

Abstract
Comprehensive studies that consolidate selective ligands, quantitative comparisons of G protein versus arrestin-2/3 coupling, together with structure-activity relationship models for G protein–coupled receptor (GPCR) systems are less commonly employed. Here we examine biased signaling at the nociceptin/orphanin FQ opioid receptor (NOPR), the most recently identified member of the opioid receptor family. Using real-time, live-cell assays, we identified the signaling profiles of several NOPR-selective ligands in upstream GPCR signaling (G protein and arrestin pathways) to determine their relative transduction coefficients and signaling bias. Complementing this analysis, we designed novel ligands on the basis of NOPR antagonist J-113,397 [(±)-1-[(3R*,4R*)-1-(cyclooctylmethyl)-3-(hydroxymethyl)-4-piperidinyl]-3-ethyl-1,3-dihydro-2H-benzimidazol-2-one] to explore structure-activity relationships. Our study shows that NOPR is capable of biased signaling, and further, the NOPR selective ligands MCOPPB [1-[1-(1-methylcyclooctyl)-4-piperidinyl]-2-(3R)-3-piperidinyl-1H-benzimidazole trihydrochloride] and NNC 63-0532 [8-(1-naphthalenylmethyl)-4-oxo-1-phenyl-1,3,8-triazaspiro[4.5]decane-3-acetic acid, methyl ester] are G protein–biased agonists. Additionally, minor structural modification of J-113,397 can dramatically shift signaling from antagonist to partial agonist activity. We explore these findings with in silico modeling of binding poses. This work is the first to demonstrate functional selectivity and identification of biased ligands at the nociceptin opioid receptor.
Introduction
Understanding of signal transduction for known receptor systems will lead to more efficient and better directed approaches for clinical application. Employing comparative studies of ligands by in vitro assays allows the calculation of signal transduction coefficients, which can then be easily applied across model systems (Rajagopal et al., 2010, 2013; Kenakin et al., 2012; Kenakin and Christopoulos, 2013; Zhou et al., 2013; van der Westhuizen et al., 2014). Further, applying ligand-receptor interaction modeling in conjunction with ligand-specific signal transduction data sets can lead to identification of the various structure-activity relationships necessary for the rational design of “ideal ligands” for a particular receptor system, thus eliciting the desired outcome. In this work, we apply these principles of receptor pharmacology to the more recently discovered nociceptin opioid receptor system (also known as ORL1, OPRL1, N/OFQ).
The nociceptin opioid receptor (NOPR) and its endogenous peptide ligand, nociceptin (N/OFQ), are widely expressed throughout the central nervous system, and show high therapeutic potential in contexts involving pain, anxiety, addiction, and cardiovascular function. These and other studies report wide-ranging behavioral outcomes, and greater divergence depending on the ligand tested, yet no reports have identified NOPR ligand bias (Mollereau et al., 1994; Reinscheid et al., 1995; Murphy et al., 1999; Mogil and Pasternak, 2001; Yamada et al., 2002; Kapusta et al., 2005; Goeldner et al., 2008; Hirao et al., 2008; Marquez et al., 2008; Reiss et al., 2008; Varty et al., 2008; Hayashi et al., 2009; Cremeans et al., 2012; Gear et al., 2014; Zhang et al., 2014). Other recent studies have characterized and described key residues involved in internalization, desensitization, and arrestin signaling of NOPR following activation with nociceptin (Corbani et al., 2004; Spampinato et al., 2007; Zhang et al., 2012). The G protein–coupled receptor (GPCR) knowledge base has recently benefitted from many well designed studies uncovering unknown characteristics and mechanisms of action, spatial-temporal dynamics, and detailed insight into structure-function relationships (Chung et al., 2011; Kahsai et al., 2011; Rasmussen et al., 2011; Bock et al., 2014; Lane et al., 2014; Motta-Mena et al., 2014). In particular, the crystal structure of NOPR was recently solved in complex with a peptide mimetic of the selective antagonist UFP-101 [[Nphe1,Arg14,Lys15]nociceptin-NH2], itself a close derivative of nociceptin (Thompson et al., 2012). However, there is still a dearth of information pertaining to the intricacies of the NOPR signal transduction system. Therefore, understanding the signal transduction capabilities of selective ligands and identifying the conformational states of biased signaling at the NOPR is critical to exploiting its potential therapeutic avenues.
Here we have examined the bifurcation of signaling at the NOPR system and propose the possible structure/function relationships of a series of commercially available and novel NOPR ligands. As part of this analysis, we designed and synthesized novel variant compounds with the selective, small-molecule, neutral antagonist J-113,397 [(±)-1-[(3R*,4R*)-1-(cyclooctylmethyl)-3-(hydroxymethyl)-4-piperidinyl]-3-ethyl-1,3-dihydro-2H-benzimidazol-2-one] as a basis. As recent GPCR studies have implicated the arrestin-recruitment conformation as a possible intermediate conformational state (Wacker et al., 2013), we first selected a zero-efficacy ligand (Chang and Bruchas, 2014), J-113,397, as our archetype structure for modification. We made several incremental modifications to the structure of J-113,397 and screened these ligands for NOPR selectivity over the δ, μ, and κ opioid receptors (Supplemental Table 1). In three NOPR-selective derivatives, we found that certain minute changes altered the putative binding pose in the orthosteric binding site of the NOPR, and were sufficient to elicit agonist activity and biased G protein signaling. Here we present a quantitative analysis of our novel NOPR-selective ligands along with commercially available small-molecule and peptide-based NOPR ligands to construct the first functional selectivity analysis of the NOPR.
Materials and Methods
3-Isobutyl-1-methylxanthine, forskolin, and coelenterazine (dissolved in 10% ethanol) were purchased from Sigma-Aldrich (St. Louis, MO). All NOPR-selective ligands: nociceptin; 1-[1-(1-methylcyclooctyl)-4-piperidinyl]-2-(3R)-3-piperidinyl-1H-benzimidazole trihydrochloride (MCOPPB trihydrochloride); 3-endo-8-[bis(2-methylphenyl)methyl]-3-phenyl-8-azabicyclo[3.2.1]octan-3-ol (SCH 221,510); 8-(1-naphthalenylmethyl)-4-oxo-1-phenyl-1,3,8-triazaspiro[4.5]decane-3-acetic acid, methyl ester (NNC 63-0532); J-113,397; N-(4-amino-2-methyl-6-quinolinyl)-2-[(4-ethylphenoxy)methyl]benzamide hydrochloride (JTC-801); and buprenorphine were purchased from Tocris Bioscience/Bio-Techne (Minneapolis, MN) and dissolved in to final 1% dimethylsulfoxide, with the exception of nociceptin (dissolved in water).
cDNA Constructs.
pGloSensor plasmid was purchased from Promega Corporation (Madison, WI). Arrestin-2-Venus and arrestin-3-Venus were generated as previously described (Vishnivetskiy et al., 2011). NOPR-Renilla luciferase 8 (Rluc8): pcDNA3 NOPR–yellow fluorescent protein (YFP) as previously described (Zhang et al., 2012) was digested with both XhoI and XbaI restriction enzymes, to remove the YFP tag. Rluc8 was polymerase chain reaction–amplified using high fidelity Taq and the following forward and reverse primers: 5′-Xho1-Rluc8 (GAC TCA CTG CTC GAG CCT GCA GGC ATG GCT T), 3′-Xba1-Rluc8 (GCT TTT AAT TAA TCT AGA GGC GCG CCG ATT ACT GC), respectively, and was ligated into digested pcDNA3 NOPR. Constructs were confirmed with DNA sequencing.
