb21, Tgfb31 |
| Regulation of actin cytoskeleton | Cdc42, Crk, Egfr, Fgf9, Fgf10, Fgf18, Fgfr11, Fgfr21, Itga5, Itgb1, Itgb8, Pdgfra1, Pdgfc1 |
| Axon guidance | Cdc42, Efna5, Efnb11, Efnb2, Gsk3b, Itgb1 |
| Endocytosis | Cdc42, Egfr, Fgfr21, Pdgfra1, Tgfbr1/Alk51, Tgfbr21 |
| Angiogenesis | Ctnnb1, Crk, Efnb11, Efnb2, Fgfr11, Fgfr21, Fzd2, Gsk3b, Grb2, Jag1, Jag2, Pdgfra1, Pdgfc1, Vegfa, Wnt5a1 |
| Family | |
| Homeobox protein | Alx11, Barx1, Alx31, Alx41, Dlx1, Dlx2, Dlx51, Gbx2, Gsc, Hoxa21, Msx11, Msx21, Pax9, Prrx11, Pitx11, Pitx2, Rax, Shox2, Vax11 |
| Tgf-beta receptor type I and II | Acvr1/Alk2, Acvr2a, Bmpr1a, Tgfbr1/Alk51, Tgfbr21 |
| Tgf-beta family | Bmp41, Bmp7, Gdf11, Inhba, Tgfb21, Tgfb31 |
| Tyrosine protein kinase | Egfr, Fgfr11, Fgfr21, Pdgfra1, Ror21, Ryk |
| Ephrin | Efna5, Efnb11, Efnb2 |
| Zinc finger protein | Gli21, Gli31, Zic31, Hic1, Snai2 |
| Forkhead protein | Foxc21, Foxd3, Foxe11, Foxf2 |
| T-box protein | Tbx11, Tbx2, Tbx221 |
| Sox transcription factor | Sox5, Sox91, Sox11 |
| Heparin-binding growth factor family member/FGF | Fgf9, Fgf10, Fgf18 |
| Sprouty | Spry1, Spry2 |
| Smad | Smad4, Smad7 |
| Integrin beta subunit | Itgb1, Itgb8 |
| Frizzled | Fzd2, Smo |
| Wnt related | Wnt5a1, Wnt9b |
| Serine-threonine protein kinase | Ilk, Mpa3k7/Tak1 |
| LIM domain containing protein | Lhx81, Lhx7, Prickle1 |
| EGF-like domain protein | Jag1, Jag2 |
Genes involved in human cleft lip and/or palate. TGF: Transforming growth factor; FGF: Fibroblast growth factor; MAPK: Mitogen-activated protein kinase; CBL: Casitas B-lineage lymphoma; EGF: Epidermal growth factor.
Figure 3.

Gene ontology analysis of genes associated with cleft palate in mice. Gene ontology analysis of genes associated with cleft palate in mice was performed using the PANTHER classification system (http://pantherdb.org). The most significantly enriched molecular function was the “transcription factor” (P = 1.2 × 10-12). A P value less than 0.05 was considered statistically significant.
To analyze mutant mice with cleft palates, the defects in palatal shelf development were divided into the following six categories (Table 3), which were modified from a previously published classification[3]. The first category is the failure of the palatal shelf formation. The gene mutations affect the initial development of the palatal shelf. The second one is the abnormal fusion of the palatal shelves and the mandible or tongue. Oral fusions between the palatal shelves and the tongue or mandible are rare. In Tbx1 (T-box 1) knockout mice, the posterior part of the palatal shelves fuse to the mandible, inhibiting the elevation of the palatal shelves[4]. The third category is the failed or delayed palatal shelf elevation. Ablation of Pax9 (paired box gene 9), Pitx1 (paired-like homeodomain 1), Gli2 (GLI family zinc finger 2), or Osr2 (Odd-skipped related transcription factor 2) in the palatal mesenchyme results in the failed palatal shelf elevation[6-9], suggesting crucial roles for these transcription factors in controlling the mesenchymal cells during palatal shelf elevation. The fourth one is the failure of the palatal shelf development after elevation. The loss of Msx1 (msh homeobox 1) and Lhx8 (LIM homeobox gene 8) and the conditional ablation of Tgfbr2 (transforming growth factor, beta receptor II) in the neural crest or Shh (sonic hedgehog) in the epithelium result in failure of the palatal shelf development[10-13]. The fifth category is the persistence of medial edge epithelial (MEE) cells. The palatal epithelia are regionally divided into three parts: oral, nasal, and MEE. The MEE cells are removed from the fusion line by epithelial cell migration, apoptosis, and epithelial-mesenchymal transdifferentiation[14]. Tgfb3 (transforming growth factor, beta 3) or Egfr (epidermal growth factor receptor) knockout mice lack the adhesive interactions between the palatal shelves because the fate of MEE cells is altered[15-18]. In the last category, the cleft palate arises as a secondary defect, due to tongue or bone anomalies during development. For example, Hoxa2 (homeobox A2) knockout mice exhibit CP, because depression of the tongue is inhibited by the abnormal attachment of the hyoglossus muscle to the greater horn of the hyoid[19,20].
Table 3.
Six categories of defects that result in cleft palate in mutant mice
| Defects | Knockout mice | |
| (1) | Failure of the palatal shelf formation (small palatal shelves) | Acvr2a[34,50], 1Fgfr2[13], 1Lhx8[11], Pitx2[126], Itga5[65], Fst[46] |
| (2) | Abnormal fusion of palatal shelves and tongue or the mandible | Jag2[70], 1Irf6[109,110], 1Tbx1[4], Fgf10[41] |
| (3) | Failure or delayed palatal shelf elevation | Pax9[6], 1Pitx1[7], 1Osr2[9], 1Gli2[8], 1Tgfb2[55], 1Pdgfc[51], Dhrs3[172] |
| (4) | Failure of the palatal shelf development after the elevation | 1Msx1[10], 1Lhx8[11], 1Tgfbr2 (Wnt1-Cre-mediated ablation)[12] |
| (5) | Persistence of medial edge epithelial cells | Apaf1[158], 1Tgfb3[18], Egfr[17], Ctnnb1 (K14-Cre-mediated ablation)[166] |
| (6) | Secondary defect | 1Hoxa2[19,20], 1Satb2[135], Acvr1/Alk2 (Wnt1-Cre-mediated ablation)[33] |
Genes involved in human cleft lip and/or palate.
There is molecular heterogeneity along the medial-lateral and anterior-posterior axes of palatal shelves. Regionally restricted expression of molecules provides distinct regulatory mechanisms for the development of palatal shelves. For instance, Msx1, Shox2 (short stature homeobox 2), Fgf10 (fibroblast growth factor 10), Bmp2 (bone morphogenetic protein 2), and Bmp4 (bone morphogenetic protein 4) are exclusively expressed in the anterior region of the palatal shelves[4,13,21,22]. The ablation of Msx1 in mice results in cell proliferation alterations in the anterior palatal mesenchyme and cleft palate[23]. Shox2 shows restricted expression patterns in the anterior palatal mesenchyme and the ablation of Shox2 in mice results in anterior cleft palates[22]. Fgf10 is also expressed in the anterior palatal mesenchymal cells and induces Shh expression through its receptor Fgfr2 (fibroblast growth factor receptor 2) in the palatal epithelium[13]. On the other hand, Pax9 is expressed in the posterior palatal shelves. Ablation of Pax9 results in cleft palates because of a palatal shelf development defect[6,21]. Even though it is known that Tbx1 induces the expression of Pax9 in the posterior part of palatal shelves[4], the mechanism of Tbx1-induced Pax9 expression during palatogenesis remains unknown. There is also molecular heterogeneity along the medial-lateral axis of the palatal shelf. For instance, Osr2 expression in the palatal shelf is characterized by a medial-lateral gradient. Loss of Osr2 results in the failure of palatal shelf elevation because of the delayed development of the medial part of palatal shelf[9].
MOLECULAR PATHOGENESIS OF CLEFT PALATES
Since most of the studies in mice focus on complete CP, the pathogenesis of other CP phenotypes is not well understood. Tbx1 is expressed in the developing palatal shelves in mice[4], highlighting the crucial function of Tbx1 in regulating palatal development. Loss of Tbx1 results in the abnormal fusion of the oral epithelia, which induces CP by preventing the elevation of palatal shelves[4]. The phenotypic variation in the Tbx1-/- palates strongly suggests that Tbx1 is involved in modifier genes and/or stochastic factors. Tgfb3-/- mice also exhibit either incomplete or complete CP[15,16]. Ablation of Shox2 results in anterior cleft palates[22]. Knockout mice of Sall3 (spalt-like transcription factor 3), which is expressed in the palatal mesenchyme, show hypoplasia of the soft palate and epiglottis[24]. These mice are unique models for studying the etiopathogenesis underlying the variety of CP phenotypes in humans.
A comprehensive list of molecules associated with CL/P in mice and their classification should provide insights into the genetic etiology of CL/P; however, the phenotype of knockout mice does not always recapitulate the phenotype in humans (Table 1). Since Table 1 includes the genes associated with tissue-specific conditional knockout mice, mutations of these genes may induce the phenotype of embryonic lethality in humans. Haploinsufficiency mutations of the TBX1 mutation are associated with CP[25]; however, heterozygous mice with Tbx1 are phenotypically normal, and Tbx1-/- mice have CP phenotypes[4], thereby suggesting a species-specific requirement for Tbx1 dosage. Mutations of the PVRL1 (poliovirus receptor-related 1 or Nectin 1) cause CL/P-ectodermal dysplasia syndrome and nonsyndromic CL/P (OMIM #225060), whereas Pvrl1-/- mice do not develop CP[26]. Lack of palatal phenotypes in mice may be a consequence of functional redundancy of Pvrl genes. Interestingly, Smad4, Smad7, Fgf9, Fgf10, and Fgf18 are involved in CP in mice (Table 1), whereas SMAD3 (OMIM *613795), FGF8 (OMIM *600483), and FGF17 (OMIM *603725) are involved in CP in humans.
