

Abstract
The epidermal growth factor (EGF)-like repeat is a common, evolutionarily conserved motif found in secreted proteins and the extracellular domain of transmembrane proteins. EGF repeats harbor six cysteine residues which form three disulfide bonds and help generate the three-dimensional structure of the EGF repeat. A subset of EGF repeats harbor consensus sequences for the addition of one or more specific O-glycans, which are initiated by O-glucose, O-fucose or O-N-acetylglucosamine. These glycans are relatively rare compared to mucin-type O-glycans. However, genetic experiments in model organisms and cell-based assays indicate that at least some of the glycosyltransferases involved in the addition of O-glycans to EGF repeats play important roles in animal development. These studies, combined with state-of-the-art biochemical and structural biology experiments have started to provide an in-depth picture of how these glycans regulate the function of the proteins to which they are linked. In this review, we will discuss the biological roles assigned to EGF repeat O-glycans and the corresponding glycosyltransferases. Since Notch receptors are the best studied proteins with biologically-relevant O-glycans on EGF repeats, a significant part of this review is devoted to the role of these glycans in the regulation of the Notch signaling pathway. We also discuss recently identified proteins other than Notch which depend on EGF repeat glycans to function properly. Several glycosyltransferases involved in the addition or elongation of O-glycans on EGF repeats are mutated in human diseases. Therefore, mechanistic understanding of the functional roles of these carbohydrate modifications is of interest from both basic science and translational perspectives.
Keywords: developmental biology, EGF repeat, Notch signaling, O-glycan, protein folding
Introduction
Glycosylation is defined as the covalent attachment of a sugar to lipids or proteins, added post- or co-translationally. This type of modification is the most common posttranslational modification of proteins and plays many roles in protein structure and function. Glycans play broad and important roles in animal development, as evidenced by various studies in model organisms and the growing list of human developmental disorders caused by mutations in components of the glycosylation machinery (Freeze et al. 2015). The two major types of glycans are N- and O-linked glycans; an N-linked glycan is attached to an asparagine residue, whereas an O-linked glycan is attached to a serine or threonine residue. Some forms of O-linked sugars are only added onto a specific sequence of amino acids; others have no defined consensus sequence yet, such as O-mannosylation and O-GalNAcylation (Breloy et al. 2008; Bennett et al. 2012). Some types of O-linked glycans are specifically found on epidermal growth factor (EGF)-like repeats (Harris and Spellman 1993; Matsuura et al. 2008). Although these glycans are relatively rare, our knowledge of their function has grown significantly within the last several years. This review will discuss the biological roles of O-linked glycans found on EGF repeats and their corresponding glycosyltransferases (Figure 1).
Fig. 1.
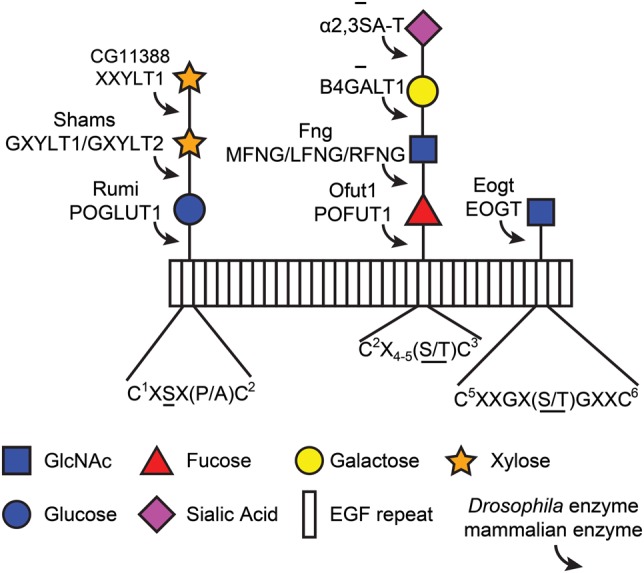
O-Linked glycans found on EGF repeats in Drosophila and mammals. Rectangles represent EGF repeats. Each modification is shown with the corresponding enzyme that adds the sugar. CG11388 is the only protein encoded by the Drosophila genome which shows a high level of homology to mammalian XXYLT1. However, its enzymatic activity remains to be verified. Addition of galactose and sialic acid to GlcNAc-fucose-O-glycans has not been observed in flies. The consensus sequence for each glycan is listed below, although the sequence for O-GlcNAc is based on a small number of confirmed sites. O-Glycans are attached to the underlined amino acid(s) in each consensus sequence. Note that a single EGF repeat can possess all three modifications (Matsuura et al. 2008). C, cysteine (the superscript numbers show the position of cysteines in the EGF repeat); X, any amino acid other than cysteine; S, serine; T, threonine; P, proline; A, alanine; Fng, Fringe; MFNG, manic fringe; LFNG, lunatic fringe; RFNG, radical fringe. This figure is available in black and white in print and in color at Glycobiology online.
EGF repeats
EGF is a small growth factor of 53 amino acids and is important for cell motility (Segall et al. 1996), proliferation (Kato et al. 1998), differentiation (Traverse et al. 1994) and survival (Arteaga 2001; Zaczek et al. 2005). It is characterized by six conserved cysteine residues that form three disulfide bonds (Savage et al. 1973; Winkler et al. 1986). Many larger proteins contain a 30–40 amino acid sequence similar to EGF, which is frequently repeated and found only in secreted proteins and the extracellular domain of transmembrane proteins. These are typically referred to as EGF-like repeats, or EGF repeats. The repeat number can vary in different proteins from one in several coagulation factors and other proteins to >300 repeats in the Drosophila cell adhesion protein Dumpy. Moreover, a subset of EGF repeats can bind calcium, which plays important roles in protein folding and proper protein–protein interactions (Fehon et al. 1990; Rebay et al. 1991; Downing et al. 1996; Rand et al. 1997). Additionally, EGF repeats can be modified with several forms of O-linked glycans, which are the focus of this review.
EGF repeats are found in functionally diverse proteins; for example, many growth factors, signaling receptors, cell adhesion molecules, plasma proteins, and extracellular matrix components contain EGF repeats (Appella et al. 1988; Mosca et al. 2012; Muriel et al. 2012; Hudak et al. 2014). Most of the functions of these EGF repeats involve mediating protein–protein interactions and trafficking. For example, the EGF repeats in the epithelial cell adhesion molecule (ep-CAM) are required for its adhesive properties between neighboring cells (Balzar et al. 2001). Additionally, thrombomodulin, a cofactor involved in preventing coagulation, requires its EGF repeats to bind to thrombin and activate protein C (Kurosawa et al. 1988; Suzuki et al. 1989). Furthermore, EGF repeats are involved in protein trafficking. For example, one of the EGF repeats of the low-density lipoprotein receptor is involved in the regulation of its recycling (Zhang et al. 2007).
Notch signaling
The O-glycans found on EGF repeats play important roles in the function of Notch receptors and have been well studied in this context. Notch signaling is a cell–cell signaling pathway that is critical for the development and adult homeostasis of animals (Kopan and Ilagan 2009; Artavanis-Tsakonas and Muskavitch 2010). Mutations in the Notch pathway components cause a variety of human diseases including cancer, vascular dementia and developmental disorders (Louvi and Artavanis-Tsakonas 2012; Penton et al. 2012; South et al. 2012). For signaling to occur, the transmembrane ligands from one cell bind the transmembrane Notch receptor in a neighboring cell, inducing the cleavage of Notch and the release of the Notch intracellular domain into the cytoplasm, where it can translocate to the nucleus and promote the expression of its target genes. Drosophila only has one Notch receptor and therefore is frequently used to study Notch signaling. However, mammals have four Notch receptors (Notch1–4) and five canonical ligands: Jagged1, Jagged2, delta-like (DLL) 1, DLL3 and DLL4. In Drosophila, the only two ligands are Delta and the Jagged homolog Serrate. The Drosophila Notch receptor and the mammalian receptors have up to 36 EGF repeats. The EGF repeats of the receptors contain all three types of O-glycans found on EGF repeats, and two (O-glucose and O-fucose) are critical for Notch function (Bruckner et al. 2000; Moloney, Shair, et al. 2000; Okajima and Irvine 2002; Sasamura et al. 2003; Shi and Stanley 2003; Acar et al. 2008; Matsuura et al. 2008; Zhou et al. 2008; Fernandez-Valdivia et al. 2011; Takeuchi et al. 2011; Lee et al. 2013).
Role of O-glucose and protein O-glucosyltransferase 1 in Notch signaling
O-Glucose is added to serine residues and was discovered nearly three decades ago on the EGF repeats of bovine blood coagulation factors VII and IX (Hase et al. 1988). O-Glucose glycans are found as a disaccharide, extended by a xylose to form xylose-glucose, or a trisaccharide (xylose-xylose-glucose). Comparison of the confirmed O-glucosylation sites in human and bovine factor VII, factor IX, protein Z and human thrombospondin revealed the consensus sequence to be C1-X-S-X-P-C2 (Nishimura et al. 1989), which was later modified to C1-X-S-X-P/A-C2 (Rana et al. 2011). Additionally, mouse Notch1, Notch2 and Drosophila Notch were found to be modified with O-glucose (Moloney, Shair, et al. 2000; Acar et al. 2008; Whitworth et al. 2010; Fernandez-Valdivia et al. 2011; Rana et al. 2011). In Drosophila Notch, 18 of 36 EGF repeats contain the consensus sequence for O-glucose, more predicted sites than any other protein. Every consensus sequence identified in mass spectrometric analysis of Drosophila Notch and mammalian Notch1 contains the sugar modification (Acar et al. 2008; Rana et al. 2011; Lee et al. 2013), indicating that the consensus sequence is highly predictive for the addition of O-glucose. However, not all of the consensus sequences in mammalian Notch1 are efficiently glucosylated (Rana et al. 2011). Later work from the Haltiwanger group showed that the folding state of the EGF repeat and the composition of the amino acids surrounding the consensus sequence affect the efficiency of O-glucosylation (Takeuchi et al. 2012).
In 2008, the corresponding O-glucosyltransferase, named Rumi, was discovered in a screen for modulators of Notch signaling in Drosophila (Acar et al. 2008). The official name for the mammalian homolog of Rumi is “protein O-glucosyltransferase 1” or POGLUT1. Rumi is a soluble endoplasmic reticulum (ER) protein with a lysine-aspartic acid-glutamic acid-leucine ER-retention motif, and is required to prevent temperature-dependent loss of Notch signaling in flies (Figure 2). Further characterization revealed that Rumi is the sole protein O-glucosyltransferase able to modify EGF repeats at the C1-X-S-X-P/A-C2 consensus sequence in both flies and mice (Acar et al. 2008; Fernandez-Valdivia et al. 2011; Takeuchi et al. 2011). O-Glucose is not required for Drosophila Notch and mammalian Notch to bind to ligands, but seems to be required for Drosophila Notch to undergo S2 cleavage (Acar et al. 2008; Fernandez-Valdivia et al. 2011; Leonardi et al. 2011). It has been proposed that the presence of the glucose residues allows Notch to undergo the conformational changes necessary to reveal the S2 cleavage site upon ligand binding (Jafar-Nejad et al. 2010; Leonardi et al. 2011), which will provide access to a disintegrin and metalloproteinase/TNFα converting enzyme/Kuzbanian proteases responsible for the S2 cleavage (Brou et al. 2000; Lieber et al. 2002).
Fig. 2.

Summary of the roles of O-glucose, xylose, O-fucose and GlcNAc in Drosophila Notch signaling. Schematic of the Notch protein in the ER, Golgi and at the cell membrane with its EGF repeat O-glucose and O-fucose glycans are shown in wild-type and various mutant backgrounds. The non-enzymatic chaperone function of Ofut1, which is not reported for its mammalian homologs, is not shown in this figure. Although a non-enzymatic activity has not been formally ruled out for the fly Rumi, O-glucose mutations in Notch recapitulate the rumi loss-of-function phenotypes in the context of Notch signaling. Therefore, rumi mutation is assumed to be equivalent to loss of O-glucose. For simplicity, only EGF repeats are drawn in the extracellular domain, and the intracellular domain is not drawn to scale. The folding of the extracellular domain is arbitrarily drawn. Since Drosophila Notch without O-glucose or O-fucose reaches the cell surface but shows a temperature-sensitive loss of signaling, the extracellular domain of Notch without either of these glycans is drawn as somewhat misfolded. However, other mechanisms might underlie the observed phenotypes. In the absence of both glycans, the Drosophila Notch is trapped in the ER, hence the misfolded schematic. For details, please see the text. It is important to note that the models proposed in this figure are strictly based on Drosophila studies. This figure is available in black and white in print and in color at Glycobiology online.
In Drosophila, all O-glucose sites contribute to Notch function in a redundant and/or additive fashion, although mutations in single sites do not significantly affect Notch signaling (Leonardi et al. 2011). In vivo structure–function analysis showed that a Notch genomic transgene in which all 18 O-glucose sites are mutated recapitulates the temperature-sensitive loss of Notch signaling observed in rumi null mutants. Transgenes harboring 16 and 10 mutations showed less severe phenotypes. Analysis of Notch transgenes harboring O-glucose mutations in smaller subsets of Notch EGF repeats showed that the O-glucose sites in EGF10–15 are more important than others. However, although the ligand-binding EGF repeats 11 and 12 reside in this region, ligand binding is not decreased upon loss of Rumi in Drosophila cells (Acar et al. 2008; Leonardi et al. 2011). Moreover, a single mutation in the O-glucosylation site of EGF12 in a Notch genomic transgene did not affect signaling (Leonardi et al. 2011). These observations are in agreement with a recent report indicating that addition of mono- or disaccharide O-glucose glycans to a fragment of human Notch1 containing EGF11–13 does not alter its binding to mammalian ligands (Taylor et al. 2014). Recent crystal structure analysis of a human Notch1 fragment containing EGF11–13 bound to a fragment of DLL4 also supports the notion that O-glucosylation of Notch EGF repeats by Rumi does not directly affect Notch-ligand binding (Luca et al. 2015). Specifically, O-glucose modifications at Rumi consensus sites in EGF12 (serine458) and EGF13 (serine496) of the human Notch1 are located away from the DLL4 binding face and cover a proline and a phenylalanine residue in each of these EGF repeats. Accordingly, it is possible that by covering hydrophobic surfaces in these EGF repeats, O-glucose residues prevent Notch aggregation as Notch molecules cluster at the cell surface and thereby allow for efficient proteolysis (Luca et al, 2015). Of note, mass spectrometric analysis of human Notch1 has recently identified an O-linked hexose attached to serine435 between cysteine residues 3 and 4 of EGF11 which does not match the consensus O-glucose or O-fucose sequence (Andrawes et al. 2013). The presence of this hexose on serine435 has also been observed in the above-mentioned crystal structure of Notch1–DLL4 (Luca et al. 2015). Curiously, this hexose is present at the binding interface between Notch1 and DLL4, suggesting that it might directly regulate binding (Luca et al. 2015). The contribution of this novel O-glycan to Notch signaling and the enzyme responsible for its addition to Notch remains to be determined.