Cell Culture and Transfections.
Human embryonic kidney (HEK) 293 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM)/F-12 media supplemented with 10% fetal bovine serum containing 1× penicillin/streptomycin (Invitrogen/Life Technologies, Grand Island, NY). HEK293 cells expressing human NOPR-YFP and GloSensor plasmid were generated as previously described (Zhang et al., 2012). Stable HEK293 cell lines expressing pcDNA3 containing NOPR-YFP were generated by transfecting HEK293 cells with 5 μg of cDNA using SuperFect (Qiagen, Valencia, CA) reagent per the manufacturer’s instructions and then placing the HEK293 cells under selective pressure with G418 (800 μg/ml) for 3 weeks. Colonies of surviving cells were selected and grown in individual 100-mm cell culture plates under 400 μg/ml of selective pressure for an additional 2–3 weeks. Fluorescence-activated cell sorting was then applied to cells for equal fluorescence between mutants and wild-type NOPR to further ensure equal receptor expression in each group. Transient transfections of NOPR-Rluc8 and Arrestin-2/3-Venus were performed 18–24 hours after cell plating using Lipofectamine 2000 (Invitrogen) according to manufacturer’s protocol.
Preparation for G Protein–Mediated cAMP Measurement.
HEK293 cells were stably transfected with both Promega’s proprietary GloSensor plasmid (optimized for room-temperature-25°C experiments) and NOPR, as previously described (Zhang et al., 2012). Cells were plated at a density of ∼100,000 cells/well on a 96-well opaque white plate 24 hours before assay. Approximately 2 hours before assay, cells were incubated at 37°C in CO2-independent media supplemented with 2% Promega GloSensor reagent. Gen 5.2 software (Biotek, Winooski, VT) was used to run the following luminescence protocol: Immediately after cells are treated with 10 μl of 10× final concentration of forskolin (10 mM) and 3-isobutyl-1-methylxanthine (1 mM), the plate was inserted into the Synergy Mx plate reader(Biotek). After a 5-second shake period, total luminescence was read every 1 or 5 minutes with the kinetic read function. The forskolin response was allowed to reach peak for 10 minutes, then cells were treated with 10 μl ligand at concentrations ranging from 10 μM to 1 pM (Table 1), and kinetic reads resumed for an additional 30 minutes. A minimum of three to four independent experiments, consisting of four replicates each, were performed for each concentration from multiple passage variations of NOPR expressing pGlo cells. Data analysis: Percent maximal cAMP response was calculated by normalizing relative luminescence units, using forskolin response as null and nociceptin response as maximum (100%).
TABLE 1.
Potencies and efficacies of NOPR ligands
Ligands were screened in both cAMP inhibition and arrestin recruitment assays. Table 1 displays potencies and efficacies (logEC50 and Emax). P values were calculated by comparison with nociceptin using unpaired two-tailed t test. Data are mean ± S.E.M. (n = 3–6 experiments, triplicate samples). Dashes indicate antagonists with no determinable EC50.
| Ligand | G protein |
Arrestin3 |
Arrestin2 |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| logEC50 | P value | Emax | P value | logEC50 | P value | Emax | P value | logEC50 | P value | Emax | P value | |
| Nociceptin | −9.69 ± 0.13 | 100 ± 4 | −6.69 ± 0.35 | 100 ± 13 | −6.40 ± 0.33 | 100 ± 6 | ||||||
| MCOPPB | −10.6 ± 0.1a | 0.009 | 105 ± 2 | 0.98 | –5.80 ± 0.20a | 0.05 | 99.3 ± 9.9 | 0.98 | −5.61 ± 0.44 | 0.33 | 123.3 ± 35.9 | 0.15 |
| SCH 221,510 | –8.37 ± 0.17a | 0.004 | 103 ± 1 | 0.99 | –5.37 ± 0.43a | 0.04 | 87.4 ± 13.4 | 0.59 | −5.83 ± 0.30 | 0.40 | 151.6 ± 16.4a | 0.0008 |
| NNC 63-0532 | –7.58 ± 0.07a | 0.0001 | 74.2 ± 3.6a | 0.0004 | — | 29.1 ± 6.9a | 0.009 | — | 57.7 ± 11.2a | 0.04 | ||
| Buprenorphine | −8.64 ± 0.15a | 0.006 | 69.3 ± 3.1a | 0.0003 | — | 25.8 ± 3.8a | 0.01 | — | 26.3 ± 24.9a | <0.0001 | ||
| JTC-801 | — | –8.94 ± 4.63a | <0.0001 | — | 1.17 ± 7.5a | 0.008 | — | 17.0 ± 8.6a | <0.0001 | |||
| J-113,397 | — | –10.3 ± 3.7a | <0.0001 | — | 5.10 ± 7.16a | 0.001 | — | 18.6 ± 16.9a | 0.0001 | |||
| RTI-816 | — | –20.5 ± 7.2a | <0.0001 | — | 9.29 ± 2.81a | 0.01 | — | 8.61 ± 10.4a | <0.0001 | |||
| RTI-819 | –7.14 ± 0.17a | 0.0003 | 74.5 ± 3.9a | 0.003 | — | 14.3 ± 3.2a | 0.004 | — | 28.0 ± 15.2a | 0.0001 | ||
| RTI-856 | –8.14 ± 0.14a | 0.001 | 76.6 ± 1.6a | 0.006 | — | 24.1 ± 4.2a | 0.01 | — | 30.9 ± 2.3a | 0.001 | ||
Values that are statistically different from nociceptin are shown with corresponding P values.
Preparation for Bioluminescence Resonance Energy Transfer.
HEK293 cells were transiently transfected with 125 ng of NOPR-Renilla luciferase8 (NOPR-Rluc8) plasmids and 1 μg of Venus-Arrestin-3 (or Venus-Arrestin-2) using Lipofectamine 2000 (Invitrogen). At 24 hours after transfection cells were reseeded in supplemented media (see Cell Culture and Transfections) without phenol red (cellgro) in triplicate (35,000–50,000 cells per well) into white, opaque, clear-bottom 96-well plates (Corning, Corning, NY). At 24 hours after plating, media was replaced with DMEM without phenol red (cellgro). Protocol: Prior to all experiments, the Synergy H1 plate reader was warmed to 37°C. Gen 5.2 Software (Biotek) was used to run the following bioluminescence resonance energy transfer (BRET) protocol. YFP fluorescence was measured from the bottom of the plate via area scan. Immediately after, cells were treated with coelenterazine-h and white plate seals (Thermo Scientific, Sunnyvale, CA) applied to the plate bottom. Plates were shaken by the reader for 5 seconds before total luminescence was measured for 1 second per well 2 times. Rluc8 and YFP (using 460/40-nm and 528/20-nm filters, respectively) were measured for 1 second every minute for 2 minutes, using the kinetic read function. To account for dilution, 10 μl of 10× ligand concentration (or vehicle) was added to each well, and plates were shaken again. Kinetic luminescence reads resumed every minute for an additional 30 minutes. Three to six experiments, of three replicates each, were performed for each ligand. Data analysis: Owing to increasing overall luminescence over time, the regression from the vehicle treated cells was subtracted from experimental raw BRET values using Prism 6.0 (GraphPad Software, San Diego, CA). The average net BRET from the reads prior to treatment with ligands was subtracted from the net BRET, to yield a baseline-corrected net BRET. The baseline-corrected net BRET between different doses was used for concentration-response curves, which were then fit to a sigmoid using Prism 6.0. Ligand-induced BRET was calculated as the BRET ratio subtracted by the average BRET of the untreated baseline. Titration: To determine the optimal donor acceptor ratio, an acceptor saturation experiment was performed. HEK293 cells were transiently transfected with 125 ng of NOPR-Rluc8 plasmids and varying amounts (0–1.5 mg) of Venus-arrestin plasmids to produce the following Rluc8-to-YFP ratios: 1:0, 1:2, 1:4, 1:6, 1:8, 1:10, and 1:12. To equalize total DNA amount for each titration, pcDNA3 was added to a final amount of 2 μg of DNA. Total fluorescence to total luminescence (F/L) is a functional output of each ratio. The net BRET, defined as raw BRET minus the BRET of cells transfected with Rluc8 only, was used to construct a saturation curve (Supplemental Fig. S1). Titration curves were fit to a hyperbola using Prism 6.0 (GraphPad Software).