Candidate genes for nonsyndromic CP in human must show a relevant spatio-temporal gene expression pattern in the developing palatal shelves, and induce a specific cleft palate phenotype when deleted[1]. Disease genes responsible for Mendelian forms of syndromic CP are also important in the etiology of nonsyndromic CP[27]. TBX1 mutations have been found in patients with incomplete CP without clinical diagnosis of del22q11.2 syndrome[25]. TBX1 is also one of the disease genes of conotruncal anomaly face syndrome (OMIM #217095), which is often associated with cleft palates, particularly submucosal CP, and bifid uvula. Tbx1-/- palatal phenotype in mice makes Tbx1 a potential candidate gene for nonsyndromic CP, especially submucosal CP and incomplete CP in humans.
RECENT ADVANCES IN PALATOGENESIS
Even though many genes associated with CP have been identified, little is known about how the environment influences gene expression in palatogenesis, and palatal phenotype. Epigenetics, such as DNA methylation and chromatin remodeling, and the microRNA (miRNA) regulation could change gene expression profiles and phenotypes. Hundreds of miRNAs, small non-coding RNAs that modulate gene expression at the post-transcriptional level are expressed in murine embryonic craniofacial tissue[28]. Conditional knockout mice of Dicer1 (dicer 1, ribonuclease type III), which regulates the generation of miRNA, resulted in disrupted palatogenesis[29], suggesting that the miRNA function may be important in mammalian palatogenesis. miR-140, which modulates BMP signaling, regulates palatogenesis in mice[30] and miR-17-92 modulates Tbx1 and Tbx3 (T-box 3) activity, resulting in orofacial clefting[31]. Interestingly, transcription of Dicer1 is regulated by TP63 (transformation related protein p63)[32], whose mutations are associated with cleft palate phenotypes (Table 1). Since genes involved in miRNA generation and individual CP genes can both be modulated by several miRNAs, it is conceivable that complex gene-miRNA interactions exist during palatogenesis. Genetically engineered mice with miRNAs, which modulate CP genes, may provide new information on the gene interactions underlying the palatogenesis. Further studies on miRNA and methylated genes involved in palatogenesis are necessary to understand the environmental factors contributing to CP.
CONCLUSION
Studies with genetically engineered mice with CP reveal the importance of regulated molecular functions in palatogenesis and provide the opportunity to discover new genes implicated in palatogenesis. However, there is still much to learn about transcriptional regulation and molecular networks in palatogenesis. The interactions between environmental/stochastic factors and genes in the etiopathogenesis of CL/P require further studies. Teratogenic effects of dioxins and retinoic acid have been reported in mice[1]. Mutant mice with CP can also be used as models to assess environmental effects or gene-environment interactions. Epithelial abnormal fusion could be one of the stochastic causes that induce a variety of CP phenotypes in mice. Understanding the palatal epithelial functions during palatogenesis may also lead to the discovery of novel therapeutic methods for CL/P.
Footnotes
Supported by The Japan Society for the Promotion of Science (JSPS) through KAKENHI grants 25670774 and 15K11004, awarded to Funato N.
Conflict-of-interest statement: The authors declare no conflicts of interest.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: April 22, 2015
First decision: May 13, 2015
Article in press: July 14, 2015
P- Reviewer: Freire-De-Lima CG, Gokul S, Yeligar SM, Zhang L S- Editor: Ji FF L- Editor: A E- Editor: Wang CH
References
- 1.Murray JC. Gene/environment causes of cleft lip and/or palate. Clin Genet. 2002;61:248–256. doi: 10.1034/j.1399-0004.2002.610402.x. [DOI] [PubMed] [Google Scholar]
- 2.Jones MC. Etiology of facial clefts: prospective evaluation of 428 patients. Cleft Palate J. 1988;25:16–20. [PubMed] [Google Scholar]
- 3.Chai Y, Maxson RE. Recent advances in craniofacial morphogenesis. Dev Dyn. 2006;235:2353–2375. doi: 10.1002/dvdy.20833. [DOI] [PubMed] [Google Scholar]
- 4.Funato N, Nakamura M, Richardson JA, Srivastava D, Yanagisawa H. Tbx1 regulates oral epithelial adhesion and palatal development. Hum Mol Genet. 2012;21:2524–2537. doi: 10.1093/hmg/dds071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mi H, Muruganujan A, Thomas PD. PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 2013;41:D377–D386. doi: 10.1093/nar/gks1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peters H, Neubüser A, Kratochwil K, Balling R. Pax9-deficient mice lack pharyngeal pouch derivatives and teeth and exhibit craniofacial and limb abnormalities. Genes Dev. 1998;12:2735–2747. doi: 10.1101/gad.12.17.2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Szeto DP, Rodriguez-Esteban C, Ryan AK, O’Connell SM, Liu F, Kioussi C, Gleiberman AS, Izpisúa-Belmonte JC, Rosenfeld MG. Role of the Bicoid-related homeodomain factor Pitx1 in specifying hindlimb morphogenesis and pituitary development. Genes Dev. 1999;13:484–494. doi: 10.1101/gad.13.4.484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mo R, Freer AM, Zinyk DL, Crackower MA, Michaud J, Heng HH, Chik KW, Shi XM, Tsui LC, Cheng SH, et al. Specific and redundant functions of Gli2 and Gli3 zinc finger genes in skeletal patterning and development. Development. 1997;124:113–123. doi: 10.1242/dev.124.1.113. [DOI] [PubMed] [Google Scholar]
- 9.Lan Y, Ovitt CE, Cho ES, Maltby KM, Wang Q, Jiang R. Odd-skipped related 2 (Osr2) encodes a key intrinsic regulator of secondary palate growth and morphogenesis. Development. 2004;131:3207–3216. doi: 10.1242/dev.01175. [DOI] [PubMed] [Google Scholar]
- 10.Satokata I, Maas R. Msx1 deficient mice exhibit cleft palate and abnormalities of craniofacial and tooth development. Nat Genet. 1994;6:348–356. doi: 10.1038/ng0494-348. [DOI] [PubMed] [Google Scholar]
- 11.Zhao Y, Guo YJ, Tomac AC, Taylor NR, Grinberg A, Lee EJ, Huang S, Westphal H. Isolated cleft palate in mice with a targeted mutation of the LIM homeobox gene lhx8. Proc Natl Acad Sci USA. 1999;96:15002–15006. doi: 10.1073/pnas.96.26.15002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ito Y, Yeo JY, Chytil A, Han J, Bringas P, Nakajima A, Shuler CF, Moses HL, Chai Y. Conditional inactivation of Tgfbr2 in cranial neural crest causes cleft palate and calvaria defects. Development. 2003;130:5269–5280. doi: 10.1242/dev.00708. [DOI] [PubMed] [Google Scholar]
- 13.Rice R, Spencer-Dene B, Connor EC, Gritli-Linde A, McMahon AP, Dickson C, Thesleff I, Rice DP. Disruption of Fgf10/Fgfr2b-coordinated epithelial-mesenchymal interactions causes cleft palate. J Clin Invest. 2004;113:1692–1700. doi: 10.1172/JCI20384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jin JZ, Ding J. Analysis of cell migration, transdifferentiation and apoptosis during mouse secondary palate fusion. Development. 2006;133:3341–3347. doi: 10.1242/dev.02520. [DOI] [PubMed] [Google Scholar]
- 15.Kaartinen V, Voncken JW, Shuler C, Warburton D, Bu D, Heisterkamp N, Groffen J. Abnormal lung development and cleft palate in mice lacking TGF-beta 3 indicates defects of epithelial-mesenchymal interaction. Nat Genet. 1995;11:415–421. doi: 10.1038/ng1295-415. [DOI] [PubMed] [Google Scholar]
- 16.Proetzel G, Pawlowski SA, Wiles MV, Yin M, Boivin GP, Howles PN, Ding J, Ferguson MW, Doetschman T. Transforming growth factor-beta 3 is required for secondary palate fusion. Nat Genet. 1995;11:409–414. doi: 10.1038/ng1295-409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miettinen PJ, Chin JR, Shum L, Slavkin HC, Shuler CF, Derynck R, Werb Z. Epidermal growth factor receptor function is necessary for normal craniofacial development and palate closure. Nat Genet. 1999;22:69–73. doi: 10.1038/8773. [DOI] [PubMed] [Google Scholar]
- 18.Taya Y, O’Kane S, Ferguson MW. Pathogenesis of cleft palate in TGF-beta3 knockout mice. Development. 1999;126:3869–3879. doi: 10.1242/dev.126.17.3869. [DOI] [PubMed] [Google Scholar]
- 19.Gendron-Maguire M, Mallo M, Zhang M, Gridley T. Hoxa-2 mutant mice exhibit homeotic transformation of skeletal elements derived from cranial neural crest. Cell. 1993;75:1317–1331. doi: 10.1016/0092-8674(93)90619-2. [DOI] [PubMed] [Google Scholar]
- 20.Barrow JR, Capecchi MR. Compensatory defects associated with mutations in Hoxa1 restore normal palatogenesis to Hoxa2 mutants. Development. 1999;126:5011–5026. doi: 10.1242/dev.126.22.5011. [DOI] [PubMed] [Google Scholar]
- 21.Hilliard SA, Yu L, Gu S, Zhang Z, Chen YP. Regional regulation of palatal growth and patterning along the anterior-posterior axis in mice. J Anat. 2005;207:655–667. doi: 10.1111/j.1469-7580.2005.00474.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu L, Gu S, Alappat S, Song Y, Yan M, Zhang X, Zhang G, Jiang Y, Zhang Z, Zhang Y, et al. Shox2-deficient mice exhibit a rare type of incomplete clefting of the secondary palate. Development. 2005;132:4397–4406. doi: 10.1242/dev.02013. [DOI] [PubMed] [Google Scholar]