RNAi-mediated knockdown of Rumi in several mammalian cell lines results in decreased Notch1 cleavage and reduced Notch target gene expression (Fernandez-Valdivia et al. 2011; Ma et al. 2011). However, because of the early lethality of Rumi (Poglut1) homozygous mutant mouse embryos (Fernandez-Valdivia et al. 2011), the in vivo role of Rumi in regulating the function of individual Notch receptors is not clear. Similar to the report on the fly Notch, mutations in single O-glucose sites do not impair ligand-mediated Notch1 activation in a cell-based signaling assay, including an O-glucose mutation in the ligand-binding EGF12. Curiously, a mutation in EGF28 significantly decreases the ability of mouse Notch1 to respond to DLL1 without affecting Jagged1-induced signaling (Rana et al. 2011). This site is not present in Drosophila Notch and in mammalian Notch receptors other than Notch1 (Jafar-Nejad et al. 2010; Fernandez-Valdivia et al. 2011), and it is not clear whether the observed effect on DLL1 signaling is due to the loss of sugar or a conformational change in EGF28.
Although Drosophila Delta harbors a Rumi consensus sequence, Rumi does not seem to be required in the signal-sending cell in flies (Acar et al. 2008). Most mammalian ligands harbor 2–4 predicted O-glucosylation sites (Jafar-Nejad et al. 2010), but it remains to be seen whether O-glucosylation plays a role in the function of mammalian Notch ligands. Finally, in addition to their well-established protein O-glucosyltransferase activity, mammalian and Drosophila Rumi also exhibit a protein O-xylosyltransferase activity towards a subset of their target EGF repeats which harbor a di-serine motif in the C1-X-S-S-P-C2 consensus sequence (Takeuchi et al. 2011). Of note, mass spectrometric analysis of a fragment of the Notch2 extracellular domain expressed in 293T cells shows that Notch2 EGF16, which contains a C1-Y-S-S-P-C2 motif, can have either an O-glucose or an O-xylose, suggesting that addition of O-xylose to Notch might be biologically relevant (Takeuchi et al. 2011). Further experiments are required to examine the biological importance of this evolutionarily conserved dual substrate specificity of Rumi.
Negative regulation of Drosophila Notch signaling by xylose-glucose-O glycans
As mentioned earlier, O-glucose in flies and mammals can be extended by one or two xylose residues to form a xylose-glucose disaccharide or a xylose-xylose-glucose trisaccharide. In humans, addition of the first xylose residue to glucose is mediated by two enzymes, glucoside xylosyltransferase (GXYLT) 1 and GXYLT2, and the extension to a trisaccharide is mediated by xyloside xylosyltransferase 1 (XXYLT1) (Sethi et al. 2010, 2012). GXYLT1 and GXYLT2 add xylose specifically to an O-glucosylated substrate, and although GXYLT1 appears more active than its counterpart based on in vitro enzymatic assays using a glucose-aglycone (glc-R) as an acceptor, no distinct specificity for either is apparent. All three xylosyltransferases are predicted to be type II transmembrane proteins typical for Golgi glycosyltransferases. However, the Bakker lab has provided strong evidence that XXYLT1 resides in the ER as a transmembrane protein (Sethi et al. 2010, 2012). Additionally, XXYLT1 is highly specific for the xylose-glucose disaccharide, as it is unable to modify substrates harboring only an O-linked xylose. The Drosophila gene that has the highest homology with human XXYLT1 is computed gene 11388 (CG11388), but whether this gene actually encodes an XXYLT enzyme remains to be determined.
Recently, our group identified and characterized the sole Drosophila GXYLT, which was named Shams (Lee et al. 2013). Surprisingly, loss of Shams leads to a gain of Notch signaling in certain developmental processes (Figure 2), in contrast to loss of Drosophila Rumi, which leads to a loss of Notch signaling in all contexts studied so far (Acar et al. 2008; Leonardi et al. 2011; Perdigoto et al. 2011). Mass spectrometric analysis of the Drosophila Notch expressed in Drosophila Schneider 2 cells indicates that xylose only exists on a subset of O-glucosylated Notch EGF repeats, namely EGF14–20. Moreover, in vivo mutational analysis of Notch indicates that EGF16–20 contains the functionally important sites of xylosylation (Lee et al. 2013). Together, these observations indicate that the activity of the Drosophila Notch can be fine-tuned by altering the distribution and structure of its O-glucose glycans. Interestingly, overexpression of human GXYLT1 in Drosophila wing results in Notch loss-of-function phenotypes but overexpression of Shams does not (Lee et al. 2013), suggesting that human GXYLT1 is a more efficient xylosyltransferase. This might explain the mass spectrometry data indicating that all the O-glucosylated EGF repeats of the mouse Notch1 are xylosylated, albeit at variable stoichiometry (Rana et al. 2011). However, the increased xylosylation of mouse Notch may also be, at least in part, due to the fact that mammals have two GXYLT enzymes (Sethi et al. 2010).
Shams is expressed at a higher level in the pupal wing than the larval wing disc, and loss of Shams results in Notch accumulation inside and at the cell surface of pupal wing cells. Moreover, pupal wing cells that express only a mutant form of Notch lacking all the functional xylosylation sites also exhibit increased cell surface expression. Accordingly, a potential mechanism for the gain of Notch signaling upon loss of Shams is increased Notch availability at the cell surface. However, it remains to be determined whether xylose also affects other steps in Notch signaling including ligand binding. Of note, EGF16–20 in Drosophila Notch harbor the xylose modification, but no specific functions have previously been assigned to these EGF repeats (Pei and Baker 2008; Yamamoto et al. 2012; Andrawes et al. 2013). The precise mechanisms underlying the regulation of Drosophila Notch signaling by xylose and the functional significance of GXYLT1/2 and XXYLT1 in mammalian Notch signaling remain to be determined.
An eye beyond Notch: Other targets of Rumi in Drosophila development
Close to 50 proteins in mammals and 14 in Drosophila are predicted to contain O-glucose modification (Fernandez-Valdivia et al. 2011; Rana et al. 2011; Haltom et al. 2014; Takeuchi and Haltiwanger 2014). Recently, our group investigated the functional significance of O-glucosylation in two other proteins with multiple consensus O-glucosylation sites, Crumbs and Eyes shut (Haltom et al. 2014). Crumbs is a highly conserved transmembrane protein required for proper apical basal polarity, regulation of organ size and epithelial tube size (Tepass et al. 1990; Laprise et al. 2010; Robinson et al. 2010). Loss of crumbs results in severe epithelial disorganization and embryonic lethality in flies (Tepass et al. 1990). Additionally, Crumbs is localized to the apical membrane of Drosophila photoreceptors and is required for Drosophila eye morphogenesis and for protecting the photoreceptors from light-induced degeneration (Izaddoost et al. 2002; Johnson et al. 2002; Pellikka et al. 2002). Eyes shut is a secreted protein with an important role in Drosophila eye morphogenesis (Husain et al. 2006; Zelhof et al. 2006). Since both of these proteins are involved in the separation of the light-sensing units of the Drosophila eye and the formation of an extracellular space between them (Gurudev et al. 2014), we will provide a brief description of this stage of Drosophila eye development here.
The compound eye of Drosophila is composed of ∼800 repetitive hexagonal units called ommatidia. Each tiny ommatidium contains eight highly polarized photoreceptors, seven of which are visible in any given cross section of the ommatidia (Figure 3). A round, electron dense organelle called the rhabdomere resides at the apical side of each photoreceptor and is composed of ∼10,000 stacks of microvilli. The purpose of the microvilli is to increase the surface area for the localization of rhodopsin, the light-sensing pigment. The photoreceptors in each ommatidium are positioned in a trapezoidal pattern. The rhabdomeres face the extracellular space at the center of the ommatidium which separates the rhabdomeres from one another and is called the inter-rhabdomeral space (IRS) (Figure 3A). The secreted protein Eyes shut is required for the rhabdomeres to separate. Eyes shut starts to be secreted into the IRS around the middle of the pupal development (PD) and continues to expand the IRS during PD (Figure 3B) (Husain et al. 2006; Zelhof et al. 2006). Separation of rhabdomeres (open rhabdom) in Drosophila and a number of other insect species allows each rhabdomere in one ommatidium to sample light from a separate point in the visual field. This is in contrast to the closed rhabdom found in many other insects like honeybees and butterflies, in which all of the rhabdomeres from the photoreceptors belonging to a given ommatidium are fused at the center of the ommatidium. Open rhabdoms and their accompanying neuronal wiring allow for an increase in light sensitivity without sacrificing resolution compared with closed rhabdoms (Braitenberg 1967; Kirschfeld 1967; Borst 2009). Remarkably, overexpression of Eyes shut is sufficient to change a closed rhabdom to an open rhabdom (Zelhof et al. 2006), highlighting the major role that a single protein can play during eye evolution.
Fig. 3.

A model for the regulation of Eyes shut and IRS formation by Rumi. (A) A scanning electron micrograph (SEM) of the adult fly eye with its 760–800 ommatidia is shown to the left. The close-up shows a number of ommatidia, with sensory bristles decorating alternating corners of each ommatidium. To the right is a transmission electron micrograph of a single ommatidium, showing seven photoreceptor cells (PRCs). SEM images are courtesy of Jessica Leonardi. (B) Schematic drawings of a developing ommatidium. At 45% PD, the apical surfaces of photoreceptors contact one another and rhabdomere formation has not started. By 70% PD, disc-shaped rhabdomeres are formed at the apical side of photoreceptors, and secretion of Eyes shut (Eys) has separated the apical surfaces of the rhabdomeres and has formed a continuous IRS. Most of the Eys protein, which is O-glucosylated by Rumi in wild-type animals, is found in the IRS. By 100% PD, rhabdomeres have assumed their round adult morphology and are well separated by an Eys-filled IRS. (C) In rumi null eyes, a significant amount of Eys, which should lack O-glucose and is likely misfolded, remains in the PRC body. At this stage, only a fraction of Eys is secreted into the IRS, which is smaller than the wild-type IRS at the same developmental time. By 100% PD, rumi mutants do not show Eys accumulation in the PRC bodies anymore and accumulate Eys in the IRS, whose size is comparable with wild-type IRS at this stage. However, the rhabdomere attachments are not resolved, and the IRS is not continuous, suggesting that rhabdomere separation needs to occur in a critical time window during development. The ommatidium schematics are adapted from Knust (2007). Circles in PRCs depict the nuclei. Glc, glucose; ZA, zonula adherens. This figure is available in black and white in print and in color at Glycobiology online.
Among the many EGF repeats that Crumbs and Eyes shut have, seven in Crumbs and five in Eyes shut contain consensus sequences for O-glucose (Figure 4). Moreover, mass spectrometric analysis of Crumbs and Eyes shut protein fragments expressed in a Drosophila cell line indicate that both proteins harbor O-glucose modifications on every consensus sequence tested, although the efficiency of O-glucosylation varies among different sites (Haltom et al. 2014; Figure 4, black circles). This raises the possibility that O-glucose is important for their function, which our group sought to investigate. Indeed, loss of Rumi or its enzymatic activity results in a failure of rhabdomeres to completely separate from the neighboring rhabdomeres during IRS development (Haltom et al. 2014), suggesting that one or more Rumi target proteins involved in IRS development require O-glucose. The IRS phenotype of rumi mutants is present even when the animals are raised at 18°C, which largely preserves the function of Notch (Leonardi et al. 2011). Moreover, rhabdomeres are fully separated in transgenic animals whose only source of Notch is from a Notch transgene mutated in all O-glucosylation sites (Haltom et al. 2014). These observations indicate that other Rumi targets must be involved in this phenotype.
Fig. 4.
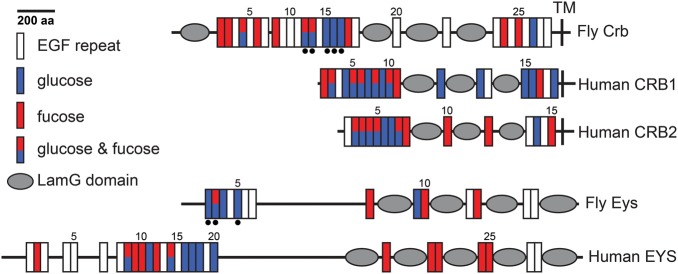
Distribution of O-glucose and O-fucose consensus sequences on fly and human Eyes shut and Crumbs proteins. Domain predictions were performed by using ScanProsite (http://prosite.expasy.org/). Black dots underneath EGF repeats indicate confirmed glycosylation sites (both O-glucose and O-fucose). Signal peptides at the N-terminal of the proteins are not marked. LamG, laminin G; TM, transmembrane domain. This figure is available in black and white in print and in color at Glycobiology online.