Calculation of Ligand Bias.
Prism 6.0 (GraphPad Software) was used to calculate transduction coefficients, (τ/KA = R), using a derivative of the Black-Leff operational model (Black and Leff, 1983; Kenakin et al., 2012; van der Westhuizen et al., 2014). It is important to note that efficacy and potency EC50 values are used for calculating transduction coefficients. Hence, it is not possible to calculate values for antagonists or ligands that do not elicit a significant response. The endogenous agonist nociceptin was used as the reference ligand, and relative bias for each ligand is ΔlogR for each pathway. Arrestin’s ΔlogR values were subtracted from G protein ΔlogR values and expressed as ΔΔlogR, demonstrating the level of signaling bias in terms of G protein.
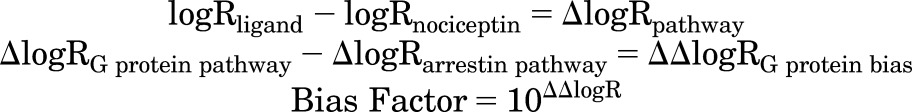 |
(1) |
Ligand Synthesis.
The synthesis of RTI-4229-816 and RTI-4229-819 are shown in Scheme 1. Reductive alkylation of 1-ethyl-3-(3-hydroxymethyl-4-piperidinyl)-1,3-dihydrobenzimidazol (1 in Scheme 1) with cycloheptanecarboxaldehyde and decalinecarboxaldehyde using silica-bound cyanoborohydride yielded RTI-4229-816 and RTI-4229-819, respectively. Reductive alkylation of 2 (in Scheme 2) with 4-isopropylcyclohexanone using sodium triacetoxyborohydride afforded RTI-4229-856.
Scheme 1.

Synthesis of RTI-816 and RTI-819. Reagents: a) Si-BH3CN (2.5 Eq), cycloheptanecarboxaldehyde (1.5 Eq), 10% HOAc/THF; b) Si-BH3CN (2.5 Eq), 1- decalinecarboxaldehyde (1.5 Eq), 10% HOAc/THF.
Scheme 2.

Synthesis of RTI-856. Reagents: a) 4-isopropylcyclohexanone, NaBH(OAc)3, THF, 25°C.
Synthesis of 1-[(3R,4R)-1-cycloheptylmethyl-3-hydroxymethyl-4-piperidinyl]-3-ethyl-1,3-dihydro-2H-benzimidazol-2-one (RTI-4229-816).
To a solution of 1-ethyl-3-(3-hydroxymethyl-4-piperidinyl)-1,3-dihydro-benzimidazol-2-one (1 in Scheme 1) (67.4 mg, 0.24 mmol) in tetrahydrofuran (THF) (2 ml) and acetic acid (0.2 ml) were added cycloheptanecarboxaldehyde (45 mg, 0.36 mmol) and silica-bound cyanoborohydride (600 mg, 0.6 mmol). The reaction mixture was stirred at room temperature for 1 day. The reaction mixture was then transferred to a 20-ml vial and 100 mg (0.35 mmol) of polymer-supported Trisamine (PS-Trisamine) was added. The vial was placed in a rotary shaker (200 rpm) for 1 day. The reaction mixture was then filtered under a pad of Celite that was rinsed with EtOAc, and the solvent was removed under reduced pressure to afford 0.13 g of product. Purification using a Redisep column (12 g) (Teledyne Isco, Lincoln, NE) eluted with 0–10% B (A = CHCl3, B = CH3OH) (0–15 minute) and 10% B (15–16 minute) afforded 86 mg (93%) of RTI-4229-816. 1H NMR (CDCl3) δ 7.37 (d, 1 H, J = 7.2 Hz), 7.14–7.03 (m, 3 H), 4.52–4.00 (m, 2 H), 3.98–3.92 (m, 2 H), 3.34– 3.33 (m, 2 H), 3.15 (d, 2 H, J = 10.8 Hz), 3.14–2.38 (m, 2 H), 2.36–2.32 (m, 1 H), 2.30 (d, 2 H, J = 6.9 Hz), 2.06–1.44 (m, 12 H), 1.34 (t, 3 H, J = 7.2 Hz), 1.30–1.10 (m, 2 H); 13C NMR (CDCl3) δ 154.67, 129.36, 121.85, 121.76, 121.43, 110.44, 106.15, 65.52, 61.74, 56.29, 53.48, 51.44, 40.35, 36.65, 36.09, 33.01, 31.52, 28.67, 28.51, 28.14, 27.17, 26.81, 26.61, 13.72; MS (APCI) 386.5 [M+H]+. Anal. Calcd. for C23H35N3O2: C, 71.65; H, 9.15; N, 10.90; Found: C, 71.65; H, 8.79; N, 11.20.
Synthesis of 1-[(3R,4R)-1-(decahydronaphthalen-1ylmethyl)-3-(hydroxymethyl)-4-piperidinyl-3-ethyl-1,3-dihydro-2H-benzimidazol-2-one (RTI-4229-819).