- 23.Zhang Z, Song Y, Zhao X, Zhang X, Fermin C, Chen Y. Rescue of cleft palate in Msx1-deficient mice by transgenic Bmp4 reveals a network of BMP and Shh signaling in the regulation of mammalian palatogenesis. Development. 2002;129:4135–4146. doi: 10.1242/dev.129.17.4135. [DOI] [PubMed] [Google Scholar]
- 24.Parrish M, Ott T, Lance-Jones C, Schuetz G, Schwaeger-Nickolenko A, Monaghan AP. Loss of the Sall3 gene leads to palate deficiency, abnormalities in cranial nerves, and perinatal lethality. Mol Cell Biol. 2004;24:7102–7112. doi: 10.1128/MCB.24.16.7102-7112.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yagi H, Furutani Y, Hamada H, Sasaki T, Asakawa S, Minoshima S, Ichida F, Joo K, Kimura M, Imamura S, et al. Role of TBX1 in human del22q11.2 syndrome. Lancet. 2003;362:1366–1373. doi: 10.1016/s0140-6736(03)14632-6. [DOI] [PubMed] [Google Scholar]
- 26.Barron MJ, Brookes SJ, Draper CE, Garrod D, Kirkham J, Shore RC, Dixon MJ. The cell adhesion molecule nectin-1 is critical for normal enamel formation in mice. Hum Mol Genet. 2008;17:3509–3520. doi: 10.1093/hmg/ddn243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jugessur A, Murray JC. Orofacial clefting: recent insights into a complex trait. Curr Opin Genet Dev. 2005;15:270–278. doi: 10.1016/j.gde.2005.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mukhopadhyay P, Brock G, Pihur V, Webb C, Pisano MM, Greene RM. Developmental microRNA expression profiling of murine embryonic orofacial tissue. Birth Defects Res A Clin Mol Teratol. 2010;88:511–534. doi: 10.1002/bdra.20684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barritt LC, Miller JM, Scheetz LR, Gardner K, Pierce ML, Soukup GA, Rocha-Sanchez SM. Conditional deletion of the human ortholog gene Dicer1 in Pax2-Cre expression domain impairs orofacial development. Indian J Hum Genet. 2012;18:310–319. doi: 10.4103/0971-6866.107984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakamura Y, Inloes JB, Katagiri T, Kobayashi T. Chondrocyte-specific microRNA-140 regulates endochondral bone development and targets Dnpep to modulate bone morphogenetic protein signaling. Mol Cell Biol. 2011;31:3019–3028. doi: 10.1128/MCB.05178-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang J, Bai Y, Li H, Greene SB, Klysik E, Yu W, Schwartz RJ, Williams TJ, Martin JF. MicroRNA-17-92, a direct Ap-2α transcriptional target, modulates T-box factor activity in orofacial clefting. PLoS Genet. 2013;9:e1003785. doi: 10.1371/journal.pgen.1003785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Su X, Chakravarti D, Cho MS, Liu L, Gi YJ, Lin YL, Leung ML, El-Naggar A, Creighton CJ, Suraokar MB, et al. TAp63 suppresses metastasis through coordinate regulation of Dicer and miRNAs. Nature. 2010;467:986–990. doi: 10.1038/nature09459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dudas M, Sridurongrit S, Nagy A, Okazaki K, Kaartinen V. Craniofacial defects in mice lacking BMP type I receptor Alk2 in neural crest cells. Mech Dev. 2004;121:173–182. doi: 10.1016/j.mod.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 34.Matzuk MM, Kumar TR, Bradley A. Different phenotypes for mice deficient in either activins or activin receptor type II. Nature. 1995;374:356–360. doi: 10.1038/374356a0. [DOI] [PubMed] [Google Scholar]
- 35.Liu W, Sun X, Braut A, Mishina Y, Behringer RR, Mina M, Martin JF. Distinct functions for Bmp signaling in lip and palate fusion in mice. Development. 2005;132:1453–1461. doi: 10.1242/dev.01676. [DOI] [PubMed] [Google Scholar]
- 36.Kouskoura T, Kozlova A, Alexiou M, Blumer S, Zouvelou V, Katsaros C, Chiquet M, Mitsiadis TA, Graf D. The etiology of cleft palate formation in BMP7-deficient mice. PLoS One. 2013;8:e59463. doi: 10.1371/journal.pone.0059463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bachiller D, Klingensmith J, Shneyder N, Tran U, Anderson R, Rossant J, De Robertis EM. The role of chordin/Bmp signals in mammalian pharyngeal development and DiGeorge syndrome. Development. 2003;130:3567–3578. doi: 10.1242/dev.00581. [DOI] [PubMed] [Google Scholar]
- 38.Ivkovic S, Yoon BS, Popoff SN, Safadi FF, Libuda DE, Stephenson RC, Daluiski A, Lyons KM. Connective tissue growth factor coordinates chondrogenesis and angiogenesis during skeletal development. Development. 2003;130:2779–2791. doi: 10.1242/dev.00505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kurihara Y, Kurihara H, Suzuki H, Kodama T, Maemura K, Nagai R, Oda H, Kuwaki T, Cao WH, Kamada N. Elevated blood pressure and craniofacial abnormalities in mice deficient in endothelin-1. Nature. 1994;368:703–710. doi: 10.1038/368703a0. [DOI] [PubMed] [Google Scholar]
- 40.Colvin JS, White AC, Pratt SJ, Ornitz DM. Lung hypoplasia and neonatal death in Fgf9-null mice identify this gene as an essential regulator of lung mesenchyme. Development. 2001;128:2095–2106. doi: 10.1242/dev.128.11.2095. [DOI] [PubMed] [Google Scholar]
- 41.Alappat SR, Zhang Z, Suzuki K, Zhang X, Liu H, Jiang R, Yamada G, Chen Y. The cellular and molecular etiology of the cleft secondary palate in Fgf10 mutant mice. Dev Biol. 2005;277:102–113. doi: 10.1016/j.ydbio.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 42.Liu Z, Xu J, Colvin JS, Ornitz DM. Coordination of chondrogenesis and osteogenesis by fibroblast growth factor 18. Genes Dev. 2002;16:859–869. doi: 10.1101/gad.965602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ohbayashi N, Shibayama M, Kurotaki Y, Imanishi M, Fujimori T, Itoh N, Takada S. FGF18 is required for normal cell proliferation and differentiation during osteogenesis and chondrogenesis. Genes Dev. 2002;16:870–879. doi: 10.1101/gad.965702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Trokovic N, Trokovic R, Mai P, Partanen J. Fgfr1 regulates patterning of the pharyngeal region. Genes Dev. 2003;17:141–153. doi: 10.1101/gad.250703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.De Moerlooze L, Spencer-Dene B, Revest JM, Hajihosseini M, Rosewell I, Dickson C. An important role for the IIIb isoform of fibroblast growth factor receptor 2 (FGFR2) in mesenchymal-epithelial signalling during mouse organogenesis. Development. 2000;127:483–492. doi: 10.1242/dev.127.3.483. [DOI] [PubMed] [Google Scholar]
- 46.Matzuk MM, Lu N, Vogel H, Sellheyer K, Roop DR, Bradley A. Multiple defects and perinatal death in mice deficient in follistatin. Nature. 1995;374:360–363. doi: 10.1038/374360a0. [DOI] [PubMed] [Google Scholar]
- 47.Homanics GE, DeLorey TM, Firestone LL, Quinlan JJ, Handforth A, Harrison NL, Krasowski MD, Rick CE, Korpi ER, Mäkelä R, et al. Mice devoid of gamma-aminobutyrate type A receptor beta3 subunit have epilepsy, cleft palate, and hypersensitive behavior. Proc Natl Acad Sci USA. 1997;94:4143–4148. doi: 10.1073/pnas.94.8.4143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee YS, Lee SJ. Regulation of GDF-11 and myostatin activity by GASP-1 and GASP-2. Proc Natl Acad Sci USA. 2013;110:E3713–E3722. doi: 10.1073/pnas.1309907110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Anderson KD, Pan L, Yang XM, Hughes VC, Walls JR, Dominguez MG, Simmons MV, Burfeind P, Xue Y, Wei Y, et al. Angiogenic sprouting into neural tissue requires Gpr124, an orphan G protein-coupled receptor. Proc Natl Acad Sci USA. 2011;108:2807–2812. doi: 10.1073/pnas.1019761108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Matzuk MM, Kumar TR, Vassalli A, Bickenbach JR, Roop DR, Jaenisch R, Bradley A. Functional analysis of activins during mammalian development. Nature. 1995;374:354–356. doi: 10.1038/374354a0. [DOI] [PubMed] [Google Scholar]
- 51.Ding H, Wu X, Boström H, Kim I, Wong N, Tsoi B, O’Rourke M, Koh GY, Soriano P, Betsholtz C, et al. A specific requirement for PDGF-C in palate formation and PDGFR-alpha signaling. Nat Genet. 2004;36:1111–1116. doi: 10.1038/ng1415. [DOI] [PubMed] [Google Scholar]
- 52.Choi SJ, Marazita ML, Hart PS, Sulima PP, Field LL, McHenry TG, Govil M, Cooper ME, Letra A, Menezes R, et al. The PDGF-C regulatory region SNP rs28999109 decreases promoter transcriptional activity and is associated with CL/P. Eur J Hum Genet. 2009;17:774–784. doi: 10.1038/ejhg.2008.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Morrison-Graham K, Schatteman GC, Bork T, Bowen-Pope DF, Weston JA. A PDGF receptor mutation in the mouse (Patch) perturbs the development of a non-neuronal subset of neural crest-derived cells. Development. 1992;115:133–142. doi: 10.1242/dev.115.1.133. [DOI] [PubMed] [Google Scholar]
- 54.Tallquist MD, Soriano P. Cell autonomous requirement for PDGFRalpha in populations of cranial and cardiac neural crest cells. Development. 2003;130:507–518. doi: 10.1242/dev.00241. [DOI] [PubMed] [Google Scholar]