In vivo mutational analysis indicates that loss of O-glucose from Crumbs cannot explain the rhabdomere attachment phenotype observed in rumi mutants. Specifically, animals homozygous for an O-glucose deficient crumbs knock-in allele with mutations in all Rumi consensus sites are viable and fertile and do not show any gross morphological defects (Haltom et al. 2014). Of note, the number of Crumbs+ puncta is increased in the photoreceptor cell body of rumi−/− animals raised at 25°C (Haltom et al. 2014), suggesting a “benign” trafficking defect in Crumbs lacking O-glucose. The Rumi consensus sequences in Crumbs are highly conserved, as human CRB1 and CRB2 contain 13 and 8 O-glucosylation sites, respectively (Haltom et al. 2014). Therefore, it is possible that O-glucosylation regulates the function of mammalian CRB proteins. Interestingly, Rumi knockout mice die at mid-gestation with embryonic phenotypes more severe than global Notch loss-of-function phenotypes, such as those reported in presenilin1 and 2, Rbpj, or Pofut1 knockouts (Oka et al. 1995; Donoviel et al. 1999; Shi and Stanley 2003; Fernandez-Valdivia et al. 2011). This suggests that Rumi regulates additional targets during mouse embryonic development. Crb1 mutant mice survive but exhibit retinal dysplasia and light-induced degeneration phenotypes (Mehalow et al. 2003; van de Pavert et al. 2004). Crb2 mutant mice exhibit severe developmental abnormalities after E7.5 and die by E10.5 (Xiao et al. 2011). Therefore, it is possible that loss of O-glucose from CRB2 contributes to the embryonic phenotypes and lethality observed in Rumi mutant mice. Further studies will be required to determine the importance of O-glucose glycans in the function of mammalian CRB proteins and in other roles of Drosophila Crumbs not examined in our studies.
Our data strongly suggest that loss of O-glucose from Eyes shut can explain the eye morphogenesis phenotype in rumi mutants (Haltom et al. 2014). Removing one copy of eyes shut strongly enhances the rumi mutant rhabdomere attachment defect. Additionally, in rumi mutant eyes, Eyes shut protein accumulates intracellularly in the pupal stage during the initial steps of rhabdomere separation, though some Eyes shut can be seen in the IRS (Figure 3C). Furthermore, expression of a mutant version of Eyes shut with mutations in the O-glucosylation sites results in its intracellular accumulation. Together, these observations strongly suggest that Eyes shut is the biologically-relevant target of Rumi during rhabdomere separation. It is noteworthy that the intracellular accumulation of Eyes shut observed in the mid-pupal stage in rumi mutants is resolved at later stages of development. However, the rhabdomere attachments persist, indicating that presence of O-glucose on Eyes shut in a critical developmental window (mid-pupal stage) is essential for full rhabdomere separation.
Temperature-shift experiments indicate that when rumi null animals are raised at higher temperatures, higher levels of Eyes shut accumulate in photoreceptors and the IRS becomes smaller. A temperature-dependent phenotype is classically seen when the defect is due to protein misfolding (Bross et al. 1999). In agreement with this notion, removing one copy of the chaperone heat shock 70-kDa protein cognate 3, which promotes proper protein folding in the ER, results in an increase in rhabdomere attachments in a rumi null background. Moreover, although total Eyes shut levels do not change in head extracts from rumi null animals raised at lower temperatures, Eyes shut levels are significantly decreased when animals are raised at higher temperatures, suggesting a temperature-dependent degradation potentially due to enhanced misfolding. In summary, the data suggest that loss of O-glucose from Eyes shut results in a temperature-dependent misfolding and therefore a decrease in Eyes shut secretion, which results in impaired rhabdomere separation.
Eyes shut has been lost in a number species during evolution, including mouse, rat and sheep (Abd El-Aziz et al. 2008). However, humans have an Eyes shut homolog (EYS) with a highly conserved domain organization compared with its Drosophila homolog and with seven predicted O-glucosylation sites (Abd El-Aziz et al. 2008; Collin et al. 2008; Haltom et al. 2014). Importantly, mutations in the human EYS cause a severe blindness disorder called autosomal recessive retinitis pigmentosa (Abd El-Aziz et al. 2008, 2010; Audo et al. 2010), and autosomal recessive cone-rod dystrophy (Katagiri et al. 2014). Additionally, single nucleotide polymorphisms in EYS are associated with statin-induced myopathy (Isackson et al. 2011). Human EYS is localized to the outer segment of the retina, which is homologous to the rhabdomere in flies, and is also expressed in the brain, spinal cord and skeletal muscle (Isackson et al. 2011). The exact function of human EYS in the eye and other tissues is unknown. It remains to be seen whether O-glucose is required for the folding, trafficking and/or function of the human EYS function as well.
A unifying theme for the function of O-glucose glycans on EGF repeats?
Notch, Crumbs and Eyes shut are the three Drosophila proteins with the highest number of O-glucosylation sites. However, the effects of loss of Rumi and O-glucose on these proteins at cell biological and functional levels are not the same. When raised at 30°C, loss of rumi phenocopies null alleles of Notch (Acar et al. 2008), indicating that O-glucosylation is essential for the function of Notch at the restrictive temperature. Eyes shut shows an intermediate requirement for Rumi, as even when rumi mutants are grown at 30°C, they still exhibit some degree of rhabdomere separation (Haltom et al. 2014), in contrast to eyes shut null mutants, in which the IRS is completely lost (Husain et al. 2006; Zelhof et al. 2006; Haltom et al. 2014). Loss of O-glucose seems to be dispensable for the function of Crumbs during rhabdomere morphogenesis when animals are raised between 18 and 25°C (Haltom et al. 2014). The effects of loss of O-glucose from Crumbs at 30°C remain unknown, because both the glucosylation-deficient knock-in allele and its wild-type progenitor strain are lethal at this temperature. Nevertheless, since the IRS defects in rumi mutants are evident even at 18°C, they cannot be explained by loss of O-glucose from Crumbs. Collectively, these observations indicate that despite the high predictive value of the C1-X-S-X-P/A-C2 consensus sequence for O-glucosylation, factors other than the number of O-glucosylation sites are involved in determining the impact of loss of O-glucose from each protein.
As mentioned before, the available data strongly suggest that Rumi is required for proper folding of Eyes shut and its adequate secretion into the extracellular space (Haltom et al. 2014). In contrast, the surface expression of Notch is increased in rumi mutants raised at the restrictive temperature (Acar et al. 2008), indicating that loss of O-glucose by itself is not enough to prevent Notch from exiting the ER (Figure 2). However, the redundant role of O-glucose and O-fucose in the ER exit of Notch (see the next section) and the worsening of the Notch loss-of-function phenotypes upon growing rumi mutants and glucosylation-defective Notch transgenes at higher temperatures strongly suggest that O-glucose also affects Notch folding (Acar et al. 2008; Leonardi et al. 2011; Ishio et al. 2015). A similar argument can be made based on the increased number of Crumbs+ puncta in rumi mutants despite lack of morphological abnormalities upon loss of O-glucose from Crumbs (Haltom et al. 2014). Based on these observations, we propose that loss of O-glucose affects the folding of all three proteins, and that the level of functional impairment that each protein shows in the absence of glucose will depend on several factors including the degree of misfolding, the level of functional redundancy between O-glucose and other O-linked glycans, and the context in which each protein functions. Of course, the available data do not allow us to rule out the possibility that each modified protein is regulated by O-glucose residues in a unique fashion.
The biological roles of O-fucose and protein O-fucosyltransferase 1
O-Fucose modifications were initially discovered in human urine in the 1970s (Hallgren et al. 1975) and were later identified on an EGF repeat on urinary-type plasminogen activator (Kentzer et al. 1990). The modification is also present on other proteins such as blood coagulation factors and Notch (Kentzer et al. 1990; Harris and Spellman 1993; Moloney, Shair, et al. 2000). The consensus sequence was identified as C2-X-X-G-G-(S/T)-C3 (Harris et al. 1992; Harris and Spellman 1993) but was later modified to C2-X-X-X-X-(S/T)-C3 (Haines and Irvine 2005). However, this sequence must be within a properly folded EGF repeat for O-fucose to be added (Wang and Spellman 1998). The mammalian protein O-fucosyltransferase 1 (POFUT1) was discovered by protein isolation and molecular cloning (Wang and Spellman 1998; Wang et al. 2001) and the Drosophila homolog was named Ofut1 (Okajima and Irvine 2002). Like Rumi, POFUT1/Ofut1 is a soluble, ER localized enzyme (Luo and Haltiwanger 2005).
Only a few proteins have been identified to contain O-fucose modifications, such as urokinase plasminogen activator, tissue type plasminogen activator, several blood coagulation factors and Notch (Kentzer et al. 1990; Harris and Spellman 1993; Moloney, Shair, et al. 2000). However, over 100 proteins contain the consensus sequence for O-fucose and are predicted to be O-fucosylated (Rampal et al. 2007). The most studied target of POFUT1/Ofut1 is the Notch receptor, which has the highest number of O-fucose consensus sites among animal proteins (Rampal et al. 2007). The first evidence that O-fucose is required for Notch signaling was the loss of Jagged1-induced Notch signaling in cells deficient in fucose (Moloney, Panin, et al. 2000; Chen et al. 2001). These observations were followed by in vivo experiments indicating that in Drosophila, Ofut1 is required for the activation of Notch signaling in all contexts studied (Okajima and Irvine 2002; Sasamura et al. 2003). Genetic experiments by the Stanley lab indicated that loss of Pofut1 in mice is embryonic lethal and causes phenotypes resembling a global loss of Notch signaling (Shi and Stanley 2003). Conditional loss of Pofut1 in several contexts shows phenotypes very similar to conditional loss of Rbpj, suggesting that Pofut1 is a global regulator of Notch signaling in mice (Okamura and Saga 2008; Tsao et al. 2009).
Both Notch and its ligands are modified with O-fucose in flies and mice (Moloney, Shair, et al. 2000; Panin et al. 2002). However, in Drosophila, Ofut1 does not seem to be required in signal-sending cells, i.e., the cells expressing Notch ligands (Okajima and Irvine 2002). Similarly, studies with Pofut1-deficient mouse embryonic stem cells show that POFUT1 is required in the signal-receiving cell (Stahl et al. 2008). These studies indicate that Notch receptors are the biologically-relevant targets of Ofut1. In contrast, mouse DLL1 with mutations in its O-fucosylation sites localizes to the cell surface and can activate Notch in neighboring cells, although it shows some intracellular accumulation as well (Muller et al. 2014). These data suggest that O-fucose is not required for the function of DLL1. Of note, this might not be true for all Notch ligands, as a recent report provides in vitro and in vivo evidence that addition of O-fucose to DLL3 and its elongation by Fringe proteins might be required for the function of DLL3 during mouse somitogenesis (Serth et al. 2015).
Analysis of cell lines and mice deficient in Pofut1 or the machinery required for the generation or the transport of GDP-fucose and rescue experiments with exogenous fucose by taking advantage of the mammalian salvage pathway for the generation of GDP-fucose provide strong evidence that POFUT1 regulates mammalian Notch signaling through its O-fucosyltransferase enzymatic activity (Smith et al. 2002; Stahl et al. 2008; Zhou et al. 2008). Furthermore, the bulk of data suggests that POFUT1 regulates mammalian Notch signaling at the level of Notch-ligand binding and is not an essential chaperone of the mammalian Notch receptors, although one in vivo study has suggested that POFUT1 is also required for the exocytic trafficking of Notch in the presomitic mesoderm and another group has reported a slight decrease in the cell-surface level of Notch receptors upon loss of POFUT1 from mammalian cells (Okamura and Saga 2008; Stahl et al. 2008; Zhou et al. 2008; Yao et al. 2011). In contrast, two non-enzymatic roles have been described for the fly Ofut1 in addition to its O-fucosyltransferase activity: promotion of the folding and ER exit of Notch as a chaperone, and promotion of endocytic trafficking of Notch (Okajima et al. 2005; Sasamura et al. 2007). As explained in the next section, Fringe glycosyltransferases add an N-acetylglucosamine (GlcNAc) residue to O-fucose on EGF repeats and play important roles in Notch signaling in flies, mice and humans (Evrard et al. 1998; Zhang and Gridley 1998; Bruckner et al. 2000; Moloney, Panin, et al. 2000; Sparrow et al. 2006). Therefore, there was little doubt that the enzymatic activity of Ofut1/POFUT1 is important for Notch signaling. However, reports on the non-enzymatic activity of Drosophila Ofut1 posed the following question: do O-fucose residues on EGF repeats play a role in Drosophila Notch signaling beyond forming an acceptor site for GlcNAc?
To determine the biological importance of Ofut1's non-enzymatic activity in flies, a mutant genomic transgene of ofut1 containing a point mutation in the GDP-fucose-binding domain was generated (Okajima et al. 2005). This transgene, along with mutations in other genes that prevent fucosylation without affecting Ofut1 levels such as an enzyme responsible for the generation of GDP-fucose called GDP-mannose 4,6-dehydratase (Gmd), was used to determine the contribution of the chaperone and enzymatic activities of Ofut1 to Notch function (Okajima et al. 2005, 2008). Phenotypic analysis of the above-mentioned transgenic and mutant fly strains indicated that the chaperone activity is the key role of Ofut1 in Notch signaling, and suggested that its enzymatic activity is only important in Fringe-dependent contexts. However, since the level of gene expression from randomly inserted transgenes can vary depending on the site of insertion into the fly genome, further studies were needed to definitively determine whether O-fucose residues on Notch have any roles other than providing a site for the addition of GlcNAc.
