

Abstract
RAS proteins are signal transduction gatekeepers that mediate cell growth, survival, and differentiation through interactions with multiple effector proteins. The RAS effector RAS- and RAB-interacting protein 1 (RIN1) activates its own downstream effectors, the small GTPase RAB5 and the tyrosine kinase Abelson tyrosine-protein kinase (ABL), to modulate endocytosis and cytoskeleton remodeling. To identify ABL substrates downstream of RAS-to-RIN1 signaling, we examined human HEK293T cells overexpressing components of this pathway. Proteomic analysis revealed several novel phosphotyrosine peptides, including Harvey rat sarcoma oncogene (HRAS)-pTyr137. Here we report that ABL phosphorylates tyrosine 137 of H-, K-, and NRAS. Increased RIN1 levels enhanced HRAS-Tyr137 phosphorylation by nearly 5-fold, suggesting that RAS-stimulated RIN1 can drive ABL-mediated RAS modification in a feedback circuit. Tyr137 is well conserved among RAS orthologs and is part of a transprotein H-bond network. Crystal structures of HRASY137F and HRASY137E revealed conformation changes radiating from the mutated residue. Although consistent with Tyr137 participation in allosteric control of HRAS function, the mutations did not alter intrinsic GTP hydrolysis rates in vitro. HRAS-Tyr137 phosphorylation enhanced HRAS signaling capacity in cells, however, as reflected by a 4-fold increase in the association of phosphorylated HRASG12V with its effector protein RAF proto-oncogene serine/threonine protein kinase 1 (RAF1). These data suggest that RAS phosphorylation at Tyr137 allosterically alters protein conformation and effector binding, providing a mechanism for effector-initiated modulation of RAS signaling.—Ting, P. Y., Johnson, C. W., Fang, C., Cao, X., Graeber, T. G., Mattos, C., Colicelli, J. Tyrosine phosphorylation of RAS by ABL allosterically enhances effector binding.
Keywords: GTPase, signal transduction, RIN1, RAF1
The vertebrate family of RAS GTPases comprises 3 members, HRAS, neuroblastoma RAS oncogene (NRAS), and Kirsten rat sarcoma oncogene (KRAS), that function as molecular switches that transduce signals from cell surface receptors to the cytoplasm. RAS is activated by the exchange of GDP for GTP, a process catalyzed by guanine nucleotide exchange factors (GEFs) (1, 2). Upon activation, RAS proteins show increased binding affinity for a diverse group of effectors that regulate cell growth, proliferation, survival, and differentiation (3). Because these effectors bind to overlapping RAS surfaces, signal output is largely determined by differences in effector expression levels, local concentration, and binding affinity (4). RAS returns to its inactive conformation by hydrolyzing GTP. RAS proteins have a low intrinsic rate of GTP hydrolysis (5), but this is greatly enhanced by GTPase-activating proteins (GAPs) such as RASA1 (also known as p120RASGAP) (6, 7). Regulation by GEFs and GAPs tightly restricts RAS activation status. Indeed, constitutive activating RAS mutations are among the most common driver mutations in human cancers (8).
A few posttranslational modifications also contribute to RAS regulation (9). All 3 RAS proteins are clipped and methylated proteolytically in conjunction with attachment of a carboxyl-terminal prenyl group (10). In HRAS, NRAS, and the KRAS-4A isoform, 1 or 2 cysteine residues near the prenyl addition site are palmitoylated (10, 11), promoting localization to the plasma membrane where receptors and upstream activators reside. Other RAS posttranslational modifications include ubiquitylation (12–14). Lys147 monoubiquitination enhances GTP loading and increases RAS affinity for the effectors RAF1 and PI3K (13). HRAS Thr144/Thr148 phosphorylation by GSK3β leads to polyubiquitination and proteasome-mediated degradation (15). KRAS-4A Ser181 phosphorylation causes dissociation from the plasma membrane, translocation to internal membranes and cell death (16, 17), and RAS Cys118 nitrosylation promotes guanine nucleotide exchange (18).
RIN1 is a RAS effector (19–21) that orchestrates downstream signals modulating receptor endocytosis, cell adhesion, and cell migration (22–25). This is achieved, in part, by RIN1 binding to and stimulating the nonreceptor tyrosine kinases Abelson tyrosine-protein kinases 1 and 2 (ABL1 and ABL2) (22, 26). RAS-RIN1-ABL complexes can be detected by several methods, including coimmunoprecipitation (22). The interaction between RIN1 and ABL is initiated by low-affinity binding between a proline-rich motif on RIN1 and the SH3 domain of ABL. ABL subsequently phosphorylates RIN1-Tyr36, which then binds the ABL-SH2 domain, resulting in a stable divalent interaction (21, 27). RIN1 binding stimulates ABL kinase activity by derepression of an autoinhibitory conformation (26). ABL tyrosine kinases preferentially phosphorylate proteins involved in cell motility, adhesion, endocytosis, and DNA damage response, and show specificity for the consensus target sequence Y-x-x-P (28).
Enhanced ABL signaling is implicated in a wide range of neoplasias (29), and BCR-ABL1 fusion proteins are the causative genetic abnormality in chronic myelogenous leukemia and many cases of acute lymphocytic leukemia (30). BCR fusion confers constitutive kinase activity, but BCR-ABL1 activity is further enhanced by RIN1’s derepression effect. RIN1 binding to BCR-ABL1 is necessary for bone marrow cell transformation to growth factor independence ex vivo, and RIN1 silencing sensitizes leukemia cells to the ABL tyrosine kinase inhibitor imatinib (31). In addition, RIN1 is upregulated and associated with poor prognosis in many tumor types, including melanoma (32), gastric adenocarcinoma (33), non–small cell lung cancer (34, 35), and bladder urothelial carcinoma (36).
To determine how RAS-to-RIN1 signaling affects ABL activity and substrate specificity, we compared phosphotyrosine peptides from cells expressing HRAS, RIN1, and ABL2. We unexpectedly identified the novel tyrosine phosphorylation site HRAS-pTyr137. Here we report that ABL can phosphorylate HRAS on Tyr137 in vitro and when overexpressed in 293T cells. Phosphorylation is enhanced by HRAS palmitoylation. HRAS-pTyr137 phosphorylation levels were also increased by RIN1 overexpression, consistent with RIN1 involvement in this posttranslational modification pathway. The contributions of Tyr137 to RAS function were investigated by structural analysis and by quantified binding of activated RAS (with or without Tyr137 phosphorylation) to the effector protein RAF1.
MATERIALS AND METHODS
Expression constructs
pcDNA3 ABL2, ABL2K319R (kinase-dead), RIN1, HRAS, and HRASG12V have previously been described (22). pcDNA3 HRASC181S/C184S palmitoylation mutants were made by amplifying HRAS with the reverse primer 5′- ATATCTCGAGTCAGGAGAGCACACACTTGCTGCTCATGCTGCCGGGGCCACTCTCATC-3′. V5-tagged HRAS was constructed by inserting annealed oligos encoding the V5 tag (5′-GGC AAA CCG ATC CCG AAT CCG CTG CTG GGC CTG GAC TCT ACC-3′). HRAS and HRASY137F fragments were generated by PCR using primers containing flanking AttB1 and AttB2 sites and cloned into pDONR221 (Life Technologies, Carlsbad, CA, USA). These fragments were then subcloned into pcDNA5-FRT/TO-3xHA-3xFLAG.
pFastBac ABL1(1-531) and ABL2 have been described previously (26). pDEST15 RASA1-C SH2 was a gift of Dr. Shawn Li (University of Western Ontario, London, ON, Canada). The R377A mutation was made by site-directed mutagenesis using forward primer 5′-GCAGTTTTCTTGTGGCGCCCTCAGATAATACTCC-3′ and a complementary reverse primer. pGEX-KG-RafRBD(1-149) was a gift of Frank McCormick (University of California, San Francisco, San Francisco, CA, USA). pProExHT-HRAS was a gift of John Kuriyan (University of California, Berkeley, Berkeley, CA, USA). The Y137E mutation was made by site-directed mutagenesis using forward primer 5′-GACCTCGCCCGAAGCGAAGGCATCCCCTACATCG-3′ and a complementary reverse primer. The Y137F mutation was made by site-directed mutagenesis using forward primer 5′-CTCGCCCGAAGCTTCGGCATCCCCTAC-3′ and a complementary reverse primer.
