

Abstract

We present a general route for the transfer of Au and Ag nanoparticles of different shapes and sizes, from water into various organic solvents. The experimental conditions for each type of nanoparticles were optimized by using a combination of thiolated poly(ethylene glycol) and a hydrophobic capping agent, such as dodecanethiol. The functionalized nanoparticles were readily transferred into organic dispersions with long-term stability (months). Such organic dispersions efficiently spread out on water, leading to self-assembly at the air/liquid interface into extended nanoparticle arrays which could in turn be transferred onto solid substrates. The dense close packing in the obtained nanoparticle monolayers results in extensive plasmon coupling, rendering them efficient substrates for surface-enhanced Raman scattering spectroscopy.
Introduction
Plasmonic nanoparticles are key building blocks in nanotechnology, with unique features that allow exploitation of light–matter interactions across different fields.1−4 Since the optical properties of plasmonic nanomaterials are directly related to their morphology, a wide range of methods have been developed for the synthesis of (mainly) water-based dispersions of noble metal nanoparticles (NPs) with well-defined morphologies and uniform sizes.5,6 Upon the assembly of suitable building blocks, nanostructures with tailored plasmonic properties can be obtained.7 Anisotropic nanoparticles are of particular interest because their plasmonic response can be tuned into the near-IR (NIR), nanostars and nanorods being prominent examples of plasmonic performance.8,9 Although the preparation of anisotropic nanoparticles with accurate size and shape control is usually carried out in water, processing them into self-assembled nanostructures can often be improved if the nanoparticles are dispersed in organic solvents.10,11 Therefore, phase transfer of nanoparticles from aqueous to organic media has become an important strategy for different applications.12 Previously reported methods were strongly limited to rather small particle dimensions (<25 nm),13−15 so additional efforts are required for larger and/or anisotropic nanoparticles. Akylamines were used to transfer Au and Ag nanoparticles with sizes up to 100 nm, octadecylamine being the most efficient at successful phase transfer.16 Tailor-made ligands such as bidentated thiols17 have also been designed to achieve this goal, as well as polymers including thiolated polystyrene,18 but the large amount of polymer required significantly hinders further application of the nanoparticles in plasmonics. PEGylation has also been employed to transfer large gold nanoparticles and nanorods, but either a common solvent19 or mechanical forces20 were needed to obtain a successful phase transfer. A method that combines thiolated poly(ethylene glycol) (PEG-SH) and dodecylamine has been recently reported21 for the transfer of spherical gold nanoparticles up to 60 nm, CTAB-capped gold nanorods, and silver nanoprims. The hydrophobic nanoparticles obtained could be subsequently coated with an amphiphilic polymer, but this phase transfer process has been reported to be rather time-consuming.
We propose herein a simple and rapid procedure based on a combination of commercially available PEG-SH and 1-dodecanethiol (DDT), as a general method to transfer a wide variety of gold and silver nanoparticles of different sizes (up to 200 nm) and shapes (spheres, nanorods, nanostars) from aqueous dispersion into chloroform. Because of the strong affinity of the thiol group to the metal particle surface, complete phase transfer can be achieved in a very short time, and particles with long-term stability are readily obtained. Although the combination of PEG-SH and DDT leads to efficient nanoparticle phase transfer, when DDT alone was used aggregation occurred. This suggests that addition of PEG-SH is a critical first step to prevent aggregation of large NPs during phase transfer, in agreement with previous reports.21,22 DDT was selected as an efficient stabilizer in nonpolar solvents because the long aliphatic chain provides hydrophobic interactions between particles (steric repulsion). Notwithstanding, complete phase transfer can also be achieved by means of other hydrophobic thiols that are soluble in chloroform, leading to dispersions that remain unaltered for several months and which can be dried and redispersed in different solvents. Additionally, hydrophobic coatings are known to facilitate self-assembly of metal nanoparticles. We exploited this property to effectively assemble plasmonic nanoparticles with various shapes into extended compact monolayers at the liquid–air interface and subsequently transfer them onto solid substrates that can be directly applied as plasmonic substrates for optical sensing applications. In particular, the obtained monolayers were used as surface-enhanced Raman scattering (SERS) substrates with great potential toward ultrasensitive (and selective) molecular detection.23−29 These novel substrates offer advantages such as high reproducibility, robustness, and reliable signal generation. Because different particle sizes, shapes and materials can be used, this versatile method for phase transfer allows the design of substrates that can be tailored for specific applications.
Materials and Methods
Materials
Milli-Q water (resistivity 18.2 MΩ·cm) was used in all experiments. Hydrogen tetrachloroaurate trihydrate (HAuCl4·3H2O, ≥99.9%), sodium citrate tribasic dihydrate (≥98%), hexadecyltrimethylammonium bromide (CTAB, ≥99%), 5-bromosalicylic acid (90%), silver nitrate (AgNO3, ≥99%), l-ascorbic acid (AA, ≥99%), tannic acid, 1-dodecanethiol (DDT, ≥98%), O-[2-(3-mercaptopropionylamino)ethyl]-O′-methylpoly(ethylene glycol) (PEG-SH, MW 5000 g/mol), 4-mercaptobenzoic acid (4-MBA, 90%), poly(isobutylene-alt-maleic anhydride) (average Mw ∼ 6000 g/mol), dodecylamine (98%), and chloroform (CHCl3, ≥99.8%) were purchased from Sigma-Aldrich. Hydrochloric acid solution (37%) was purchased from Panreac. All glassware was washed with aqua regia, rinsed 3-fold with Milli-Q water, and dried before use.
Synthesis of Nanoparticles
Gold nanoparticle seed solution (Au seed) comprised an aqueous colloid (average diameter 13 ± 3 nm) that was prepared by adding 5 mL of 1 wt % sodium citrate solution to 95 mL of boiling 0.5 mM HAuCl4 under vigorous stirring. After 15 min of boiling, the solution was cooled down to room temperature and then stored at 4 °C for long-term storage. Spherical gold nanoparticles (AuNPs) with average diameters of 30 ± 4 nm (Au 30), 50 ± 5 nm (Au 50), and 100 ± 10 nm (Au 100) were synthesized according to a reported seeded growth method.30 As-synthesized AuNPs featured LSPR bands centered at 521, 530, and 555 nm, respectively, and they were used without further purification. AuNPs of 204 ± 8 nm with LSPR peaks at 567 nm (quadrupole) and ∼796 nm (dipole) were purchased from BBI solutions and washed using several centrifugation steps (870 g; 10 min) before use.
Gold nanostars (AuNSs) of different sizes were synthesized by a modified seed-mediated growth method.31 Different amounts of citrate-stabilized gold nanoparticle seed solution were added to 10 mL of 0.25 mM HAuCl4 containing 10 μL of 1.0 M HCl in a 20 mL glass vial at room temperature under moderate stirring. Quickly, 100 μL of AgNO3 (3 mM) and 50 μL of ascorbic acid (100 mM) were simultaneously added to the above solution, which rapidly turned from light red to blue or greenish-black, depending on the added amount of Au0. Specifically, 1 mL, 500 μL, and 100 μL of gold nanoparticle seeds were added to obtain small (sAuNS), middle (mAuNS), and big gold nanostars (bAuNS) with LSPR bands at 725, 770, and 884 nm and core/overall sizes of 23 ± 2 nm/34 ± 4 nm, 35 ± 3 nm/63 ± 7 nm, and 56 ± 4 nm/98 ± 7 nm, respectively. Immediately after growth, the solutions were stirred with PEG-SH (see PEG-SH concentrations in Table 1) for 15 min, washed by centrifugation at 1190g, 25 min, 10 °C, and redispersed in water.