To a solution of 1-ethyl-3-(3-hydroxymethyl)-4-piperidinyl)-1,3-dihydro-benzimidazol-2-one (1 in Scheme 1) (46.5 mg, 0.17 mmol) in THF (1 ml) and acetic acid (0.1 ml) were added 1-decalinecarboxaldehyde (42 mg, 0.25 mmol) and silica-bound cyanoborohydride (420 mg, 0.42 mmol). The reaction mixture was stirred at room temperature for 1 day. The reaction mixture was then transferred to a 20 ml vial and 100 mg (0.35 mmol) of PS-Trisamine was added. The vial was placed in a rotary shaker (200 rpm) for 1 day. The reaction mixture was then filtered through a pad of Celite that was rinsed with EtOAc, and the solvent was removed under reduced pressure to afford 0.11 g of product. Purification using a Redisep column (12 g) eluted with 0–10% B (A = CHCl3, B = CH3OH) (0–15 minute) and 10% B (15–16 minute) afforded 43 mg (60%) of RTI-4229-819. 1H NMR (CDCl3) δ 7.35–7.04 (m, 4 H), 4.40 (bs, 1 H), 4.00–3.88 (m, 5 H), 3.60–3.35 (m, 1 H), 3.34 (s, 2 H), 3.30–3.00 (m, 3 H), 2.80–1.45 (m, 26 H), 1.32 (t, 3 H, J = 7.2 Hz), 1.29–1.22 (m, 2 H), 0.93–0.88 (m, 4 H); 13C NMR (CDCl3) δ 154.74, 129.40, 121.77, 121.38, 121.04, 110.43, 108.13, 107.59, 61.98, 56.01, 55.28, 52.31, 51.73, 46.03, 36.82, 36.28, 35.79, 35.69, 35.23, 35.09, 34.42, 31.09, 29.82, 29.71, 28.50, 27.35, 26.72, 26.63, 26.53, 21.05, 13.73; MS (APCI) 426.5 [M+H]+. Anal. Calcd. for C26H39N3O2: C, 73.37; H, 9.24; N, 9.87; Found: C, 73.07; H, 9.16 N, 9.95.
Synthesis of 1-ethyl-3-{1-[4-(1-methylethyl)cyclohexyl]piperidin-4-yl}-1,3-dihydro-2H-benzimidazol-2-one hydrochloride (RTI-4229-856).
4-Isopropylcyclohexanone, NaBH(OAc)3 and HOAc were added sequentially to a solution of 1-(piperidin-4-yl)-3-ethyl-1,3-dihydro-2-one (2 in Scheme 2) in THF at 25°C and the mixture was stirred at 25°C overnight, diluted with EtOAc (10 ml), and quenched with aqueous NaOH (1N, 10 ml). The layers were separated and the aqueous layer was extracted with EtOAc (2 × 10 ml). The combined organic extracts were washed with brine, dried (Na2SO4), and concentrated under reduced pressure. The residue was purified on a silica gel column using pressure column chromatography (CH2Cl2→CMA80) to provide 62% of RTI-4229-856 freebase. 1H NMR (CDCl3) δ 7.39-7.27 (m, 1H), 7.07-6.98 (m, 3H), 4.41-4.36 (m, 1H), 3.94 (q, 2H, J = 7.2 Hz), 3.20-3.05 (m, 2H), 2.60-2.25 (m, 5H), 2.00-1.83 (m, 1H), 1.81-1.58 (m, 7H), 1.44-1.41 (m, 3H), 1.33 (t, 3H, J = 7.2 Hz), 1.20-1.10 (m, 1H), 0.90 (d, 6H, J = 6.9 Hz); 13C NMR (CDCl3) δ 153.9, 129.6, 121.0, 110.4, 110.2, 107.8, 64.6, 51.7, 49.7, 49.2, 44.2, 42.0, 36.2, 33.0, 29.8,, 26.0, 21.1, 20.3, 14.0. RTI-4229-856 was obtained as a pale yellow solid by treating the free base with HCl (1 M in ether) and evaporating the solvent. mp 184–186°C; Anal. Calcd. for C23H36ClN3O•0.5H2O: C, 66.56; H, 8.99; N, 10.12; Found: C, 66.86; H, 9.06; N, 9.71.
Results
Real-Time cAMP Signaling Analysis Reveals Distinct G Protein Pathway Pharmacology of NOPR Ligands.
We employed the previously described GloSensor assay (Zhang et al., 2012; Tsvetanova and von Zastrow, 2014) to quantify real-time downstream G protein signaling of a collection of well known, highly selective NOPR ligands (Fig. 1A; Table 1) by measuring the Gαi protein-induced inhibition of cAMP accumulation. Our impetus was to select ligands that have been previously shown to be highly NOPR-selective over other opioid and GPCR receptor subtypes, and that comprise a diverse array of chemical structures in order to provide better insights into structure-activity relationships and relative signaling profiles resulting from diverse functional groups. Our results show clearly distinguishable rank orders of potency (Fig. 1B) and statistically significant differences in the efficacies of partial agonists and antagonists (Fig. 1C; Table 1).
Fig. 1.

NOPR cAMP signaling profiles showing rank order potencies and efficacies. (A) Structures of NOPR-selective ligands used in this study. (B) Concentration-response curves of NOPR agonists displaying rank order of potency. (C) Maximum efficacy of NOPR ligands showing full agonists (nociceptin, MCOPPB, SCH 221,510); partial agonists (†NNC 63-0532 and buprenorphine); and antagonist, JTC-801. All points are mean ± S.E.M. (n = 3–6, triplicate samples). n.s., not significant.
The small molecule MCOPPB was the most potent agonist tested, approximately 10-fold more potent than nociceptin and 100-fold more potent than SCH 221,510. MCOPPB also shows full agonist efficacy, comparable to the endogenous reference ligand nociceptin, and full agonist SCH 221,510 (Hirao et al., 2008; Varty et al., 2008). Additionally, buprenorphine and NNC 63-0532 exhibit partial agonist efficacy (67.01 ± 2.99% and 71.78 ± 3.45%, respectively) and relatively lower potency, in congruence with previous studies, with NNC 63-0532 being the least potent NOPR-selective agonist tested (Table 1). These results establish relative pharmacological properties of G protein–mediated signaling for NOPR-selective ligands and allows for determination of rank orders of potency and efficacy in comparison with the reference endogenous ligand, nociceptin (Table 1).
Synthesized Derivatives Demonstrate Antagonist/Agonist Structure Activity Relationships.
Using the small-molecule NOPR-selective neutral antagonist J-113,397 (Fig. 2A) as the archetype ligand structure, we made minute modifications to the “message” moiety of the ligand (Zaveri et al., 2013) and tested three derivatives that showed high NOPR-selectivity (Supplemental Table 1) to explore the structure-activity relationships. In RTI-816, the N-cyclooctylmethyl is replaced with a N-cycloheptylmethyl, and the ligand remained an antagonist in our screen, with weak inverse agonist activity in this NOPR expression system. However, when the cyclo-octylmethyl is replaced with a decahydronaphthalen-1-methyl as in RTI-819, the signaling dramatically shifted from an unbiased antagonist to partial G protein agonist (Fig. 2C). These data suggest the replacement of the N-cyclo-octylmethyl with an isopropylcyclohexane in RTI-856 does not change the partial agonist efficacy of the ligand; however, it should be noted that the loss of the alcohol from the piperidine could be responsible for the increase in potency seen in RTI-856 over RTI-819 (Fig. 2B).
Fig. 2.

cAMP Inhibition profiles of novel NOPR ligands. (A) Structures of J-113,397 and novel NOPR ligands synthesized in this study. Compound J-113,397 is a potent and selective antagonist for NOPR. RTI-816 and RTI-819 are analogs of J-113,397, where the N-cyclo-octylmethyl on the piperidine ring of J-113,397 has been replaced by N-cycloheptylmethyl and decahydronaphthalen-1-methyl groups, respectively. Compounds RTI-856 and MCOPPB are also similar in structure to J-113,397, but differ by more than just changes in the N-substituent on the piperidine ring. (B) Concentration-response curves of novel synthesized NOPR-selective agonists. Both agonists RTI-819 and RTI-856 exhibit similar efficacies but have an ∼10-fold difference in potency. (C) Compound RTI-816 shows antagonist profile similar to its archetype J-113,397, and neither are statistically different from vehicle control. Both agonists RTI-819 and RTI-856 show partial agonist activity. (†versus nociceptin, P ≤ 0.05, one-way analysis of variance, Dunnett’s post-hoc). All points are mean ± S.E.M. (n = 3–6, triplicate samples). n.s., not significant.