Protein expression and purification
ABL1 and ABL2 were produced in Sf9 insect cells and purified as described previously (26). GST-tagged RASA1-SH2C, RASA1-SH2CR377A and RAF1-RAS binding domain (RBD) were expressed in Escherichia coli strain BL21 and purified as follows. Bacteria were grown at 37°C to optical density of 0.6, induced with 1 mM isopropyl β-d-1-thiogalactopyranoside and grown at 37°C for 3 h. Cultures were pelleted and then resuspended in wash buffer (20 mM Tris pH 8.0, 250 mM NaCl, 10% glycerol, 0.01% Triton X-100, protease inhibitors) and sonicated. Lysates were clarified by centrifugation at 15,000 g for 20 min at 4°C. Supernatant was rotated with glutathione-Sepharose beads (GE Life Sciences, Pittsburgh, PA, USA) for 1 h at 4°C. Beads were washed over a column 4 times with wash buffer and then eluted with increasing concentrations of reduced glutathione (5–40 mM). Proteins were dialyzed in 50 mM Tris, pH 8, 50 mM NaCl, 1 mM DTT, and 10% glycerol. Protein concentrations were determined by Bradford assay, and aliquots were frozen at −80°C until use.
His-tagged HRAS, HRASY137F, and HRASY137E were expressed in BL21 bacteria and purified as follows. Bacteria were grown to optical density of 2.5 and then diluted 1:1 with fresh Luria-Bertani medium. Cultures were induced with 1 mM isopropyl β-d-1-thiogalactopyranoside and grown at 18°C overnight. After pelleting, cells were resuspended in lysis buffer (2.65 mM NaH2PO4, 47.35 mM Na2HPO4, 500 mM NaCl, 20 mM imidazole) and sonicated. Lysates were clarified by centrifugation at 15,000 g for 30 min at 4°C. Supernatants were rotated with nickel-NTA beads (Qiagen, Venlo, The Netherlands) for 1 h at 4°C. Beads were washed over a column 4 times with lysis buffer and then eluted with increasing concentrations of imidazole (150–500 mM). Proteins were dialyzed in 25 mM Tris, pH 8.0, and 50 mM NaCl and stored as described above.
Cell culture and reagents
HEK293T cells were cultured in DMEM (Media Tech, Manassas, VA, USA) with 10% fetal bovine serum (FBS; Thermo Fisher Scientific, Waltham, MA, USA) and 1% penicillin streptomycin (Life Technologies). All transfections were performed using Polyfect (Qiagen). Imatinib and dasatinib treatments were performed using 500 nM drug for 12 h, followed by an additional 500 nM dose and incubation for 30 min before lysing in Nonidet P-40 (NP-40) buffer.
Flp-In T-REX-293 cells (Life Technologies) expressing 3xHA-3xFLAG-HRAS and 3xHA-3xFLAG-HRASY137F were generated using the Flp-In system (Life Technologies) according to the manufacturer’s directions.
Immunoprecipitation and Immunoblotting
Antibodies used and their sources were ABL1 1:1000 (sc-131; Santa Cruz Biotechnology, Santa Cruz, CA, USA), ABL2 1:1000 (sc-6356; Santa Cruz Biotechnology), FLAG 1:3000 (F1804; Sigma-Aldrich, St. Louis, MO, USA), phosphotyrosine 4G10 1:1000 (05-321; EMD Millipore, Billerica, MA, USA), RAS 1:10,000 (EP1125Y; Novus Bio), RIN1 1:1000 (mouse mAb, clone C9E11) (24), V5 1:3000 (R960-25; Life Technologies). A peptide encoding pTyr137 (Ac-AQDLARSpY137GIPYI-Ahx-C-amide) was used to generate anti-RAS-pTyr137 rabbit polyclonal antibodies (1:500; 21st Century Biochemicals, Marlboro, MA, USA) that were used. Antibodies were purified from bleeds by incubation with protein A beads. To eliminate antibodies that bind unphosphorylated RAS or residues flanking Tyr137, antibodies were negatively selected using an unphosphorylated peptide (Ac-AQDLARSY137GIPYI-Ahx-C-amide) and then negatively selected again over bacterially purified GST-HRAS(1-166). Secondary antibodies included sheep-anti-mouse-horseradish peroxidase (HRP) 1:3000 (NA931; Amersham Biosciences, Piscataway, NJ, USA), goat-anti-rabbit-HRP 1:3000 (4741506; Kirkegaard and Perry, Gaithersburg, MD, USA), goat-anti-rabbit-IRDye 800 1:5000 (926-32211; Li-Cor Biosciences, Lincoln, NE, USA) and goat-anti-mouse-IRDye 680 1:5000 (926-32220; Li-Cor Biosciences).
Mass spectrometry and phosphopeptide identification by fragmentation spectra sequencing and chromatography alignment
HEK 293T cells were cultured in DMEM with 10% FBS and 1% penicillin/streptomycin. Cells were transfected with pcDNA3 RIN1, pcDNA3 ABL2, and/or pcDNA3 HRASG12V using Polyfect (Qiagen). Cells were lysed by sonication in urea buffer (8 M urea, 50 mM Tris-HCl, pH 7.5, 1 mM vanadate). Phosphotyrosine peptides were immunoprecipitated with antiphosphotyrosine antibodies (clone 4G10; Millipore) using 2 × 108 cells as described previously (37).
Phosphorylated peptides were analyzed by liquid chromatography-mass spectrometry/mass spectrometry using an autosampler coupled with Nano2DLC pump (Eksigent, Framingham, MA, USA) and LTQ-Orbitrap (Thermo Fisher Scientific). The samples were loaded onto an analytical column (10 cm × 75 μm inner diameter) packed with 5 μm Integrafit Proteopep2 300 Å C18 (New Objective, Woburn, MA, USA). Peptides were eluted into the mass spectrometer using a HPLC gradient of 5−40% buffer B in 45 min followed by a quick gradient of 40−90% buffer B in 10 min, where buffer A contains 0.1% formic acid in water and buffer B contains 0.1% formic acid in acetonitrile (Ultima Gold; Thermo Fisher Scientific). Mass spectra were collected in positive ion mode using the Orbitrap for parent mass determination and the LTQ for data-dependent MS/MS acquisition of the top 5 most abundant peptides. Each sample was analyzed twice (replicate runs), and in each run, one-half of the sample was injected.
MS/MS fragmentation spectra were searched with SEQUEST (version v.27, rev. 12; Thermo Fisher Scientific) against a database containing the human International Protein Index protein database (ftp://ftp.ebi.ac.uk/pub/databases/IPI). Search parameters included carboxyamidomethylation of cysteine as static modification. Dynamic modifications included phosphorylation on tyrosine and oxidation on methionine. Results derived from database searching were filtered using the following criteria: Xcorr > 1.0(+1), 1.5(+2), 2(+3); peptide probability score < 0.001; deltaCn > 0.1; and mass accuracy < 5 parts per million with Bioworks, version 3.2 (Thermo Electron Corp., Madison, WI, USA). We estimated the false-positive rate of sequence assignments at 0.5% on the basis of a composite target-reversed decoy database search strategy (38). The Ascore algorithm was used to more accurately localize the phosphate on the peptide (http://ascore.med.harvard.edu) (39).
As is common in data-dependent MS2 fragmentation sequencing, some peptides identified by sequencing in one sample may not be sequenced or identified in another sample even if the peak is present. Peptide peaks sequenced in some samples but not in others were located in the remaining samples by aligning the chromatogram elution profiles by means of a dynamic time warping algorithm (40). An extended explanation of the strategy used in this work, and example performance results, can be found in the supporting information of Zimman et al. (37).
In vitro kinase assays
Purified ABL and GST-HRAS were incubated in a kinase buffer (100 mM NaCl, 10 mM Tris, pH 7.5, 1 mM DTT, 10 mM MgCl2, 500 μM ATP, 100 μM Na3VO4, 100 μM NaF) for 15 min at 30°C. To load HRAS with GDP or GTPγS, purified GST-HRAS was incubated with 5 mM DTT, 4 mM EDTA, and ×10 M excess of nucleotide on ice. After 1.5 h, 10 mM MgCl2 was added and the reaction was incubated on ice for an additional 30 min.
RAS peptide pull-down assays
Biotinylated RAS peptides were synthesized by 21st Century Biochemicals and encoded pTyr137 (Biot-Ahx-LARS(pY)GIPFIE-amide) or Y137F (Biot-Ahx-LARSFGIPFIE-amide) surrounded by flanking sequences. For the pull-down assays, 10 μg peptide was mixed with 10 μg GST-RASA1-SH2C in 300 μl NP-40 buffer and rotated at 4°C for 4 h; 50 μl of 50% avidin slurry then was added to each tube and rotated for 2 h at 4°C. Beads were washed 4 times with NP-40 buffer and then boiled in SDS-PAGE sample buffer.