The Matsuno group recently sought to address this issue by generating a knock-in allele of the enzymatic null ofut1 (Ishio et al. 2015). They found that embryos homozygous for the mutant allele ofutR245A knock-in, which lacks enzymatic function, exhibit a severe, temperature-sensitive neurogenic phenotype, indicative of loss of Notch signaling. Since fringe mutants do not show this phenotype, the data indicate an important role for O-fucose in Drosophila Notch signaling independent of its function as an acceptor for GlcNAc. It is important to note that animals homozygous for the ofut14R6 null allele show a severe neurogenic phenotype even at lower temperatures (Sasamura et al. 2003), unlike ofutR245A knock-in animals, which only show a neurogenic phenotype at the restrictive (high) temperature (Ishio et al. 2015). This indicates that at least when flies are grown at lower temperatures, Ofut1 plays an important non-enzymatic role in Drosophila Notch signaling, unlike POFUT1 which does not seem to play such a role in mammalian Notch signaling. Additionally, animals deficient in enzymes required in the GDP-fucose synthesis pathway show a temperature-sensitive neurogenic phenotype, further supporting the notion that the temperature-sensitive neurogenic phenotype observed in ofutR245A knock-in animals is due to the loss of Ofut1's enzymatic function. In animals homozygous for null alleles of rumi or for the ofutR245A knock-in allele, the lateral inhibition defect and the loss of wing margin are only observed when the larvae are raised at 28°–30°C (Acar et al. 2008; Ishio et al. 2015). However, in animals double mutant for rumi and ofutR245A knock-in, these defects arise even when raised at 18°C, indicating that O-fucose and O-glucose monosaccharides have a redundant role in the function of the Notch receptor in these contexts (Ishio et al. 2015). Loss of O-fucose does not decrease surface localization of Notch at 30°C but causes Notch to accumulate intracellularly, similar to what has been reported in rumi mutants (Acar et al. 2008; Ishio et al. 2015). However, simultaneous loss of rumi and the enzymatic activity of Ofut1 results in a severe decrease in the cell surface levels of Notch and accumulation of Notch in the ER (Ishio et al. 2015), strongly suggesting that O-glucose and O-fucose play a redundant role in the ER exit of the Drosophila Notch receptor, likely through promoting proper Notch folding (Figure 2).
O-glucose and O-fucose are added to properly folded EGF repeats, suggesting that these sugars are not required initially for proper folding of EGF repeats (Wang and Spellman 1998; Takeuchi et al. 2012). How can one reconcile these observations with the role suggested for these sugars in the folding of their modified proteins (Notch and Eys) in Drosophila (Haltom et al. 2014; Ishio et al. 2015)? A potential clue might come from a recent elegant study by the Haltiwanger group on a noncanonical form of ER quality control mediated by glucose-fucose-O disaccharides on thrombospondin repeats (TSRs) (Vasudevan et al. 2015). According to this report, instead of adding the carbohydrates to unfolded TSRs to help them fold, protein O-fucosyltransferase 2 and β3-glucosyltransferase add the O-fucose and the subsequent glucose to properly folded TSRs (Kozma et al. 2006; Luo, Koles, et al. 2006; Luo, Nita-Lazar, et al. 2006) to stabilize the folded structure and thereby to drive the folding equilibrium forward. Whether O-linked glycans on EGF repeats play a similar role in the quality control of proteins like Notch and Eys in flies remains to be examined. As mentioned above, the evidence is lacking for an essential non-enzymatic role of POFUT1 in mammalian Notch signaling, strongly suggesting that the chaperone function described for the Drosophila Ofut1 is not a conserved function of this protein. Further experiments are required to determine whether EGF repeat O-glycans play a redundant role in the ER exit of mammalian Notch or other modified proteins similar to the role reported for these sugars in the ER exit of the Drosophila Notch (Ishio et al. 2015).
In addition to Notch, a second biologically-relevant target of POFUT1 has been identified in mammals. POFUT1 has been shown to add an O-fucose residue to EGF4 of the mammalian AGRIN, an extracellular matrix protein essential for neuromuscular junction (Kim et al. 2008). O-Fucosylation modulates the ability of AGRIN to induce the clustering of nicotinic acetylcholine receptors at the postsynaptic membrane of the mammalian neuromuscular junction (Kim et al. 2008). Interestingly, loss of POFUT1 results in a gain of AGRIN function, such that without O-fucose, AGRIN recruits more acetylcholine receptors to the postsynaptic membrane, resulting in higher acetylcholine receptor clustering. Together with our recent work on the role of Rumi in the Drosophila eye (Haltom et al. 2014), this study shows that Rumi and POFUT1 each have targets other than the Notch receptors. However, so far Notch is the only protein known to be regulated by both of these enzymes. It is noteworthy that recent studies have identified mutations in POFUT1 and POGLUT1 in Dowling-Degos disease, which is a rare, autosomal dominant form of skin disease characterized by altered pigmentation and other skin lesions (Li et al. 2013; Basmanav et al. 2014). It remains to be seen whether these phenotypes result from a decrease in the enzymatic activity of these enzymes or from a reduction in a yet-to-be-determined non-enzymatic role, and whether they are caused by altered Notch signaling, which is a known regulator of skin homeostasis and pigmentation (Moriyama et al. 2006; Kumano et al. 2008; Nowell and Radtke 2013), or by impaired activity of a different common target of these enzymes.
Fringe proteins enable Notch to distinguish between different ligands
The O-fucose on Notch can be extended to form a disaccharide, trisaccharide or tetrasaccharide. Fringe (Fng) proteins are a group of enzymes that extend the O-fucose modification to GlcNAcβ1–3Fucose-O to generate the disaccharide. Fng was first discovered to be required for dorsal–ventral boundary specification in the Drosophila wing. Loss of fng results in a loss of wing tissue, but juxtaposition of cells with and without Fng function leads to ectopic margin formation, suggesting that Fng is involved in a boundary-specific cell–cell communication (Irvine and Wieschaus 1994; Kim et al. 1995). Fng was later shown to modulate Notch signaling (Fleming et al. 1997; Panin et al. 1997; Klein and Arias 1998; Bruckner et al. 2000; Moloney, Panin, et al. 2000). Addition of GlcNAc to O-fucose by Fng results in sensitization of the Drosophila Notch to Delta-mediated signaling and inhibition of Serrate-mediated Notch signaling at the level of ligand binding (Figure 2) (Fleming et al. 1997; Panin et al. 1997; Bruckner et al. 2000; Moloney, Panin, et al. 2000; Okajima et al. 2003).
The same basic mechanism applies to mammalian systems, however, with more complexity. Mammals have four Notch receptors, five Notch ligands and three Fng homologs: manic fringe (MFNG), lunatic fringe (LFNG) and radical fringe (RFNG). Although biochemically all three Fng homologs have the same enzymatic function, the story is rather different in vivo. The Fng homologs can affect Notch signaling differently depending on which ligand binds Notch. Shown by co-culture and luciferase assays, LFNG promotes DLL1-mediated Notch1 signaling and suppresses Jagged1-induced Notch1 signaling (Hicks et al. 2000; Yang et al. 2004). However, LFNG has been reported to enhance Notch2 signaling in response to both DLL1 and Jag1 (Hicks et al. 2000). Additionally, MFNG inhibits Jagged1-induced Notch1 signaling (Hicks et al. 2000; Moloney, Panin, et al. 2000; Chen et al. 2001; Yang et al. 2004) and promotes DLL1-induced Notch1 signaling, although more weakly than LFNG (Yang et al. 2005). RFNG promotes DLL1-induced Notch1 signaling strongly, and interestingly also promotes signaling by Jagged1 (Yang et al. 2005). The differential effects of mammalian FNG proteins on signaling mediated by various Notch-ligand pairs might in part result from the preference of each FNG protein to extend O-fucose on a different set of Notch EGF repeats. Similar to the effect of the Drosophila Fng on Delta-mediated signaling, promotion of DLL-mediate Notch1 signaling by mammalian FNG proteins correlates well with increased Notch1-ligand binding mediated by FNG proteins (Yang et al. 2005). However, even though FNG proteins decrease Jagged1-mediated Notch1 signaling, they do not decrease Notch1-Jagged1 binding (Yang et al. 2005).
The above-mentioned functions of Fng are in the context of trans-activation of Notch, during which ligand from a neighboring cell binds to Notch and activates it. However, until recently, it remained unknown whether Fng proteins function in the context of cis inhibition. Cis inhibition is the binding of Notch to its ligand in the same cell, resulting in the sequestration of the Notch receptor and the ligand, thereby making them unavailable to engage in trans signaling (de Celis and Bray 1997; Micchelli et al. 1997; Becam et al. 2010; Sprinzak et al. 2010). The Elowitz group sought to determine whether Fng played a role on Notch in the context of cis inhibition (LeBon et al. 2014). Using an elegant quantitative co-culture system with a fluorescent-based Notch reporter and constructs capable of inducible expression of fluorescently tagged DLL1 and Jagged1, they showed that when any of the three Fng homologs are expressed, cis interactions between Notch1 and DLL1 are stronger. However, expression of MFNG or LFNG in cells expressing Notch1 and Jagged1 decreases the level of cis interactions between them. In contrast, expression of RFNG enhances cis inhibition between Notch1 and Jagged1, similar to its role in Notch1-DLL1 cis inhibition. These observations are supported by in vivo fly genetic experiments in the developing wing. This study supports a model in which MFNG, LFNG and their Drosophila homolog regulate cis and trans interactions of Notch with each class of ligands in the same direction: they promote both trans and cis interactions of Notch with DLL1/Delta, and they decrease both trans and cis interactions of Notch with Jagged1/Serrate (LeBon et al. 2014).
The mechanism of the differential response of Notch to different ligands when modified by GlcNAc has just begun to be elucidated. The Handford lab performed in vitro experiments to determine whether the effects of Fng on Notch signaling are due to structural changes in the ligand-binding domain or due to a difference in ligand affinity upon addition of GlcNAc to O-fucose. The group generated a human Notch1 fragment (EGF11–13), which contains the ligand-binding domain, and assessed the effect of different glycosylation states on this fragment's ability to bind to Jagged1, DLL1 and DLL4 ligands (Taylor et al. 2014). They found that addition of GlcNAc to O-fucose on EGF12 in this fragment substantially enhanced its binding to both DLL1 and Jagged1. These glycan modifications did not affect binding between the Notch fragment and DLL4, possibly because the affinity between these two is high even without sugars (Andrawes et al. 2013). However, crystal structure analysis indicated that addition of GlcNAc-fucose-O to EGF12 does not induce a conformational change in the EGF11–13 fragment. These results provide strong evidence that addition of GlcNAc to EGF12 of Notch promotes DLL1-Notch signaling by directly increasing the affinity of these two proteins. However, because of the discrepancy between the inhibitory effect of LFNG on Jagged1-mediated Notch signaling and the increased binding between Jagged1 and EGF11–13 upon addition of GlcNAc, the authors propose that GlcNAc modifications on other EGF repeats must function to inhibit Jagged1-Notch1 signaling.
Recently, the Garcia lab reported the co-crystal structure of a glycosylated version of human Notch1-EGF11–13 bound to a fragment from DLL4 (Luca et al. 2015). To facilitate crystallization, they performed in vitro mutagenesis and generated DLL4 variants with increased affinity for the human Notch1 fragment. This study demonstrated that the O-fucose residue on EGF12 directly contacts DLL4 and serves as a surrogate amino acid. Moreover, modeling of the GlcNAc residue that would be attached to this O-fucose strongly suggests that the GlcNAc residue on EGF12 contacts amino acids from both Notch1 and DLL4 and thereby can directly contribute to Notch1–DLL4 interaction (Luca et al. 2015). These observations provide an example of a posttranslational modification directly involved in specific interaction between a signaling receptor and its ligand.
Although extensive work has been done to elucidate the effect of each mammalian FNG homolog on Notch function, only LFNG seems to be important for mouse development. LFNG is broadly expressed in the developing mouse embryo, including in the presomitic mesoderm, rhombomeres 3 and 5, developing ear, retina and spinal cord, and is required for proper skeletal development (Cohen et al. 1997; Johnston et al. 1997; Evrard et al. 1998). Mice mutant for Lfng show a severe disorganization of the axial skeleton and reduced viability and fertility, although some survive to adulthood (Evrard et al. 1998; Zhang and Gridley 1998). MFNG is expressed in the neural tube, head, cranial nerves, dorsal root ganglia and otic vesicle (Cohen et al. 1997; Johnston et al. 1997). RFNG is expressed in the developing limb bud, head, anterior neural tube and branchial arches (Cohen et al. 1997; Johnston et al. 1997). However, neither MFNG nor RFNG appear to be required for viability and fertility in mice (Moran et al. 1999, 2009; Zhang et al. 2002). Moreover, mutations in human LFNG cause spondylocostal dysostosis (Sparrow et al. 2006), but to our knowledge, no diseases have been associated with loss-of-function mutations in MFNG or RFNG. Of note, a recent report provides strong evidence that MFNG plays an oncogenic role in a form of breast cancer associated with poor prognosis (Zhang et al. 2015).
FNG proteins play important roles in the immune system. Notch1 signaling is required in the thymus to suppress B-cell development and promote T-cell development (Radtke et al. 1999; Stanley and Guidos 2009). This process seems to require the function of LFNG, as loss of LFNG from fetal liver hematopoietic stem cells results in defective T-cell development, especially in competitive repopulation experiments (Visan et al. 2006). FNG proteins also play a key role in the development of marginal zone (MZ) B cells in the spleen. Although neither LFNG nor MFNG is essential for MZ B-cell development in the spleen, loss of each of them compromises the competitive ability of MZ precursors to generate B cells in mixed chimeras (Tan et al. 2009). Moreover, loss of both proteins results in a significant decrease in the number of MZ B cells even in non-competitive chimeras (Tan et al. 2009). These observations indicate that LFNG and MFNG cooperate to promote MZ B-cell development.