Table 1. Phase Transfer Conditions.
| nanoparticle | [NP]/M | PEG-SH molecules per nm2 | molecules of DDT per nm2 | centrifugation after phase transfer |
|---|---|---|---|---|
| Au seeds | 4.8 × 10–9 | 0.8 | 148 | 3900g; 30 min; 20 °C |
| Au 30 | 9.5 × 10–10 | 0.6 | 150 | 2430g; 15 min; 20 °C |
| Au 50 | 2.0 × 10–10 | 0.6 | 155 | 1550g; 15 min; 20 °C |
| Au 100 | 2.0 × 10–11 | 0.6 | 165 | 600g; 15 min; 20 °C |
| Au 200 | 6.2 × 10–13 | 1.4 | 125 | 470g; 10 min; 20 °C |
| Ag 30 | 7.8 × 10–11 | 0.9 | 113 | 1550g; 15 min; 20 °C |
| Ag 50 | 2.5 × 10–11 | 1.0 | 130 | 1190g; 15 min; 20 °C |
| Ag 100 | 3.9 × 10–11 | 2.1 | 204 | 870g; 15 min; 20 °C |
| AuNRs | 3.3 × 10–10 | 1.4 | 133 | 2430g; 15 min; 20 °C |
| sAuNSsa | 3.5 × 10–10 | 1.4 | 155 | 1730g; 25 min; 10 °C |
| mAuNSsa | 6.5 × 10–11 | 1.4 | 155 | 1320g; 25 min; 10 °C |
| bAuNSsa | 8 × 10–12 | 1.4 | 155 | 970g; 25 min; 10 °C |
The diameter of small, middle, and big AuNSs was estimated considering the diameter of a sphere of 30, 50, and 100 nm, respectively.
Single-crystalline gold nanorods (AuNRs) (61 ± 5 nm length; 15 ± 2 nm width) were synthesized through a seed-mediated method involving the prereduction of HAuCl4 with salicylic acid.32,33 Nanorods with an absorbance maximum at 735 nm were obtained within 4 h after seed addition. The AuNRs were then washed by centrifugation (4760g, 40 min, 29 °C), and the pellet was redispersed in 0.05 M CTAB to remove excess reactants.
Silver nanoparticles (AgNPs) with average diameters of 28 ± 6 nm (Ag 30), 51 ± 8 nm (Ag 50), and 94 ± 12 nm (Ag 100) were synthesized following a published procedure.34 The prepared AgNPs displayed LSPR bands centered at 420, 439, and 487 nm. The nanoparticle solutions were centrifuged after synthesis (7870g, 20 °C, 40 min for Ag 30 and Ag 50, 20 min for Ag 100) to remove the excess of tannic acid used in the synthesis.
Phase Transfer
Gold and silver nanoparticles of different shapes and sizes were transferred from water to chloroform. The aqueous nanoparticle colloids were first stabilized with a suitable amount of PEG-SH, calculated to be between 0.6 and 2.1 molecules per nm2 depending of each particle type (Table 1). AuNPs and AuNSs were PEGylated directly without purification from the synthesis, whereas AuNRs and AgNPs were washed twice by centrifugation (4760g, 40 min, 29 °C) and redispersed in water to remove excess of CTAB. Subsequently, a DDT solution (estimated to provide between 113 and 204 molecules per nm2 of NP surface) in CHCl3 was added to the aqueous phase containing the NPs. Phase transfer occurred after vigorous stirring for periods between 15 min and 1 h for different nanoparticles. The transfer was assisted by addition of 20–40 μL of concentrated HCl to 5 mL of NPs solution (see Table 1) for AuNPs, AuNRs, and AgNPs.17 Upon transfer to CHCl3, the organosols were purified by centrifugation and washing to remove free DDT. Precipitation was facilitated by adding ethanol to the chloroform solution (1:5), and centrifugation conditions were optimized for each particle type (Table 1). For stability studies and further use, all samples were stored at 4 °C.
Self-Assembly at the Air/Liquid Interface and Fabrication of Plasmonic Substrates
The dispersions of plasmonic nanoparticles in chloroform were dried under a gentle flow of dry nitrogen. The dry nanoparticles were then redispersed in an ethanol:hexane solvent mixture at 1:4 volume ratio to facilitate monolayer formation at the air/water interface. Ethanol was added first, immediately followed by hexane. The resulting dispersions were immersed in an ultrasonic bath for 5 s to ensure the absence of aggregates prior to spreading at the air/water interface. Plastic Petri dishes with a diameter of ca. 3 cm were used as water containers. For self-assembly of the nanoparticles at the air/water interface, the nanoparticle dispersions were gently spread dropwise on top of the surface of pure water. Upon evaporation of the organic solvents, the water surface was covered by a monolayer of nanoparticles. In each case, the concentration of the dispersion was adjusted to obtain complete coverage of the water surface with a single monolayer of the nanoparticles. The monolayer was then transferred onto a substrate by gently touching the monolayer with the substrate parallel to the surface, which is known as “horizontal lifting” or the “Langmuir–Schaeffer” technique. Transfer onto transmission electron microscopy (TEM) grids was performed to acquire TEM images; transfer onto glass substrates was performed for UV–vis spectroscopy and SERS. Glass slides (24 × 24 mm2 Menzel-Gläser, Thermo Scientific, Germany) were thoroughly cleaned by sonication in water with soap, ethanol, and acetone, 15 min for each solvent. After this cleaning procedure, the glass slides were thoroughly rinsed with Milli-Q ultrapure water and gently blow-dried with a stream of N2 gas. Substrates were stored in sealed Petri dishes.
Sample Preparation for SERS
Prior to using self-assembled monolayers for SERS experiments, the organic ligands were removed from the nanoparticle surface by UV/ozone cleaning (UV/ozone ProCleaner, BioforceNanoscience) for 1 h. The substrates were cut into equal pieces with a surface area of approximately 5 × 2.5 mm2 and immediately incubated in 300 μL of a 10 μM 4-MBA aqueous solution for 1 h, freshly prepared from a 1 mM stock in ethanol. The samples were then extensively washed with Milli-Q water to remove unbound 4-MBA molecules and dried under moderate nitrogen flow; SERS spectra were collected, typically within 1–5 h. In particular, AgNP assemblies were measured immediately after drying to avoid oxidation.
Polymer Coating
For the polymer coating, 2.5 mL of mAuNS solution ([NP] ∼ 1.5 × 10–10 M) in CHCl3 was mixed with the prepared amphiphilic polymer (PMA) dissolved in CHCl3 (VP = 10 μL, cP = 0.05 M, Rp/area = 150 nm–2) in a 25 mL round-bottom flask. The mixture was stirred, and the solvent was slowly evaporated. The resulting solid film containing the NPs was dissolved in 28 mM sodium borate buffer at pH = 12 (SBB 12). After polymer coating the particles were purified, concentrated, and characterized by different techniques.