Silver staining was performed by fixing the gel in 40% ethanol and 10% acetic acid while shaking for 1 h. After draining the fixing solution, sensitizing solution (30% ethanol, 0.2% sodium thiosulfate, 6.8% sodium acetate) was added and the gel shaken for 30 min. The gel then was washed 3 times with water. Silver nitrate solution (0.25% silver nitrate in water) was added, and the gel was shaken for 20 min. After washing twice with water, developing solution (2.5% sodium carbonate, 0.015% formaldehyde) was added and the gel was shaken until a brown, smoky precipitate appeared. The solution then was replaced with fresh developer solution, continuing until the desired intensity of spots was achieved, at which time the developer solution was drained and 5% acetic acid stop solution was added for 10 min. The gel then was washed 3 times with water (5 min each time), and the image was acquired.
GTP hydrolysis assays
The single turnover hydrolysis rate experiments were done using C-terminally truncated HRAS, HRASY137E, and HRASY137F (residues 1-166) (41). In brief, a 5 µM concentration of wild-type and mutant HRAS were loaded with 50 mM [γ–32P]GTP for 5 min at 37°C [buffers in Kearney et al. (41)]. The nucleotide-exchanged protein was then diluted 5-fold with hydrolysis buffer preheated to 30°C, bringing protein and nucleotide concentrations to 1 µM and 10 nM, respectively. Reactions proceeded for 100 min. Counts per minute was converted to femtomoles of inorganic phosphate [HPO32− (Pi)] and self-normalized to 100%. Hydrolysis reaction half-time (t1/2) was determined using the curve-fitting program ProFit (http://www.quansoft.com), and kobs was determined as the reciprocal of t1/2. Reactions including Raf were done in the presence 5-fold excess Raf1-RBD, at 5 µM.
Crystallization and structure determination of Tyr137 mutants
C-terminal truncated HRAS, HRASY137E, and HRASY137F (residues 1–166) were purified as published previously (41), and the bound GDP was exchanged for the nucleotide analog guanylyl-imidodiphosphate (GppNHp) in stabilization buffer (20 mM HEPES, 50 mM NaCl, 20 mM MgCl2, and either 10 or 1 mM DTT) (42). Protein was concentrated, flash-frozen, and stored at −80°C. Reagents and materials for crystallization were purchased from Hampton Research, Inc., Aliso Viejo, CA, USA. Crystals grew in 24 well plates with reservoir volumes ranging from 425 to 625 µl. HRASY137E was crystallized in hanging drops containing 2 µl protein at a concentration of 12.4 mg/ml in stabilization buffer and 2 µl of reservoir solution consisting of 152 mM Ca(OAc)2, 24.8% polyethylene glycol 3350, and 4.8% stabilization buffer at pH 7.5. For HRASY137F crystallization, the protein was at a concentration 20.7 mg/ml and the reservoir contained 139 mM Ca(OAc)2, 22.6% polyethylene glycol 3350, and 13% stabilization buffer at pH 7.5. Both proteins crystallized with symmetry of the space group P3221. Data for HRASY137E and HRASY137F were collected using a Rigaku X-ray generator (Tokyo, Japan) at 100 K with X-ray wavelength at 1.54 Å. Data were processed with HKL2000 (43). The coordinates with Protein Data Bank (PDB) code 1CTQ were used for phasing with the molecular replacement method, followed by refinement and model building using the Phenix (python-based hierarchial environment for integraged xtallography) suite of programs (44) and with Coot (an open-source model-building program) (45).
RAF1-RBD pull-down assay
HEK293T cells were plated at 6 × 105 cells/well in a 6 well plate. At 24 h after plating, cells were transfected with pcDNA3 ABL2 and pcDNA3 HRASG12V. At 48 h posttransfection, cells were lysed in 20 mM Tris pH 7.4, 150 mM NaCl, 5 mM MgCl2, 1% Triton X-100, 1 mM DTT. Cells were incubated on ice for 5 min, and then the lysate was clarified by centrifuging for 10 min at 15,000 g. A sample of whole-cell lysate (WCL) was reserved for immunoblotting. Meanwhile, 4 μg of purified Raf1-RBD was incubated with 30 μl glutathione-Sepharose in lysis buffer and rotated for 1 h at 4°C. Beads were washed twice with lysis buffer to remove excess Raf1-RBD. Clarified lysate was added to the beads and incubated for 1 h at 4°C. Beads were washed 3 times in lysis buffer and then resuspended in SDS-PAGE sample buffer and boiled.
RESULTS
ABL phosphorylates HRAS on tyrosine 137
To better understand how RAS-to-RIN1 signal transduction regulates ABL activity and substrate specificity, we used proteomic analysis to compare phosphopeptide profiles from cells transfected with ABL2+RIN1 or ABL2+RIN1+HRASG12V (constitutively active RAS). Cells transfected with ABL2 alone served as a baseline control. The ABL2 isoform of ABL was used because it colocalizes with RIN1 in the cytoplasm (ABL1 is primarily nuclear). We detected the previously unreported phosphopeptide HRAS-pTyr137 in samples from cells transfected with ABL2, RIN1, and HRASG12V constructs but not in matched cells without HRASG12V (Table 1).
TABLE 1.
Mass spectrometry analysis of tyrosine-phosphorylated peptides
| Substrate | pTyr site | A | AR | ARR |
|---|---|---|---|---|
| RIN1 | 36 | 8.2 × 106 | 3.8 × 108 | 1.4 × 108 |
| FKBP4 | 220 | 9.3 × 106 | 7.5 × 107 | 5.1 × 107 |
| HRAS | 137 | — | — | 1.3 × 109 |
| HRAS | 96 | — | 1.4 × 104 | 2.2 × 106 |
| ENO2 | 43 | 1.8 × 107 | 4.7 × 107 | 1.3 × 107 |
HEK293T cells were transfected with ABL2 (A), ABL2 + RIN1 (AR), or ABL2 + RIN1 + HRAS (ARR) and phosphotyrosine peptides were enriched and analyzed by mass spectrometry. The most enriched peptides are listed. The data are representative of 3 independent experiments.
We hypothesized that Tyr137 phosphorylation could serve as a feedback modification of HRAS for several reasons. First, the signal intensity suggested that phosphorylation at this site is relatively efficient. Second, the residues surrounding HRAS-Tyr137 conform to the ABL target site consensus sequence Y-x-x-P (Fig. 1A), consistent with being a direct ABL substrate. Third, of 8 tyrosines conserved in mammalian, fly, and worm RAS proteins, Tyr137 is the only one not also found in yeast (Fig. 1A), consistent with Tyr137 serving as a phospho-regulatory site that evolved in conjunction with eukaryotic tyrosine kinases. Our data (Table 1) also included the previously validated ABL substrates RIN1-pTyr36 and ENO2-pTyr43, as well as a novel ABL-dependent phosphopeptide, FKBP4-pTyr220. HRAS-pTyr96, which can be phosphorylated by the closely related kinase SRC in vitro (46), also appeared in our data set.
Figure 1.
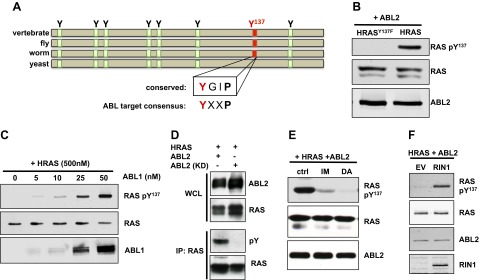
ABL phosphorylates HRAS on Tyr137. A) Schematic alignment of tyrosine residues in vertebrate, fly, worm, and yeast RAS. B) RAS-pTyr137 phosphospecific antibody was used to immunoblot extracts of HEK293T cells transfected with ABL2 and HRAS or HRASY137F. The immunoblot image is representative of 5 independent experiments. C) In vitro kinase assay performed with 500 nM purified HRAS and increasing concentrations of ABL1. D–F) HEK293T cell lysates analyzed for tyrosine-phosphorylated HRAS. D) Cells expressed HRAS and ABL2 or a kinase-dead mutant, ABL2K319R (ABL2 KD). E) Cells expressed HRAS and ABL2 and were treated with imatinib (IM) or dasatinib (DA). The immunoblot experiment is representative of 3 independent experiments. F) Cells expressed HRAS, ABL2, and empty vector (EV) or RIN1. All immunoblot images are representative of 2 independent experiments unless otherwise noted.
We next generated a phosphospecific antibody to HRAS-pTyr137 and tested its specificity using 293T cells overexpressing ABL2 with HRAS or HRASY137F. The antibody recognized wild-type HRAS but not the HRAS mutant with phenylalanine at position 137 (Fig. 1B). This antibody was also used to quantify in vitro kinase assays, demonstrating that purified HRAS is phosphorylated in an ABL-dependent manner. This result (Fig. 1C) is consistent with HRAS serving as a direct ABL substrate.