In mammals, the extension of GlcNAc-fucose disaccharide to a trisaccharide results in the formation of Galactose-GlcNAc-Fucose, which is present on mammalian Notch1 (Moloney, Panin, et al. 2000; Moloney, Shair, et al. 2000). This trisaccharide can further be extended to the tetrasaccharide, sialic acid-galactose-GlcNAc-fucose, which was first observed on human clotting factor IX (Nishimura et al. 1992; Harris et al. 1993). In mammals, addition of Galactose to GlcNAc can be catalyzed by β1,4-galactosyltransferase (B4GALT) enzymes (Lee et al. 2001). On Notch EGF repeats, this modification seems to be primarily mediated by one of these enzymes, B4GALT1 (Chen et al. 2001). Loss of the B4galt1 gene in mice is semi-lethal, and homozygous mice exhibit growth retardation, skeletal defects, impaired wound healing, endocrine insufficiency, abnormal differentiation of intestinal villi and increased proliferation of skin cells and cells of the small intestine (Asano et al. 1997; Lu et al. 1997; Mori et al. 2004; Chen et al. 2006). Although B4galt1 null embryos showed normal somitogenesis at embryonic (E) day 9.5, the mutant mice had an extra lumbar vertebra at E18.5 (Chen et al. 2006). The mutant mice also misexpress a number of Notch pathway components similar to Lfng mutant mice (Evrard et al. 1998; Zhang and Gridley 1998; Zhang et al. 2002; Chen et al. 2006). Only 20% of B4galt1 null mice survive past 16 weeks (Asano et al. 1997). Therefore, B4galt1 is not required for early embryonic development, but is required for late embryonic development and survival after birth. The relatively mild phenotypes of B4galt1 null mice might be due to contribution of other B4GALTs to Notch signaling in a redundant fashion (Stanley 2007).
Extension of GlcNAc-Fucose disaccharide has not been observed in Drosophila. However, analysis of O-glycans from Drosophila embryos by Tiemeyer lab has identified a novel branched O-fucose trisaccharide which harbors a GlcNAc-β1–3 (the same position to which Fng adds GlcNAc) and a glucuronic acid β1–4 directly linked to O-fucose (Aoki et al. 2008). In this study, O-glycans were released from proteins before analysis. Therefore, it is not clear whether this glycan exists on EGF repeats or another protein module. However, the prevalence of this branched form of O-fucose trisaccharide shows a clear correlation with the level of Fng both in wing imaginal disc and in embryonic extracts, suggesting that it might have functional relevance to Drosophila Notch signaling. Identification of the enzyme responsible for this modification will be required to test this hypothesis.
Evidence suggests that the terminal Galactose is important for Notch signaling, but the sialic acid appears to be dispensable. In Chinese hamster ovary (CHO) cells deficient in the transporter required for cytidine 5′-monophosphate-sialic acid to enter the Golgi, loss of the sialic acid does not affect the usual inhibition of Jagged1-Notch signaling or the enhancement of DLL1-Notch1 signaling by LFNG (Chen et al. 2001; Hou et al. 2012). However, in CHO cells deficient in galactose, neither LFNG nor MFNG are able to inhibit Jagged1-induced Notch signaling, and LFNG is unable to enhance DLL1-induced Notch signaling. MFNG is still able to enhance DLL1-induced Notch signaling in the absence of galactose (Hou et al. 2012). Since addition of galactose to the GlcNAc-fucose-O glycan on EGF12 did not alter the binding of the EGF11–13 fragment of the human Notch1 to Jagged1 and DLL1 (Taylor et al. 2014), these results suggest that addition of galactose to one or more Notch EGF repeats outside of the core ligand-binding domain is required for Fng proteins to modulate Notch signaling in some contexts.
O-GlcNAc on EGF repeats: Cell adhesion and maybe Notch
While searching for O-linked glycans on the extracellular domain of the Drosophila Notch, the Okajima group unexpectedly discovered O-GlcNAc modification on EGF20 (Matsuura et al. 2008). They performed western blots on fragments of Notch with an O-GlcNAc antibody (CTD110.6) to search for other EGF repeats that may have the modification and found that the EGF1–10 and EGF22–31 fragments, but not EGF6–10, contain the O-GlcNAc modification. The signal is nearly eliminated in these fragments when treated with β-N-acetylhexosaminidase, indicating that O-GlcNAc is attached to its recipient amino acid in a β-anomeric configuration. This modification was later found on the EGF repeats of other proteins such as Drosophila Delta, Serrate and Dumpy (Matsuura et al. 2008; Sakaidani et al. 2011; Muller et al. 2013), the latter of which is a large 2.5 MDa extracellular matrix protein with 308 EGF repeats (Wilkin et al. 2000). Comparison of sites with a confirmed O-GlcNAc has suggested the putative consensus sequence C5XXGX(S/T)GXXC6 (Alfaro et al. 2012). As more modified sites are identified, it is possible that this sequence will be refined, like those of O-glucose and O-fucose.
In 2011, the Okajima group identified the enzyme responsible for the addition of O-GlcNAc on EGF repeats in Drosophila and called it EGF domain-specific O-GlcNAc transferase or Eogt (Sakaidani et al. 2011). Eogt resides in the ER and is conserved in mice (Sakaidani et al. 2012). The maternal contribution of the Drosophila Eogt is required for embryonic development in a Notch-independent manner. Eogt mutant flies that receive the maternal component die between the second instar and early third instar larval stages. The mutant larvae display cuticle defects and defects in tracheal morphology similar to those observed in animals that lack dumpy (Prout et al. 1997; Wilkin et al. 2000; Sakaidani et al. 2011). Additionally, loss of Eogt or Dumpy in the wing results in wing blistering that is independent of integrin function. Moreover, Dumpy requires Eogt enzymatic activity and the O-GlcNAc modification to function. These results highlight the importance of O-GlcNAc modification on Dumpy (Sakaidani et al. 2011).
Although O-GlcNAc is found on Notch, Eogt mutants do not show Notch mutant phenotypes (Sakaidani et al. 2011). To determine whether other proteins are involved in the Eogt null phenotype and require O-GlcNAc to function, the Stanley and Jenny laboratories collaborated to perform genetic interaction experiments with genes that were likely to play a role in the wing adhesion phenotype of Eogt mutants (Muller et al. 2013). Removal of one copy of an integrin in an Eogt knockdown wing did not alter the phenotype, but removal of one copy of wing blister, which encodes laminin α chain, enhanced the wing blistering phenotype. Surprisingly, although loss of Eogt did not recapitulate Notch mutant phenotypes, removing one copy of Notch or Notch pathway components in Eogt-knockdown animals suppressed the wing blistering phenotype. This may be due to the loss of O-GlcNAc from Notch. However, dumpy alleles interact with the γ-secretase Presenilin, which is crucial for Notch pathway activation (Mahoney et al. 2006). Therefore, the genetic interaction may not be due to the loss of O-GlcNAc from Notch, but instead it is possible that Dumpy and Notch both contribute to wing adhesion.
Dumpy genetically interacts with components of the pyrimidine synthesis pathway, and feeding pyrimidine synthesis inhibitors to dumpy mutant flies reverses the defects in wing shape (Rizki and Rizki 1965; Stroman 1974; Blass and Hunt 1980). Therefore, the authors sought to determine whether components of the pyrimidine synthesis pathway genetically interact with Eogt. Reduction of genes that contribute to the synthesis of UMP/pyrimidine suppresses the wing blistering caused by Eogt knockdown (Muller et al. 2013). Consistent with this, the authors hypothesize that reducing the dosage of genes responsible for pyrimidine degradation should enhance the wing blistering phenotype, which proves to be correct, and such a genetic interaction is lethal. Collectively, these data lead to the possibility that increased pyrimidine synthesis, such as increased Uracil, could cause the wing blistering phenotype in Eogt mutants.
Less is known about the mammalian EOGT. Okajima and colleagues identified and confirmed the biochemical function of mouse EOGT and found that the enzyme is expressed in all tissues examined (Sakaidani et al. 2012). Additionally, mammalian EOGT reaches optimal function when divalent cations, especially Mn2+, are accessible, similar to the Drosophila enzyme (Sakaidani et al. 2011, 2012). Transgenic expression of the mouse and human EOGT was able to rescue the wing blistering phenotypes in Eogt knockdown fly wings, indicating enzymatic and functional conservation between fly and mammalian EOGT proteins (Sakaidani et al. 2012; Muller et al. 2013). Moreover, although mammals lack a Dumpy homolog, O-GlcNAc has been identified on the extracellular domain of a number of mammalian proteins including Notch1 and Notch2 (Alfaro et al. 2012; Sakaidani et al. 2012). Importantly, EOGT mutations have recently been identified in patients with autosomal recessive Adams-Oliver syndrome (AOS) (Shaheen et al. 2013; Cohen et al. 2014). AOS is a rare disorder characterized by aplasia cutis congenita (vertex scalp defects) and terminal transverse limb defects (Burton et al. 1976; Bonafede and Beighton 1979). Biochemical experiments indicate that even those AOS-causing EOGT mutations that do not affect the expression level of EOGT impair its ability to add O-GlcNAc to Notch, strongly suggesting that a defect in the enzymatic activity of EOGT underlies this disease (Ogawa et al. 2015). Mutations in several Notch pathway components have been described in AOS, such as heterozygous mutations in NOTCH1 and RBPJ (recombination signal binding protein for immunoglobulin kappa J region), which encodes the primary nuclear effector of the Notch pathway (Hassed et al. 2012; Stittrich et al. 2014). These observations indicate that EOGT is important for mammalian biology and strongly suggest that O-GlcNAc is important for mammalian Notch signaling, although further studies are required to test these hypotheses.
Funding
This work was supported by grants from the NIH/NIGMS (R01GM084135), the Mizutani Foundation for Glycoscience (grant #110071) and the March of Dimes Foundation (Cell Lineage and Differentiation Research Grant #1-FY10-501). Our work is also supported by the Grace Wilsey Foundation.
Conflict of interest statement
None declared.
Abbreviations
B4galt1, β1,4-galactosyltransferase 1; CG11388, computed gene 11388; CHO, Chinese hamster ovary; CRB, crumbs; DLL, delta-like; EGF, epidermal growth factor-like; EOGT, EGF domain-specific O-GlcNAc transferase; ER, endoplasmic reticulum; ep-CAM, epithelial cell adhesion molecule; EYS, eyes shut homolog; fng, fringe; GalNAc, N-acetylgalactosamine; GDP, guanosine diphosphate; GlcNAc, N-acetylglucosamine; GXYLT, glucoside xylosyltransferase; IRS, inter-rhabdomeral space; LFNG, lunatic fringe; MFNG, manic fringe; MZ, marginal zone; Ofut1, the Drosophila protein O-fucosyltransferase 1; POFUT1, the mammalian protein O-fucosyltransferase 1; POGLUT1, protein O-glucosyltransferase 1; RBPJ, recombination signal binding protein for immunoglobulin kappa J region; RFNG, radical fringe.
Acknowledgements
We thank Tom V. Lee for comments on the manuscript, Jessica Leonardi for the SEM images used in Figure 3A, and Robert Haltiwanger for stimulating discussions. We apologize to colleagues whose work on EGF repeat O-glycans has not been cited here due to space limitations.