Characterization
TEM images were collected with a JEOL JEM-1400PLUS transmission electron microscope operating at 120 kV, using carbon-coated 400 square mesh copper grids. Scanning electron microscopy (SEM) images were obtained using an ESEM Quanta250 FEG (FEI, The Netherlands). UV–vis optical extinction spectra were recorded using an Agilent 8453 UV–vis diode-array spectrophotometer and an Agilent Cary 5000 UV–vis–NIR spectrophotometer. 1H NMR spectra were acquired using a Bruker 500 MHz spectrometer (see Supporting Information for details). SERS experiments were performed with a confocal Raman microscope (Renishaw InVia) equipped with a motorized scan stage, two Peltier-cooled CCD detectors, and three excitation lasers of 532, 633, and 785 nm (maximum output powers of 41, 13, and 157 mW, respectively). The laser beam was focused onto the solid sample surface under ambient air conditions through a 100× objective with a numerical aperture of 0.85 and set to effective irradiation powers of 120 μW (at 532 nm), 100 μW (at 633 nm), and 180 μW (at 785 nm) as measured by a photodiode power sensor (PD300-3W, Ophir) or through an immersion 40× objective with NA of 0.8 and set effective to powers of 250 μW (at 532 nm), 110 μW (at 633 nm), and 1200 μW (at 785 nm) when working in solution. The scattered light was collected with an integration time of 1 s. All SERS spectra presented here were averaged over 100 single spectra measured at different points on the substrate. Typically, a 20 × 20 μm2 area was chosen, and single spectra were collected with a point distance of 2 μm in x- and y-direction (10 × 10 spectra). The averaged spectra were baseline corrected using the Wire 3.4 software. Two-dimensional SERS maps were generated by plotting the baseline corrected intensity of the 4-MBA ring stretch mode at 1078 cm–1 as a function of the position on the defined grid.
Results and Discussion
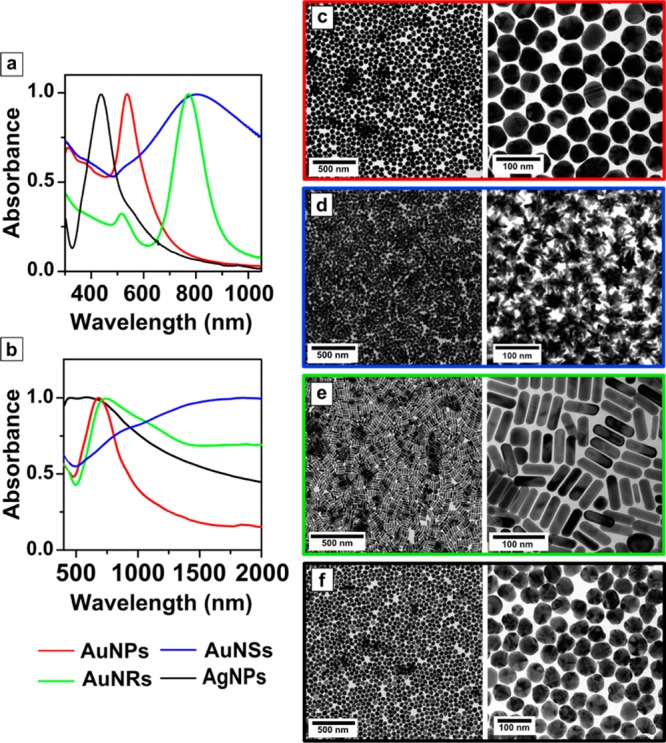
Complete transfer of Au and Ag nanoparticles of different shapes and sizes was achieved using DDT as hydrophobic capping agent, with PEG-SH as prestabilizer (see Materials and Methods section for details). In order to confirm the presence of both ligands on the particle surface upon transfer into chloroform, NMR analysis was carried out (Figure S1A,B). Comparison of the 1H NMR spectra of the functionalized nanoparticles with those of solutions of both ligands confirms that the ligands are attached to the nanoparticles, as indicated by the observed chemical shifts and signal broadening35,36 and the perfect fit in the assignment of the number of protons for DDT. Importantly, the peaks for functional groups located near the metal are not observed, confirming the absence of free ligands. Analysis of the NMR spectra indicated a small decrease in the PEG/DDT ratio upon phase transfer and washing (see complete discussion in the Supporting Information). Successful phase transfer was additionally indicated by the concomitant transfer of red, blue, and yellow colors for AuNPs (and NRs), AuNSs, and AgNPs, respectively, from water to chloroform (Figure 1).
Figure 1.

Phase transfer of PEGylated Au 50 (a), mAuNSs (b), AuNRs (c), and Ag 100 (d) from water (upper phase) into a solution of DDT in chloroform (lower phase). Left and right tubes show the same samples before and after phase transfer. The time lapse between both pictures was 1 hour in all cases.
On the other hand, both the red-shift of the LSPR band without broadening and the well-separated particles found in TEM images confirmed the absence of aggregation as well as the preservation of particle size and shape upon phase transfer (Figure 2 and Figures S2–S4). The strong affinity between the thiol group and the metallic surfaces was essential to ensure rapid phase transfer, so that all the nanoparticles were transferred from water into chloroform within less than 1 h, just 15 min in the case of AuNSs. Prestabilization with a small amount of PEG-SH was needed in all cases to avoid aggregation and facilitate the transfer of single (nonaggregated) particles. Nevertheless, DDT, which provides hydrophobicity to the particles, was added in excess to guarantee the maximum surface coating and therefore the complete phase transfer to organic media. Besides, addition of HCl further facilitated the process, in agreement with previous reports.17,37
Figure 2.
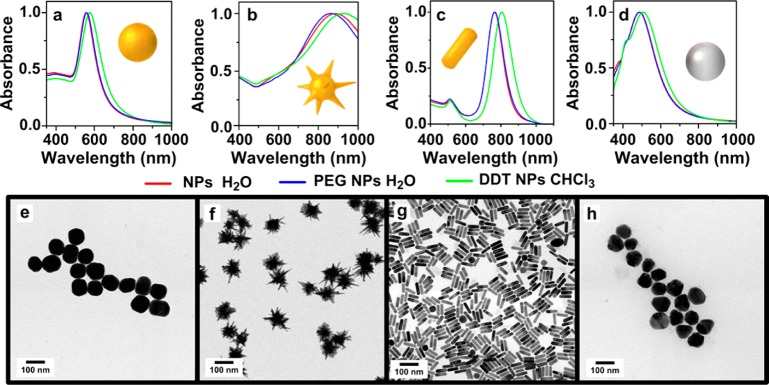
Vis–NIR spectra of NPs in water (red lines), after adding PEG-SH in water (blue lines) and after coating with PEG/DDT and transfer into CHCl3 (green lines). TEM images for Au 100 (a, e), bAuNSs (b, f), AuNRs (c, g), and Ag 100 (d, h).
