

Abstract
We have reported that xeroderma pigmentosum group A (Xpa) gene-knockout mice [Xpa (−/−) mice] are deficient in nucleotide excision repair (NER) and highly sensitive to UV-induced skin carcinogenesis. Although xeroderma pigmentosum group A patients show growth retardation, immature sexual development, and neurological abnormalities as well as a high incidence of UV-induced skin tumors, Xpa (−/−) mice were physiologically and behaviorally normal. In the present study, we kept Xpa (−/−) mice for two years under specific pathogen-free (SPF) conditions and found that the testis diminished in an age-dependent manner, and degenerating seminiferous tubules and no spermatozoa were detected in the 24-month old Xpa (−/−) mice. In addition, a higher incidence of spontaneous tumorigenesis was observed in the 24-month old Xpa (−/−) mice compared to Xpa (+/+) controls. Xpa (−/−) mice provide a useful model for investigating the aging and internal tumor formation in XP-A patients.
Keywords: Xeroderma pigmentosum (XP), Nucleotide excision repair (NER), Spermatogenesis, Spontaneous tumorigenesis, Knockout mice
1. Introduction
Xeroderma pigmentosum (XP) is an autosomal recessive disease characterized by an extreme hypersensitivity to sunlight, a predisposition to skin cancer on sun-exposed areas, and progressive neurological abnormalities. Cells from XP patients show hypersensitivity to killing by UV-irradiation. There are eight genetic complementation groups in XP; XP-A through XP-G and a variant (XP-V). The primary defect in XP-A through XP-G resides in an early step of nucleotide excision repair (NER) while XP-V has a normal NER process but a defect in translesion DNA synthesis. To date, the genes responsible for NER and translesion synthesis associated XP have been identified (XPA-XPG and XPV, respectively), and core NER reactions have been reconstituted using purified proteins including XPA-XPG proteins (1). We have made Xpa gene-knockout mice [Xpa (−/−) mice] by the insertion of neo gene into the exon 4. The mice were defective in NER and highly susceptible to UVB- or DMBA-induced skin tumorigenesis, verifying that Xpa (−/−) mice are a good animal model with which to study UVB-induced skin tumorigenesis in XP-A patients (2). To elucidate the molecular events involved in UVB-induced skin tumorigenesis in Xpa (−/−) mice, we have established five cell lines derived from skin cancers induced in UVB-irradiated Xpa (−/−) mice (3). In spite of a complete loss of NER activity, the skin cancer cell lines were resistant to killing by UV-irradiation. Further, three of these cell lines were deficient in UV-induced G1/S cell cycle checkpoint and mismatch repair activity, suggesting that a mismatch repair-defect is involved in the UVB-induced skin carcinogenesis in Xpa (−/−) mice (3, and our unpublished data). Using Xpa (−/−) mice containing the rpsL-reporter gene, we found that the frequency of UVB-induced mutations such as the CC to TT tandem transition was higher than in wild-type mice (4, 5). We also found that UVB-induced immunosuppression was enhanced in Xpa (−/−) mice (6).
In addition to photosensitivity and a high incidence of sun-induced skin cancer, most XP-A patients show growth retardation, immature sexual development, and neurological abnormalities such as microcephaly and ataxia in the first 7 years after birth (7, 8). Some XP patients also show a high incidence of spontaneous tumorigenesis (8, 9). Endogenous DNA lesions that are repaired by NER but not by other pathways, which include the 8,5′-cyclopurine deoxynucleosides (cPu) (10, 11), lipid aldehyde derived DNA lesions such as M1G (12,13,14) and possibly others (15), are likely to be responsible for these pathologic effects.
In the present study, we kept Xpa (−/−) mice for 24 months under specific pathogen-free (SPF) conditions, and found that they exhibited an impairment of spermatogenesis and higher incidence of spontaneous tumorigenesis later in life. We discuss the possible mechanistic explanations for these observations.