When wild-type ABL2 was replaced in the cell assay with a kinase dead mutant of ABL2 (ABL2K319R), HRAS phosphorylation could not be detected (Fig. 1D). Treatment of cells with the ABL tyrosine kinase inhibitors imatinib and dasatinib markedly decreased HRAS-pTyr137 (Fig. 1E), arguing against the contribution of other kinases. All 3 RAS isoforms could be phosphorylated at Tyr137 by ABL2 (Supplemental Fig. 1A), and both ABL isoforms could phosphorylate HRAS (Supplemental Fig. 1B), indicating a functional conservation across isoforms. It is noteworthy that RIN1 overexpression significantly increased HRAS-pTyr137 (Fig. 1F), suggesting that RAS-RIN1-mediated ABL stimulation feeds back to regulate RAS by tyrosine phosphorylation.
ABL can phosphorylate both active and inactive HRAS
We next considered whether guanine nucleotide binding (i.e., activation state) might influence the efficiency of HRAS phosphorylation. HEK293T cells were transfected with ABL2 and wild-type HRAS, which is primarily GDP-bound due to the slow intrinsic rate of GDP release following hydrolysis (5), or oncogenic HRASG12V, which hydrolyzes GTP more slowly than wild-type and is insensitive to GAPs, leading to constitutively elevated levels of the GTP-bound form (47). Both wild-type HRAS and HRASG12V were phosphorylated on Tyr137 by ABL2 (Fig. 2A). Likewise, in an in vitro kinase assay, HRAS could be phosphorylated regardless of whether it was loaded with GDP or the nonhydrolyzable GTP analog GTPγS (Fig. 2B). This indicated that the 2 distinct nucleotide-bound conformations of RAS can serve as ABL substrates.
Figure 2.

ABL2 can phosphorylate GDP- and GTP-bound HRAS. A) HEK293T cells transfected with ABL2 and wild-type HRAS (primarily GDP-bound) or HRASG12V (primarily GTP-bound), with corresponding Y137F controls. The immunoblot images are representative of 2 independent experiments. B) In vitro kinase assay with 200 nM ABL2 and 2 μM HRAS loaded with GDP or GTPγS.
HRAS-Tyr137 phosphorylation is enhanced by RAS palmitoylation
HRAS palmitoylation on cysteines 181 and 184 mediates stable attachment to the plasma membrane. To examine the effect of acylation on HRAS phosphorylation, we treated cells with 2-bromopalmitate, an inhibitor of protein palmitoylation enzymes (48). Blocking palmitoylation resulted in decreased HRAS-pTyr137 levels (Fig. 3A). To rule out the possibility that broad inhibition of palmitoylation might influence HRAS phosphorylation indirectly, we created a palmitoylation-defective mutant HRASC181S/C184S. When expressed with ABL2, the palmitoylation-defective mutant exhibited significantly less phosphorylation compared with wild-type HRAS (Fig. 3B, C). Together, our results suggest that HRAS palmitoylation facilitates Tyr137 phosphorylation. This could, of course, be attributed to reduced HRAS plasma membrane localization rather than the absence of palmitate per se.
Figure 3.
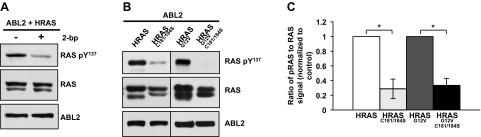
Palmitoylation enhances HRAS-Tyr137 phosphorylation. A) Lysates from 293T cells transfected with ABL2 and HRAS and treated with 100 μM 2-bromopalmitate (2-bp) for 24 h were immunoblotted for HRAS-Tyr137, total RAS, and ABL2. B) Representative immunoblot of 293T cells transfected with HRAS or the palmitoylation mutant HRASC181S/C184S, in wild-type and G12V background. C) Three independent experiments, performed as described in (B), were quantified and averaged. The ratio of RAS-pTyr137/RAS (without C-terminal mutations) was normalized to 1 for each experiment. *P < 0.0008 (2-tailed equal variant Student’s t test).
HRAS-Tyr137 mutations alter RAF1-dependent intrinsic hydrolysis
Because Tyr137 was implicated previously as part of an allosteric hot spot on HRAS (49), we considered whether changes at position 137 might alter the rate of GTP hydrolysis, a property fundamental to RAS regulation. RAS proteins have a relatively weak GTPase activity that is enhanced in cells by GAP proteins. Several oncogenic RAS mutations, such as RASG12V, lead to significantly slower rates of intrinsic hydrolysis as well as reduced responsiveness to GAPs, resulting in persistence of the GTP-bound active RAS conformation (50). We determined intrinsic hydrolysis rate constants (k) in single turnover assays for HRAS and 2 mutants with alterations of the position 137 side chain: HRASY137F and HRASY137E (all RAS constructs used in these experiments contained residues 1–166; i.e., all but the C-terminal 23 amino acids). Neither mutation produced a significant change in rate compared with HRAS in this GTPase assay (Fig. 4A).
Figure 4.
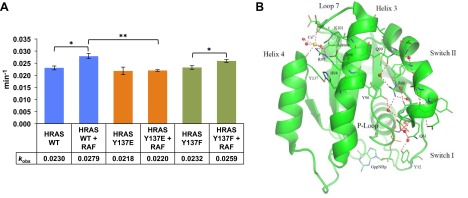
GTP hydrolysis rate constants and the influence of effector binding. A) Histogram of kobs for intrinsic hydrolysis of GTP for wild-type and Tyr137 mutants in the absence and presence of RAF1-RBD. Columns represent average values of 4 independent single-turnover reactions with standard error represented as T-lines. Half-boxes represent Student's t tests between indicated kobs averages. *P < 0.05, **P < 0.005. B) The R state structure for wild-type HRAS is shown in green (PDB code 3K8Y). Binding of Ca2+ and acetate at the allosteric site near Arg97 promotes a water-mediated hydrogen-bonding network that stabilizes switch II in a precatalytic conformation by positioning Gln61 in the active site. In this structure, Tyr32 is stabilized by crystal contacts in a conformation similar to that found in the RAS/RAF-RBD complex (PDB code 4G0N). Residues in the network are depicted in stick as is the nucleotide analog GppNHp. The Ca2+ ion is shown in yellow, oxygen atoms in red, nitrogen atoms in blue, and the phosphorous atoms of the nucleotide are in orange. Water molecules are represented as red spheres and H-bonding interactions are shown in red dashed lines.
RAS switch I (residues 30–37) and switch II (residues 60–75) segments are disordered prior to effector binding (51). Interaction with the RAF1-RBD, which binds to RAS primarily at switch I, imposes a stable switch I fold and switch II remains disordered (52, 53). Helices 3 and 4 are allosterically connected to switch II through a water-mediated H-bonding network leading from the allosteric site containing Tyr137 to the catalytic residue Gln61 (Fig. 4B). This network promotes a disordered-to-ordered transition in switch II, completing the active site (52). Therefore, we performed hydrolysis measurements in the presence of RAF1-RBD. Both wild-type RAS and HRASY137F showed significant hydrolysis rate increases in the presence of RAF1-RBD, relative to the uncomplexed protein (Fig. 4A). In contrast, the catalytic rate for HRASY137E did not change upon RAF1-RBD binding.
HRAS-Tyr137 mutations result in long-range conformation effects
To better understand the possible consequences of Tyr137 phosphorylation and its effect on HRAS biochemistry, we purified and crystallized HRASY137E and HRASY137F (residues 1–166) bound to a nonhydrolyzable GTP analog, GppNHp. HRASY137E and HRASY137F crystallized with symmetry of the space group P3221 and with one molecule in the asymmetric unit. X-ray data collection and structure refinement statistics are shown in Table 2. The tyrosyl moiety of Tyr137 of HRAS packs along the aliphatic portion of the Arg97 side chain and participates in a hydrophobic pocket beneath the allosteric site while simultaneously forming an H-bond with His94. Thus, in HRAS, Tyr137 packs in the protein core and at the same time bridges helices 3 and 4.
TABLE 2.