Ligand-Induced Arrestin Recruitment Using BRET Reveals Signaling Bias and Kinetic Differences.
To directly quantify the magnitude of ligand-induced arrestin interaction with the NOP receptor, we employed a real-time BRET assay system to measure the direct interaction between NOPR and both arrestin-2 and arrestin-3 (Bertrand et al., 2002). We used transient transfections consisting of the human NOP receptor with the Renilla reniformis luciferase (Rluc) energy-transfer donor fused in frame to the C-terminal domain, together with arrestin-2-Venus or arrestin-3-Venus energy-transfer acceptor proteins (Gimenez et al., 2012). Coupling of NOPR to arrestin-3 was detected as an increase in the BRET signal, and indicative of arrestin-mediated signaling initiation (Figs. 3A, 4A, and 4C). Receptor and arrestin expression stoichiometry was carefully optimized to ensure ideal conditions (titers) of the receptor and both arrestin-2 and arrestin-3 to yield high dynamic range of signal-to-noise (Supplemental Fig. S1). These BRET data yielded concentration-response curves and EC50s for each NOPR agonist, as well as maximum efficacy in arrestin-3 versus arrestin-2 recruitment. In general, most NOPR agonists showed higher efficacy in arrestin-3 recruitment over arrestin-2 (Fig. 4), consistent with our prior analysis of NOPR regulation pathways (Zhang et al., 2012). Importantly, our system also allows for temporal resolution of recruitment and strikingly showed that nociceptin and MCOPPB induced maximum arrestin association with the receptor within 2–3 minutes (Fig. 5, A and C). However, the agonist SCH 221,510 showed a significant prolonged response in time-to-peak for arrestin-3 and -2 recruitment in comparison with the other agonists, including the endogenous ligand nociceptin (Fig. 5, B and D). Interestingly, SCH 221,510 also uniquely recruited arrestin-3 and -2 at equal potency and efficacy (Fig. 4, B and D; Table 1). This is not consistent with the endogenous ligand nociceptin (Fig. 4, A–D), and consequently SCH 221,510 shows an arrestin-2 bias relative to nociceptin (Table 2).
Fig. 3.

Quantification of ligand-induced arrestin recruitment at NOPR. (A) BRET constructs were used to assess recruitment of arrestins to NOPR upon activation. Rluc8 luminescence (460 nm) is capable of exciting the Venus-tag on arrestin-2/3 resulting in a distinct emission wavelength (528 nm). Ligand-induced BRET between NOPR-Rluc8 and indicates arrestin recruitment to the receptor. Differences in recruitment level can be measured quantitatively by increase in fluorescence over luminescence. (B) Normalized concentration response curves for arrestin-3 recruitment to NOPR show rank order of potency for tested agonists. All points are mean ± S.E.M., n = 3–6, triplicate samples.
Fig. 4.

Arrestin recruitment profiles of NOPR ligands. (A) Despite the differences in efficacy, normalized CRCs of nociceptin-induced arrestin recruitment virtually overlap, showing similar potencies. (B and D) Maximum NOPR ligand-induced arrestin-3 and -2 recruitment shown as net BRET. MCOPPB and SCH 221,510 show full agonist activity and are not significantly different from nociceptin, while all other ligands tested show no significant difference from vehicle controls but are statistically different from nociceptin (†). Ligand-induced arrestin-2 recruitment is similar to arrestin-3, except SCH 221,510, which is significantly different from both nociceptin and vehicle control. (C) Concentration-response curves showing different maximum efficacies, but similar potencies in nociceptin-induced arrestin-2/3 recruitment. All points are mean ± S.E.M. (***P ≤ 0.001, one-way analysis of variance, Tukey’s least significant difference post-hoc test, n = 3–6, triplicate samples). n.s., not significant.
Fig. 5.

Ligand-induced arrestin recruitment kinetics. (A and C) Quantified time-to-peak for arrestin recruitment show that SCH 221,510 recruits both arrestins significantly more slowly than either nociceptin or MCOPPB. (B and D) Representative time traces showing temporal recruitment of arrestin by each ligand. Nociceptin and MCOPPB share similar time-to-peak recruitment, whereas SCH 221,510 exhibits a more shallow slope. All points are mean ± S.E.M. (***P ≤ 0.001, ****P ≤ 0.0001, one-way analysis of variance, Tukey’s least significant difference post-hoc test, n = 3–6, triplicate samples).
TABLE 2.
Calculated transduction coefficients of NOPR ligands
Prism 6.0 was used to calculate transduction coefficients, (τ/KA = R), using a derivative of the Black-Leff operational model (Kenakin et al., 2012; van der Westhuizen et al., 2014). The endogenous agonist nociceptin was used as the reference ligand, and relative bias for each ligand is ΔlogR for each pathway. Arrestin ΔlogRs were subtracted from G protein ΔlogRs, and arrestin-2 from arrestin-3 ΔlogRs, then expressed as ΔΔlogR, demonstrating the level of signaling bias in terms of G protein or arrestin-3. P values were calculated by comparison with nociceptin using an unpaired two-tailed t test. Bias factor = 10ΔΔlogR. Data are mean ± S.E.M. (n = 3–6 experiments, triplicate samples). Dashes denote antagonists for which signal cannot be calculated.
| G-protein |
Arrestin3 |
Arrestin2 |
G versus Arr3 |
G versus Arr2 |
Arr3 versus Arr2 |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ligand | logR | ΔlogR | P value | logR | logR | P value | logR | ΔlogR | P value | ΔΔlogR | P value | Bias Factor | ΔΔlogR | P value | Bias Factor | ΔΔlogR | P value | Bias Factor |
| Nociceptin | 9.55 ± 0.10 | 0.00 ± 0.14 | — | 6.78 ± 0.19 | 0.00 ± 0.23 | — | 6.35 ± 0.24 | 0.00 ± 0.34 | — | 0.00 ± 0.27 | — | 1.00 | 0.00 ± 0.36 | 1.00 | 0.00 ± 0.41 | 1.00 | ||
| MCOPPB | 10.6 ± 0.1 | 1.01 ± 0.14a | 0.007 | 6.24 ± 0.19 | −0.54 ± 0.22 | 0.17 | 5.84 ± 0.21 | −0.52 ± 0.32 | 0.33 | 1.55 ± 0.27a | 0.02 | 35.5 | 1.52 ± 0.35a | 0.04 | 33.1 | 0.02 ± 0.39 | 0.96 | 1.05 |
| SCH 221,510 | 7.68 ± 0.08 | −1.88 ± 0.13a | 0.0007 | 5.55 ± 0.24 | −1.23 ± 0.27a | 0.03 | 5.94 ± 0.17 | −0.42 ± 0.30 | 0.41 | −0.65 ± 0.30 | 0.18 | 0.22 | −1.47 ± 0.32a | 0.04 | 0.03 | −0.81 ± 0.40 | 0.23 | 0.15 |
| NNC 63-0532 | 8.57 ± 0.10 | −1.00 ± 0.14a | 0.007 | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — |
| Buprenorphine | 8.35 ± 0.10 | −1.22 ± 0.14a | 0.004 | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — |
| RTI-819 | 7.20 ± 0.10 | −2.36 ± 0.14a | 0.0003 | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — |
| RTI-856 | 8.11 ± 0.10 | −1.45 ± 0.14a | 0.002 | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — |
Values that are statistically different from nociceptin are shown with corresponding P values.