2. Materials and methods
2.1. Mutant mice
The Xpa gene-knocked out mice [Xpa (−/−) mice] were generated by the insertion of neo gene into the exon 4 of mouse Xpa gene (2), and had a chimeric genetic background of CBA/C57BL6/CD-1. W/Wv mice are compound heterozygotes for W and Wv (W-viable) mutations in the KIT gene that encodes a receptor tyrosine kinase. These mutations are known to cause a defect in tyrosine kinase activity and induce pleiotropic effects, such as sterility, macrocytic anemia, and depletion of melanocytes and mast cells. Severe impairment of fertility in W/Wv male mice is due to the almost total absence of germ cells in the gonads. W/Wv mice are widely used in the germ cell-deficient testis model (16, 17, 18). The mice were kept under specific pathogen-free (SPF) conditions for an extended period. All animals were housed in a controlled environment at 20–26°C and fed on a CE-2 diet (CLEA Japan Inc., Tokyo, Japan) and sterilized water ad libitum. The humidity of the room was 45–70%. The room lights were turned on at AM 7:00 and off at PM 7:00.
2.2. RNA extraction and Northern blotting
For the Northern blot analysis of mouse tissues, a mouse MTN blot membrane (Clontech), containing 2 μg of poly (A)+ RNA per track, was used. For the Northern blot analysis of mouse testes, freshly removed mouse testes (8-day, 16-day, and 10-week-old C57BL/6 and W/Wv mice) were weighed and homogenized in Isogen (Nippongene, Tokyo, Japan). Total RNA was extracted according to the manufacturer’s recommendations, quantified by measuring optical density, separated by electrophoresis on a 1% formaldehyde gel, and transferred to a nitrocellulose membrane. After UV-crosslinking, the nitrocellulose membrane was prehybridized in 50% formamide, 4x SSC, 5x Denhardt’s solution, 0.2% SDS, and 120 μg/ml denatured sonicated salmon sperm DNA at 42°C for 2 h and hybridized under the same conditions for 24 h with a [32P]-labeled full-length Xpa cDNA probe that was made by the random primer method. The membrane was washed with 0.1x SSC, 0.5% SDS at 55°C for 30 min. Autoradiography and quantification of the hybridized signals were carried out with a Molecular Imager (Bio-Rad GS525). C57BL/6 mice and W/Wv mice were purchased from Japan SLC Inc., Shizuoka, Japan.
2.3. Fractionation of normal testicular cells and histologic examination of testis
For the fractionation of testicular cells, four testes were collected from 10-week-old C57BL/6 mice, and the tunica albuginea was removed. The seminiferous tubules were placed in Eagle’s minimal essential medium (MEM) containing 0.02M Hepes and 0.1% collagenase (Wako, Osaka, Japan), gently unraveled with forceps, and incubated at 33°C for 30 min. After the addition of PBS containing 1mM EDTA, the tubule suspension was transferred into a conical tube and kept standing for 5 min to precipitate tubule fragments. The supernatant containing separated cells was filtrated through a nylon mesh Cell Strainer (Becton Dickinson Co., New Jersey, USA) and centrifuged at 600x g for 10 min. The precipitate was used as a Leydig cell fraction. The remaining tubules were re-incubated in MEM containing collagenase at 33°C for 15min and then dispersed by gentle pipetting a few times in PBS containing 1mM EDTA to remove residual Leydig cells. Tubules were then transferred to a plastic Petri dish, cut into small fragments with a knife, transferred to a 50ml conical tube, and washed by pipetting in PBS containing 1mM EDTA. The conical tube was kept standing for 5 min and the supernatant fraction was used as a germ cell fraction. The remaining sedimented tubules were vigorously pipetted a few times. Then, the sample was kept standing for 5 min. The supernatant fraction containing mainly the remaining germ cells was discarded. The sedimented sample was used as a tubule fraction (containing mainly Sertoli cells).