- 55.Sanford LP, Ormsby I, Gittenberger-de Groot AC, Sariola H, Friedman R, Boivin GP, Cardell EL, Doetschman T. TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development. 1997;124:2659–2670. doi: 10.1242/dev.124.13.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dudas M, Kim J, Li WY, Nagy A, Larsson J, Karlsson S, Chai Y, Kaartinen V. Epithelial and ectomesenchymal role of the type I TGF-beta receptor ALK5 during facial morphogenesis and palatal fusion. Dev Biol. 2006;296:298–314. doi: 10.1016/j.ydbio.2006.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li WY, Dudas M, Kaartinen V. Signaling through Tgf-beta type I receptor Alk5 is required for upper lip fusion. Mech Dev. 2008;125:874–882. doi: 10.1016/j.mod.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xu X, Han J, Ito Y, Bringas P, Urata MM, Chai Y. Cell autonomous requirement for Tgfbr2 in the disappearance of medial edge epithelium during palatal fusion. Dev Biol. 2006;297:238–248. doi: 10.1016/j.ydbio.2006.05.014. [DOI] [PubMed] [Google Scholar]
- 59.Stalmans I, Lambrechts D, De Smet F, Jansen S, Wang J, Maity S, Kneer P, von der Ohe M, Swillen A, Maes C, et al. VEGF: a modifier of the del22q11 (DiGeorge) syndrome? Nat Med. 2003;9:173–182. doi: 10.1038/nm819. [DOI] [PubMed] [Google Scholar]
- 60.Mima J, Koshino A, Oka K, Uchida H, Hieda Y, Nohara K, Kogo M, Chai Y, Sakai T. Regulation of the epithelial adhesion molecule CEACAM1 is important for palate formation. PLoS One. 2013;8:e61653. doi: 10.1371/journal.pone.0061653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Holmberg J, Clarke DL, Frisén J. Regulation of repulsion versus adhesion by different splice forms of an Eph receptor. Nature. 2000;408:203–206. doi: 10.1038/35041577. [DOI] [PubMed] [Google Scholar]
- 62.Compagni A, Logan M, Klein R, Adams RH. Control of skeletal patterning by ephrinB1-EphB interactions. Dev Cell. 2003;5:217–230. doi: 10.1016/s1534-5807(03)00198-9. [DOI] [PubMed] [Google Scholar]
- 63.Dravis C, Henkemeyer M. Ephrin-B reverse signaling controls septation events at the embryonic midline through separate tyrosine phosphorylation-independent signaling avenues. Dev Biol. 2011;355:138–151. doi: 10.1016/j.ydbio.2011.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yu H, Smallwood PM, Wang Y, Vidaltamayo R, Reed R, Nathans J. Frizzled 1 and frizzled 2 genes function in palate, ventricular septum and neural tube closure: general implications for tissue fusion processes. Development. 2010;137:3707–3717. doi: 10.1242/dev.052001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bader BL, Rayburn H, Crowley D, Hynes RO. Extensive vasculogenesis, angiogenesis, and organogenesis precede lethality in mice lacking all alpha v integrins. Cell. 1998;95:507–519. doi: 10.1016/s0092-8674(00)81618-9. [DOI] [PubMed] [Google Scholar]
- 66.Liang D, Wang X, Mittal A, Dhiman S, Hou SY, Degenhardt K, Astrof S. Mesodermal expression of integrin α5β1 regulates neural crest development and cardiovascular morphogenesis. Dev Biol. 2014;395:232–244. doi: 10.1016/j.ydbio.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Aszodi A, Hunziker EB, Brakebusch C, Fässler R. Beta1 integrins regulate chondrocyte rotation, G1 progression, and cytokinesis. Genes Dev. 2003;17:2465–2479. doi: 10.1101/gad.277003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhu J, Motejlek K, Wang D, Zang K, Schmidt A, Reichardt LF. beta8 integrins are required for vascular morphogenesis in mouse embryos. Development. 2002;129:2891–2903. doi: 10.1242/dev.129.12.2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Humphreys R, Zheng W, Prince LS, Qu X, Brown C, Loomes K, Huppert SS, Baldwin S, Goudy S. Cranial neural crest ablation of Jagged1 recapitulates the craniofacial phenotype of Alagille syndrome patients. Hum Mol Genet. 2012;21:1374–1383. doi: 10.1093/hmg/ddr575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jiang R, Lan Y, Chapman HD, Shawber C, Norton CR, Serreze DV, Weinmaster G, Gridley T. Defects in limb, craniofacial, and thymic development in Jagged2 mutant mice. Genes Dev. 1998;12:1046–1057. doi: 10.1101/gad.12.7.1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zaritsky JJ, Eckman DM, Wellman GC, Nelson MT, Schwarz TL. Targeted disruption of Kir2.1 and Kir2.2 genes reveals the essential role of the inwardly rectifying K(+) current in K(+)-mediated vasodilation. Circ Res. 2000;87:160–166. doi: 10.1161/01.res.87.2.160. [DOI] [PubMed] [Google Scholar]
- 72.Song L, Li Y, Wang K, Wang YZ, Molotkov A, Gao L, Zhao T, Yamagami T, Wang Y, Gan Q, et al. Lrp6-mediated canonical Wnt signaling is required for lip formation and fusion. Development. 2009;136:3161–3171. doi: 10.1242/dev.037440. [DOI] [PubMed] [Google Scholar]
- 73.Schwabe GC, Trepczik B, Süring K, Brieske N, Tucker AS, Sharpe PT, Minami Y, Mundlos S. Ror2 knockout mouse as a model for the developmental pathology of autosomal recessive Robinow syndrome. Dev Dyn. 2004;229:400–410. doi: 10.1002/dvdy.10466. [DOI] [PubMed] [Google Scholar]
- 74.Halford MM, Armes J, Buchert M, Meskenaite V, Grail D, Hibbs ML, Wilks AF, Farlie PG, Newgreen DF, Hovens CM, et al. Ryk-deficient mice exhibit craniofacial defects associated with perturbed Eph receptor crosstalk. Nat Genet. 2000;25:414–418. doi: 10.1038/78099. [DOI] [PubMed] [Google Scholar]
- 75.Zvaritch E, Depreux F, Kraeva N, Loy RE, Goonasekera SA, Boncompagni S, Kraev A, Gramolini AO, Dirksen RT, Franzini-Armstrong C, et al. An Ryr1I4895T mutation abolishes Ca2+ release channel function and delays development in homozygous offspring of a mutant mouse line. Proc Natl Acad Sci USA. 2007;104:18537–18542. doi: 10.1073/pnas.0709312104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Krakowiak PA, Wassif CA, Kratz L, Cozma D, Kovárová M, Harris G, Grinberg A, Yang Y, Hunter AG, Tsokos M, et al. Lathosterolosis: an inborn error of human and murine cholesterol synthesis due to lathosterol 5-desaturase deficiency. Hum Mol Genet. 2003;12:1631–1641. doi: 10.1093/hmg/ddg172. [DOI] [PubMed] [Google Scholar]
- 77.Huang X, Litingtung Y, Chiang C. Ectopic sonic hedgehog signaling impairs telencephalic dorsal midline development: implication for human holoprosencephaly. Hum Mol Genet. 2007;16:1454–1468. doi: 10.1093/hmg/ddm096. [DOI] [PubMed] [Google Scholar]
- 78.Jeong J, Mao J, Tenzen T, Kottmann AH, McMahon AP. Hedgehog signaling in the neural crest cells regulates the patterning and growth of facial primordia. Genes Dev. 2004;18:937–951. doi: 10.1101/gad.1190304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sang L, Miller JJ, Corbit KC, Giles RH, Brauer MJ, Otto EA, Baye LM, Wen X, Scales SJ, Kwong M, et al. Mapping the NPHP-JBTS-MKS protein network reveals ciliopathy disease genes and pathways. Cell. 2011;145:513–528. doi: 10.1016/j.cell.2011.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fu J, Ivy Yu HM, Maruyama T, Mirando AJ, Hsu W. Gpr177/mouse Wntless is essential for Wnt-mediated craniofacial and brain development. Dev Dyn. 2011;240:365–371. doi: 10.1002/dvdy.22541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yamaguchi TP, Bradley A, McMahon AP, Jones S. A Wnt5a pathway underlies outgrowth of multiple structures in the vertebrate embryo. Development. 1999;126:1211–1223. doi: 10.1242/dev.126.6.1211. [DOI] [PubMed] [Google Scholar]
- 82.Carroll TJ, Park JS, Hayashi S, Majumdar A, McMahon AP. Wnt9b plays a central role in the regulation of mesenchymal to epithelial transitions underlying organogenesis of the mammalian urogenital system. Dev Cell. 2005;9:283–292. doi: 10.1016/j.devcel.2005.05.016. [DOI] [PubMed] [Google Scholar]
- 83.Ferretti E, Li B, Zewdu R, Wells V, Hebert JM, Karner C, Anderson MJ, Williams T, Dixon J, Dixon MJ, et al. A conserved Pbx-Wnt-p63-Irf6 regulatory module controls face morphogenesis by promoting epithelial apoptosis. Dev Cell. 2011;21:627–641. doi: 10.1016/j.devcel.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Uz E, Alanay Y, Aktas D, Vargel I, Gucer S, Tuncbilek G, von Eggeling F, Yilmaz E, Deren O, Posorski N, et al. Disruption of ALX1 causes extreme microphthalmia and severe facial clefting: expanding the spectrum of autosomal-recessive ALX-related frontonasal dysplasia. Am J Hum Genet. 2010;86:789–796. doi: 10.1016/j.ajhg.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Beverdam A, Brouwer A, Reijnen M, Korving J, Meijlink F. Severe nasal clefting and abnormal embryonic apoptosis in Alx3/Alx4 double mutant mice. Development. 2001;128:3975–3986. doi: 10.1242/dev.128.20.3975. [DOI] [PubMed] [Google Scholar]
- 86.Reilly PT, Afzal S, Gorrini C, Lui K, Bukhman YV, Wakeham A, Haight J, Ling TW, Cheung CC, Elia AJ, et al. Acidic nuclear phosphoprotein 32kDa (ANP32)B-deficient mouse reveals a hierarchy of ANP32 importance in mammalian development. Proc Natl Acad Sci USA. 2011;108:10243–10248. doi: 10.1073/pnas.1106211108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Schmahl J, Raymond CS, Soriano P. PDGF signaling specificity is mediated through multiple immediate early genes. Nat Genet. 2007;39:52–60. doi: 10.1038/ng1922. [DOI] [PubMed] [Google Scholar]