In these experiments, our ligand screen displayed a similar rank order of potency (Fig. 3B) consistent with an ∼1000-fold decrease in arrestin EC50 compared with the EC50 for cAMP inhibition for all agonists of both pathways (Table 1) in this study, but not exclusive to this ligand portfolio (unpublished data). Interestingly, the small-molecule MCOPPB, which exhibited the highest potency in the G protein–signaling assay, was markedly less potent in arrestin recruitment and uniquely showed an additional ∼100-fold decrease in potency for arrestin coupling compared with G protein activation (Fig. 6, A and B). MCOPPB shows a concentration-dependent or potency bias, as the ligand is a full agonist in both signaling pathways but distinguishes itself in its potency at G protein versus arrestin signaling. Additionally, the maximum efficacy data demonstrate that the small-molecule NNC 63-0532 is not sufficiently efficacious in promoting arrestin recruitment (Fig. 4, B and D) but acts as a partial agonist in the G protein–signaling pathway (Fig. 1C). Potencies and efficacies for all ligands were determined where applicable, since a concentration response cannot be determined for neutral antagonists (Table 1). Furthermore, while both J-113,397 derivatives are shown to be partial G protein agonists, RTI-819 and RTI-856 very weakly induced arrestin-2 and -3 recruitment (Fig. 4, B and D). These data suggest that the NOP receptor is capable of functional selectivity and can do so through multiple modalities of G protein signaling or arrestin-3/2 engagement.
Fig. 6.

Potency shift and calculated transduction coefficients. (A) Comparison of pathway potency shifts (ΔlogEC50) shows a unique ∼100-fold shift (lower) in potency for agonist MCOPPB that is not conserved for nociceptin or SCH 221,510. (B) Bias plot showing signaling bias in terms of G protein signaling for NOPR ligands. After combining individual pathway biases (Supplemental Fig. S3), MCOPPB consequently shows a significant G protein bias, while SCH 221,510 resolves no overall pathway bias. All points are mean ± S.E.M. (*P ≤ 0.05, compared with reference ligand).
Determination of Ligand-Receptor Transduction Coefficients.
Calculating signal transduction coefficients for ligands tested against a reference ligand can allow one to assume transduction properties of said ligands in unstudied systems (Rajagopal et al., 2010; Zhou et al., 2013). Here we employed the widely used Black-Leff operational model to directly quantify a ligand’s relative agonism, or transduction coefficient (τ/KA or R) (Black and Leff, 1983; Kenakin et al., 2012; van der Westhuizen et al., 2014). We selected the endogenous ligand nociceptin as the reference ligand, a natural choice that is further justified by its full agonist activity in both signaling outputs. We calculated transduction coefficients in both signaling outputs, and determined the level of agonism at each pathway relative to the reference ligand as Δlog(τ/KA), expressing the signaling capacity of a ligand in a given pathway, relative to the reference ligand. We then generated the comparative signaling pathway bias factor for each ligand, expressed as ΔΔlog(τ/KA). We expressed the bias factor calculations in terms of G protein signaling, as the only observed biased ligands were G protein biased in this initial screen of available and new NOPR ligands (Fig. 6B; Table 2). Furthermore, differences in transduction seen in other ligands relative to the reference ligand should be system-independent and as such are predicted to hold their relative differences or similarities across model systems (Kenakin and Christopoulos, 2013; van der Westhuizen et al., 2014).
As expected, the biased ligands that we previously observed to exhibit G protein bias in cAMP-inhibition and arrestin-recruitment assays show distinct G protein biases upon application of the operational model, with MCOPPB being the most effective in G protein signal transduction, and SCH 221,510 following in rank order. All other G protein partial agonists, including the novel partial agonists RTI-819 and RTI-856, show bias toward G protein signaling owing to their very weak recruitment of arrestins (Fig. 4, B and D). Of note, buprenorphine also shows a G protein bias, although its nonselectivity for MOPR and KOPR must be noted for its application in other systems. Ligands that showed agonism of both pathways were analyzed for ΔΔlogR to show their G protein bias compared with nociceptin (Fig. 6B; Table 2). These analyses confirm the observed pharmacology that showed G protein bias in NOPR ligands but provide a universally applicable, quantitative description of functionally selective commercially available ligands at NOPR.
Discussion
In this work, we have constructed an in-depth analysis of variable ligand-directed signaling at the NOPR. This work is the first to identify biased signaling at the NOPR, and in two types of functional selectivity: potency bias and complete bias. These findings suggest new studies involving NOPR, whereby investigators can further design functionally selective compounds for NOPR and other opioid-related GPCRs.
We observed ligands that quantitatively exhibit ligand bias in more than one distinct form. Although this distinction is clear in terms of experimental data, how this may arise in terms of theoretical receptor conformation is more speculative. These differences may be rooted in degrees of stabilization of the arrestin-recruiting receptor conformation. Alternatively, the observed distinctions may not in fact be different phenomena but could be explained by the potency of each ligand in the G protein–signaling arm, obfuscating a shared characteristic. Nevertheless, they remain different in terms of functional applications, since the low potency ligand makes arrestin recruitment occur at unrealistic and physiologically irrelevant concentrations. Importantly, this difference is independent of the aforementioned interassay potency difference of ∼1000-fold seen even in ligands not included in this study (unpublished data). This universal difference in interassay potency may be attributable to the amplification of signaling output or sensitivity of each assay. Whereas the BRET-based arrestin-recruitment assay is essentially a direct measure of recruitment, the G protein–output assay is a measure of an amplified tertiary downstream messenger, resulting in signal amplification. However, in our system this seems less probable, because of the close agreement of observed G protein–signaling potencies with previously published binding affinities and GTPγS data (Supplemental Table 2, includes references). A more likely scenario for this discrepancy in potency is the converse, in that the BRET-based arrestin-recruitment assay is less sensitive in detecting signaling by comparison. We chose the BRET assay over other methods specifically because it allows for quantitative direct measurement of recruitment to the receptor without amplification; however, it is still limited by signal-to-noise ratio and detection limits, which require optimization (Supplemental Fig. S1). Furthermore, this discrepancy in potencies may be compounded by the lack of arrestin recruitment during low levels of receptor activation, since arrestin recruitment is known to be the main mechanism of receptor desensitization and in some cases may not be recruited at detectable levels until a threshold level of G protein receptor activation/saturation and G protein–coupled receptor kinase phosphorylation is reached. This seems to be the case for NNC 63-0532, RTI-819, and RTI-856, as their partial agonist activity mirrors their inability to robustly recruit arrestin.