The obtained hydrophobic nanoparticles displayed a remarkable colloidal stability in chloroform, which was monitored by recording the corresponding vis–NIR spectra for 2 months. As shown in Figure 3 and Figures S2–S4 no trace of aggregation or sedimentation of the particles could be observed, either in the first step (prestabilization with PEG-SH) or in the second one (transfer into chloroform via DDT adsorption). Note that the stability of AgNPs is comparable to that of AuNPs while in solution, but after drying on solid substrates oxidation of AgNPs was found to occur, as indicated by morphological changes (Figure S5). Regarding the stability of AuNSs, upon storage in water the tips were observed to become more rounded as the LSPR band significantly blue-shifted over time, in agreement with previous reports, but after PEG/DDT capping and phase transfer into chloroform, no LSPR shift was observed and the tips remained sharp, which is likely due to surface passivation through Au–S bonds38 (Figure S6).
Figure 3.
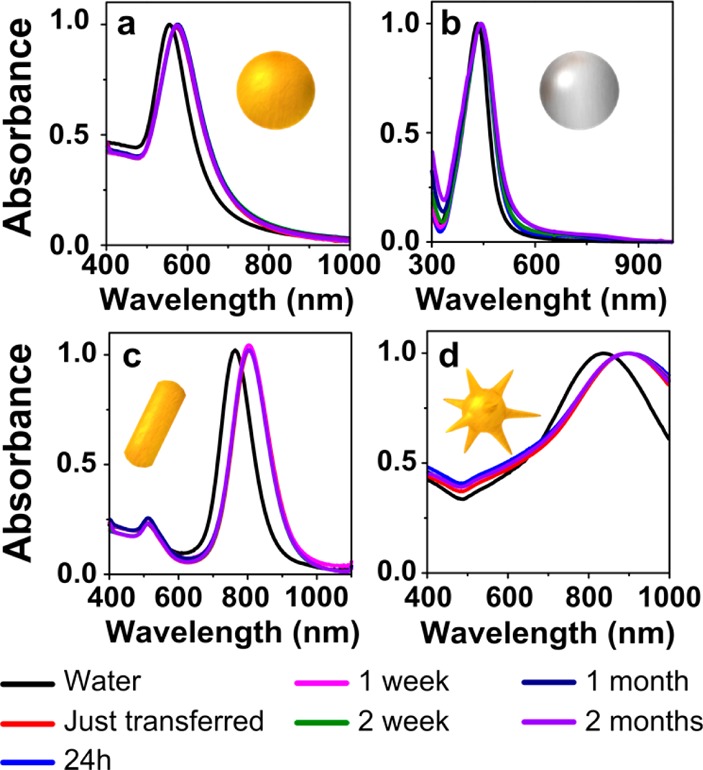
Normalized vis–NIR spectra in water (black lines) and in chloroform (colored lines), after selected periods of time as labeled, for Au 100 (a), Ag 50 (b), AuNRs (c), and bAuNSs (d).
Once the nanoparticles have been transferred into chloroform they can be washed by centrifugation, dried, and redispersed in different organic solvents. As an example, we show in Figure S7 vis–NIR spectra of AuNRs and mAuNSs dispersed in various solvents. The registered LSPR shifts are in agreement with the respective refractive indexes, except for hexane, where the particles present lower stability due to the lack of solubility of PEG-SH. PEG can be easily dissolved in organic solvents that display hydrogen bond donating capacity such as chloroform, but this does not apply to hexane.19 Interestingly, when reducing the amount of PEG-SH and keeping a constant DDT concentration, the colloidal stability in hexane or other nonpolar solvents such as toluene can be notably improved, as confirmed by less broadened LSPR bands. A different strategy to improve NP solubility includes the selection of different thiolated hydrophobic molecules. For example, combining 11-mercaptoundecanol with DDT in a 1:1 ratio and adding a shorter PEG-SH molecule (750 Da), the solubility and stability of particles in slightly polar solvents such as ethyl acetate can be significantly increased. Other thiolated ligands soluble in chloroform were tested (not shown), confirming the wide versatility of the method presented here.
An ethanol:hexane mixture (1:4) was used as spreading solvent. This mixture of solvents allowed us to obtain extensive nanoparticle monolayers, which were then transferred onto solid substrates (Figure 4).39−41 It should be noted that solvents with larger spreading coefficients, e.g. mixtures of chloroform and isopropanol, might be beneficial for spreading of the nanoparticles. In all cases, long-range order was achieved, with dense nanoparticle packing and small interparticle gaps.
Figure 4.
(a) Vis–NIR spectra of different plasmonic nanoparticles dispersed in ethanol:hexane (1:4). (b) Vis–NIR spectra of plasmonic nanoparticle monolayers transferred onto a transparent glass substrate. (c–f) TEM images of Au 50 (c), mAuNSs (d), AuNRs (e), and Ag 50 (f) monolayers upon transfer onto TEM grids.
High-magnification TEM images from monolayers transferred onto carbon-coated copper grids show high packing density over large areas (Figure 4). As expected, such short interparticle distances lead to plasmon coupling, which is reflected in significant red-shift and broadening of LSPR bands (Figure 4b and Figure S8). Plasmon coupling has been demonstrated to enhance the plasmonic features in 2D SERS-active platforms.42 In the case of AuNSs, dense packing leads to a high degree of spike interdigitation, which is rarely achieved using chemical immobilization methods,43 and leads to a particularly large extent of plasmon coupling as compared to nanospheres. Remarkably, these monolayers present several advantages for the fabrication of plasmonic substrates, such as readily achieved homogeneous large areas on the square centimeter range, while “coffee ring” or similar local effects are avoided. Formation of dense monolayers was therefore achieved through a simple experimental procedure for both phase transfer and assembly, which can be scaled up to arbitrarily large areas by simply selecting the appropriate amount of nanoparticles (Figure S9).
Since phase transfer, self-assembly, and cleaning processes are identical for all nanoparticles, regardless of size, shape, and composition, we can readily compare their relative performances as SERS substrates. We selected 4-mercaptobenzoic acid (4-MBA) as a covalently binding probe molecule, at a concentration of 10 μM, to probe the SERS activity using three different laser excitation wavelengths of 532, 633, and 785 nm. Figure 5a as well as Figures S10 and S11 depict SERS spectra obtained with illumination at 785 nm, showing in all cases the characteristic vibrational fingerprint of (mainly deprotonated) 4-MBA, as assigned in previous works.44−47 The most prominent peaks at 1078 and 1587 cm–1 correspond to excitation of ring breathing and axial ring deformation modes.44,46 Several peaks with lower intensities were observed at 1484, 1180, 1142, 720, and 696 cm–1 and can be assigned as the 19a and 9b modes (based on the nomenclature for benzene),48,49 in-plane CH bending,46,49 out-of-plane ring hydrogen wagging, and a mixture of OCO bending, in-plane ring compression, and C–S stretching, respectively.46 The broad bands around 1400 and 845 cm–1 correspond to the COO– symmetric stretch mode and the bending mode, respectively.44,46,50 An additional high signal around 920 cm–1, which was exclusively registered from the Ag 50 sample, originates from organic residues formed during synthesis and plasma cleaning.
Figure 5.