The testes were examined microscopically. Each testis was fixed in Bouin’s solution and embedded in paraffin. Two to 3 sections from each testis stained with hematoxylin and eosin were examined. For the measurement of tubule diameters, a minimum of 30 tubules per testis were examined and the frequency distribution was determined. Only round, not oval-shaped, cross-sections of seminiferous tubules were counted. We separated more than 50 seminiferous tubules of each testis into 5 categories: 0-Empty tubules, I-Sertoli cells only, II-Sertoli cells and spermatogonia, III- Sertoli cells, spermatogonia, and spermatocytes, and IV-normal tubules. Germ cells were differentially counted by use of the criteria of Leblond and Clermonts (19) and of Oakberg (20). The epididymis was homogenized in phosphate-buffered saline, to count the number of spermatozoa using a Neubauer hemocytometer.
2.4. Determination of organ weight and tumor sampling
1. The mice were sacrificed by cervical dislocation and organs including brain (cutting level was from olfactory bulb to the end of cerebellum), lung, heart, liver, spleen, kidneys, and testes were removed and weighed. The spontaneous tumors and organs were fixed in 4% paraformaldehyde and embedded in paraffin. Paraffin sections 5 μm thick were prepared and stained with hematoxylin and eosin for light microscopic examination. Tumor-free mice, based pathological examinations were used to analyze organ weight and spermatogenesis. All experiments were conducted in accordance with the Institutional Guidelines for Osaka University.
2.5. Hormonal Assay
Blood was collected by orbital sinus puncture under ether anesthesia. Testosterone concentrations were measured using a total testosterone kit (Diagnostic Products Corporation, Los Angeles, USA). FSH and LH concentrations were measured using a rat FSH and LH kit from Amersham Pharmacia Biotech (Little Chalfont, Buckinghamshire, UK). Statistical differences were examined using the Mann-Whitney test.
3. Results
3.1. Reduction of organ and body weights in Xpa (−/−) mice
We have reported that Xpa (−/−) mice develop normally and are physiologically and behaviorally indistinguishable from Xpa (+/+) mice. However, a comparison of body weights at various ages up to two years revealed a reduction in the weights of Xpa (−/−) mice compared with the Xpa (+/+) littermates in males (p<0.05), but no difference in females (Fig. 1A and 1B). It is known that some XP-A patients show growth retardation and have smaller organs (kidney, brain, testis, etc) than normal individuals (our unpublished data). Therefore, we measured the weight of several organs (liver, heart, spleen, kidney, lung, testis, ovary, and brain) and examined their histopathology in the Xpa (−/−) and Xpa (+/+) mice at the ages of 3, 6, 12, and 24 months.
Fig. 1. Body weight and relative weight ratio of organs in the Xpa (−/−) and Xpa (+/+) mice.


(A). Body weight of male Xpa (−/−) and Xpa (+/+) mice. Body weight was significantly reduced in the Xpa (−/−) mice when compared with the Xpa (+/+) mice (p<0.01:11–20, 100, and 104-week-old Xpa (−/−) mice and p<0.05: 10, 24–32, and 96-week-old-Xpa (−/−) mice). The numbers of animals examined were 20 and 32 in the Xpa (−/−) and Xpa (+/+) mice, respectively. Body weight of mice was measured weekly at 10 – 104 weeks of age. (B). Body weight of female Xpa (−/−) and Xpa (+/+) mice. The numbers of animals examined were 28 and 32 in the Xpa (−/−) and Xpa (+/+) mice, respectively. (C-G). Relative weight ratio (organ weight/body weight) of brain, kidney and testis in the Xpa (−/−) and Xpa (+/+) mice. (C). Relative weight ratio of brain in male Xpa (−/−) and Xpa (+/+) mice. Solid circles (green) indicate the mean values of relative weight ratio of brain in Xpa (−/−) mice, while solid triangles (red) indicate those in Xpa (+/+) mice. The relative weight ratio of brain of male Xpa (−/−) mice was significantly small when compared with the Xpa (+/+) mice at the age of 12 months (#p<0.05). (D). Relative weight ratio of brain in female Xpa (−/−) and Xpa (+/+) mice. The relative weight ratio of brain in female Xpa (−/−) mice was significantly small when compared with that in female Xpa (+/+) mice at the age of 12 months (*p<0.0005). (E). Relative weight ratio of kidney in male Xpa (−/−) and Xpa (+/+) mice. Kidney in Xpa (−/−) mice was significantly small when compared with that in Xpa (+/+) mice at age of 3 months (*p<0.0005). (F). Relative weight ratio of kidney in female Xpa (−/−) and Xpa (+/+) mice. Kidney of female Xpa (−/−) mice was significantly small when compared with that in Xpa (+/+) mice at ages of 6 months and 12 months (+p<0.01, *p<0.0005). (G). Relative weight ratio of testis of Xpa (−/−) and Xpa (+/+) mice. The combined weight of both testes is shown. The relative weight ratio of testis in Xpa (−/−) mice was significantly small when compared with that in Xpa (+/+) mice after 3 month-old (#p<0.05, *p<0.0005).