In this collection of well known NOPR-selective ligands, and novel antagonist derivatives, we were unable to find any ligands that displayed a dramatic signaling bias for arrestin recruitment, with the exception of SCH 221,510 which shows modest arrestin-2 bias relative to the other compounds in our assays (Fig. 6B; Supplemental Fig. S3; Table 2). All other tested agonists, unbiased or biased, transduced their signals through the G protein–signaling pathway. The lack of arrestin-biased ligand discovery seems to agree with the large proportion of published biased GPCR ligands that is predominately G protein biased, and further suggest that arrestin recruitment to the receptor may not be truly independent of G protein activation but may be consequential in the context of the receptor–G protein activation, and conformationally additive to the activated receptor/G protein complex. This could explain the observed G protein agonism as the first stage of agonism and resolve the G protein–biased ligands as having less propensity for driving the conformation toward full or unbiased agonism to varying degrees. However, future studies with discrete point mutations in NOPR and inverse agonist ligands designed from this working model are needed to further examine these possibilities.
The importance of the results obtained in this study include not only the novelty of identifying biased signaling in the NOP receptor system but also the utility of the data that transcend system limitations and should apply to other model systems for NOPR. Using relative, quantitative comparisons of ligands to a reference ligand should eliminate “system bias” as well as “observational bias” and yield a similar relative signal transduction in another model system of interest, such as behavioral or clinical studies.
The relative signaling profiles extrapolated from our ligand collection containing distinct potencies, efficacies, and types of agonism inform structure-based strategies of functional NOPR modulation. In addition to identifying biased signaling at NOPR, we also identify several commercially available biased ligands that are highly selective for NOPR. Further, in this work we elucidate some previously unknown ligand structure-activity relationships through the synthesis of novel NOPR ligands. Still, additional work is needed to fully characterize NOPR functional selectivity, including characterizing additional signaling pathways and their implications in behavior, and understanding the variable receptor interactions with biased ligands, but we hope these findings are informative for future pharmacological investigations of opioid GPCRs.
Supplementary Material
Acknowledgments
The authors thank Emilie Smith, Cong Liu, and Jim Thomas from RTI International for synthesizing novel RTI compounds; William Planer for BRET cDNA constructs; and Nancy Zhang, Gina Migneco, and Robert Gereau IV for helpful discussion (Washington University). The authors thank the additional members of the Carroll and Bruchas laboratories for helpful support.
Abbreviations
- BRET
bioluminescence resonance energy transfer
- GPCR
G protein–coupled receptor
- J-113,397
(±)-1-[(3R*,4R*)-1-(cyclooctylmethyl)-3-(hydroxymethyl)-4-piperidinyl]-3-ethyl-1,3-dihydro-2H-benzimidazol-2-one
- JTC-801
N-(4-amino-2-methyl-6-quinolinyl)-2-[(4-ethylphenoxy)methyl]benzamide hydrochloride
- MCOPPB
1-[1-(1-methylcyclooctyl)-4-piperidinyl]-2-(3R)-3-piperidinyl-1H-benzimidazole trihydrochloride
- NNC 63-0532
8-(1-naphthalenylmethyl)-4-oxo-1-phenyl-1,3,8-triazaspiro[4.5]decane-3-acetic acid, methyl ester
- NOPR
nociceptin opioid receptor
- SCH 221,510
3-endo-8-[bis(2-methylphenyl)methyl]-3-phenyl-8-azabicyclo[3.2.1]octan-3-ol
- Rluc
Renilla luciferase
- THF
tetrahydrofuran
- UFP-101
[Nphe1,Arg14,Lys15]nociceptin-NH2
- YFP
yellow fluorescent protein
Authorship Contributions
Participated in research design: Chang, Mascarella, Carroll, Bruchas.
Conducted experiments: Chang, Mascarella, Spangler, Navarro.
Contributed new reagents or analytic tools: Chang, Mascarella, Spangler, Navarro, Gurevich, Carroll, Bruchas.
Performed data analysis: Chang, Mascarella, Spangler, Bruchas.
Wrote or contributed to the writing of the manuscript: Chang, Mascarella, Spangler, Carroll, Bruchas.
Footnotes
This work was supported by the National Institutes of Health National Institute on Drug Abuse [R21-034929 and T32-DA007261].
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.
References
- Bertrand L, Parent S, Caron M, Legault M, Joly E, Angers S, Bouvier M, Brown M, Houle B, Ménard L. (2002) The BRET2/arrestin assay in stable recombinant cells: a platform to screen for compounds that interact with G protein-coupled receptors (GPCRS). J Recept Signal Transduct Res 22:533–541. [DOI] [PubMed] [Google Scholar]
- Black JW, Leff P. (1983) Operational models of pharmacological agonism. Proc R Soc Lond B Biol Sci 220:141–162. [DOI] [PubMed] [Google Scholar]
- Bock A, Chirinda B, Krebs F, Messerer R, Bätz J, Muth M, Dallanoce C, Klingenthal D, Tränkle C, Hoffmann C, et al. (2014) Dynamic ligand binding dictates partial agonism at a G protein-coupled receptor. Nat Chem Biol 10:18–20. [DOI] [PubMed] [Google Scholar]
- Chang SD, Bruchas MR. (2014) Functional selectivity at GPCRs: new opportunities in psychiatric drug discovery. Neuropsychopharmacology 39:248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung KY, Rasmussen SGF, Liu T, Li S, DeVree BT, Chae PS, Calinski D, Kobilka BK, Woods VL, Jr, Sunahara RK. (2011) Conformational changes in the G protein Gs induced by the β2 adrenergic receptor. Nature 477:611–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbani M, Gonindard C, Meunier J-C. (2004) Ligand-regulated internalization of the opioid receptor-like 1: a confocal study. Endocrinology 145:2876–2885. [DOI] [PubMed] [Google Scholar]
- Cremeans CM, Gruley E, Kyle DJ, Ko M-C. (2012) Roles of μ-opioid receptors and nociceptin/orphanin FQ peptide receptors in buprenorphine-induced physiological responses in primates. J Pharmacol Exp Ther 343:72–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gear RW, Bogen O, Ferrari LF, Green PG, Levine JD. (2014) NOP receptor mediates anti-analgesia induced by agonist-antagonist opioids. Neuroscience 257:139–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimenez LE, Kook S, Vishnivetskiy SA, Ahmed MR, Gurevich EV, Gurevich VV. (2012) Role of receptor-attached phosphates in binding of visual and non-visual arrestins to G protein-coupled receptors. J Biol Chem 287:9028–9040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goeldner C, Reiss D, Wichmann J, Meziane H, Kieffer BL, Ouagazzal A-M. (2008) Nociceptin receptor impairs recognition memory via interaction with NMDA receptor-dependent mitogen-activated protein kinase/extracellular signal-regulated kinase signaling in the hippocampus. J Neurosci 28:2190–2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi S, Hirao A, Imai A, Nakamura H, Murata Y, Ohashi K, Nakata E. (2009) Novel non-peptide nociceptin/orphanin FQ receptor agonist, 1-[1-(1-Methylcyclooctyl)-4-piperidinyl]-2-[(3R)-3-piperidinyl]-1H-benzimidazole: design, synthesis, and structure-activity relationship of oral receptor occupancy in the brain for orally potent antianxiety drug. J Med Chem 52:610–625. [DOI] [PubMed] [Google Scholar]