- 88.Abdel-Wahab O, Gao J, Adli M, Dey A, Trimarchi T, Chung YR, Kuscu C, Hricik T, Ndiaye-Lobry D, Lafave LM, et al. Deletion of Asxl1 results in myelodysplasia and severe developmental defects in vivo. J Exp Med. 2013;210:2641–2659. doi: 10.1084/jem.20131141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Miletich I, Yu WY, Zhang R, Yang K, Caixeta de Andrade S, Pereira SF, Ohazama A, Mock OB, Buchner G, Sealby J, et al. Developmental stalling and organ-autonomous regulation of morphogenesis. Proc Natl Acad Sci USA. 2011;108:19270–19275. doi: 10.1073/pnas.1112801108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Aizawa R, Yamada A, Suzuki D, Iimura T, Kassai H, Harada T, Tsukasaki M, Yamamoto G, Tachikawa T, Nakao K, et al. Cdc42 is required for chondrogenesis and interdigital programmed cell death during limb development. Mech Dev. 2012;129:38–50. doi: 10.1016/j.mod.2012.02.002. [DOI] [PubMed] [Google Scholar]
- 91.Bosman EA, Penn AC, Ambrose JC, Kettleborough R, Stemple DL, Steel KP. Multiple mutations in mouse Chd7 provide models for CHARGE syndrome. Hum Mol Genet. 2005;14:3463–3476. doi: 10.1093/hmg/ddi375. [DOI] [PubMed] [Google Scholar]
- 92.Sperry ED, Hurd EA, Durham MA, Reamer EN, Stein AB, Martin DM. The chromatin remodeling protein CHD7, mutated in CHARGE syndrome, is necessary for proper craniofacial and tracheal development. Dev Dyn. 2014;243:1055–1066. doi: 10.1002/dvdy.24156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bentham J, Michell AC, Lockstone H, Andrew D, Schneider JE, Brown NA, Bhattacharya S. Maternal high-fat diet interacts with embryonic Cited2 genotype to reduce Pitx2c expression and enhance penetrance of left-right patterning defects. Hum Mol Genet. 2010;19:3394–3401. doi: 10.1093/hmg/ddq251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kasper LH, Boussouar F, Boyd K, Xu W, Biesen M, Rehg J, Baudino TA, Cleveland JL, Brindle PK. Two transactivation mechanisms cooperate for the bulk of HIF-1-responsive gene expression. EMBO J. 2005;24:3846–3858. doi: 10.1038/sj.emboj.7600846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Qiu M, Bulfone A, Ghattas I, Meneses JJ, Christensen L, Sharpe PT, Presley R, Pedersen RA, Rubenstein JL. Role of the Dlx homeobox genes in proximodistal patterning of the branchial arches: mutations of Dlx-1, Dlx-2, and Dlx-1 and -2 alter morphogenesis of proximal skeletal and soft tissue structures derived from the first and second arches. Dev Biol. 1997;185:165–184. doi: 10.1006/dbio.1997.8556. [DOI] [PubMed] [Google Scholar]
- 96.Acampora D, Merlo GR, Paleari L, Zerega B, Postiglione MP, Mantero S, Bober E, Barbieri O, Simeone A, Levi G. Craniofacial, vestibular and bone defects in mice lacking the Distal-less-related gene Dlx5. Development. 1999;126:3795–3809. doi: 10.1242/dev.126.17.3795. [DOI] [PubMed] [Google Scholar]
- 97.Depew MJ, Liu JK, Long JE, Presley R, Meneses JJ, Pedersen RA, Rubenstein JL. Dlx5 regulates regional development of the branchial arches and sensory capsules. Development. 1999;126:3831–3846. doi: 10.1242/dev.126.17.3831. [DOI] [PubMed] [Google Scholar]
- 98.Chen CM, Behringer RR. Ovca1 regulates cell proliferation, embryonic development, and tumorigenesis. Genes Dev. 2004;18:320–332. doi: 10.1101/gad.1162204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Xu PX, Adams J, Peters H, Brown MC, Heaney S, Maas R. Eya1-deficient mice lack ears and kidneys and show abnormal apoptosis of organ primordia. Nat Genet. 1999;23:113–117. doi: 10.1038/12722. [DOI] [PubMed] [Google Scholar]
- 100.Iida K, Koseki H, Kakinuma H, Kato N, Mizutani-Koseki Y, Ohuchi H, Yoshioka H, Noji S, Kawamura K, Kataoka Y, et al. Essential roles of the winged helix transcription factor MFH-1 in aortic arch patterning and skeletogenesis. Development. 1997;124:4627–4638. doi: 10.1242/dev.124.22.4627. [DOI] [PubMed] [Google Scholar]
- 101.Teng L, Mundell NA, Frist AY, Wang Q, Labosky PA. Requirement for Foxd3 in the maintenance of neural crest progenitors. Development. 2008;135:1615–1624. doi: 10.1242/dev.012179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.De Felice M, Ovitt C, Biffali E, Rodriguez-Mallon A, Arra C, Anastassiadis K, Macchia PE, Mattei MG, Mariano A, Schöler H, et al. A mouse model for hereditary thyroid dysgenesis and cleft palate. Nat Genet. 1998;19:395–398. doi: 10.1038/1289. [DOI] [PubMed] [Google Scholar]
- 103.Wang T, Tamakoshi T, Uezato T, Shu F, Kanzaki-Kato N, Fu Y, Koseki H, Yoshida N, Sugiyama T, Miura N. Forkhead transcription factor Foxf2 (LUN)-deficient mice exhibit abnormal development of secondary palate. Dev Biol. 2003;259:83–94. doi: 10.1016/s0012-1606(03)00176-3. [DOI] [PubMed] [Google Scholar]
- 104.Byrd NA, Meyers EN. Loss of Gbx2 results in neural crest cell patterning and pharyngeal arch artery defects in the mouse embryo. Dev Biol. 2005;284:233–245. doi: 10.1016/j.ydbio.2005.05.023. [DOI] [PubMed] [Google Scholar]
- 105.Huang X, Goudy SL, Ketova T, Litingtung Y, Chiang C. Gli3-deficient mice exhibit cleft palate associated with abnormal tongue development. Dev Dyn. 2008;237:3079–3087. doi: 10.1002/dvdy.21714. [DOI] [PubMed] [Google Scholar]
- 106.Yamada G, Mansouri A, Torres M, Stuart ET, Blum M, Schultz M, De Robertis EM, Gruss P. Targeted mutation of the murine goosecoid gene results in craniofacial defects and neonatal death. Development. 1995;121:2917–2922. doi: 10.1242/dev.121.9.2917. [DOI] [PubMed] [Google Scholar]
- 107.Yanagisawa H, Clouthier DE, Richardson JA, Charité J, Olson EN. Targeted deletion of a branchial arch-specific enhancer reveals a role of dHAND in craniofacial development. Development. 2003;130:1069–1078. doi: 10.1242/dev.00337. [DOI] [PubMed] [Google Scholar]
- 108.Carter MG, Johns MA, Zeng X, Zhou L, Zink MC, Mankowski JL, Donovan DM, Baylin SB. Mice deficient in the candidate tumor suppressor gene Hic1 exhibit developmental defects of structures affected in the Miller-Dieker syndrome. Hum Mol Genet. 2000;9:413–419. doi: 10.1093/hmg/9.3.413. [DOI] [PubMed] [Google Scholar]
- 109.Ingraham CR, Kinoshita A, Kondo S, Yang B, Sajan S, Trout KJ, Malik MI, Dunnwald M, Goudy SL, Lovett M, et al. Abnormal skin, limb and craniofacial morphogenesis in mice deficient for interferon regulatory factor 6 (Irf6) Nat Genet. 2006;38:1335–1340. doi: 10.1083/ng1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Richardson RJ, Dixon J, Malhotra S, Hardman MJ, Knowles L, Boot-Handford RP, Shore P, Whitmarsh A, Dixon MJ. Irf6 is a key determinant of the keratinocyte proliferation-differentiation switch. Nat Genet. 2006;38:1329–1334. doi: 10.1038/ng1894. [DOI] [PubMed] [Google Scholar]
- 111.Böse J, Gruber AD, Helming L, Schiebe S, Wegener I, Hafner M, Beales M, Köntgen F, Lengeling A. The phosphatidylserine receptor has essential functions during embryogenesis but not in apoptotic cell removal. J Biol. 2004;3:15. doi: 10.1186/jbiol10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Thomas T, Corcoran LM, Gugasyan R, Dixon MP, Brodnicki T, Nutt SL, Metcalf D, Voss AK. Monocytic leukemia zinc finger protein is essential for the development of long-term reconstituting hematopoietic stem cells. Genes Dev. 2006;20:1175–1186. doi: 10.1101/gad.1382606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Denaxa M, Sharpe PT, Pachnis V. The LIM homeodomain transcription factors Lhx6 and Lhx7 are key regulators of mammalian dentition. Dev Biol. 2009;333:324–336. doi: 10.1016/j.ydbio.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hsu CY, Chang NC, Lee MW, Lee KH, Sun DS, Lai C, Chang AC. LUZP deficiency affects neural tube closure during brain development. Biochem Biophys Res Commun. 2008;376:466–471. doi: 10.1016/j.bbrc.2008.08.170. [DOI] [PubMed] [Google Scholar]
- 115.Verzi MP, Agarwal P, Brown C, McCulley DJ, Schwarz JJ, Black BL. The transcription factor MEF2C is required for craniofacial development. Dev Cell. 2007;12:645–652. doi: 10.1016/j.devcel.2007.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Jin JZ, Ding J. Analysis of Meox-2 mutant mice reveals a novel postfusion-based cleft palate. Dev Dyn. 2006;235:539–546. doi: 10.1002/dvdy.20641. [DOI] [PubMed] [Google Scholar]
- 117.Meester-Smoor MA, Vermeij M, van Helmond MJ, Molijn AC, van Wely KH, Hekman AC, Vermey-Keers C, Riegman PH, Zwarthoff EC. Targeted disruption of the Mn1 oncogene results in severe defects in development of membranous bones of the cranial skeleton. Mol Cell Biol. 2005;25:4229–4236. doi: 10.1128/MCB.25.10.4229-4236.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Toyo-oka K, Hirotsune S, Gambello MJ, Zhou ZQ, Olson L, Rosenfeld MG, Eisenman R, Hurlin P, Wynshaw-Boris A. Loss of the Max-interacting protein Mnt in mice results in decreased viability, defective embryonic growth and craniofacial defects: relevance to Miller-Dieker syndrome. Hum Mol Genet. 2004;13:1057–1067. doi: 10.1093/hmg/ddh116. [DOI] [PubMed] [Google Scholar]