For the analysis of organ weights in the male Xpa (+/+) mice, numbers of animals examined were 25, 25, 23, and 15 at the ages of 3 months, 6 months, 12 months, and 24 months, respectively. While in the male Xpa (−/−) mice, 16, 25, 26, and 15 mice were examined at the ages of 3 months, 6 months, 12 months, and 24 months, respectively. In the case of female Xpa (+/+) mice, numbers examined were 25, 25, 26, and 16 at the ages of 3 months, 6 months, 12 months, and 24 months, respectively. While in the female Xpa (−/−) mice, 29, 25, 25, and 14 mice were examined at the ages of 3 months, 6 months, 12 months, and 24 months, respectively. Error bars show SEM. Statistical differences were examined using the Mann-Whitney test. For the organ weight analyses, mice without tumors were examined.
Organ weights were normalized by body weights, and the relative weight ratio of brain, kidney and testis in male and female mice was shown in Fig. 1C, 1D, 1E, 1F, 1G. In the male mice, the relative weight ratio of brain, kidney and testis was significantly decreased in the Xpa (−/−) mice when compared with that in the Xpa (+/+) mice (Fig. 1C, 1E, 1G) [kidney (p<0.01) at 3, 6, 12, and 24 months; testes at 3 months (p<0.05) and at 6, 12, 24 months (p<0.01); brain (p<0.05) at 12 months]. Interestingly, the relative weight ratio of testes in the Xpa (−/−) mice decreased markedly in an age-dependent manner while only slightly decreased in the Xpa (+/+) mice. Decreased organ weight in male Xpa (−/−) mice was also observed in heart, lung, and liver at some ages [heart (p<0.05) at 3 and 24 months; lung (p<0.05) at 3 and 6 months; liver (p<0.01) at 24 months] (data not shown). In the female mice, the relative weight ratio of some organs in the Xpa (−/−) mice was also significantly smaller as compared with that in the Xpa (+/+) mice (Fig. 1D and 1F, and data not shown) [kidney (p<0.01) at 6 and 12 months; brain (p<0.05) at 12 months; spleen (p<0.05) at 6 months; liver (p<0.01) at 12 months; lung (p<0.01) at 12 months; heart (p<0.01) at 12 months old]. We do not have an explanation for gender specific differences in organ and body weight in Xpa (−/−) mice as compared to Xpa (+/+) mice. The difference may be due to a hormonal effect between male and female mice.
In view of the reduced organ weights in the Xpa (−/−) mice, we looked for histopathological changes in hematoxylin and eosin stained organs. However, we detected no histopathological abnormalities in the liver, heart, lung, spleen, kidney, and brain in the Xpa (−/−) mice. In addition, the ovaries appeared normal and contained ova and follicles, sometimes with signs of cyclic changes, indicating that pituitary regulation functions normally in the Xpa (−/−) mice.
As the Xpa (−/−) mice had a small brain at 12 months old, we examined whether they show neurological abnormalities by conducting behavioral tests such as the “foot-print test” to identify an ataxic gait, “circling behavior” check for a balance-abnormality, and “tremors” in the 24-month old Xpa (−/−) mice. Our results indicated that the Xpa (−/−) mice behaved normally in these tests (data not shown). We also checked the calvaria, because abnormal thickening of calvarial bones was observed in XP-A patients (7). We could not find any difference in the calvaria between Xpa (−/−) and Xpa (+/+) mice (data not shown).