(a) Averaged SERS spectra of 4-MBA (10 μM) on Au 100 (b), AuNRs (c), Ag 50 (d), and bAuNSs (e). The corresponding SERS maps for the signal at 1078 cm–1 (red-shaded area in (a) as a function of the deviation from the average intensity dI (in %), together with representative SEM images of the assemblies before analyte incubation. The black SERS spectrum in (a) corresponds to the average of three 4-MBA spectra on the AuNR sample measured with a portable Raman spectrometer.
The highest SERS signal was observed from AuNR (Figure 5c) and AuNP films (Figure 5b), where the intensity was found to increase with increasing sphere diameter (Figure S10). The SERS performance of AgNP and in particular AuNS monolayers (Figure 5d,e) was significantly lower, with intensities 1 order of magnitude smaller than those from AuNP and AuNR films. This behavior can be explained taking into account that the highest enhancement is obtained on substrates with LSPR bands located between excitation the scattered frequencies.51 At an excitation wavelength of 785 nm, this condition complies best with the AuNP and AuNR samples (Figure 4b and Figure S8).
The SERS signal intensity also depends on several additional surface specific factors, e.g., analyte binding affinity, presence of surfactants,52−54 or contaminants and surface defects.55 Additionally, potential applications require reliability of the measured signal as a key factor. Because of the relatively large laser spot size (>1 μm2), defects on the atomic scale do not play a significant role, but shape imperfections, a small degree of disorder within the assembly, and remaining organic residues may cause significant point-to-point intensity fluctuations. To avoid large errors and to statistically improve the signal, we averaged SERS spectra over 100 points located on a 20 × 20 μm2 grid. To probe the local homogeneity, we analyzed intensity fluctuations of single-point spectra presented as SERS maps in Figure 5 as well as Figures S10 and S11. The lowest signal variation from the averaged value (dI in %) was observed for the AuNR sample with 12% (Figure 5c). The AuNP samples showed fluctuations ranging from 15% to 34% for increasing particle diameters (Figure 5b and Figure S10), whereas the SERS signal from small, middle, and big AuNSs was found to scatter within ca. 30% (Figure 5e and Figure S11). The lowest degree of homogeneity was found for Ag 50 (Figure 5d), with signal deviations up to 50%. These results are directly related to the degree of disorder within the assemblies as well as with larger interparticle contact area for smaller nanoparticles. The relatively high disorder of the Ag 50 substrate accompanied by the presence of organics which could prevent the effective binding of the analyte are likely to be responsible for the high signal fluctuations on the Ag substrate. Ag particles are additionally sensitive to oxidation, as discussed above (Figure S5), so the formation of a thin oxide layer can affect the SERS performance. To probe homogeneity over large areas, SERS maps were recorded from at least three different areas distributed over the substrate. The intrasample average signal variation did not exceed 5% for AuNRs but amounted 7%, 10%, and 13% for Au 30, 50, and 100, respectively. In small, middle, and big AuNSs the SERS signal fluctuated less than 15%. Reproducibility of the SERS response was studied by repeating sample preparation with different nanoparticle batches, finding average batch-to-batch intensity fluctuations lower than 5% for AuNRs and 15% for AuNPs. When 4-MBA SERS spectra were recorded using 532 and 633 nm excitations, different results were obtained, as at 532 nm the most intense signals were obtained from Ag 50, reaching intensities 2 orders of magnitude higher than those from AuNP substrates (Figure S12e). In line with the SERS electromagnetic mechanism, recorded intensities were lower from Ag 50 at 633 nm but higher from AuNPs (Figure S12f). AuNS substrates did not show SERS activity when excited with both high energy laser lines.
As additional advantages of this system, we show that detection can be performed directly from solution and using a portable Raman spectrometer. Thus, we measured SERS from substrates immersed in 10 μM 4-MBA solutions. Exemplarily, the spectrum and map for the AuNR sample are shown in Figure S13. The average SERS intensity (at 785 nm) only varies by 2.5%. For other nanoparticle assemblies we recorded in solution the same average intensity deviations and point-to-point fluctuations as those measured in air. Applicability outside the laboratory will also require using easy to handle standard analytical equipment for rapid detection instead of sophisticated scientific setups. We measured 4-MBA on the AuNR substrate using a portable Raman spectrometer (BWTEK, i-Raman-785S) working at a fixed 785 nm excitation wavelength. Plaser and tint were adjusted so that the 4-MBA SERS intensities in both setups were of the same order of magnitude without damaging the substrate, e.g., through high power exposition. To reach these conditions, Plaser = 19 mW measured at sample and tint = 1 s were needed (laser spot size of about 80 μm). We thus measured at three randomly chosen points distributed over the whole surface, and the average is shown in Figure 5a, with a standard deviation of 7%.
SERS enhancement factors (EF) were calculated by means of the frequently used equation56
where ISERS and IRaman represent the 4-MBA signal intensities recorded from the probed number of molecules absorbed on the substrate (NSERS) and from the number of molecules within the scattering volume (Nbulk) of a solid powder, respectively. For determination of the scattering volume from the bulk material the depth of field approach presented by Khan et al.57 was used, resulting in Nbulk (785 nm) ≈ 1013. NSERS (785 nm) was estimated to be 108 assuming formation of a 4-MBA monolayer (more details in Supporting Information). IRaman and ISERS were measured with 95 and 3517 cps, respectively, leading to an EF (785 nm) of approximately 3.7 × 106 in the case of the AuNR substrate. This value is of the same order of magnitude as previously reported values.57
We finally show that the obtained nanoparticle organosols can also be used for secondary coating with an amphiphilic polymer that has been previously reported as suitable for biological applications.21,58 After removal of excess DDT by centrifugation of the dispersion in chloroform, the reported polymer coating procedure (cf. Materials and Methods and Supporting Information section 9) was applied using dodecylamine modified polyisobutylene-alt-maleic anhydride (PMA). The hydrophopic nanoparticles were thus wrapped with the polymer via hydrophobic interactions between the side chains of the polymer (dodecylamine) and the NP ligands. The particles could then be transferred into water, where the charged polymer shell provides sufficient stability in biofluids. Representative vis–NIR spectra and TEM images (Figure 6a,b) of PMA-coated AuNSs confirm the colloidal stability of the particles upon polymer coating. TEM images of negatively stained, polymer coated Au NSs confirm the presence of an organic layer wrapping the particles in water. A white shell surrounding the NPs can be clearly appreciated for PMA-coated particles (Figure 6d and Figure S16), whereas noncoated NSs (Figure 6c) do not display the characteristic white halo.
Figure 6.