References
- Abd El-Aziz MM, Barragan I, O'Driscoll CA, Goodstadt L, Prigmore E, Borrego S, Mena M, Pieras JI, El-Ashry MF, Safieh LA et al. . 2008. EYS, encoding an ortholog of Drosophila spacemaker, is mutated in autosomal recessive retinitis pigmentosa. Nat Genet. 40:1285–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abd El-Aziz MM, O'Driscoll CA, Kaye RS, Barragan I, El-Ashry MF, Borrego S, Antinolo G, Pang CP, Webster AR, Bhattacharya SS. 2010. Identification of novel mutations in the ortholog of drosophila eyes shut gene (EYS) causing autosomal recessive retinitis pigmentosa. Invest Ophthalmol Vis Sci. 51:4266–4272. [DOI] [PubMed] [Google Scholar]
- Acar M, Jafar-Nejad H, Takeuchi H, Rajan A, Ibrani D, Rana NA, Pan H, Haltiwanger RS, Bellen HJ. 2008. Rumi is a CAP10 domain glycosyltransferase that modifies Notch and is required for Notch signaling. Cell. 132:247–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfaro JF, Gong CX, Monroe ME, Aldrich JT, Clauss TR, Purvine SO, Wang Z, Camp DG 2nd, Shabanowitz J, Stanley P et al. . 2012. Tandem mass spectrometry identifies many mouse brain O-GlcNAcylated proteins including EGF domain-specific O-GlcNAc transferase targets. Proc Natl Acad Sci USA. 109:7280–7285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrawes MB, Xu X, Liu H, Ficarro SB, Marto JA, Aster JC, Blacklow SC. 2013. Intrinsic selectivity of Notch 1 for Delta-like 4 over Delta-like 1. J Biol Chem. 288:25477–25489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki K, Porterfield M, Lee SS, Dong B, Nguyen K, McGlamry KH, Tiemeyer M. 2008. The diversity of O-linked glycans expressed during Drosophila melanogaster development reflects stage- and tissue-specific requirements for cell signaling. J Biol Chem. 283:30385–30400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appella E, Weber IT, Blasi F. 1988. Structure and function of epidermal growth factor-like regions in proteins. FEBS Lett. 231:1–4. [DOI] [PubMed] [Google Scholar]
- Artavanis-Tsakonas S, Muskavitch MA. 2010. Notch: The past, the present, and the future. Curr Top Dev Biol. 92:1–29. [DOI] [PubMed] [Google Scholar]
- Arteaga CL. 2001. The epidermal growth factor receptor: From mutant oncogene in nonhuman cancers to therapeutic target in human neoplasia. J Clin Oncol. 19:32S–40S. [PubMed] [Google Scholar]
- Asano M, Furukawa K, Kido M, Matsumoto S, Umesaki Y, Kochibe N, Iwakura Y. 1997. Growth retardation and early death of beta-1,4-galactosyltransferase knockout mice with augmented proliferation and abnormal differentiation of epithelial cells. EMBO J. 16:1850–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audo I, Sahel JA, Mohand-Said S, Lancelot ME, Antonio A, Moskova-Doumanova V, Nandrot EF, Doumanov J, Barragan I, Antinolo G et al. . 2010. EYS is a major gene for rod-cone dystrophies in France. Hum Mutat. 31:E1406–E1435. [DOI] [PubMed] [Google Scholar]
- Balzar M, Briaire-de Bruijn IH, Rees-Bakker HA, Prins FA, Helfrich W, de Leij L, Riethmuller G, Alberti S, Warnaar SO, Fleuren GJ et al. . 2001. Epidermal growth factor-like repeats mediate lateral and reciprocal interactions of Ep-CAM molecules in homophilic adhesions. Mol Cell Biol. 21:2570–2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basmanav FB, Oprisoreanu AM, Pasternack SM, Thiele H, Fritz G, Wenzel J, Grosser L, Wehner M, Wolf S, Fagerberg C et al. . 2014. Mutations in POGLUT1, encoding protein O-glucosyltransferase 1, cause autosomal-dominant Dowling-Degos disease. Am J Hum Genet. 94:135–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becam I, Fiuza UM, Arias AM, Milan M. 2010. A role of receptor Notch in ligand cis-inhibition in Drosophila. Curr Biol. 20:554–560. [DOI] [PubMed] [Google Scholar]
- Bennett EP, Mandel U, Clausen H, Gerken TA, Fritz TA, Tabak LA. 2012. Control of mucin-type O-glycosylation: A classification of the polypeptide GalNAc-transferase gene family. Glycobiology. 22:736–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blass DH, Hunt DM. 1980. Pyrimidine biosynthesis in the dumpy mutants of Drosophila melanogaster. Mol Gen Genet. 178:437–442. [DOI] [PubMed] [Google Scholar]
- Bonafede RP, Beighton P. 1979. Autosomal dominant inheritance of scalp defects with ectrodactyly. Am J Med Genet. 3:35–41. [DOI] [PubMed] [Google Scholar]
- Borst A. 2009. Drosophila's view on insect vision. Curr Biol. 19:R36–R47. [DOI] [PubMed] [Google Scholar]
- Braitenberg V. 1967. Patterns of projection in the visual system of the fly. I. Retina-lamina projections. Exp Brain Res. 3:271–298. [DOI] [PubMed] [Google Scholar]
- Breloy I, Schwientek T, Gries B, Razawi H, Macht M, Albers C, Hanisch FG. 2008. Initiation of mammalian O-mannosylation in vivo is independent of a consensus sequence and controlled by peptide regions within and upstream of the alpha-dystroglycan mucin domain. J Biol Chem. 283:18832–18840. [DOI] [PubMed] [Google Scholar]
- Bross P, Corydon TJ, Andresen BS, Jorgensen MM, Bolund L, Gregersen N. 1999. Protein misfolding and degradation in genetic diseases. Hum Mutat. 14:186–198. [DOI] [PubMed] [Google Scholar]
- Brou C, Logeat F, Gupta N, Bessia C, LeBail O, Doedens JR, Cumano A, Roux P, Black RA, Israel A. 2000. A novel proteolytic cleavage involved in Notch signaling: The role of the disintegrin-metalloprotease TACE. Mol Cell. 5:207–216. [DOI] [PubMed] [Google Scholar]
- Bruckner K, Perez L, Clausen H, Cohen S. 2000. Glycosyltransferase activity of Fringe modulates Notch-Delta interactions. Nature. 406:411–415. [DOI] [PubMed] [Google Scholar]
- Burton BK, Hauser L, Nadler HL. 1976. Congenital scalp defects with distal limb anomalies: Report of a family. J Med Genet. 13:466–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Lu L, Shi S, Stanley P. 2006. Expression of Notch signaling pathway genes in mouse embryos lacking beta4galactosyltransferase-1. Gene Expr Patterns. 6:376–382. [DOI] [PubMed] [Google Scholar]
- Chen J, Moloney DJ, Stanley P. 2001. Fringe modulation of Jagged1-induced Notch signaling requires the action of beta 4galactosyltransferase-1. Proc Natl Acad Sci USA. 98:13716–13721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen B, Bashirullah A, Dagnino L, Campbell C, Fisher WW, Leow CC, Whiting E, Ryan D, Zinyk D, Boulianne G et al. . 1997. Fringe boundaries coincide with Notch-dependent patterning centres in mammals and alter Notch-dependent development in Drosophila. Nat Genet. 16:283–288. [DOI] [PubMed] [Google Scholar]
- Cohen I, Silberstein E, Perez Y, Landau D, Elbedour K, Langer Y, Kadir R, Volodarsky M, Sivan S, Narkis G et al. . 2014. Autosomal recessive Adams-Oliver syndrome caused by homozygous mutation in EOGT, encoding an EGF domain-specific O-GlcNAc transferase. Eur J Hum Genet. 22:374–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collin RW, Littink KW, Klevering BJ, van den Born LI, Koenekoop RK, Zonneveld MN, Blokland EA, Strom TM, Hoyng CB, den Hollander AI et al. . 2008. Identification of a 2 Mb human ortholog of Drosophila eyes shut/spacemaker that is mutated in patients with retinitis pigmentosa. Am J Hum Genet. 83:594–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Celis JF, Bray S. 1997. Feed-back mechanisms affecting Notch activation at the dorsoventral boundary in the Drosophila wing. Development. 124:3241–3251. [DOI] [PubMed] [Google Scholar]
- Donoviel DB, Hadjantonakis AK, Ikeda M, Zheng H, Hyslop PS, Bernstein A. 1999. Mice lacking both presenilin genes exhibit early embryonic patterning defects. Genes Dev. 13:2801–2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downing AK, Knott V, Werner JM, Cardy CM, Campbell ID, Handford PA. 1996. Solution structure of a pair of calcium-binding epidermal growth factor-like domains: Implications for the Marfan syndrome and other genetic disorders. Cell. 85:597–605. [DOI] [PubMed] [Google Scholar]
- Evrard YA, Lun Y, Aulehla A, Gan L, Johnson RL. 1998. Lunatic fringe is an essential mediator of somite segmentation and patterning. Nature. 394:377–381. [DOI] [PubMed] [Google Scholar]
- Fehon RG, Kooh PJ, Rebay I, Regan CL, Xu T, Muskavitch MA, Artavanis-Tsakonas S. 1990. Molecular interactions between the protein products of the neurogenic loci Notch and Delta, two EGF-homologous genes in Drosophila. Cell. 61:523–534. [DOI] [PubMed] [Google Scholar]
- Fernandez-Valdivia R, Takeuchi H, Samarghandi A, Lopez M, Leonardi J, Haltiwanger RS, Jafar-Nejad H. 2011. Regulation of the mammalian Notch signaling and embryonic development by the protein O-glucosyltransferase Rumi. Development. 138:1925–1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming RJ, Gu Y, Hukriede NA. 1997. Serrate-mediated activation of Notch is specifically blocked by the product of the gene fringe in the dorsal compartment of the Drosophila wing imaginal disc. Development. 124:2973–2981. [DOI] [PubMed] [Google Scholar]
- Freeze HH, Eklund EA, Ng BG, Patterson MC. 2015. Neurological aspects of human glycosylation disorders. Annu Rev Neurosci. 38:105–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurudev N, Yuan M, Knust E. 2014. Chaoptin, prominin, eyes shut and crumbs form a genetic network controlling the apical compartment of Drosophila photoreceptor cells. Biol Open. 3:332–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haines N, Irvine KD. 2005. Functional analysis of Drosophila beta1,4-N-acetlygalactosaminyltransferases. Glycobiology. 15:335–346. [DOI] [PubMed] [Google Scholar]
- Hallgren P, Lundblad A, Svensson S. 1975. A new type of carbohydrate-protein linkage in a glycopeptide from normal human urine. J Biol Chem. 250:5312–5314. [PubMed] [Google Scholar]
- Haltom AR, Lee TV, Harvey BM, Leonardi J, Chen YJ, Hong Y, Haltiwanger RS, Jafar-Nejad H. 2014. The protein O-glucosyltransferase Rumi modifies eyes shut to promote rhabdomere separation in Drosophila. PLoS Genet. 10:e1004795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RJ, Ling VT, Spellman MW. 1992. O-linked fucose is present in the first epidermal growth factor domain of factor XII but not protein C. J Biol Chem. 267:5102–5107. [PubMed] [Google Scholar]
- Harris RJ, Spellman MW. 1993. O-linked fucose and other post-translational modifications unique to EGF modules. Glycobiology. 3:219–224. [DOI] [PubMed] [Google Scholar]
- Harris RJ, van Halbeek H, Glushka J, Basa LJ, Ling VT, Smith KJ, Spellman MW. 1993. Identification and structural analysis of the tetrasaccharide NeuAc alpha(2-->6)Gal beta(1-->4)GlcNAc beta(1-->3)Fuc alpha 1-->O-linked to serine 61 of human factor IX. Biochemistry. 32:6539–6547. [DOI] [PubMed] [Google Scholar]
- Hase S, Kawabata S, Nishimura H, Takeya H, Sueyoshi T, Miyata T, Iwanaga S, Takao T, Shimonishi Y, Ikenaka T. 1988. A new trisaccharide sugar chain linked to a serine residue in bovine blood coagulation factors VII and IX. J Biochem. 104:867–868. [DOI] [PubMed] [Google Scholar]
- Hassed SJ, Wiley GB, Wang S, Lee JY, Li S, Xu W, Zhao ZJ, Mulvihill JJ, Robertson J, Warner J et al. . 2012. RBPJ mutations identified in two families affected by Adams-Oliver syndrome. Am J Hum Genet. 91:391–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks C, Johnston SH, diSibio G, Collazo A, Vogt TF, Weinmaster G. 2000. Fringe differentially modulates Jagged1 and Delta1 signalling through Notch1 and Notch2. Nat Cell Biol. 2:515–520. [DOI] [PubMed] [Google Scholar]
- Hou X, Tashima Y, Stanley P. 2012. Galactose differentially modulates lunatic and manic fringe effects on Delta1-induced NOTCH signaling. J Biol Chem. 287:474–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudak CS, Gulyaeva O, Wang Y, Park SM, Lee L, Kang C, Sul HS. 2014. Pref-1 marks very early mesenchymal precursors required for adipose tissue development and expansion. Cell Rep. 8:678–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husain N, Pellikka M, Hong H, Klimentova T, Choe KM, Clandinin TR, Tepass U. 2006. The agrin/perlecan-related protein eyes shut is essential for epithelial lumen formation in the Drosophila retina. Dev Cell. 11:483–493. [DOI] [PubMed] [Google Scholar]
- Irvine KD, Wieschaus E. 1994. Fringe, a Boundary-specific signaling molecule, mediates interactions between dorsal and ventral cells during Drosophila wing development. Cell. 79:595–606. [DOI] [PubMed] [Google Scholar]
- Isackson PJ, Ochs-Balcom HM, Ma C, Harley JB, Peltier W, Tarnopolsky M, Sripathi N, Wortmann RL, Simmons Z, Wilson JD et al. . 2011. Association of common variants in the human eyes shut ortholog (EYS) with statin-induced myopathy: Evidence for additional functions of EYS. Muscle Nerve. 44:531–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishio A, Sasamura T, Ayukawa T, Kuroda J, Ishikawa HO, Aoyama N, Matsumoto K, Gushiken T, Okajima T, Yamakawa T et al. . 2015. O-Fucose monosaccharide of drosophila notch has a temperature-sensitive function and cooperates with O-glucose glycan in notch transport and notch signaling activation. J Biol Chem. 290:505–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izaddoost S, Nam SC, Bhat MA, Bellen HJ, Choi KW. 2002. Drosophila Crumbs is a positional cue in photoreceptor adherens junctions and rhabdomeres. Nature. 416:178–183. [DOI] [PubMed] [Google Scholar]
- Jafar-Nejad H, Leonardi J, Fernandez-Valdivia R. 2010. Role of glycans and glycosyltransferases in the regulation of Notch signaling. Glycobiology. 20:931–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson K, Grawe F, Grzeschik N, Knust E. 2002. Drosophila crumbs is required to inhibit light-induced photoreceptor degeneration. Curr Biol. 12:1675–1680. [DOI] [PubMed] [Google Scholar]