- Hirao A, Imai A, Sugie Y, Yamada Y, Hayashi S, Toide K. (2008) Pharmacological characterization of the newly synthesized nociceptin/orphanin FQ-receptor agonist 1-[1-(1-methylcyclooctyl)-4-piperidinyl]-2-[(3R)-3-piperidinyl]-1H-benzimidazole as an anxiolytic agent. J Pharmacol Sci 106:361–368. [DOI] [PubMed] [Google Scholar]
- Kahsai AW, Xiao K, Rajagopal S, Ahn S, Shukla AK, Sun J, Oas TG, Lefkowitz RJ. (2011) Multiple ligand-specific conformations of the β2-adrenergic receptor. Nat Chem Biol 7:692–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapusta DR, Burmeister MA, Calo’ G, Guerrini R, Gottlieb HB, Kenigs VA. (2005) Functional selectivity of nociceptin/orphanin FQ peptide receptor partial agonists on cardiovascular and renal function. J Pharmacol Exp Ther 314:643–651. [DOI] [PubMed] [Google Scholar]
- Kenakin T, Christopoulos A. (2013) Signalling bias in new drug discovery: detection, quantification and therapeutic impact. Nat Rev Drug Discov 12:205–216. [DOI] [PubMed] [Google Scholar]
- Kenakin T, Watson C, Muniz-Medina V, Christopoulos A, Novick S. (2012) A simple method for quantifying functional selectivity and agonist bias. ACS Chem Neurosci 3:193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane JR, Donthamsetti P, Shonberg J, Draper-Joyce CJ, Dentry S, Michino M, Shi L, López L, Scammells PJ, Capuano B, et al. (2014) A new mechanism of allostery in a G protein-coupled receptor dimer. Nat Chem Biol 10:745–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquez P, Nguyen AT, Hamid A, Lutfy K. (2008) The endogenous OFQ/N/ORL-1 receptor system regulates the rewarding effects of acute cocaine. Neuropharmacology 54:564–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogil JS, Pasternak GW. (2001) The molecular and behavioral pharmacology of the orphanin FQ/nociceptin peptide and receptor family. Pharmacol Rev 53:381–415. [PubMed] [Google Scholar]
- Mollereau C, Parmentier M, Mailleux P, Butour JL, Moisand C, Chalon P, Caput D, Vassart G, Meunier JC. (1994) ORL1, a novel member of the opioid receptor family. Cloning, functional expression and localization. FEBS Lett 341:33–38. [DOI] [PubMed] [Google Scholar]
- Motta-Mena LB, Reade A, Mallory MJ, Glantz S, Weiner OD, Lynch KW, Gardner KH. (2014) An optogenetic gene expression system with rapid activation and deactivation kinetics. Nat Chem Biol 10:196–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy NP, Lee Y, Maidment NT. (1999) Orphanin FQ/nociceptin blocks acquisition of morphine place preference. Brain Res 832:168–170. [DOI] [PubMed] [Google Scholar]
- Rajagopal S, Rajagopal K, Lefkowitz RJ. (2010) Teaching old receptors new tricks: biasing seven-transmembrane receptors. Nat Rev Drug Discov 9:373–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopal S, Bassoni DL, Campbell JJ, Gerard NP, Gerard C, Wehrman TS. (2013) Biased agonism as a mechanism for differential signaling by chemokine receptors. J Biol Chem 288:35039–35048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SGF, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, et al. (2011) Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature 477:549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinscheid RK, Nothacker HP, Bourson A, Ardati A, Henningsen RA, Bunzow JR, Grandy DK, Langen H, Monsma FJ, Jr, Civelli O. (1995) Orphanin FQ: a neuropeptide that activates an opioidlike G protein-coupled receptor. Science 270:792–794. [DOI] [PubMed] [Google Scholar]
- Reiss D, Wichmann J, Tekeshima H, Kieffer BL, Ouagazzal A-M. (2008) Effects of nociceptin/orphanin FQ receptor (NOP) agonist, Ro64-6198, on reactivity to acute pain in mice: comparison to morphine. Eur J Pharmacol 579:141–148. [DOI] [PubMed] [Google Scholar]
- Spampinato S, Baiula M, Calienni M. (2007) Agonist-regulated internalization and desensitization of the human nociceptin receptor expressed in CHO cells. Curr Drug Targets 8:137–146. [DOI] [PubMed] [Google Scholar]
- Thompson AA, Liu W, Chun E, Katritch V, Wu H, Vardy E, Huang X-P, Trapella C, Guerrini R, Calo G, et al. (2012) Structure of the nociceptin/orphanin FQ receptor in complex with a peptide mimetic. Nature 485:395–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsvetanova NG, von Zastrow M. (2014) Spatial encoding of cyclic AMP signaling specificity by GPCR endocytosis. Nat Chem Biol 10:1061–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Westhuizen ET, Breton B, Christopoulos A, Bouvier M. (2014) Quantification of ligand bias for clinically relevant β2-adrenergic receptor ligands: implications for drug taxonomy. Mol Pharmacol 85:492–509. [DOI] [PubMed] [Google Scholar]
- Varty GB, Lu SX, Morgan CA, Cohen-Williams ME, Hodgson RA, Smith-Torhan A, Zhang H, Fawzi AB, Graziano MP, Ho GD, et al. (2008) The anxiolytic-like effects of the novel, orally active nociceptin opioid receptor agonist 8-[bis(2-methylphenyl)methyl]-3-phenyl-8-azabicyclo[3.2.1]octan-3-ol (SCH 221510). J Pharmacol Exp Ther 326:672–682. [DOI] [PubMed] [Google Scholar]
- Vishnivetskiy SA, Gimenez LE, Francis DJ, Hanson SM, Hubbell WL, Klug CS, Gurevich VV. (2011) Few residues within an extensive binding interface drive receptor interaction and determine the specificity of arrestin proteins. J Biol Chem 286:24288–24299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker D, Wang C, Katritch V, Han GW, Huang X-P, Vardy E, McCorvy JD, Jiang Y, Chu M, Siu FY, et al. (2013) Structural features for functional selectivity at serotonin receptors. Science 340:615–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada H, Nakamoto H, Suzuki Y, Ito T, Aisaka K. (2002) Pharmacological profiles of a novel opioid receptor-like1 (ORL(1)) receptor antagonist, JTC-801. Br J Pharmacol 135:323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaveri NT, Jiang F, Olsen C, Polgar WE, Toll L. (2013) Designing bifunctional NOP receptor-mu opioid receptor ligands from NOP receptor-selective scaffolds. Part I. Bioorg Med Chem Lett 23:3308–3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang NR, Planer W, Siuda ER, Zhao H-C, Stickler L, Chang SD, Baird MA, Cao Y-Q, Bruchas MR. (2012) Serine 363 is required for nociceptin/orphanin FQ opioid receptor (NOPR) desensitization, internalization, and arrestin signaling. J Biol Chem 287:42019–42030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Simpson-Durand CD, Standifer KM. (2014) Nociceptin/orphanin FQ peptide receptor antagonist JTC-801 reverses pain and anxiety symptoms in a rat model of post-traumatic stress disorder. Br J Pharmacol 172:571–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Lovell KM, Frankowski KJ, Slauson SR, Phillips AM, Streicher JM, Stahl E, Schmid CL, Hodder P, Madoux F, et al. (2013) Development of functionally selective, small molecule agonists at kappa opioid receptors. J Biol Chem 288:36703–36716. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.