3.2. Impairment of spermatogenesis in Xpa (−/−) mice
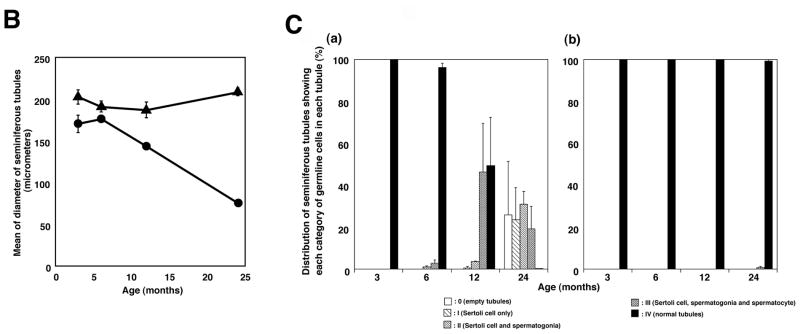
Consistent with the findings that 12 % of adult XP patients show immature sexual development (8), we found that the weight of the testis in the Xpa (−/−) mice decreased in an age-dependent manner while no decrease was observed in the Xpa (+/+) mice. Therefore, we examined the testis in detail in the Xpa (−/−) and Xpa (+/+) mice at various ages. The relative weight ratio of the testis in the Xpa (−/−) mice diminished even at 3 months, and at 24 months old, was reduced to one-third of that in the Xpa (+/+) mice. A marked reduction in the weight of the testis paralleled the pronounced overall regression of spermatogenesis (Fig. 2A). Male Xpa (−/−) mice were fertile until about 30 weeks of age. Histological analysis at the age of 3 months revealed that the testes of Xpa (−/−) mice had normal spermatogenesis although they were small (Fig. 2Aa, b). At the age of 6 months, the Xpa (−/−) mice had a few degenerative seminiferous tubules of various diameters containing Sertoli cells and spermatogonia (categories II), or Sertoli cells, spermatogonia, and spermatocytes (categories III) (Fig. 2Ac, d and Fig. 2Ca). At 12 months of age, half of the seminiferous tubules in the Xpa (−/−) mice had spermatogenic epithelia with vacuoles, disarrays of germ cells, and hyperplasia of Leydig cells (categories II and III) (Fig. 2Ae, f and Fig. 2Ca). The rest were normal. At 24 months, all of the seminiferous tubules showed abnormal spermatogenesis (Fig. 2Ag, h), belonging to category 0, I, II or III. No category IV (normal) tubules were detected (Fig. 2Ca). On the other hand, almost all the seminiferous tubules belonged to category IV (normal) in the Xpa (+/+) mice even at the age of 24 months (Fig. 2Ag and Fig. 2Cb). In addition, all of the seminiferous tubules in the Xpa (−/−) mice at 12 and 24 months showed hyperplasia of Leydig cells (Fig. 2Af, h).
Fig. 2. Impaired spermatogenesis in the Xpa (−/−) mice.