- 119.Winograd J, Reilly MP, Roe R, Lutz J, Laughner E, Xu X, Hu L, Asakura T, vander Kolk C, Strandberg JD, et al. Perinatal lethality and multiple craniofacial malformations in MSX2 transgenic mice. Hum Mol Genet. 1997;6:369–379. doi: 10.1093/hmg/6.3.369. [DOI] [PubMed] [Google Scholar]
- 120.Shi W, Bain AL, Schwer B, Al-Ejeh F, Smith C, Wong L, Chai H, Miranda MS, Ho U, Kawaguchi M, et al. Essential developmental, genomic stability, and tumour suppressor functions of the mouse orthologue of hSSB1/NABP2. PLoS Genet. 2013;9:e1003298. doi: 10.1371/journal.pgen.1003298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Feldhahn N, Ferretti E, Robbiani DF, Callen E, Deroubaix S, Selleri L, Nussenzweig A, Nussenzweig MC. The hSSB1 orthologue Obfc2b is essential for skeletogenesis but dispensable for the DNA damage response in vivo. EMBO J. 2012;31:4045–4056. doi: 10.1038/emboj.2012.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ross AP, Mansilla MA, Choe Y, Helminski S, Sturm R, Maute RL, May SR, Hozyasz KK, Wójcicki P, Mostowska A, et al. A mutation in mouse Pak1ip1 causes orofacial clefting while human PAK1IP1 maps to 6p24 translocation breaking points associated with orofacial clefting. PLoS One. 2013;8:e69333. doi: 10.1371/journal.pone.0069333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zhang B, Jain S, Song H, Fu M, Heuckeroth RO, Erlich JM, Jay PY, Milbrandt J. Mice lacking sister chromatid cohesion protein PDS5B exhibit developmental abnormalities reminiscent of Cornelia de Lange syndrome. Development. 2007;134:3191–3201. doi: 10.1242/dev.005884. [DOI] [PubMed] [Google Scholar]
- 124.Takihara Y, Tomotsune D, Shirai M, Katoh-Fukui Y, Nishii K, Motaleb MA, Nomura M, Tsuchiya R, Fujita Y, Shibata Y, et al. Targeted disruption of the mouse homologue of the Drosophila polyhomeotic gene leads to altered anteroposterior patterning and neural crest defects. Development. 1997;124:3673–3682. doi: 10.1242/dev.124.19.3673. [DOI] [PubMed] [Google Scholar]
- 125.Lanctôt C, Moreau A, Chamberland M, Tremblay ML, Drouin J. Hindlimb patterning and mandible development require the Ptx1 gene. Development. 1999;126:1805–1810. doi: 10.1242/dev.126.9.1805. [DOI] [PubMed] [Google Scholar]
- 126.Lu MF, Pressman C, Dyer R, Johnson RL, Martin JF. Function of Rieger syndrome gene in left-right asymmetry and craniofacial development. Nature. 1999;401:276–278. doi: 10.1038/45797. [DOI] [PubMed] [Google Scholar]
- 127.Joo JH, Lee YJ, Munguba GC, Park S, Taxter TJ, Elsagga MY, Jackson MR, Oh SP, Sugrue SP. Role of Pinin in neural crest, dorsal dermis, and axial skeleton development and its involvement in the regulation of Tcf/Lef activity in mice. Dev Dyn. 2007;236:2147–2158. doi: 10.1002/dvdy.21243. [DOI] [PubMed] [Google Scholar]
- 128.Bjork BC, Turbe-Doan A, Prysak M, Herron BJ, Beier DR. Prdm16 is required for normal palatogenesis in mice. Hum Mol Genet. 2010;19:774–789. doi: 10.1093/hmg/ddp543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Martin JF, Bradley A, Olson EN. The paired-like homeo box gene MHox is required for early events of skeletogenesis in multiple lineages. Genes Dev. 1995;9:1237–1249. doi: 10.1101/gad.9.10.1237. [DOI] [PubMed] [Google Scholar]
- 130.Metzis V, Courtney AD, Kerr MC, Ferguson C, Rondón Galeano MC, Parton RG, Wainwright BJ, Wicking C. Patched1 is required in neural crest cells for the prevention of orofacial clefts. Hum Mol Genet. 2013;22:5026–5035. doi: 10.1093/hmg/ddt353. [DOI] [PubMed] [Google Scholar]
- 131.Schwab KR, Patterson LT, Hartman HA, Song N, Lang RA, Lin X, Potter SS. Pygo1 and Pygo2 roles in Wnt signaling in mammalian kidney development. BMC Biol. 2007;5:15. doi: 10.1186/1741-7007-5-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Voronina VA, Kozlov S, Mathers PH, Lewandoski M. Conditional alleles for activation and inactivation of the mouse Rx homeobox gene. Genesis. 2005;41:160–164. doi: 10.1002/gene.20109. [DOI] [PubMed] [Google Scholar]
- 133.Mann MB, Hodges CA, Barnes E, Vogel H, Hassold TJ, Luo G. Defective sister-chromatid cohesion, aneuploidy and cancer predisposition in a mouse model of type II Rothmund-Thomson syndrome. Hum Mol Genet. 2005;14:813–825. doi: 10.1093/hmg/ddi075. [DOI] [PubMed] [Google Scholar]
- 134.Aberg T, Cavender A, Gaikwad JS, Bronckers AL, Wang X, Waltimo-Sirén J, Thesleff I, D’Souza RN. Phenotypic changes in dentition of Runx2 homozygote-null mutant mice. J Histochem Cytochem. 2004;52:131–139. doi: 10.1177/002215540405200113. [DOI] [PubMed] [Google Scholar]
- 135.Dobreva G, Chahrour M, Dautzenberg M, Chirivella L, Kanzler B, Fariñas I, Karsenty G, Grosschedl R. SATB2 is a multifunctional determinant of craniofacial patterning and osteoblast differentiation. Cell. 2006;125:971–986. doi: 10.1016/j.cell.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 136.Britanova O, Depew MJ, Schwark M, Thomas BL, Miletich I, Sharpe P, Tarabykin V. Satb2 haploinsufficiency phenocopies 2q32-q33 deletions, whereas loss suggests a fundamental role in the coordination of jaw development. Am J Hum Genet. 2006;79:668–678. doi: 10.1086/508214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Shamblott MJ, Bugg EM, Lawler AM, Gearhart JD. Craniofacial abnormalities resulting from targeted disruption of the murine Sim2 gene. Dev Dyn. 2002;224:373–380. doi: 10.1002/dvdy.10116. [DOI] [PubMed] [Google Scholar]
- 138.Parada C, Li J, Iwata J, Suzuki A, Chai Y. CTGF mediates Smad-dependent transforming growth factor β signaling to regulate mesenchymal cell proliferation during palate development. Mol Cell Biol. 2013;33:3482–3493. doi: 10.1128/MCB.00615-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Papangeli I, Scambler PJ. Tbx1 genetically interacts with the transforming growth factor-β/bone morphogenetic protein inhibitor Smad7 during great vessel remodeling. Circ Res. 2013;112:90–102. doi: 10.1161/CIRCRESAHA.112.270223. [DOI] [PubMed] [Google Scholar]
- 140.Murray SA, Oram KF, Gridley T. Multiple functions of Snail family genes during palate development in mice. Development. 2007;134:1789–1797. doi: 10.1242/dev.02837. [DOI] [PubMed] [Google Scholar]
- 141.Smits P, Li P, Mandel J, Zhang Z, Deng JM, Behringer RR, de Crombrugghe B, Lefebvre V. The transcription factors L-Sox5 and Sox6 are essential for cartilage formation. Dev Cell. 2001;1:277–290. doi: 10.1016/s1534-5807(01)00003-x. [DOI] [PubMed] [Google Scholar]
- 142.Bi W, Huang W, Whitworth DJ, Deng JM, Zhang Z, Behringer RR, de Crombrugghe B. Haploinsufficiency of Sox9 results in defective cartilage primordia and premature skeletal mineralization. Proc Natl Acad Sci USA. 2001;98:6698–6703. doi: 10.1073/pnas.111092198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Mori-Akiyama Y, Akiyama H, Rowitch DH, de Crombrugghe B. Sox9 is required for determination of the chondrogenic cell lineage in the cranial neural crest. Proc Natl Acad Sci USA. 2003;100:9360–9365. doi: 10.1073/pnas.1631288100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Sock E, Rettig SD, Enderich J, Bösl MR, Tamm ER, Wegner M. Gene targeting reveals a widespread role for the high-mobility-group transcription factor Sox11 in tissue remodeling. Mol Cell Biol. 2004;24:6635–6644. doi: 10.1128/MCB.24.15.6635-6644.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kasberg AD, Brunskill EW, Steven Potter S. SP8 regulates signaling centers during craniofacial development. Dev Biol. 2013;381:312–323. doi: 10.1016/j.ydbio.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Coré N, Caubit X, Metchat A, Boned A, Djabali M, Fasano L. Tshz1 is required for axial skeleton, soft palate and middle ear development in mice. Dev Biol. 2007;308:407–420. doi: 10.1016/j.ydbio.2007.05.038. [DOI] [PubMed] [Google Scholar]
- 147.Jerome LA, Papaioannou VE. DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1. Nat Genet. 2001;27:286–291. doi: 10.1038/85845. [DOI] [PubMed] [Google Scholar]
- 148.Zirzow S, Lüdtke TH, Brons JF, Petry M, Christoffels VM, Kispert A. Expression and requirement of T-box transcription factors Tbx2 and Tbx3 during secondary palate development in the mouse. Dev Biol. 2009;336:145–155. doi: 10.1016/j.ydbio.2009.09.020. [DOI] [PubMed] [Google Scholar]
- 149.Pauws E, Hoshino A, Bentley L, Prajapati S, Keller C, Hammond P, Martinez-Barbera JP, Moore GE, Stanier P. Tbx22null mice have a submucous cleft palate due to reduced palatal bone formation and also display ankyloglossia and choanal atresia phenotypes. Hum Mol Genet. 2009;18:4171–4179. doi: 10.1093/hmg/ddp368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Dixon J, Brakebusch C, Fässler R, Dixon MJ. Increased levels of apoptosis in the prefusion neural folds underlie the craniofacial disorder, Treacher Collins syndrome. Hum Mol Genet. 2000;9:1473–1480. doi: 10.1093/hmg/9.10.1473. [DOI] [PubMed] [Google Scholar]