- Johnston SH, Rauskolb C, Wilson R, Prabhakaran B, Irvine KD, Vogt TF. 1997. A family of mammalian Fringe genes implicated in boundary determination and the Notch pathway. Development. 124:2245–2254. [DOI] [PubMed] [Google Scholar]
- Katagiri S, Akahori M, Hayashi T, Yoshitake K, Gekka T, Ikeo K, Tsuneoka H, Iwata T. 2014. Autosomal recessive cone-rod dystrophy associated with compound heterozygous mutations in the EYS gene. Doc Ophthalmol. 128:211–217. [DOI] [PubMed] [Google Scholar]
- Kato Y, Tapping RI, Huang S, Watson MH, Ulevitch RJ, Lee JD. 1998. Bmk1/Erk5 is required for cell proliferation induced by epidermal growth factor. Nature. 395:713–716. [DOI] [PubMed] [Google Scholar]
- Kentzer EJ, Buko A, Menon G, Sarin VK. 1990. Carbohydrate composition and presence of a fucose-protein linkage in recombinant human pro-urokinase. Biochem Biophys Res Commun. 171:401–406. [DOI] [PubMed] [Google Scholar]
- Kim ML, Chandrasekharan K, Glass M, Shi S, Stahl MC, Kaspar B, Stanley P, Martin PT. 2008. O-fucosylation of muscle agrin determines its ability to cluster acetylcholine receptors. Mol Cell Neurosci. 39:452–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Irvine KD, Carroll SB. 1995. Cell recognition, signal induction, and symmetrical gene activation at the dorsal-ventral boundary of the developing Drosophila wing. Cell. 82:795–802. [DOI] [PubMed] [Google Scholar]
- Kirschfeld K. 1967. [The projection of the optical environment on the screen of the rhabdomere in the compound eye of the Musca]. Exp Brain Res. 3:248–270. [DOI] [PubMed] [Google Scholar]
- Klein T, Arias AM. 1998. Interactions among Delta, Serrate and Fringe modulate Notch activity during Drosophila wing development. Development. 125:2951–2962. [DOI] [PubMed] [Google Scholar]
- Knust E. 2007. Photoreceptor morphogenesis and retinal degeneration: Lessons from Drosophila. Curr Opin Neurobiol. 17:541–547. [DOI] [PubMed] [Google Scholar]
- Kopan R, Ilagan MX. 2009. The canonical Notch signaling pathway: Unfolding the activation mechanism. Cell. 137:216–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozma K, Keusch JJ, Hegemann B, Luther KB, Klein D, Hess D, Haltiwanger RS, Hofsteenge J. 2006. Identification and characterization of abeta1,3-glucosyltransferase that synthesizes the Glc-beta1,3-Fuc disaccharide on thrombospondin type 1 repeats. J Biol Chem. 281:36742–36751. [DOI] [PubMed] [Google Scholar]
- Kumano K, Masuda S, Sata M, Saito T, Lee SY, Sakata-Yanagimoto M, Tomita T, Iwatsubo T, Natsugari H, Kurokawa M et al. . 2008. Both Notch1 and Notch2 contribute to the regulation of melanocyte homeostasis. Pigment Cell Melanoma Res. 21:70–78. [DOI] [PubMed] [Google Scholar]
- Kurosawa S, Stearns DJ, Jackson KW, Esmon CT. 1988. A 10-kDa cyanogen bromide fragment from the epidermal growth factor homology domain of rabbit thrombomodulin contains the primary thrombin binding site. J Biol Chem. 263:5993–5996. [PubMed] [Google Scholar]
- Laprise P, Paul SM, Boulanger J, Robbins RM, Beitel GJ, Tepass U. 2010. Epithelial polarity proteins regulate Drosophila tracheal tube size in parallel to the luminal matrix pathway. Curr Biol. 20:55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBon L, Lee TV, Sprinzak D, Jafar-Nejad H, Elowitz MB. 2014. Fringe proteins modulate Notch-ligand cis and trans interactions to specify signaling states. Elife. 3:e02950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TV, Sethi MK, Leonardi L, Rana NA, Buettner FF, Haltiwanger RS, Bakker H, Jafar-Nejad H. 2013. Negative regulation of Notch signaling by xylose. PLoS Genet. 9:e1003547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Sundaram S, Shaper NL, Raju TS, Stanley P. 2001. Chinese hamster ovary (CHO) cells may express six beta 4-galactosyltransferases (beta 4GalTs). Consequences of the loss of functional beta 4GalT-1, beta 4GalT-6, or both in CHO glycosylation mutants. J Biol Chem. 276:13924–13934. [DOI] [PubMed] [Google Scholar]
- Leonardi J, Fernandez-Valdivia R, Li YD, Simcox AA, Jafar-Nejad H. 2011. Multiple O-glucosylation sites on Notch function as a buffer against temperature-dependent loss of signaling. Development. 138:3569–3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Cheng R, Liang J, Yan H, Zhang H, Yang L, Li C, Jiao Q, Lu Z, He J et al. . 2013. Mutations in POFUT1, encoding protein O-fucosyltransferase 1, cause generalized Dowling-Degos disease. Am J Hum Genet. 92:895–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieber T, Kidd S, Young MW. 2002. Kuzbanian-mediated cleavage of Drosophila Notch. Genes Dev. 16:209–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louvi A, Artavanis-Tsakonas S. 2012. Notch and disease: A growing field. Semin Cell Dev Biol. 23:473–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Q, Hasty P, Shur BD. 1997. Targeted mutation in beta1,4-galactosyltransferase leads to pituitary insufficiency and neonatal lethality. Dev Biol. 181:257–267. [DOI] [PubMed] [Google Scholar]
- Luca VC, Jude KM, Pierce NW, Nachury MV, Fischer S, Garcia KC. 2015. Structural biology. Structural basis for Notch1 engagement of Delta-like 4. Science. 347:847–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, Haltiwanger RS. 2005. O-Fucosylation of notch occurs in the endoplasmic reticulum. J Biol Chem. 280:11289–11294. [DOI] [PubMed] [Google Scholar]
- Luo Y, Koles K, Vorndam W, Haltiwanger RS, Panin VM. 2006. Protein O-fucosyltransferase 2 adds O-fucose to thrombospondin type 1 repeats. J Biol Chem. 281:9393–9399. [DOI] [PubMed] [Google Scholar]
- Luo Y, Nita-Lazar A, Haltiwanger RS. 2006. Two distinct pathways for O-fucosylation of epidermal growth factor-like or thrombospondin type 1 repeats. J Biol Chem. 281:9385–9392. [DOI] [PubMed] [Google Scholar]
- Ma W, Du J, Chu Q, Wang Y, Liu L, Song M, Wang W. 2011. hCLP46 regulates U937 cell proliferation via Notch signaling pathway. Biochem Biophys Res Commun. 408:84–88. [DOI] [PubMed] [Google Scholar]
- Mahoney MB, Parks AL, Ruddy DA, Tiong SY, Esengil H, Phan AC, Philandrinos P, Winter CG, Chatterjee R, Huppert K et al. . 2006. Presenilin-based genetic screens in Drosophila melanogaster identify novel notch pathway modifiers. Genetics. 172:2309–2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura A, Ito M, Sakaidani Y, Kondo T, Murakami K, Furukawa K, Nadano D, Matsuda T, Okajima T. 2008. O-Linked N-acetylglucosamine is present on the extracellular domain of notch receptors. J Biol Chem. 283:35486–35495. [DOI] [PubMed] [Google Scholar]
- Mehalow AK, Kameya S, Smith RS, Hawes NL, Denegre JM, Young JA, Bechtold L, Haider NB, Tepass U, Heckenlively JR et al. . 2003. CRB1 is essential for external limiting membrane integrity and photoreceptor morphogenesis in the mammalian retina. Hum Mol Genet. 12:2179–2189. [DOI] [PubMed] [Google Scholar]
- Micchelli CA, Rulifson EJ, Blair SS. 1997. The function and regulation of cut expression on the wing margin of Drosophila: Notch, Wingless and a dominant negative role for Delta and Serrate. Development. 124:1485–1495. [DOI] [PubMed] [Google Scholar]
- Moloney DJ, Panin VM, Johnston SH, Chen J, Shao L, Wilson R, Wang Y, Stanley P, Irvine KD, Haltiwanger RS et al. . 2000. Fringe is a glycosyltransferase that modifies Notch. Nature. 406:369–375. [DOI] [PubMed] [Google Scholar]
- Moloney DJ, Shair LH, Lu FM, Xia J, Locke R, Matta KL, Haltiwanger RS. 2000. Mammalian Notch1 is modified with two unusual forms of O-linked glycosylation found on epidermal growth factor-like modules. J Biol Chem. 275:9604–9611. [DOI] [PubMed] [Google Scholar]
- Moran JL, Levorse JM, Vogt TF. 1999. Limbs move beyond the radical fringe. Nature. 399:742–743. [DOI] [PubMed] [Google Scholar]
- Moran JL, Shifley ET, Levorse JM, Mani S, Ostmann K, Perez-Balaguer A, Walker DM, Vogt TF, Cole SE. 2009. Manic fringe is not required for embryonic development, and fringe family members do not exhibit redundant functions in the axial skeleton, limb, or hindbrain. Dev Dyn. 238:1803–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori R, Kondo T, Nishie T, Ohshima T, Asano M. 2004. Impairment of skin wound healing in beta-1,4-galactosyltransferase-deficient mice with reduced leukocyte recruitment. Am J Pathol. 164:1303–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriyama M, Osawa M, Mak SS, Ohtsuka T, Yamamoto N, Han H, Delmas V, Kageyama R, Beermann F, Larue L et al. . 2006. Notch signaling via Hes1 transcription factor maintains survival of melanoblasts and melanocyte stem cells. J Cell Biol. 173:333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosca TJ, Hong W, Dani VS, Favaloro V, Luo L. 2012. Trans-synaptic Teneurin signalling in neuromuscular synapse organization and target choice. Nature. 484:237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller R, Jenny A, Stanley P. 2013. The EGF repeat-specific O-GlcNAc-transferase Eogt Interacts with Notch signaling and pyrimidine metabolism pathways in Drosophila. PLoS ONE. 8:e62835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller J, Rana NA, Serth K, Kakuda S, Haltiwanger RS, Gossler A. 2014. O-Fucosylation of the notch ligand mDLL1 by POFUT1 is dispensable for ligand function. PLoS ONE. 9:e88571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muriel JM, Dong C, Vogel BE. 2012. Distinct regions within fibulin-1D modulate interactions with hemicentin. Exp Cell Res. 318:2543–2547. [DOI] [PubMed] [Google Scholar]
- Nishimura H, Kawabata S, Kisiel W, Hase S, Ikenaka T, Takao T, Shimonishi Y, Iwanaga S. 1989. Identification of a disaccharide (Xyl-Glc) and a trisaccharide (Xyl2-Glc) O-glycosidically linked to a serine residue in the first epidermal growth factor-like domain of human factors VII and IX and protein Z and bovine protein Z. J Biol Chem. 264:20320–20325. [PubMed] [Google Scholar]
- Nishimura H, Takao T, Hase S, Shimonishi Y, Iwanaga S. 1992. Human factor IX has a tetrasaccharide O-glycosidically linked to serine 61 through the fucose residue. J Biol Chem. 267:17520–17525. [PubMed] [Google Scholar]
- Nowell C, Radtke F. 2013. Cutaneous Notch signaling in health and disease. Cold Spring Harb Perspect Med. 3:a017772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa M, Sawaguchi S, Kawai T, Nadano D, Matsuda T, Yagi H, Kato K, Furukawa K, Okajima T. 2015. Impaired O-linked N-acetylglucosaminylation in the endoplasmic reticulum by mutated epidermal growth factor (EGF) domain-specific O-linked N-acetylglucosamine transferase found in Adams-Oliver syndrome. J Biol Chem. 290:2137–2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka C, Nakano T, Wakeham A, de la Pompa JL, Mori C, Sakai T, Okazaki S, Kawaichi M, Shiota K, Mak TW et al. . 1995. Disruption of the mouse RBP-J kappa gene results in early embryonic death. Development. 121:3291–3301. [DOI] [PubMed] [Google Scholar]
- Okajima T, Irvine KD. 2002. Regulation of notch signaling by O-linked fucose. Cell. 111:893–904. [DOI] [PubMed] [Google Scholar]
- Okajima T, Reddy B, Matsuda T, Irvine KD. 2008. Contributions of chaperone and glycosyltransferase activities of O-fucosyltransferase 1 to Notch signaling. BMC Biol. 6:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okajima T, Xu A, Irvine KD. 2003. Modulation of notch-ligand binding by protein O-fucosyltransferase 1 and fringe. J Biol Chem. 278:42340–42345. [DOI] [PubMed] [Google Scholar]
- Okajima T, Xu A, Lei L, Irvine KD. 2005. Chaperone activity of protein O-fucosyltransferase 1 promotes notch receptor folding. Science. 307:1599–1603. [DOI] [PubMed] [Google Scholar]
- Okamura Y, Saga Y. 2008. Pofut1 is required for the proper localization of the Notch receptor during mouse development. Mech Dev. 125:663–673. [DOI] [PubMed] [Google Scholar]
- Panin VM, Papayannopoulos V, Wilson R, Irvine KD. 1997. Fringe modulates Notch-ligand interactions. Nature. 387:908–912. [DOI] [PubMed] [Google Scholar]
- Panin VM, Shao L, Lei L, Moloney DJ, Irvine KD, Haltiwanger RS. 2002. Notch ligands are substrates for protein O-fucosyltransferase-1 and Fringe. J Biol Chem. 277:29945–29952. [DOI] [PubMed] [Google Scholar]
- Pei Z, Baker NE. 2008. Competition between Delta and the Abruptex domain of Notch. BMC Dev Biol. 8:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellikka M, Tanentzapf G, Pinto M, Smith C, McGlade CJ, Ready DF, Tepass U. 2002. Crumbs, the Drosophila homologue of human CRB1/RP12, is essential for photoreceptor morphogenesis. Nature. 416:143–149. [DOI] [PubMed] [Google Scholar]
- Penton AL, Leonard LD, Spinner NB. 2012. Notch signaling in human development and disease. Semin Cell Dev Biol. 23:450–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perdigoto CN, Schweisguth F, Bardin AJ. 2011. Distinct levels of Notch activity for commitment and terminal differentiation of stem cells in the adult fly intestine. Development. 138:4585–4595. [DOI] [PubMed] [Google Scholar]
- Prout M, Damania Z, Soong J, Fristrom D, Fristrom JW. 1997. Autosomal mutations affecting adhesion between wing surfaces in Drosophila melanogaster. Genetics. 146:275–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radtke F, Wilson A, Stark G, Bauer M, van Meerwijk J, MacDonald HR, Aguet M. 1999. Deficient T cell fate specification in mice with an induced inactivation of Notch1. Immunity. 10:547–558. [DOI] [PubMed] [Google Scholar]
- Rampal R, Luther KB, Haltiwanger RS. 2007. Notch signaling in normal and disease States: Possible therapies related to glycosylation. Curr Mol Med. 7:427–445. [DOI] [PubMed] [Google Scholar]