X-ray data collection and structure refinement statistics
| Statistic | HRASY137E | HRASY137F |
|---|---|---|
| Data collection | ||
| PDB entry | 4XVQ | 4XVR |
| Wavelength (Å) | 1.5418 | 1.5418 |
| Space group | P3221 | P3221 |
| Cell dimensions | ||
| a, b, c (Å) | 39.05, 39.05, 158.74 | 39.23, 39.23, 157.20 |
| α, β, γ (deg) | 90, 90, 120 | 90, 90, 120 |
| Total reflections | 58,204 (1780) | 89,345 (9534) |
| Unique reflections | 11,267 (890) | 9711 (944) |
| Multiplicity | 5.2 (2.0) | 9.2 (10.1) |
| Rsym | 0.054 (0.318) | 0.062 (0.971) |
| I/σ | 29.65 (5.54) | 36.42 (8.42) |
| Completeness (%) | 93.49 (74.92) | 99.55 (99.58) |
| Redundancy | 2.9 (1.3) | 5.1 (5.4) |
| Wilson B-factor | 29.45 | 39.75 |
| Refinement | ||
| Resolution (Å) | 28.49–1.89 (1.95–1.89) | 33.98–2.03 (2.10–2.03) |
| Rwork | 0.2025 (0.2736) | 0.2218 (0.2936) |
| Rfree | 0.2536 (0.3157) | 0.2857 (0.4187) |
| Average B-factor | 29.6 | 42.2 |
| No. atoms | ||
| Protein | 1272 | 1238 |
| Water | 82 | 42 |
| Ligands | 40 | 34 |
| Total | 1394 | 1314 |
| No. molecules | ||
| Protein | 1 | 1 |
| Ligands | 2 | 1 |
| Metals | 2 | 2 |
| RMSD | ||
| Bond length (Å) | 0.008 | 0.008 |
| Bond angle (deg) | 1.19 | 1.13 |
| Ramachandran (%) | 99 | 96 |
In the HRASY137E structure (Fig. 5A), the aliphatic portion of Arg97 drops into a cavity occupied by the Tyr137 phenyl ring in wild-type HRAS, and the Arg97 guanidinium group forms a salt bridge with the Glu137 side chain. At the same time, Lys101 swings down so that its amino group resides in the Arg97 guanidinium group position of wild-type HRAS. The repositioned Lys101 also forms a salt bridge with Glu137. The Y137E substitution causes the side chain of His94 in helix 3 to become somewhat disordered with B-factors around 45 A2. Helix 4 shifts away from helix 3 to accommodate this reorganization. Wild-type and mutant active sites superimpose well, even though the N-terminal end of switch II is disordered in the mutant. The allosteric site contains a water molecule within H-bonding distance from both the side chain of Glu137 and its backbone carbonyl group.
Figure 5.
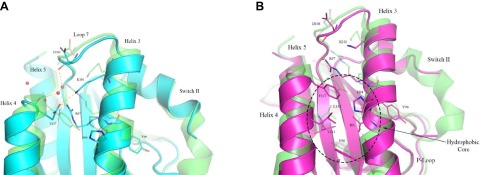
Crystal structures HRASY137E and HRASY137F bound to GppNHp focused on the allosteric site. The mutant structures are superimposed on the wild-type HRAS, shown in green (PDB code 3K8Y). Residues in the allosteric site are depicted in stick, with oxygen atoms in red, and nitrogen atoms in blue. Water molecules are represented as red spheres and H-bonds are in red dashed lines. A) HRASY137E is shown in cyan. B) HRASY137F is shown in purple. Note the cluster of hydrophobic residues that includes Phe137.
When RAF is bound to RAS, we expect intrinsic hydrolysis to be controlled by an allosteric mechanism (52) whereby a shift of helix 3 and loop 7 increases switch II stability. This helix 3 shift is described as a transition from a disordered switch II (T state) to an ordered switch II (R state), where switch I is already stabilized by RAF1 binding (54). The shift from T to R state in HRAS requires that Arg97 extend out toward the solvent to reduce packing in the hydrophobic pocket and allow helix 3 room to shift toward helix 4. In HRASY137E the aliphatic portion of Arg97 reaches deeper into the hydrophobic core than in the wild-type structure (Fig. 5A), potentially keeping helix 3/loop7 shifted toward switch II, which is poorly ordered as a consequence. Our results suggest that HRASY137E shifts the conformational equilibrium toward the T state due to the repositioning of the Arg97 side chain, with its aliphatic portion interacting with the protein core between helices 3 and 4 and its guanidinium group making a salt bridge with Glu137. This is consistent with a reduced rate constant compared with wild-type in single-turnover hydrolysis assays in the presence of RAF1-RBD (Fig. 4A).
In the RASY137F mutant (Fig. 5B), Phe137 is repositioned closer to the hydrophobic core consisting of Lys133, Ile93, Phe90, and Val113. His94 becomes part of this core, with its ring roughly perpendicular to the phenyl ring of Phe137, although electron density for this residue is not continuous for the side chain. Arg97 shifts to stack over Phe137, but unlike its position in the HRASY137E mutant, it does not drop into the protein core. HRASY137F is similar to HRASY137E in that Lys101 is near the Arg97 guanidinium position found in the wild-type structure and with respect to a poorly ordered switch II. However, the phenylalanine substitution effect on the allosteric site causes less perturbation than the glutamate substitution. It appears that the HRASY137F may better accommodate the helix 3 shift needed for intrinsic hydrolysis, as Phe137 maintains the same conformation as Tyr137 in HRAS, and the rest of the allosteric site and helix 3 superimpose nicely with HRAS. This is consistent with our kinetic data for HRASY137F, which showed little difference in the single turnover hydrolysis rate compared with HRAS (Fig. 4A). Comparison of the 2 mutant structures suggests that the GTPase rate change in effector-bound HRASY137E is due to a more constrained helix 3 associated with an allosteric switch mechanism.
Although it was not possible to obtain sufficient quantities of the phosphorylated protein for structure analysis, information from wild-type and mutant structures provide insights into the possible consequences of Tyr137 phosphorylation. This bulky, highly charged covalent modification would most likely interact with Arg97 and/or with Lys101, as seen for the Y137E mutant. However, unlike the mutant, the large size of the modified side chain would be expected to completely prevent the shift in helix 3 toward helix 4 associated with hydrolysis in the presence of RAF (54, 55). Therefore, RASTyr137 phosphorylation is expected to attenuate GTP hydrolysis activity and might enhance effector binding.
RAF1-RBD shows increased binding to HRAS-pTyr137 compared with unphosphorylated HRAS
To examine the consequence of tyrosine phosphorylation on HRAS signal transduction, we probed its binding to RAF1 using 293T cells overexpressing both ABL2 and HRASG12V. A portion of the WCL was reserved and the remaining material was incubated with purified GST-RAF1-RBD, which includes an RBD that interacts preferentially with activated (GTP-bound) RAS. In this protocol, activated RAS in a cell extract binds to RAF1-RBD in solution and the complex is pulled down using glutathione-Sepharose beads. Inactive (GDP-bound) RAS has much lower affinity for the RBD construct. An equal ratio of RAF1-RBD pull-down material to WCL was run in separate lanes on an SDS-PAGE gel and immunoblotted for total RAS or RASpTyr137. If tyrosine phosphorylation of RAS had no effect on hydrolysis rate or RAF1 binding, we would expect to see the same signal ratio (RBD pull down/WCL) for both RAS and RASpTyr137 immunoblots (Fig. 6A). However, the immunoblots showed a 3.9-fold increase in this ratio when using the anti-RASpTyr137 probe compared with the anti-RAS (total RAS) probe (Fig. 6B). This is most easily explained by preferential binding of RAF1 to RASG12VpTyr137 compared with unphosphorylated RASG12V (Fig. 6B).
Figure 6.

Tyr137-phosphorylated HRAS shows increased binding to Raf1-RBD than unphosphorylated HRAS. A) Schematic of experimental design in which 293T cells transfected with ABL2 and HRASG12V are lysed. Part of the WCL is reserved, and the remaining lysate is incubated with GST-Raf1-RBD for a pull-down assay. Equal ratios of pull-down assay to WCL are examined by immunoblot against RAS and RASpTyr137. If pTyr137 has no effect on Raf1 binding, the ratio of pull down to WCL would be expected to be the same whether analyzed anti-RAS or anti-RASpTyr137. B) Raf1-RBD pull down as described in (A). Results were averaged from 5 independent experiments. P = 0.001 (2-tailed equal variant Student’s t test).
We used activated RASG12V to obtain a detectable ratio of GTP-bound RAS in our lysates, as wild-type RAS is found almost entirely in the GDP-bound state in cells. Figure 6B indeed shows some unphosphorylated RASG12V in complex with RAF-RBD. The significantly higher amount of RASG12V pTyr137 bound to RAF1-RBD in Fig. 6B is consistent with a higher proportion of cellular RASG12V pTyr137 in the GTP-bound state, compared with unphosphorylated RASG12V. This could be explained by a phosphorylation-induced decrease in the intrinsic hydrolysis rate or the RAF1-accelerated hydrolysis rate. This interpretation is consistent with data from the HRASY137E mutant (Fig. 4A), in addition to the rate reduction attributable to the G12V mutation alone (55). Although we have been unable to purify sufficient amounts of phosphorylated RAS to test this directly, our results are consistent with analyses of HRAS mutant structures that support an allosteric role for Tyr137 and suggest that a phosphate group at this position should lead to a significant decrease in the GTP hydrolysis rate. However, we cannot exclude the possibility that Tyr137 phosphorylation allosterically enhances RAS affinity for RAF1-RBD.