- 151.Brewer S, Feng W, Huang J, Sullivan S, Williams T. Wnt1-Cre-mediated deletion of AP-2alpha causes multiple neural crest-related defects. Dev Biol. 2004;267:135–152. doi: 10.1016/j.ydbio.2003.10.039. [DOI] [PubMed] [Google Scholar]
- 152.Yang A, Schweitzer R, Sun D, Kaghad M, Walker N, Bronson RT, Tabin C, Sharpe A, Caput D, Crum C, et al. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature. 1999;398:714–718. doi: 10.1038/19539. [DOI] [PubMed] [Google Scholar]
- 153.Bertuzzi S, Hindges R, Mui SH, O’Leary DD, Lemke G. The homeodomain protein vax1 is required for axon guidance and major tract formation in the developing forebrain. Genes Dev. 1999;13:3092–3105. doi: 10.1101/gad.13.23.3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Nimura K, Ura K, Shiratori H, Ikawa M, Okabe M, Schwartz RJ, Kaneda Y. A histone H3 lysine 36 trimethyltransferase links Nkx2-5 to Wolf-Hirschhorn syndrome. Nature. 2009;460:287–291. doi: 10.1038/nature08086. [DOI] [PubMed] [Google Scholar]
- 155.Kurima K, Hertzano R, Gavrilova O, Monahan K, Shpargel KB, Nadaraja G, Kawashima Y, Lee KY, Ito T, Higashi Y, et al. A noncoding point mutation of Zeb1 causes multiple developmental malformations and obesity in Twirler mice. PLoS Genet. 2011;7:e1002307. doi: 10.1371/journal.pgen.1002307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Jiang Z, Zhu L, Hu L, Slesnick TC, Pautler RG, Justice MJ, Belmont JW. Zic3 is required in the extra-cardiac perinodal region of the lateral plate mesoderm for left-right patterning and heart development. Hum Mol Genet. 2013;22:879–889. doi: 10.1093/hmg/dds494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Yang Y, Mahaffey CL, Bérubé N, Frankel WN. Interaction between fidgetin and protein kinase A-anchoring protein AKAP95 is critical for palatogenesis in the mouse. J Biol Chem. 2006;281:22352–22359. doi: 10.1074/jbc.M603626200. [DOI] [PubMed] [Google Scholar]
- 158.Cecconi F, Alvarez-Bolado G, Meyer BI, Roth KA, Gruss P. Apaf1 (CED-4 homolog) regulates programmed cell death in mammalian development. Cell. 1998;94:727–737. doi: 10.1016/s0092-8674(00)81732-8. [DOI] [PubMed] [Google Scholar]
- 159.Dowdle WE, Robinson JF, Kneist A, Sirerol-Piquer MS, Frints SG, Corbit KC, Zaghloul NA, van Lijnschoten G, Mulders L, Verver DE, et al. Disruption of a ciliary B9 protein complex causes Meckel syndrome. Am J Hum Genet. 2011;89:94–110. doi: 10.1016/j.ajhg.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Atasoy D, Schoch S, Ho A, Nadasy KA, Liu X, Zhang W, Mukherjee K, Nosyreva ED, Fernandez-Chacon R, Missler M, et al. Deletion of CASK in mice is lethal and impairs synaptic function. Proc Natl Acad Sci USA. 2007;104:2525–2530. doi: 10.1073/pnas.0611003104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Yan Y, Frisén J, Lee MH, Massagué J, Barbacid M. Ablation of the CDK inhibitor p57Kip2 results in increased apoptosis and delayed differentiation during mouse development. Genes Dev. 1997;11:973–983. doi: 10.1101/gad.11.8.973. [DOI] [PubMed] [Google Scholar]
- 162.Zhang P, Liégeois NJ, Wong C, Finegold M, Hou H, Thompson JC, Silverman A, Harper JW, DePinho RA, Elledge SJ. Altered cell differentiation and proliferation in mice lacking p57KIP2 indicates a role in Beckwith-Wiedemann syndrome. Nature. 1997;387:151–158. doi: 10.1038/387151a0. [DOI] [PubMed] [Google Scholar]
- 163.Li Q, Lu Q, Hwang JY, Büscher D, Lee KF, Izpisua-Belmonte JC, Verma IM. IKK1-deficient mice exhibit abnormal development of skin and skeleton. Genes Dev. 1999;13:1322–1328. doi: 10.1101/gad.13.10.1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Park TJ, Boyd K, Curran T. Cardiovascular and craniofacial defects in Crk-null mice. Mol Cell Biol. 2006;26:6272–6282. doi: 10.1128/MCB.00472-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Lin C, Fisher AV, Yin Y, Maruyama T, Veith GM, Dhandha M, Huang GJ, Hsu W, Ma L. The inductive role of Wnt-β-Catenin signaling in the formation of oral apparatus. Dev Biol. 2011;356:40–50. doi: 10.1016/j.ydbio.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.He F, Xiong W, Wang Y, Li L, Liu C, Yamagami T, Taketo MM, Zhou C, Chen Y. Epithelial Wnt/β-catenin signaling regulates palatal shelf fusion through regulation of Tgfβ3 expression. Dev Biol. 2011;350:511–519. doi: 10.1016/j.ydbio.2010.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Okano J, Kimura W, Papaionnou VE, Miura N, Yamada G, Shiota K, Sakai Y. The regulation of endogenous retinoic acid level through CYP26B1 is required for elevation of palatal shelves. Dev Dyn. 2012;241:1744–1756. doi: 10.1002/dvdy.23862. [DOI] [PubMed] [Google Scholar]
- 168.Keber R, Motaln H, Wagner KD, Debeljak N, Rassoulzadegan M, Ačimovič J, Rozman D, Horvat S. Mouse knockout of the cholesterogenic cytochrome P450 lanosterol 14alpha-demethylase (Cyp51) resembles Antley-Bixler syndrome. J Biol Chem. 2011;286:29086–29097. doi: 10.1074/jbc.M111.253245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Wassif CA, Zhu P, Kratz L, Krakowiak PA, Battaile KP, Weight FF, Grinberg A, Steiner RD, Nwokoro NA, Kelley RI, et al. Biochemical, phenotypic and neurophysiological characterization of a genetic mouse model of RSH/Smith--Lemli--Opitz syndrome. Hum Mol Genet. 2001;10:555–564. doi: 10.1093/hmg/10.6.555. [DOI] [PubMed] [Google Scholar]
- 170.Fitzky BU, Moebius FF, Asaoka H, Waage-Baudet H, Xu L, Xu G, Maeda N, Kluckman K, Hiller S, Yu H, et al. 7-Dehydrocholesterol-dependent proteolysis of HMG-CoA reductase suppresses sterol biosynthesis in a mouse model of Smith-Lemli-Opitz/RSH syndrome. J Clin Invest. 2001;108:905–915. doi: 10.1172/JCI12103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Adams MK, Belyaeva OV, Wu L, Kedishvili NY. The retinaldehyde reductase activity of DHRS3 is reciprocally activated by retinol dehydrogenase 10 to control retinoid homeostasis. J Biol Chem. 2014;289:14868–14880. doi: 10.1074/jbc.M114.552257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Billings SE, Pierzchalski K, Butler Tjaden NE, Pang XY, Trainor PA, Kane MA, Moise AR. The retinaldehyde reductase DHRS3 is essential for preventing the formation of excess retinoic acid during embryonic development. FASEB J. 2013;27:4877–4889. doi: 10.1096/fj.13-227967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Caruana G, Bernstein A. Craniofacial dysmorphogenesis including cleft palate in mice with an insertional mutation in the discs large gene. Mol Cell Biol. 2001;21:1475–1483. doi: 10.1128/MCB.21.5.1475-1483.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Zhang Z, Wlodarczyk BJ, Niederreither K, Venugopalan S, Florez S, Finnell RH, Amendt BA. Fuz regulates craniofacial development through tissue specific responses to signaling factors. PLoS One. 2011;6:e24608. doi: 10.1371/journal.pone.0024608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Schaeper U, Vogel R, Chmielowiec J, Huelsken J, Rosario M, Birchmeier W. Distinct requirements for Gab1 in Met and EGF receptor signaling in vivo. Proc Natl Acad Sci USA. 2007;104:15376–15381. doi: 10.1073/pnas.0702555104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Asada H, Kawamura Y, Maruyama K, Kume H, Ding RG, Kanbara N, Kuzume H, Sanbo M, Yagi T, Obata K. Cleft palate and decreased brain gamma-aminobutyric acid in mice lacking the 67-kDa isoform of glutamic acid decarboxylase. Proc Natl Acad Sci USA. 1997;94:6496–6499. doi: 10.1073/pnas.94.12.6496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Condie BG, Bain G, Gottlieb DI, Capecchi MR. Cleft palate in mice with a targeted mutation in the gamma-aminobutyric acid-producing enzyme glutamic acid decarboxylase 67. Proc Natl Acad Sci USA. 1997;94:11451–11455. doi: 10.1073/pnas.94.21.11451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Li JP, Gong F, Hagner-McWhirter A, Forsberg E, Abrink M, Kisilevsky R, Zhang X, Lindahl U. Targeted disruption of a murine glucuronyl C5-epimerase gene results in heparan sulfate lacking L-iduronic acid and in neonatal lethality. J Biol Chem. 2003;278:28363–28366. doi: 10.1074/jbc.C300219200. [DOI] [PubMed] [Google Scholar]
- 179.Miyaoka Y, Tanaka M, Imamura T, Takada S, Miyajima A. A novel regulatory mechanism for Fgf18 signaling involving cysteine-rich FGF receptor (Cfr) and delta-like protein (Dlk) Development. 2010;137:159–167. doi: 10.1242/dev.041574. [DOI] [PubMed] [Google Scholar]
- 180.Saxton TM, Cheng AM, Ong SH, Lu Y, Sakai R, Cross JC, Pawson T. Gene dosage-dependent functions for phosphotyrosine-Grb2 signaling during mammalian tissue morphogenesis. Curr Biol. 2001;11:662–670. doi: 10.1016/s0960-9822(01)00198-1. [DOI] [PubMed] [Google Scholar]
- 181.Liu KJ, Arron JR, Stankunas K, Crabtree GR, Longaker MT. Chemical rescue of cleft palate and midline defects in conditional GSK-3beta mice. Nature. 2007;446:79–82. doi: 10.1038/nature05557. [DOI] [PubMed] [Google Scholar]