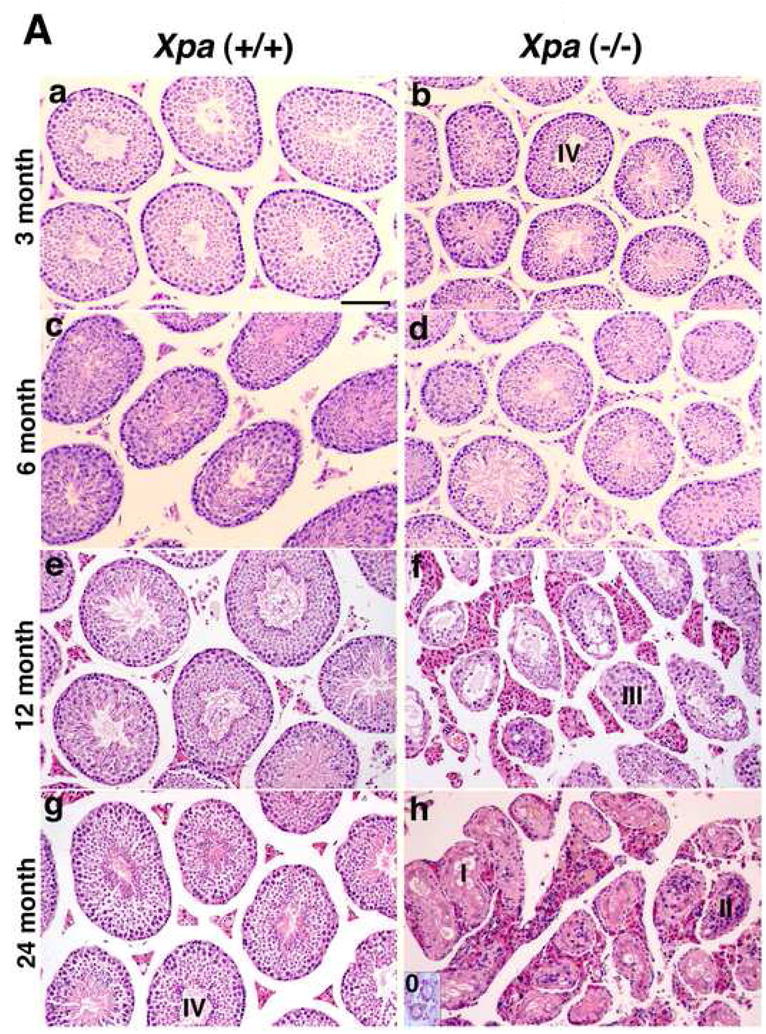
(A). Histology of testis in the Xpa (+/+) and Xpa (−/−) mice.
Histological sections were prepared as described in the Materials and methods 2.3. and stained with hematoxylin and eosin. The panels to the left (a, c, e, g) show the testis of Xpa (+/+) mice, while the panels to the right (b, d, f, h) show that of Xpa (−/−) mice. (a), (b): 3-month-old mice; (c), (d): 6-month-old mice; (e), (f): 12-month-old mice; (g), (h): 24-month-old mice. Bar in (a) corresponds to 100μm. Seminiferous tubules were separated into 5 categories (see Materials and methods 2.3). 0-Empty tubules, I-Sertoli cells only, II-Sertoli cells and spermatogonia, III- Sertoli cells, spermatogonia, and spermatocytes, and IV-normal tubules. Note that in (a) and (b), the diameter of seminiferous tubules in the 3-month-old Xpa (−/−) mice is shorter than in the Xpa (+/+) littermates; in (d), the diameters of seminiferous tubules in the Xpa (−/−) mice are very variable; in (f), the testis displayed grossly dysmorphic seminiferous tubules (vacuolated tubules, disarrays of germ cells) with hyperplasia of Leydig cells; in (h), the testis displayed completely disrupted seminiferous tubules, and empty tubules were shown in the inset of Fig. 2Ah. (B). The diameter of seminiferous tubules in the Xpa (+/+) and Xpa (−/−) mice. The mean and SEM of diameters are indicated. Closed triangle: Xpa (+/+) mice; closed circle: Xpa (−/−) mice. Note that the diameter of seminiferous tubules in the Xpa (−/−) mice decreased in an age-dependent manner. (C). Classification of the seminiferous tubules into 5 categories depending on the germline cells and the chronological distribution of each category. (a) testis of Xpa (−/−) mice; (b) testis of Xpa (+/+) mice, □ category 0 (empty tubules) [Fig. 2A, h, inset];  category I (Sertoli cells only) [Fig. 2A, h];
category I (Sertoli cells only) [Fig. 2A, h];  category II (Sertoli cells and spermatogonia) [Fig. 2A, h];
category II (Sertoli cells and spermatogonia) [Fig. 2A, h];  category III (Sertoli cells, spermatogonia, and spermatocytes) [Fig. 2A, f]; ■ category IV (normal tubules) [Fig. 2Ab, g]. The mean value and SEM of the frequency of each category are indicated. Degenerative seminiferous tubules (category 0, I, II, and III) gradually increased in the Xpa (−/−) mice in an age-dependent manner. For the classification analysis, mice without tumors were examined.