HRAS-pTyr137 binds to RASA1-SH2
Many signal transduction pathways use tyrosine phosphorylation as a mechanism to increase binding affinity for SH2 domain partners. Therefore, we considered the possibility that RAS tyrosine phosphorylation conferred new interaction capabilities, and searched for SH2 domains that might bind RAS-pTyr137. We used a scoring matrix-assisted ligand identification (SMALI) program to predict binders based on pTyr137 flanking sequences (56). Intriguingly, the top hit from this nonbiased approach was the carboxyl-terminal SH2 domain of RASA1, a GAP that stimulates GTP hydrolysis by RAS proteins.
We examined whether HRAS-pTyr137 binds to the RASA1 C-terminal SH2 domain using biotinylated RAS peptides and a RASA1 SH2-C domain purified from bacteria. A peptide encoding pTyr137 and flanking RAS sequences efficiently pulled down RASA1 SH2-C, but a mutant peptide with phenylalanine in position 137 was unable to do so (Fig. 7A). As a control, we used a RASA1 SH2-C arginine mutation (R377A) known to impair phosphotyrosine binding (57). Consistent with dependence on a standard SH2 docking arrangement, the pTyr137 peptide pulled down significantly less RASA1 SH2-CR377A than wild-type RASA1 SH2-C (Fig. 7A). Using a reciprocal experimental design, GST-RASA1 SH2-C pulled down more HRAS than HRASY137F from cell lysate (Fig. 7B). These results are consistent with RASA1 SH2-C binding to tyrosine phosphorylated HRAS and suggested a conditional association between RASA1 and tyrosine phosphorylated RAS proteins.
Figure 7.

HRAS-pTyr137 binds to RASA1 C-terminal SH2 domain. A) Biotinylated peptide pull-down of RASA1-SH2C. Top: Biotinylated pTyr137 or Y137F control peptide were incubated with RASA1-SH2C or RASA1-SH2CR377A and pulled down with avidin beads. Bottom: RASA1-SH2C loading control. Samples were resolved by SDS-PAGE and visualized by silver staining. B) HEK293T cells were transfected with ABL2 and HRAS or HRASY137F. Cell extract was incubated with 2.5 μg purified GST-RASA1-SH2C and pulled down with glutathione-Sepharose beads.
DISCUSSION
Despite more than 3 decades of intense research into RAS protein structure and function, relatively little is known about posttranslational modifications that contribute to the regulation of this paradigmatic small GTPase. Here we report the tyrosine phosphorylation of HRAS-Tyr137 by ABL kinases in vitro and in cultured human cells. This posttranslational modification is enhanced by overexpression of RIN1, a direct RAS effector and ABL activator expressed in many epithelial, hematopoietic and central nervous system cells (58–60). The RIN1 carboxyl-terminal domain binds to activated RAS and the RIN1 amino-terminal region binds to ABL tyrosine kinases, allowing RIN1 to stably connect RAS with ABL proteins in cultured cells (22). We also note that RAS-Tyr137 resides in a consensus ABL target site conserved in species that encode tyrosine kinases. These data suggest that RIN1 facilitates RAS tyrosine phosphorylation by bringing it in close proximity to an ABL tyrosine kinase catalytic domain. Modulation of RAS tyrosine phosphorylation by RIN1 would represent a novel mechanism for RAS regulation whereby effector binding can result in feedback phosphorylation (Fig. 8).
Figure 8.

HRAS phosphorylation model. RIN1 mediates phosphorylation of RAS by convening ABL and RAS in close proximity. RIN1 binds to ABL through a proline-rich domain (PxxP) and pTyr36 binding to ABL SH3 and SH2, respectively. RIN1 binds to RAS through a C-terminal RAS association domain (RA).
To better understand the biologic effects of tyrosine phosphorylation, we searched for endogenous RAS proteins with this modification. We focused our search on cells that express RAS, RIN1, and ABL proteins, and examined diverse stimulatory conditions as well as cancer cell lines with constitutively active RAS or ABL proteins. Our inability to detect endogenous RAS-pTyr137 by immunoblot could indicate that phosphorylation requires RAS overexpression, or it may simply reflect the relatively low fraction of RAS protein subject to this modification. Regulatory tyrosine phosphorylations are known to be both infrequent and transient, comprising only about 0.04% of total protein phosphorylation in cells, with tyrosine kinase oncogenes increasing this to about 0.3% (61). We were also unable to purify a sufficient quantity of HRAS-pTyr137 for direct biochemical or structural analysis.
Analysis of RAS-Tyr137 mutations revealed several perturbations that could explain the role of phosphorylation at this site. A conservative Tyr-to-Phe mutation had no effect on single turnover hydrolysis rate, which was consistent with only minimal structure perturbations in HRASY137F relative to wild-type HRAS. A more disruptive Tyr-to Glu mutation, which introduces a negative charge, significantly blunted the enhancing effect of RBD binding on GTP hydrolysis rate. Structural analysis revealed allosteric site changes resulting in a partial disruption of the mechanism that leads to an ordered active site in the context of the RAS:RAF1 complex (53, 62). This impairment in allosteric modulation is consistent with the observed reduction in effector-induced hydrolysis rate. Although glutamate is the natural amino acid residue that most closely resembles it, a phosphate group could have a larger effect because of its bulk and −2 charge. Phosphorylation of Tyr137 would be expected to exacerbate the effects observed in the RASY137E mutant and severely impair or even abolish the ability of RAS to hydrolyze GTP. If phosphorylation indeed reduces hydrolysis and prolongs the GTP-bound state, this would explain the fact that HRASG12V-pTyr137 was pulled down more efficiently than HRASG12V by the RAF1-RBD effector construct. It remains possible, however, that Tyr137 phosphorylation triggers allosteric changes in GTP-bound RAS that enhance its affinity for RAF1-RBD or that cellular factors contribute to a stronger association of HRASG12V-pTyr137 with its effector proteins.
Tyr137 was previously identified as part of an allosteric hot spot on HRAS (49). Both the main chain and side chain of Tyr137 participate in a hydrogen bond network that stretches across the molecule to stabilize switch II in the crystal structure of HRAS with calcium and acetate bound at the allosteric site (Fig. 4B) (52). Allosteric modulation of the active site has been linked to the RAS/RAF/MEK/ERK pathway and is thought to be important for enhancing GTP hydrolysis by RAS when complexed with RAF and bound to Ca2+, which coordinates directly with the backbone carbonyl group of Tyr137 (52–55). Phosphorylation of Tyr137 appears to weaken intrinsic hydrolysis, consequently promoting signaling through RAF. Intriguingly, HRAS-pTyr137 binding to the C-terminal SH2 domain of RASA1, a GAP for RAS, suggests another mechanism by which phosphorylation could modulate RAS signaling. An intriguing possibility is that a RASpTyr137::RASA1 interaction could influence RASA1 activity and cause a temporary signal inflection. This suggests a regulatory circuit in which Tyr137 phosphorylation of activated RAS could hamper intrinsic GTP hydrolysis by impairing the allosteric switch mechanism in addition to modifying the contribution of cellular GAPs.
The data presented here support the hypothesis that residues remote from the active site, such as Tyr137, can allosterically modulate RAS function. The crystal structures of HRASY137E and HRASY137F, together with biochemical experiments, provide insight on the mechanism through which RAS posttranslational modifications at the allosteric site could function as triggers that influence nucleotide hydrolysis and effector binding at this important signaling hub.