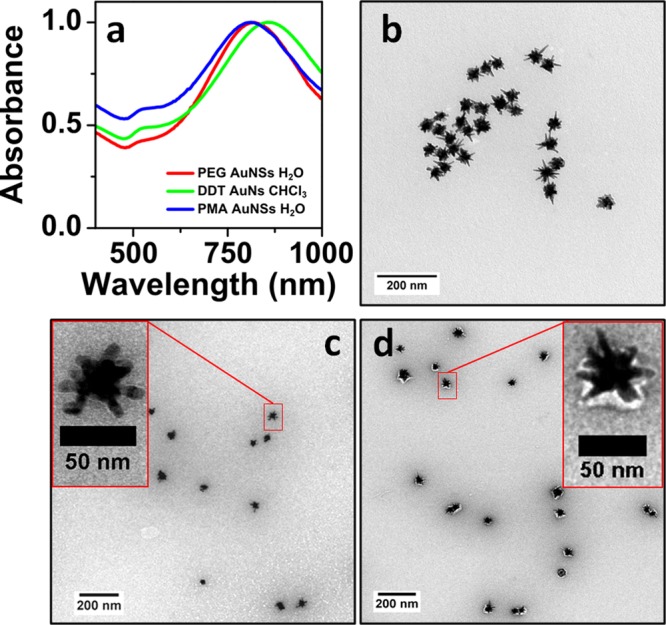
(a) UV–vis spectra of PEG-SH stabilized Au NSs in water (red), after transfer into CHCl3 with PEG/DDT (green), and again in water after PMA coating (blue). (b–d) TEM images of PMA-coated Au NSs (b), negatively stained bare Au NSs (c), and negatively stained polymer coated Au NSs (d).
Conclusions
A general route for transferring plasmonic nanoparticles from aqueous dispersions into chloroform and other organic solvents has been developed, which can be applied to Ag and Au nanoparticles of arbitrary sizes and shapes, including spheres, nanorods, and nanostars. The transferred particles are stable in chloroform for months and can be readily dried, purified, and redispersed in various organic solvents. This efficient phase transfer procedure is based on functionalization of the nanoparticles’ surface with a mixture of PEG-SH and DDT. While DDT provides the required hydrophobicity for the nanoparticles to reach the organic phase, PEG-SH provides enough stability to avoid aggregation during phase transfer. Using ethanol:hexane 1:4 mixtures as spreading solvent, homogeneous extended self-assembled nanoparticle monolayers were obtained on the centimeter scale. Whereas the self-assembly of plasmonic nanoparticles at liquid interfaces has been traditionally restricted due to limited size of nanoparticles available in organic solvents, limited stability, and complicated experimental procedures, the method proposed here allows us to obtain plasmonic nanoparticles in organic solvents with no restriction in size, shape, or surface chemical composition, readily forming nanoparticle assemblies. The close packing of the plasmonic nanoparticles favors plasmon coupling, thus providing enhanced plasmonic features. We compared the SERS activity between the different plasmonic substrates fabricated by detection of 4-MBA, finding that AuNP and AuNR self-assembled monolayers displayed optimum SERS performance in air and in solution for 785 and 633 nm excitation lasers, with high signal reproducibility, robustness, and moderate point-to-point fluctuations. We also showed that high-end experimental setups are not necessary to obtain intense and reproducible SERS signals from our assemblies. Additionally, the obtained hydrophobic nanoparticles proved suitable for subsequent polymer coating, thereby broadening potential applications due to the obtained high stability under biological conditions.
Acknowledgments
Funding is acknowledged from the European Commission (Grant #310445-2 SAVVY) and the European Research Council (ERC Advanced Grant #267867 Plasmaquo). L.M.L.-M. acknowledges funding from by the Deanship of Scientific Research, Research Centre, College of Science, and the DSFP Program, King Saud University. J.J.G.-C. acknowledges the Ministry of Economy and Competitiveness for a Juan de la Cierva fellowship (#JCI-2012-12517). The authors acknowledge Andrea La Porta and Sergey M. Novikov for the SEM characterization and Marco Möller for useful discussion on TEM images.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.langmuir.5b01838.
Figures S1–S16 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Webb J. A.; Bardhan R. Emerging Advances in Nanomedicine with Engineered Gold Nanostructures. Nanoscale 2014, 6, 2502–2530 10.1039/c3nr05112a. [DOI] [PubMed] [Google Scholar]
- Anker J. N.; Hall W. P.; Lyandres O.; Shah N. C.; Zhao J.; Van Duyne R. P. Biosensing with Plasmonic Nanosensors. Nat. Mater. 2008, 7, 442–453 10.1038/nmat2162. [DOI] [PubMed] [Google Scholar]
- Atwater H. A.; Polman A. Plasmonics for Improved Photovoltaic Devices. Nat. Mater. 2010, 9, 205–213 10.1038/nmat2629. [DOI] [PubMed] [Google Scholar]
- Zijlstra P.; Chon J. W. M.; Gu M. Five-Dimensional Optical Recording Mediated by Surface Plasmons in Gold Nanorods. Nature 2009, 459, 410–413 10.1038/nature08053. [DOI] [PubMed] [Google Scholar]
- Gong J.; Li G.; Tang Z. Self-Assembly of Noble Metal Nanocrystals: Fabrication, Optical Property, and Application. Nano Today 2012, 7, 564–585 10.1016/j.nantod.2012.10.008. [DOI] [Google Scholar]
- Grzelczak M.; Pérez-Juste J.; Mulvaney P.; Liz-Marzán L. M. Shape Control in Gold Nanoparticle Synthesis. Chem. Soc. Rev. 2008, 37, 1783. 10.1039/b711490g. [DOI] [PubMed] [Google Scholar]
- Liz-Marzán L. M. Tailoring Surface Plasmons through the Morphology and Assembly of Metal Nanoparticles. Langmuir 2006, 22, 32–41 10.1021/la0513353. [DOI] [PubMed] [Google Scholar]
- Li N.; Zhao P.; Astruc D. Anisotropic Gold Nanoparticles: Synthesis, Properties, Applications, and Toxicity. Angew. Chem., Int. Ed. 2014, 53, 1756–1789 10.1002/anie.201300441. [DOI] [PubMed] [Google Scholar]
- Guerrero-Martínez A.; Barbosa S.; Pastoriza-Santos I.; Liz-Marzán L. M. Nanostars Shine Bright for You. Curr. Opin. Colloid Interface Sci. 2011, 16, 118–127 10.1016/j.cocis.2010.12.007. [DOI] [Google Scholar]
- Yoshida A.; Imazu K.; Li X.; Okamoto K.; Tamada K. Spectroscopic Properties of Multilayered Gold Nanoparticle 2D Sheets. Langmuir 2012, 28, 17153–17158 10.1021/la303533f. [DOI] [PubMed] [Google Scholar]
- Zhou J.; Liu H.; Wang T.; Li Y.; Zhang J.; Lu Z.; Fu Y.; Li F. Adjusting the Inter-Particle Spacing of a Nanoparticle Array at the Sub-Nanometre Scale by Thermal Annealing. Chem. Commun. 2014, 50, 14547–14549 10.1039/C4CC06912A. [DOI] [PubMed] [Google Scholar]
- Yang J.; Lee J. Y.; Ying J. Y. Phase Transfer and Its Applications in Nanotechnology. Chem. Soc. Rev. 2011, 40, 1672. 10.1039/B916790K. [DOI] [PubMed] [Google Scholar]