- Rana NA, Nita-Lazar A, Takeuchi H, Kakuda S, Luther KB, Haltiwanger RS. 2011. O-glucose trisaccharide is present at high but variable stoichiometry at multiple sites on mouse Notch1. J Biol Chem. 286:31623–31637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand MD, Lindblom A, Carlson J, Villoutreix BO, Stenflo J. 1997. Calcium binding to tandem repeats of EGF-like modules. Expression and characterization of the EGF-like modules of human Notch-1 implicated in receptor-ligand interactions. Protein Sci. 6:2059–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebay I, Fleming RJ, Fehon RG, Cherbas L, Cherbas P, Artavanis-Tsakonas S. 1991. Specific EGF repeats of Notch mediate interactions with Delta and Serrate: Implications for Notch as a multifunctional receptor. Cell. 67:687–699. [DOI] [PubMed] [Google Scholar]
- Rizki RM, Rizki TM. 1965. Morphogenetic effects of 6-azauracil and 6-azauridine. Science. 150:222–223. [DOI] [PubMed] [Google Scholar]
- Robinson BS, Huang J, Hong Y, Moberg KH. 2010. Crumbs regulates Salvador/Warts/Hippo signaling in Drosophila via the FERM-domain protein expanded. Curr Biol. 20:582–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaidani Y, Ichiyanagi N, Saito C, Nomura T, Ito M, Nishio Y, Nadano D, Matsuda T, Furukawa K, Okajima T. 2012. O-Linked-N-acetylglucosamine modification of mammalian Notch receptors by an atypical O-GlcNAc transferase Eogt1. Biochem Biophys Res Commun. 419:14–19. [DOI] [PubMed] [Google Scholar]
- Sakaidani Y, Nomura T, Matsuura A, Ito M, Suzuki E, Murakami K, Nadano D, Matsuda T, Furukawa K, Okajima T. 2011. O-Linked-N-acetylglucosamine on extracellular protein domains mediates epithelial cell-matrix interactions. Nat Commun. 2:583. [DOI] [PubMed] [Google Scholar]
- Sasamura T, Ishikawa HO, Sasaki N, Higashi S, Kanai M, Nakao S, Ayukawa T, Aigaki T, Noda K, Miyoshi E et al. . 2007. The O-fucosyltransferase O-fut1 is an extracellular component that is essential for the constitutive endocytic trafficking of Notch in Drosophila. Development. 134:1347–1356. [DOI] [PubMed] [Google Scholar]
- Sasamura T, Sasaki N, Miyashita F, Nakao S, Ishikawa HO, Ito M, Kitagawa M, Harigaya K, Spana E, Bilder D et al. . 2003. Neurotic, a novel maternal neurogenic gene, encodes an O-fucosyltransferase that is essential for Notch-Delta interactions. Development. 130:4785–4795. [DOI] [PubMed] [Google Scholar]
- Savage CR Jr, Hash JH, Cohen S. 1973. Epidermal growth factor. Location of disulfide bonds. J Biol Chem. 248:7669–7672. [PubMed] [Google Scholar]
- Segall JE, Tyerech S, Boselli L, Masseling S, Helft J, Chan A, Jones J, Condeelis J. 1996. EGF stimulates lamellipod extension in metastatic mammary adenocarcinoma cells by an actin-dependent mechanism. Clin Exp Metastasis. 14:61–72. [DOI] [PubMed] [Google Scholar]
- Serth K, Schuster-Gossler K, Kremmer E, Hansen B, Marohn-Kohn B, Gossler A. 2015. O-Fucosylation of DLL3 is required for its function during somitogenesis. PLoS ONE. 10:e0123776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethi MK, Buettner FF, Ashikov A, Krylov VB, Takeuchi H, Nifantiev NE, Haltiwanger RS, Gerardy-Schahn R, Bakker H. 2012. Molecular cloning of a xylosyltransferase that transfers the second xylose to O-glucosylated epidermal growth factor repeats of notch. J Biol Chem. 287:2739–2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethi MK, Buettner FF, Krylov VB, Takeuchi H, Nifantiev NE, Haltiwanger RS, Gerardy-Schahn R, Bakker H. 2010. Identification of glycosyltransferase 8 family members as xylosyltransferases acting on O-glucosylated notch epidermal growth factor repeats. J Biol Chem. 285:1582–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaheen R, Aglan M, Keppler-Noreuil K, Faqeih E, Ansari S, Horton K, Ashour A, Zaki MS, Al-Zahrani F, Cueto-Gonzalez AM et al. . 2013. Mutations in EOGT confirm the genetic heterogeneity of autosomal-recessive Adams-Oliver syndrome. Am J Hum Genet. 92:598–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi S, Stanley P. 2003. Protein O-fucosyltransferase 1 is an essential component of Notch signaling pathways. Proc Natl Acad Sci USA. 100:5234–5239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PL, Myers JT, Rogers CE, Zhou L, Petryniak B, Becker DJ, Homeister JW, Lowe JB. 2002. Conditional control of selectin ligand expression and global fucosylation events in mice with a targeted mutation at the FX locus. J Cell Biol. 158:801–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- South AP, Cho RJ, Aster JC. 2012. The double-edged sword of Notch signaling in cancer. Semin Cell Dev Biol. 23:458–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparrow DB, Chapman G, Wouters MA, Whittock NV, Ellard S, Fatkin D, Turnpenny PD, Kusumi K, Sillence D, Dunwoodie SL. 2006. Mutation of the LUNATIC FRINGE gene in humans causes spondylocostal dysostosis with a severe vertebral phenotype. Am J Hum Genet. 78:28–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprinzak D, Lakhanpal A, Lebon L, Santat LA, Fontes ME, Anderson GA, Garcia-Ojalvo J, Elowitz MB. 2010. Cis-interactions between Notch and Delta generate mutually exclusive signalling states. Nature. 465:86–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl M, Uemura K, Ge C, Shi S, Tashima Y, Stanley P. 2008. Roles of Pofut1 and O-fucose in mammalian Notch signaling. J Biol Chem. 283:13638–13651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley P. 2007. Regulation of Notch signaling by glycosylation. Curr Opin Struct Biol. 17:530–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley P, Guidos CJ. 2009. Regulation of Notch signaling during T- and B-cell development by O-fucose glycans. Immunol Rev. 230:201–215. [DOI] [PubMed] [Google Scholar]
- Stittrich AB, Lehman A, Bodian DL, Ashworth J, Zong Z, Li H, Lam P, Khromykh A, Iyer RK, Vockley JG et al. . 2014. Mutations in NOTCH1 cause Adams-Oliver syndrome. Am J Hum Genet. 95:275–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroman P. 1974. Pyrimidine-sensitive drosophila wing mutants: Withered (whd), tilt (tt) and dumpy (dp). Hereditas. 78:157–168. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Hayashi T, Nishioka J, Kosaka Y, Zushi M, Honda G, Yamamoto S. 1989. A domain composed of epidermal growth factor-like structures of human thrombomodulin is essential for thrombin binding and for protein C activation. J Biol Chem. 264:4872–4876. [PubMed] [Google Scholar]
- Takeuchi H, Fernandez-Valdivia RC, Caswell DS, Nita-Lazar A, Rana NA, Garner TP, Weldeghiorghis TK, Macnaughtan MA, Jafar-Nejad H, Haltiwanger RS. 2011. Rumi functions as both a protein O-glucosyltransferase and a protein O-xylosyltransferase. Proc Natl Acad Sci USA. 108:16600–16605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi H, Haltiwanger RS. 2014. Significance of glycosylation in Notch signaling. Biochem Biophys Res Commun. 453:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi H, Kantharia J, Sethi MK, Bakker H, Haltiwanger RS. 2012. Site-specific O-glucosylation of the epidermal growth factor-like (EGF) repeats of notch: Efficiency of glycosylation is affected by proper folding and amino acid sequence of individual EGF repeats. J Biol Chem. 287:33934–33944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan JB, Xu K, Cretegny K, Visan I, Yuan JS, Egan SE, Guidos CJ. 2009. Lunatic and manic fringe cooperatively enhance marginal zone B cell precursor competition for delta-like 1 in splenic endothelial niches. Immunity. 30:254–263. [DOI] [PubMed] [Google Scholar]
- Taylor P, Takeuchi H, Sheppard D, Chillakuri C, Lea SM, Haltiwanger RS, Handford PA. 2014. Fringe-mediated extension of O-linked fucose in the ligand-binding region of Notch1 increases binding to mammalian Notch ligands. Proc Natl Acad Sci USA. 111:7290–7295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tepass U, Theres C, Knust E. 1990. Crumbs encodes an EGF-like protein expressed on apical membranes of Drosophila epithelial cells and required for organization of epithelia. Cell. 61:787–799. [DOI] [PubMed] [Google Scholar]
- Traverse S, Seedorf K, Paterson H, Marshall CJ, Cohen P, Ullrich A. 1994. EGF triggers neuronal differentiation of PC12 cells that overexpress the EGF receptor. Curr Biol. 4:694–701. [DOI] [PubMed] [Google Scholar]
- Tsao PN, Vasconcelos M, Izvolsky KI, Qian J, Lu J, Cardoso WV. 2009. Notch signaling controls the balance of ciliated and secretory cell fates in developing airways. Development. 136:2297–2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Pavert SA, Kantardzhieva A, Malysheva A, Meuleman J, Versteeg I, Levelt C, Klooster J, Geiger S, Seeliger MW, Rashbass P et al. . 2004. Crumbs homologue 1 is required for maintenance of photoreceptor cell polarization and adhesion during light exposure. J Cell Sci. 117:4169–4177. [DOI] [PubMed] [Google Scholar]
- Vasudevan D, Takeuchi H, Johar SS, Majerus E, Haltiwanger RS. 2015. Peters plus syndrome mutations disrupt a noncanonical ER quality-control mechanism. Curr Biol. 25:286–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visan I, Tan JB, Yuan JS, Harper JA, Koch U, Guidos CJ. 2006. Regulation of T lymphopoiesis by Notch1 and Lunatic fringe-mediated competition for intrathymic niches. Nat Immunol. 7:634–643. [DOI] [PubMed] [Google Scholar]
- Wang Y, Shao L, Shi S, Harris RJ, Spellman MW, Stanley P, Haltiwanger RS. 2001. Modification of epidermal growth factor-like repeats with O-fucose. Molecular cloning and expression of a novel GDP-fucose protein O-fucosyltransferase. J Biol Chem. 276:40338–40345. [DOI] [PubMed] [Google Scholar]
- Wang Y, Spellman MW. 1998. Purification and characterization of a GDP-fucose:polypeptide fucosyltransferase from Chinese hamster ovary cells. J Biol Chem. 273:8112–8118. [DOI] [PubMed] [Google Scholar]
- Whitworth GE, Zandberg WF, Clark T, Vocadlo DJ. 2010. Mammalian Notch is modified by D-Xyl-alpha1-3-D-Xyl-alpha1-3-D-Glc-beta1-O-Ser: Implementation of a method to study O-glucosylation. Glycobiology. 20:287–299. [DOI] [PubMed] [Google Scholar]
- Wilkin MB, Becker MN, Mulvey D, Phan I, Chao A, Cooper K, Chung HJ, Campbell ID, Baron M, MacIntyre R. 2000. Drosophila dumpy is a gigantic extracellular protein required to maintain tension at epidermal-cuticle attachment sites. Curr Biol. 10:559–567. [DOI] [PubMed] [Google Scholar]
- Winkler ME, Bringman T, Marks BJ. 1986. The purification of fully active recombinant transforming growth factor alpha produced in Escherichia coli. J Biol Chem. 261:13838–13843. [PubMed] [Google Scholar]
- Xiao Z, Patrakka J, Nukui M, Chi L, Niu D, Betsholtz C, Pikkarainan T, Vainio S, Tryggvason K. 2011. Deficiency in crumbs homolog 2 (Crb2) affects gastrulation and results in embryonic lethality in mice. Dev Dyn. 240:2646–2656. [DOI] [PubMed] [Google Scholar]
- Yamamoto S, Charng WL, Rana NA, Kakuda S, Jaiswal M, Bayat V, Xiong B, Zhang K, Sandoval H, David G et al. . 2012. A mutation in EGF repeat-8 of Notch discriminates between Serrate/Jagged and Delta family ligands. Science. 338:1229–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Klein R, Tian X, Cheng HT, Kopan R, Shen J. 2004. Notch activation induces apoptosis in neural progenitor cells through a p53-dependent pathway. Dev Biol. 269:81–94. [DOI] [PubMed] [Google Scholar]
- Yang LT, Nichols JT, Yao C, Manilay JO, Robey EA, Weinmaster G. 2005. Fringe glycosyltransferases differentially modulate Notch1 proteolysis induced by Delta1 and Jagged1. Mol Biol Cell. 16:927–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao D, Huang Y, Huang X, Wang W, Yan Q, Wei L, Xin W, Gerson S, Stanley P, Lowe JB et al. . 2011. Protein O-fucosyltransferase 1 (Pofut1) regulates lymphoid and myeloid homeostasis through modulation of Notch receptor ligand interactions. Blood. 117:5652–5662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaczek A, Brandt B, Bielawski KP. 2005. The diverse signaling network of EGFR, HER2, HER3 and HER4 tyrosine kinase receptors and the consequences for therapeutic approaches. Histol Histopathol. 20:1005–1015. [DOI] [PubMed] [Google Scholar]
- Zelhof AC, Hardy RW, Becker A, Zuker CS. 2006. Transforming the architecture of compound eyes. Nature. 443:696–699. [DOI] [PubMed] [Google Scholar]
- Zhang S, Chung WC, Wu G, Egan SE, Miele L, Xu K. 2015. Manic fringe promotes a claudin-low breast cancer phenotype through Notch-mediated PIK3CG induction. Cancer Res. 75:1936–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, Gridley T. 1998. Defects in somite formation in lunatic fringe-deficient mice. Nature. 394:374–377. [DOI] [PubMed] [Google Scholar]
- Zhang DW, Lagace TA, Garuti R, Zhao Z, McDonald M, Horton JD, Cohen JC, Hobbs HH. 2007. Binding of proprotein convertase subtilisin/kexin type 9 to epidermal growth factor-like repeat A of low density lipoprotein receptor decreases receptor recycling and increases degradation. J Biol Chem. 282:18602–18612. [DOI] [PubMed] [Google Scholar]
- Zhang N, Norton CR, Gridley T. 2002. Segmentation defects of Notch pathway mutants and absence of a synergistic phenotype in lunatic fringe/radical fringe double mutant mice. Genesis. 33:21–28. [DOI] [PubMed] [Google Scholar]
- Zhou L, Li LW, Yan Q, Petryniak B, Man Y, Su C, Shim J, Chervin S, Lowe JB. 2008. Notch-dependent control of myelopoiesis is regulated by fucosylation. Blood. 112:308–319. [DOI] [PMC free article] [PubMed] [Google Scholar]