Supplementary Material
Acknowledgments
The authors thank Drs. James Wohlschlegel and Ajay Vashisht for helpful discussions and insight into RAS binding partners, Dr. Greg Buhrman for initial guidance in performing the hydrolysis rates experiments, and Dr. Paul Swartz for help with data collection for the RASY137E mutant. The authors also thank the following colleagues for generously sharing key reagents: Dr. Shawn Li (pDEST15 RASA1-C SH2), Dr. John Kuriyan (pProExHT-HRAS), and Dr. Frank McCormick [pGEX-KG-RafRBD(1–149)]. This work was supported by the U.S. National Institutes of Health (NIH) National Cancer Institute (NCI) Grant CA136699 (to J.C.); the Ruth L. Kirschstein National Research Service Award Grant GM007185 (to P.T.); the Whitcome Training Fellowship (to P.T.); the U.S. National Science Foundation Grant MCB-1244203 (to C.M.); NIH NCI Grant CA168585 (to T.G.G.); American Cancer Society Research Scholar Award RSG-12-257-01-TBE (to T.G.G.); U.S. National Center for Advancing Translational Sciences Grant UL1TR000124 (to T.G.G.), and University of California, Los Angeles (UCLA) Jonsson Comprehensive Cancer Center for support of the Molecular Screening Shared Resource. The coordinates and structure factors for the X-ray crystal structures have been deposited in the PDB with the following codes: HRASY137E-GppNHp, PDB code 4XVQ; and HRASY137F-GppNHp, PDB code 4XVR.
Glossary
- ABL1/2
Abelson tyrosine-protein kinase 1/2
- FBS
fetal bovine serum
- GAP
GTPase-activating protein
- GEF
guanine nucleotide exchange factor
- GppNHp
guanylyl-imidodiphosphate
- HRAS
Harvey rat sarcoma oncogene
- HRP
horseradish peroxidase
- KRAS
Kirsten rat sarcoma oncogene
- NP-40
Nonidet P-40
- NRAS
neuroblastoma RAS oncogene
- PDB
Protein Data Bank
- RAF1
RAF proto-oncogene serine/threonine protein kinase 1
- RASA1
RAS p21 protein activator (GTPase activating protein) 1
- RIN1
RAS and RAB interacting protein 1
- RBD
RAS binding domain
- WCL
whole-cell lysate
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
REFERENCES
- 1.Chardin P., Camonis J. H., Gale N. W., van Aelst L., Schlessinger J., Wigler M. H., Bar-Sagi D. (1993) Human Sos1: a guanine nucleotide exchange factor for Ras that binds to GRB2. Science 260, 1338–1343 [DOI] [PubMed] [Google Scholar]
- 2.Quilliam L. A., Rebhun J. F., Castro A. F. (2002) A growing family of guanine nucleotide exchange factors is responsible for activation of Ras-family GTPases. Prog. Nucleic Acid Res. Mol. Biol. 71, 391–444 [DOI] [PubMed] [Google Scholar]
- 3.Colicelli J. (2004) Human RAS superfamily proteins and related GTPases. Sci. STKE 2004, RE13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wittinghofer A., Herrmann C. (1995) Ras-effector interactions, the problem of specificity. FEBS Lett. 369, 52–56 [DOI] [PubMed] [Google Scholar]
- 5.Neal S. E., Eccleston J. F., Hall A., Webb M. R. (1988) Kinetic analysis of the hydrolysis of GTP by p21N-ras. The basal GTPase mechanism. J. Biol. Chem. 263, 19718–19722 [PubMed] [Google Scholar]
- 6.Trahey M., McCormick F. (1987) A cytoplasmic protein stimulates normal N-ras p21 GTPase, but does not affect oncogenic mutants. Science 238, 542–545 [DOI] [PubMed] [Google Scholar]
- 7.Ahmadian M. R., Stege P., Scheffzek K., Wittinghofer A. (1997) Confirmation of the arginine-finger hypothesis for the GAP-stimulated GTP-hydrolysis reaction of Ras. Nat. Struct. Biol. 4, 686–689 [DOI] [PubMed] [Google Scholar]
- 8.Prior I. A., Lewis P. D., Mattos C. (2012) A comprehensive survey of Ras mutations in cancer. Cancer Res. 72, 2457–2467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ahearn I. M., Haigis K., Bar-Sagi D., Philips M. R. (2012) Regulating the regulator: post-translational modification of RAS. Nat. Rev. Mol. Cell Biol. 13, 39–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hancock J. F., Magee A. I., Childs J. E., Marshall C. J. (1989) All ras proteins are polyisoprenylated but only some are palmitoylated. Cell 57, 1167–1177 [DOI] [PubMed] [Google Scholar]
- 11.Buss J. E., Sefton B. M. (1986) Direct identification of palmitic acid as the lipid attached to p21ras. Mol. Cell. Biol. 6, 116–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu L., Lubkov V., Taylor L. J., Bar-Sagi D. (2010) Feedback regulation of Ras signaling by Rabex-5-mediated ubiquitination. Curr. Biol. 20, 1372–1377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sasaki A. T., Carracedo A., Locasale J. W., Anastasiou D., Takeuchi K., Kahoud E. R., Haviv S., Asara J. M., Pandolfi P. P., Cantley L. C. (2011) Ubiquitination of K-Ras enhances activation and facilitates binding to select downstream effectors. Sci. Signal. 4, ra13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jura N., Scotto-Lavino E., Sobczyk A., Bar-Sagi D. (2006) Differential modification of Ras proteins by ubiquitination. Mol. Cell 21, 679–687 [DOI] [PubMed] [Google Scholar]
- 15.Jeong W.-J., Yoon J., Park J.-C., Lee S.-H., Lee S.-H., Kaduwal S., Kim H., Yoon J.-B., Choi K.-Y. (2012) Ras stabilization through aberrant activation of Wnt/β-catenin signaling promotes intestinal tumorigenesis. Sci. Signal. 5, ra30 [DOI] [PubMed] [Google Scholar]
- 16.Bivona T. G., Quatela S. E., Bodemann B. O., Ahearn I. M., Soskis M. J., Mor A., Miura J., Wiener H. H., Wright L., Saba S. G., Yim D., Fein A., Pérez de Castro I., Li C., Thompson C. B., Cox A. D., Philips M. R. (2006) PKC regulates a farnesyl-electrostatic switch on K-Ras that promotes its association with Bcl-XL on mitochondria and induces apoptosis. Mol. Cell 21, 481–493 [DOI] [PubMed] [Google Scholar]
- 17.Sung P. J., Tsai F. D., Vais H., Court H., Yang J., Fehrenbacher N., Foskett J. K., Philips M. R. (2013) Phosphorylated K-Ras limits cell survival by blocking Bcl-xL sensitization of inositol trisphosphate receptors. Proc. Natl. Acad. Sci. USA 110, 20593–20598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Williams J. G., Pappu K., Campbell S. L. (2003) Structural and biochemical studies of p21Ras S-nitrosylation and nitric oxide-mediated guanine nucleotide exchange. Proc. Natl. Acad. Sci. USA 100, 6376–6381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Y., Colicelli J. (2001) RAS interaction with effector target RIN1. Methods Enzymol. 332, 139–151 [DOI] [PubMed] [Google Scholar]
- 20.Wang Y., Waldron R. T., Dhaka A., Patel A., Riley M. M., Rozengurt E., Colicelli J. (2002) The RAS effector RIN1 directly competes with RAF and is regulated by 14-3-3 proteins. Mol. Cell. Biol. 22, 916–926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Han L., Wong D., Dhaka A., Afar D., White M., Xie W., Herschman H., Witte O., Colicelli J. (1997) Protein binding and signaling properties of RIN1 suggest a unique effector function. Proc. Natl. Acad. Sci. USA 94, 4954–4959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hu H., Bliss J. M., Wang Y., Colicelli J. (2005) RIN1 is an ABL tyrosine kinase activator and a regulator of epithelial-cell adhesion and migration. Curr. Biol. 15, 815–823 [DOI] [PubMed] [Google Scholar]
- 23.Hu H., Milstein M., Bliss J. M., Thai M., Malhotra G., Huynh L. C., Colicelli J. (2008) Integration of transforming growth factor beta and RAS signaling silences a RAB5 guanine nucleotide exchange factor and enhances growth factor-directed cell migration. Mol. Cell. Biol. 28, 1573–1583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Balaji K., Mooser C., Janson C. M., Bliss J. M., Hojjat H., Colicelli J. (2012) RIN1 orchestrates the activation of RAB5 GTPases and ABL tyrosine kinases to determine the fate of EGFR. J. Cell Sci. 125, 5887–5896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Balaji K., Colicelli J. (2013) RIN1 regulates cell migration through RAB5 GTPases and ABL tyrosine kinases. Commun. Integr. Biol. 6, e25421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cao X., Tanis K. Q., Koleske A. J., Colicelli J. (2008) Enhancement of ABL kinase catalytic efficiency by a direct binding regulator is independent of other regulatory mechanisms. J. Biol. Chem. 283, 31401–31407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Afar D. E., Han L., McLaughlin J., Wong S., Dhaka A., Parmar K., Rosenberg N., Witte O. N., Colicelli J. (1997) Regulation of the oncogenic activity of BCR-ABL by a tightly bound substrate protein RIN1. Immunity 6, 773–782 [DOI] [PubMed] [Google Scholar]