- Underwood S.; Mulvaney P. Effect of the Solution Refractive Index on the Color of Gold Colloids. Langmuir 1994, 10, 3427–3430 10.1021/la00022a011. [DOI] [Google Scholar]
- Vijaya Sarathy K.; Kulkarni G. U.; Rao C. N. R. A Novel Method of Preparing Thiol-Derivatised Nanoparticles of Gold, Platinum and Silver Forming Superstructures. Chem. Commun. 1997, 537–538 10.1039/a700738h. [DOI] [Google Scholar]
- Li L.; Leopold K.; Schuster M. Comparative Study of Alkylthiols and Alkylamines for the Phase Transfer of Gold Nanoparticles from an Aqueous Phase to N-Hexane. J. Colloid Interface Sci. 2013, 397, 199–205 10.1016/j.jcis.2013.01.062. [DOI] [PubMed] [Google Scholar]
- Wang X.; Xu S.; Zhou J.; Xu W. A Rapid Phase Transfer Method for Nanoparticles Using Alkylamine Stabilizers. J. Colloid Interface Sci. 2010, 348, 24–28 10.1016/j.jcis.2010.03.068. [DOI] [PubMed] [Google Scholar]
- Lista M.; Liu D. Z.; Mulvaney P. Phase Transfer of Noble Metal Nanoparticles to Organic Solvents. Langmuir 2014, 30, 1932–1938 10.1021/la404569h. [DOI] [PubMed] [Google Scholar]
- Goulet P. J. G.; Bourret G. R.; Lennox R. B. Facile Phase Transfer of Large, Water-Soluble Metal Nanoparticles to Nonpolar Solvents. Langmuir 2012, 28, 2909–2913 10.1021/la2038894. [DOI] [PubMed] [Google Scholar]
- Alkilany A. M.; Yaseen A. I. B.; Park J.; Eller J. R.; Murphy C. J. Facile Phase Transfer of Gold Nanoparticles from Aqueous Solution to Organic Solvents with Thiolated Poly(ethylene Glycol). RSC Adv. 2014, 4, 52676–52679 10.1039/C4RA11928B. [DOI] [Google Scholar]
- Liu M.; Law W.-C.; Kopwitthaya A.; Liu X.; Swihart M. T.; Prasad P. N. Exploring the Amphiphilicity of PEGylated Gold Nanorods: Mechanical Phase Transfer and Self-Assembly. Chem. Commun. 2013, 49, 9350. 10.1039/c3cc45103h. [DOI] [PubMed] [Google Scholar]
- Soliman M. G.; Pelaz B.; Parak W. J.; del Pino P. Phase Transfer and Polymer Coating Methods toward Improving the Stability of Metallic Nanoparticles for Biological Applications. Chem. Mater. 2015, 27, 990–997 10.1021/cm5043167. [DOI] [Google Scholar]
- Yuan H.; Fales A. M.; Vo-Dinh T. TAT Peptide-Functionalized Gold Nanostars: Enhanced Intracellular Delivery and Efficient NIR Photothermal Therapy Using Ultralow Irradiance. J. Am. Chem. Soc. 2012, 134, 11358–11361 10.1021/ja304180y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kneipp K.; Kneipp H.; Itzkan I.; Dasari R. R.; Feld M. S. Ultrasensitive Chemical Analysis by Raman Spectroscopy. Chem. Rev. 1999, 99, 2957–2976 10.1021/cr980133r. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Lorenzo L.; Álvarez-Puebla R. A.; Pastoriza-Santos I.; Mazzucco S.; Stéphan O.; Kociak M.; Liz-Marzán L. M.; García de Abajo F. J. Zeptomol Detection Through Controlled Ultrasensitive Surface-Enhanced Raman Scattering. J. Am. Chem. Soc. 2009, 131, 4616–4618 10.1021/ja809418t. [DOI] [PubMed] [Google Scholar]
- Qian K.; Yang L.; Li Z.; Liu J. A New-Type Dynamic SERS Method for Ultrasensitive Detection. J. Raman Spectrosc. 2013, 44, 21–28 10.1002/jrs.4137. [DOI] [Google Scholar]
- Wu Z.; Liu Y.; Zhou X.; Shen A.; Hu J. A “Turn-off” SERS-Based Detection Platform for Ultrasensitive Detection of Thrombin Based on Enzymatic Assays. Biosens. Bioelectron. 2013, 44, 10–15 10.1016/j.bios.2013.01.006. [DOI] [PubMed] [Google Scholar]
- Domenici F.; Bizzarri A. R.; Cannistraro S. SERS-Based Nanobiosensing for Ultrasensitive Detection of the p53 Tumor Suppressor. Int. J. Nanomed. 2011, 6, 2033–2042 10.2147/IJN.S23845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J.; Li Y.; Zhang M.; Li Z.; Yan T.; Qian W. Ultrasensitive Surface-Enhanced Raman Scattering Detection of Alkaline Phosphatase. Anal. Methods 2014, 6, 9168–9172 10.1039/C4AY01885K. [DOI] [Google Scholar]
- Zheng J.; Hu Y.; Bai J.; Ma C.; Li J.; Li Y.; Shi M.; Tan W.; Yang R. Universal Surface-Enhanced Raman Scattering Amplification Detector for Ultrasensitive Detection of Multiple Target Analytes. Anal. Chem. 2014, 86, 2205–2212 10.1021/ac404004m. [DOI] [PubMed] [Google Scholar]
- Bastús N. G.; Comenge J.; Puntes V. Kinetically Controlled Seeded Growth Synthesis of Citrate-Stabilized Gold Nanoparticles of up to 200 nm: Size Focusing versus Ostwald Ripening. Langmuir 2011, 27, 11098–11105 10.1021/la201938u. [DOI] [PubMed] [Google Scholar]
- Yuan H.; Khoury C. G.; Hwang H.; Wilson C. M.; Grant G. A.; Vo-Dinh T. Gold Nanostars: Surfactant-Free Synthesis, 3D Modelling, and Two-Photon Photoluminescence Imaging. Nanotechnology 2012, 23, 075102. 10.1088/0957-4484/23/7/075102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye X.; Jin L.; Caglayan H.; Chen J.; Xing G.; Zheng C.; Doan-Nguyen V.; Kang Y.; Engheta N.; Kagan C. R.; et al. Improved Size-Tunable Synthesis of Monodisperse Gold Nanorods through the Use of Aromatic Additives. ACS Nano 2012, 6, 2804–2817 10.1021/nn300315j. [DOI] [PubMed] [Google Scholar]
- Scarabelli L.; Grzelczak M.; Liz-Marzán L. M. Tuning Gold Nanorod Synthesis through Prereduction with Salicylic Acid. Chem. Mater. 2013, 25, 4232–4238 10.1021/cm402177b. [DOI] [Google Scholar]
- Bastús N. G.; Merkoçi F.; Piella J.; Puntes V. Synthesis of Highly Monodisperse Citrate-Stabilized Silver Nanoparticles of up to 200 nm: Kinetic Control and Catalytic Properties. Chem. Mater. 2014, 26, 2836–2846 10.1021/cm500316k. [DOI] [Google Scholar]