- 182.Bullock SL, Fletcher JM, Beddington RS, Wilson VA. Renal agenesis in mice homozygous for a gene trap mutation in the gene encoding heparan sulfate 2-sulfotransferase. Genes Dev. 1998;12:1894–1906. doi: 10.1101/gad.12.12.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Keady BT, Samtani R, Tobita K, Tsuchya M, San Agustin JT, Follit JA, Jonassen JA, Subramanian R, Lo CW, Pazour GJ. IFT25 links the signal-dependent movement of Hedgehog components to intraflagellar transport. Dev Cell. 2012;22:940–951. doi: 10.1016/j.devcel.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Grashoff C, Aszódi A, Sakai T, Hunziker EB, Fässler R. Integrin-linked kinase regulates chondrocyte shape and proliferation. EMBO Rep. 2003;4:432–438. doi: 10.1038/sj.embor.embor801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Sohaskey ML, Yu J, Diaz MA, Plaas AH, Harland RM. JAWS coordinates chondrogenesis and synovial joint positioning. Development. 2008;135:2215–2220. doi: 10.1242/dev.019950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Jacoby M, Cox JJ, Gayral S, Hampshire DJ, Ayub M, Blockmans M, Pernot E, Kisseleva MV, Compère P, Schiffmann SN, et al. INPP5E mutations cause primary cilium signaling defects, ciliary instability and ciliopathies in human and mouse. Nat Genet. 2009;41:1027–1031. doi: 10.1038/ng.427. [DOI] [PubMed] [Google Scholar]
- 187.Brugmann SA, Allen NC, James AW, Mekonnen Z, Madan E, Helms JA. A primary cilia-dependent etiology for midline facial disorders. Hum Mol Genet. 2010;19:1577–1592. doi: 10.1093/hmg/ddq030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Song Z, Liu C, Iwata J, Gu S, Suzuki A, Sun C, He W, Shu R, Li L, Chai Y, et al. Mice with Tak1 deficiency in neural crest lineage exhibit cleft palate associated with abnormal tongue development. J Biol Chem. 2013;288:10440–10450. doi: 10.1074/jbc.M112.432286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Yumoto K, Thomas PS, Lane J, Matsuzaki K, Inagaki M, Ninomiya-Tsuji J, Scott GJ, Ray MK, Ishii M, Maxson R, et al. TGF-β-activated kinase 1 (Tak1) mediates agonist-induced Smad activation and linker region phosphorylation in embryonic craniofacial neural crest-derived cells. J Biol Chem. 2013;288:13467–13480. doi: 10.1074/jbc.M112.431775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Kowalczyk MS, Hughes JR, Babbs C, Sanchez-Pulido L, Szumska D, Sharpe JA, Sloane-Stanley JA, Morriss-Kay GM, Smoot LB, Roberts AE, et al. Nprl3 is required for normal development of the cardiovascular system. Mamm Genome. 2012;23:404–415. doi: 10.1007/s00335-012-9398-y. [DOI] [PubMed] [Google Scholar]
- 191.Ferrante MI, Zullo A, Barra A, Bimonte S, Messaddeq N, Studer M, Dollé P, Franco B. Oral-facial-digital type I protein is required for primary cilia formation and left-right axis specification. Nat Genet. 2006;38:112–117. doi: 10.1038/ng1684. [DOI] [PubMed] [Google Scholar]
- 192.Lu S, Lu LY, Liu MF, Yuan QJ, Sham MH, Guan XY, Huang JD. Cerebellar defects in Pdss2 conditional knockout mice during embryonic development and in adulthood. Neurobiol Dis. 2012;45:219–233. doi: 10.1016/j.nbd.2011.08.006. [DOI] [PubMed] [Google Scholar]
- 193.Keller P, Tremml G, Rosti V, Bessler M. X inactivation and somatic cell selection rescue female mice carrying a Piga-null mutation. Proc Natl Acad Sci USA. 1999;96:7479–7483. doi: 10.1073/pnas.96.13.7479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Kinoshita M, Era T, Jakt LM, Nishikawa S. The novel protein kinase Vlk is essential for stromal function of mesenchymal cells. Development. 2009;136:2069–2079. doi: 10.1242/dev.026435. [DOI] [PubMed] [Google Scholar]
- 195.Imuta Y, Nishioka N, Kiyonari H, Sasaki H. Short limbs, cleft palate, and delayed formation of flat proliferative chondrocytes in mice with targeted disruption of a putative protein kinase gene, Pkdcc (AW548124) Dev Dyn. 2009;238:210–222. doi: 10.1002/dvdy.21822. [DOI] [PubMed] [Google Scholar]
- 196.Yang T, Jia Z, Bryant-Pike W, Chandrasekhar A, Murray JC, Fritzsch B, Bassuk AG. Analysis of PRICKLE1 in human cleft palate and mouse development demonstrates rare and common variants involved in human malformations. Mol Genet Genomic Med. 2014;2:138–151. doi: 10.1002/mgg3.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Ng JM, Vrieling H, Sugasawa K, Ooms MP, Grootegoed JA, Vreeburg JT, Visser P, Beems RB, Gorgels TG, Hanaoka F, et al. Developmental defects and male sterility in mice lacking the ubiquitin-like DNA repair gene mHR23B. Mol Cell Biol. 2002;22:1233–1245. doi: 10.1128/MCB.22.4.1233-1245.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Jin YR, Turcotte TJ, Crocker AL, Han XH, Yoon JK. The canonical Wnt signaling activator, R-spondin2, regulates craniofacial patterning and morphogenesis within the branchial arch through ectodermal-mesenchymal interaction. Dev Biol. 2011;352:1–13. doi: 10.1016/j.ydbio.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Nam JS, Park E, Turcotte TJ, Palencia S, Zhan X, Lee J, Yun K, Funk WD, Yoon JK. Mouse R-spondin2 is required for apical ectodermal ridge maintenance in the hindlimb. Dev Biol. 2007;311:124–135. doi: 10.1016/j.ydbio.2007.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Insolera R, Shao W, Airik R, Hildebrandt F, Shi SH. SDCCAG8 regulates pericentriolar material recruitment and neuronal migration in the developing cortex. Neuron. 2014;83:805–822. doi: 10.1016/j.neuron.2014.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Wojcik SM, Katsurabayashi S, Guillemin I, Friauf E, Rosenmund C, Brose N, Rhee JS. A shared vesicular carrier allows synaptic corelease of GABA and glycine. Neuron. 2006;50:575–587. doi: 10.1016/j.neuron.2006.04.016. [DOI] [PubMed] [Google Scholar]
- 202.Oh WJ, Westmoreland JJ, Summers R, Condie BG. Cleft palate is caused by CNS dysfunction in Gad1 and Viaat knockout mice. PLoS One. 2010;5:e9758. doi: 10.1371/journal.pone.0009758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Yang X, Kilgallen S, Andreeva V, Spicer DB, Pinz I, Friesel R. Conditional expression of Spry1 in neural crest causes craniofacial and cardiac defects. BMC Dev Biol. 2010;10:48. doi: 10.1186/1471-213X-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Welsh IC, Hagge-Greenberg A, O’Brien TP. A dosage-dependent role for Spry2 in growth and patterning during palate development. Mech Dev. 2007;124:746–761. doi: 10.1016/j.mod.2007.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Alkuraya FS, Saadi I, Lund JJ, Turbe-Doan A, Morton CC, Maas RL. SUMO1 haploinsufficiency leads to cleft lip and palate. Science. 2006;313:1751. doi: 10.1126/science.1128406. [DOI] [PubMed] [Google Scholar]
- 206.Qu X, Pan Y, Carbe C, Powers A, Grobe K, Zhang X. Glycosaminoglycan-dependent restriction of FGF diffusion is necessary for lacrimal gland development. Development. 2012;139:2730–2739. doi: 10.1242/dev.079236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Cui C, Chatterjee B, Lozito TP, Zhang Z, Francis RJ, Yagi H, Swanhart LM, Sanker S, Francis D, Yu Q, et al. Wdpcp, a PCP protein required for ciliogenesis, regulates directional cell migration and cell polarity by direct modulation of the actin cytoskeleton. PLoS Biol. 2013;11:e1001720. doi: 10.1371/journal.pbio.1001720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Li SW, Prockop DJ, Helminen H, Fässler R, Lapveteläinen T, Kiraly K, Peltarri A, Arokoski J, Lui H, Arita M. Transgenic mice with targeted inactivation of the Col2 alpha 1 gene for collagen II develop a skeleton with membranous and periosteal bone but no endochondral bone. Genes Dev. 1995;9:2821–2830. doi: 10.1101/gad.9.22.2821. [DOI] [PubMed] [Google Scholar]
- 209.Costell M, Gustafsson E, Aszódi A, Mörgelin M, Bloch W, Hunziker E, Addicks K, Timpl R, Fässler R. Perlecan maintains the integrity of cartilage and some basement membranes. J Cell Biol. 1999;147:1109–1122. doi: 10.1083/jcb.147.5.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Arikawa-Hirasawa E, Watanabe H, Takami H, Hassell JR, Yamada Y. Perlecan is essential for cartilage and cephalic development. Nat Genet. 1999;23:354–358. doi: 10.1038/15537. [DOI] [PubMed] [Google Scholar]
- 211.Masago Y, Hosoya A, Kawasaki K, Kawano S, Nasu A, Toguchida J, Fujita K, Nakamura H, Kondoh G, Nagata K. The molecular chaperone Hsp47 is essential for cartilage and endochondral bone formation. J Cell Sci. 2012;125:1118–1128. doi: 10.1242/jcs.089748. [DOI] [PubMed] [Google Scholar]
- 212.Rainger J, van Beusekom E, Ramsay JK, McKie L, Al-Gazali L, Pallotta R, Saponari A, Branney P, Fisher M, Morrison H, et al. Loss of the BMP antagonist, SMOC-1, causes Ophthalmo-acromelic (Waardenburg Anophthalmia) syndrome in humans and mice. PLoS Genet. 2011;7:e1002114. doi: 10.1371/journal.pgen.1002114. [DOI] [PMC free article] [PubMed] [Google Scholar]