- 28.Colicelli J. (2010) ABL tyrosine kinases: evolution of function, regulation, and specificity. Sci. Signal. 3, re6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Greuber E. K., Smith-Pearson P., Wang J., Pendergast A. M. (2013) Role of ABL family kinases in cancer: from leukaemia to solid tumours. Nat. Rev. Cancer 13, 559–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wong S., Witte O. N. (2004) The BCR-ABL story: bench to bedside and back. Annu. Rev. Immunol. 22, 247–306 [DOI] [PubMed] [Google Scholar]
- 31.Thai M., Ting P. Y., McLaughlin J., Cheng D., Müschen M., Witte O. N., Colicelli J. (2011) ABL fusion oncogene transformation and inhibitor sensitivity are mediated by the cellular regulator RIN1. Leukemia 25, 290–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fang P., Zhao Z., Tian H., Zhang X. (2012) RIN1 exhibits oncogenic property to suppress apoptosis and its aberrant accumulation associates with poor prognosis in melanoma. Tumour Biol. 33, 1511–1518 [DOI] [PubMed] [Google Scholar]
- 33.Yu H.-F., Zhao G., Ge Z.-J., Wang D.-R., Chen J., Zhang Y., Zha T.-Z., Zhang K., Zhang M., Tan Y.-F., Zhou S.-J., Jiang C. (2012) High RIN1 expression is associated with poor prognosis in patients with gastric adenocarcinoma. Tumour Biol. 33, 1557–1563 [DOI] [PubMed] [Google Scholar]
- 34.Wang Q., Gao Y., Tang Y., Ma L., Zhao M., Wang X. (2012) Prognostic significance of RIN1 gene expression in human non-small cell lung cancer. Acta Histochem. 114, 463–468 [DOI] [PubMed] [Google Scholar]
- 35.Tomshine J. C., Severson S. R., Wigle D. A., Sun Z., Beleford D. A. T., Shridhar V., Horazdovsky B. F. (2009) Cell proliferation and epidermal growth factor signaling in non-small cell lung adenocarcinoma cell lines are dependent on Rin1. J. Biol. Chem. 284, 26331–26339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shan G.-Y., Zhang Z., Chen Q.-G., Yu X.-Y., Liu G.-B., Kong C.-Z. (2012) Overexpression of RIN1 associates with tumor grade and progression in patients of bladder urothelial carcinoma. Tumour Biol. 33, 847–855 [DOI] [PubMed] [Google Scholar]
- 37.Zimman A., Chen S. S., Komisopoulou E., Titz B., Martínez-Pinna R., Kafi A., Berliner J. A., Graeber T. G. (2010) Activation of aortic endothelial cells by oxidized phospholipids: a phosphoproteomic analysis. J. Proteome Res. 9, 2812–2824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Elias J. E., Haas W., Faherty B. K., Gygi S. P. (2005) Comparative evaluation of mass spectrometry platforms used in large-scale proteomics investigations. Nat. Methods 2, 667–675 [DOI] [PubMed] [Google Scholar]
- 39.Beausoleil S. A., Villén J., Gerber S. A., Rush J., Gygi S. P. (2006) A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol. 24, 1285–1292 [DOI] [PubMed] [Google Scholar]
- 40.Prakash A., Mallick P., Whiteaker J., Zhang H., Paulovich A., Flory M., Lee H., Aebersold R., Schwikowski B. (2006) Signal maps for mass spectrometry-based comparative proteomics. Mol. Cell. Proteomics 5, 423–432 [DOI] [PubMed] [Google Scholar]
- 41.Kearney B. M., Johnson C. W., Roberts D. M., Swartz P., Mattos C. (2014) DRoP: a water analysis program identifies Ras-GTP-specific pathway of communication between membrane-interacting regions and the active site. J. Mol. Biol. 426, 611–629 [DOI] [PubMed] [Google Scholar]
- 42.Buhrman G., de Serrano V., Mattos C. (2003) Organic solvents order the dynamic switch II in Ras crystals. Structure 11, 747–751 [DOI] [PubMed] [Google Scholar]
- 43.Otwinowski Z., Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. In Methods in Enzymology (Carter C. W. Jr., and Sweet R. M., eds.), pp. 307–326, Academic Press, New York: [DOI] [PubMed] [Google Scholar]
- 44.Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 46.Bunda S., Heir P., Srikumar T., Cook J. D., Burrell K., Kano Y., Lee J. E., Zadeh G., Raught B., Ohh M. (2014) Src promotes GTPase activity of Ras via tyrosine 32 phosphorylation. Proc. Natl. Acad. Sci. USA 111, E3785–E3794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang K., DeClue J. E., Vass W. C., Papageorge A. G., McCormick F., Lowy D. R. (1990) Suppression of c-ras transformation by GTPase-activating protein. Nature 346, 754–756 [DOI] [PubMed] [Google Scholar]
- 48.Webb Y., Hermida-Matsumoto L., Resh M. D. (2000) Inhibition of protein palmitoylation, raft localization, and T cell signaling by 2-bromopalmitate and polyunsaturated fatty acids. J. Biol. Chem. 275, 261–270 [DOI] [PubMed] [Google Scholar]
- 49.Buhrman G., O’Connor C., Zerbe B., Kearney B. M., Napoleon R., Kovrigina E. A., Vajda S., Kozakov D., Kovrigin E. L., Mattos C. (2011) Analysis of binding site hot spots on the surface of Ras GTPase. J. Mol. Biol. 413, 773–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gibbs J. B., Sigal I. S., Poe M., Scolnick E. M. (1984) Intrinsic GTPase activity distinguishes normal and oncogenic ras p21 molecules. Proc. Natl. Acad. Sci. USA 81, 5704–5708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ito Y., Yamasaki K., Iwahara J., Terada T., Kamiya A., Shirouzu M., Muto Y., Kawai G., Yokoyama S., Laue E. D., Wälchli M., Shibata T., Nishimura S., Miyazawa T. (1997) Regional polysterism in the GTP-bound form of the human c-Ha-Ras protein. Biochemistry 36, 9109–9119 [DOI] [PubMed] [Google Scholar]
- 52.Buhrman G., Holzapfel G., Fetics S., Mattos C. (2010) Allosteric modulation of Ras positions Q61 for a direct role in catalysis. Proc. Natl. Acad. Sci. USA 107, 4931–4936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fetics S. K., Guterres H., Kearney B. M., Buhrman G., Ma B., Nussinov R., Mattos C. (2015) Allosteric effects of the oncogenic RasQ61L mutant on Raf-RBD. Structure 23, 505–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Johnson C. W., Mattos C. (2013) The allosteric switch and conformational states in Ras GTPase affected by small molecules. Enzymes 33(Pt A), 41–67 [DOI] [PubMed] [Google Scholar]
- 55.Buhrman G., Kumar V. S. S., Cirit M., Haugh J. M., Mattos C. (2011) Allosteric modulation of Ras-GTP is linked to signal transduction through RAF kinase. J. Biol. Chem. 286, 3323–3331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li L., Wu C., Huang H., Zhang K., Gan J., Li S. S.-C. (2008) Prediction of phosphotyrosine signaling networks using a scoring matrix-assisted ligand identification approach. Nucleic Acids Res. 36, 3263–3273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lock P., Casagranda F., Dunn A. R. (1999) Independent SH2-binding sites mediate interaction of Dok-related protein with RasGTPase-activating protein and Nck. J. Biol. Chem. 274, 22775–22784 [DOI] [PubMed] [Google Scholar]
- 58.Han L., Colicelli J. (1995) A human protein selected for interference with Ras function interacts directly with Ras and competes with Raf1. Mol. Cell. Biol. 15, 1318–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dhaka A., Costa R. M., Hu H., Irvin D. K., Patel A., Kornblum H. I., Silva A. J., O’Dell T. J., Colicelli J. (2003) The RAS effector RIN1 modulates the formation of aversive memories. J. Neurosci. 23, 748–757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dzudzor B., Huynh L., Thai M., Bliss J. M., Nagaoka Y., Wang Y., Ch’ng T. H., Jiang M., Martin K. C., Colicelli J. (2010) Regulated expression of the Ras effector Rin1 in forebrain neurons. Mol. Cell. Neurosci. 43, 108–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hunter T., Sefton B. M. (1980) Transforming gene product of Rous sarcoma virus phosphorylates tyrosine. Proc. Natl. Acad. Sci. USA 77, 1311–1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Johnson C. W., Mattos C. (2013) The allosteric switch and conformational states in Ras GTPase affected by small moleculesl In The Enzymes (Tamanoi F., ed.), pp. 42–67, Elsevier, Amsterdam, The Netherlands [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.