- Hostetler M. J.; Wingate J. E.; Zhong C.-J.; Harris J. E.; Vachet R. W.; Clark M. R.; Londono J. D.; Green S. J.; Stokes J. J.; Wignall G. D.; et al. Alkanethiolate Gold Cluster Molecules with Core Diameters from 1.5 to 5.2 nm: Core and Monolayer Properties as a Function of Core Size. Langmuir 1998, 14, 17–30 10.1021/la970588w. [DOI] [Google Scholar]
- Zelakiewicz B. S.; de Dios A. C.; Tong 13C NMR Spectroscopy of 13C1-Labeled Octanethiol-Protected Au Nanoparticles: Shifts, Relaxations, and Particle-Size Effect. J. Am. Chem. Soc. 2003, 125, 18–19 10.1021/ja028302j. [DOI] [PubMed] [Google Scholar]
- Zhao S.-Y.; Chen S.-H.; Wang S.-Y.; Li D.-G.; Ma H.-Y. Preparation, Phase Transfer, and Self-Assembled Monolayers of Cubic Pt Nanoparticles. Langmuir 2002, 18, 3315–3318 10.1021/la011262x. [DOI] [Google Scholar]
- Vega M. M.; Bonifacio A.; Lughi V.; Marsi S.; Carrato S.; Sergo V. Long-Term Stability of Surfactant-Free Gold Nanostars. J. Nanopart. Res. 2014, 16, 1–6 10.1007/s11051-014-2729-z. [DOI] [Google Scholar]
- Dong A.; Chen J.; Vora P. M.; Kikkawa J. M.; Murray C. B. Binary Nanocrystal Superlattice Membranes Self-Assembled at the Liquid-Air Interface. Nature 2010, 466, 474–477 10.1038/nature09188. [DOI] [PubMed] [Google Scholar]
- Giner-Casares J. J.; Brezesinski G.; Möhwald H. Langmuir Monolayers as Unique Physical Models. Curr. Opin. Colloid Interface Sci. 2014, 19, 176–182 10.1016/j.cocis.2013.07.006. [DOI] [Google Scholar]
- Scarabelli L.; Coronado-Puchau M.; Giner-Casares J. J.; Langer J.; Liz-Marzán L. M. Monodisperse Gold Nanotriangles: Size Control, Large-Scale Self-Assembly, and Performance in Surface-Enhanced Raman Scattering. ACS Nano 2014, 8, 5833–5842 10.1021/nn500727w. [DOI] [PubMed] [Google Scholar]
- Giner-Casares J. J.; Liz-Marzán L. M. Plasmonic Nanoparticles in 2D for Biological Applications: Toward Active Multipurpose Platforms. Nano Today 2014, 9, 365–377 10.1016/j.nantod.2014.05.004. [DOI] [Google Scholar]
- Shiohara A.; Langer J.; Polavarapu L.; Liz-Marzán L. M. Solution Processed Polydimethylsiloxane/gold Nanostar Flexible Substrates for Plasmonic Sensing. Nanoscale 2014, 6, 9817–9823 10.1039/C4NR02648A. [DOI] [PubMed] [Google Scholar]
- Michota A.; Bukowska J. Surface-Enhanced Raman Scattering (SERS) of 4-Mercaptobenzoic Acid on Silver and Gold Substrates. J. Raman Spectrosc. 2003, 34, 21–25 10.1002/jrs.928. [DOI] [Google Scholar]
- Talley C. E.; Jusinski L.; Hollars C. W.; Lane S. M.; Huser T. Intracellular pH Sensors Based on Surface-Enhanced Raman Scattering. Anal. Chem. 2004, 76, 7064–7068 10.1021/ac049093j. [DOI] [PubMed] [Google Scholar]
- Bishnoi S. W.; Rozell C. J.; Levin C. S.; Gheith M. K.; Johnson B. R.; Johnson D. H.; Halas N. J. All-Optical Nanoscale pH Meter. Nano Lett. 2006, 6, 1687–1692 10.1021/nl060865w. [DOI] [PubMed] [Google Scholar]
- Pallaoro A.; Braun G. B.; Reich N. O.; Moskovits M. Mapping Local pH in Live Cells Using Encapsulated Fluorescent SERS Nanotags. Small 2010, 6, 618–622 10.1002/smll.200901893. [DOI] [PubMed] [Google Scholar]
- Varsányi G.Assignments for Vibrational Spectra of Seven Hundred Benzene Derivatives; Wiley: New York, 1974. [Google Scholar]
- Kwon Y. J.; Son D. H.; Ahn S. J.; Kim M. S.; Kim K. Vibrational Spectroscopic Investigation of Benzoic Acid Adsorbed on Silver. J. Phys. Chem. 1994, 98, 8481–8487 10.1021/j100085a030. [DOI] [Google Scholar]
- Suh J. S.; Kim J. Three Distinct Geometries of Surface-Adsorbed Carboxylate Groups. J. Raman Spectrosc. 1998, 29, 143–148. [DOI] [Google Scholar]
- McFarland A. D.; Young M. A.; Dieringer J. A.; Van Duyne R. P. Wavelength-Scanned Surface-Enhanced Raman Excitation Spectroscopy. J. Phys. Chem. B 2005, 109, 11279–11285 10.1021/jp050508u. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Lorenzo L.; Álvarez-Puebla R. A.; de Abajo F. J. G.; Liz-Marzán L. M. Surface Enhanced Raman Scattering Using Star-Shaped Gold Colloidal Nanoparticles. J. Phys. Chem. C 2010, 114167336–7340 10.1021/jp909253w. [DOI] [Google Scholar]
- Alvarez-Puebla R. A.; Liz-Marzán L. M. Traps and Cages for Universal SERS Detection. Chem. Soc. Rev. 2012, 41, 43–51 10.1039/C1CS15155J. [DOI] [PubMed] [Google Scholar]
- Tebbe M.; Maennel M.; Fery A.; Pazos-Perez N.; Alvarez-Puebla R. A. Organized Solid Thin Films of Gold Nanorods with Different Sizes for Surface-Enhanced Raman Scattering Applications. J. Phys. Chem. C 2014, 118, 28095–28100 10.1021/jp5086685. [DOI] [Google Scholar]
- Litorja M.; Haynes C. L.; Haes A. J.; Jensen T. R.; Van Duyne R. P. Surface-Enhanced Raman Scattering Detected Temperature Programmed Desorption: Optical Properties, Nanostructure, and Stability of Silver Film over SiO2 Nanosphere Surfaces. J. Phys. Chem. B 2001, 105, 6907–6915 10.1021/jp010333y. [DOI] [Google Scholar]
- Cai W. B.; Ren B.; Li X. Q.; She C. X.; Liu F. M.; Cai X. W.; Tian Z. Q. Investigation of Surface-Enhanced Raman Scattering from Platinum Electrodes Using a Confocal Raman Microscope: Dependence of Surface Roughening Pretreatment. Surf. Sci. 1998, 406, 9–22 10.1016/S0039-6028(97)01030-3. [DOI] [Google Scholar]
- Khan M. A.; Hogan T. P.; Shanker B. Surface-Enhanced Raman Scattering from Gold-Coated Germanium Oxide Nanowires. J. Raman Spectrosc. 2008, 39, 893–900 10.1002/jrs.1931. [DOI] [Google Scholar]
- Zhang F.; Lees E.; Amin F.; Rivera Gil P.; Yang F.; Mulvaney P.; Parak W. J. Polymer-Coated Nanoparticles: A Universal Tool for Biolabelling Experiments. Small 2011, 7, 3113–3127 10.1002/smll.201100608. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.