category III (Sertoli cells, spermatogonia, and spermatocytes) [Fig. 2A, f]; ■ category IV (normal tubules) [Fig. 2Ab, g]. The mean value and SEM of the frequency of each category are indicated. Degenerative seminiferous tubules (category 0, I, II, and III) gradually increased in the Xpa (−/−) mice in an age-dependent manner. For the classification analysis, mice without tumors were examined.
We also examined the number of spermatozoa from epididymis. The sperm number in the 24-month-old Xpa (+/+) mice was 3.9 ± 0.7 (×106) cells/ml, but no spermatozoa were found in the 24-month-old Xpa (−/−) mice. Consistent with these findings, the serum follicle-stimulating hormone (FSH) level (ng/ml) in the 24-month-old Xpa (−/−) mice was significantly elevated when compared with that in Xpa (+/+) mice [Xpa (+/+) vs. Xpa (−/−) = 23.6 ± 7 (N=5) vs. 54.2 ± 14 (N=8) (mean ± SD) (P<0.05)]. Serum testosterone and luteinizing hormone (LH) levels in the 24-month-old Xpa (−/−) mice were the same as those in the Xpa (+/+) mice (data not shown). These results suggested that the loss of germ cells in the testis of Xpa (−/−) mice resulted in the elevation of serum FSH in the Xpa (−/−) mice.
3.3. Expression of Xpa gene in mouse organs and different testis cell types
Our results indicated that the decrease in organ weight in the Xpa (−/−) mice was dependent on the type of organ. We examined whether the level of Xpa expression correlates with the decrease. The Xpa mRNA was detected as a 1.0–1.1kb transcript by Northern blot analysis. The expression of the Xpa gene was strong in heart, kidney, liver, and testis, the weights of which were diminished in the Xpa (−/−) mice (Fig. 3A, data not shown). These results suggested that there is some correlation between the level of Xpa mRNA and the reduction in organ weight, although there are exceptions such as brain and liver.
Fig. 3. Expression of Xpa mRNA in mouse organs.

(A). The mouse Xpa cDNA probe was hybridized with 2 μg of poly (A)+ RNA from each mouse organ as indicated (Clontech). β-actin cDNA was used as a control probe (lower). (B). Expression of Xpa mRNA in the testis of normal mice at various ages, in the testis of W/Wv mutant mice, and in the fractions of testicular germ cells and somatic cells. Northern blots containing 30 μg of total RNA from testes of 8 day-, 16 day-, and 10 week-old mice, and germ cell-deficient mice (W/Wv) were hybridized with the probes of Xpa (upper) and β-actin (lower). Fractions of germ cells, Leydig cells, and Sertoli cells were prepared for Northern blot analyses as described in Materials and methods. Thirty micrograms of total RNA from each fraction was electrophoresed and blotted to nitrocellulose filters and hybridized with 32P-labeled Xpa cDNA. All filters were re-hybridized with β-actin cDNA as a control.
Xpa mRNA was expressed in the testis of 8-day-old, 16-day-old, and adult mice. In addition, the mRNA was detected in the testis of W/Wv mice that have no spermatogenesis. Xpa mRNA was detected in the fractions of Leydig cells and Sertoli cells as well as spermatogenic cells (Fig. 3B). These results indicate that the Xpa gene is expressed in the testis in which spermatogenesis has not occurred yet, and is also expressed in the testicular somatic cells. Taken together, these results suggested that both spermatogenic cells and testicular somatic cells express the Xpa mRNA. However, while spermatogonia and spermatocyte production and differentiation cannot continue normally without Xpa throughout the life span of mice, Leydig cells show increased proliferation in the absence of Xpa.
3.4. High incidence of spontaneous tumorigenesis in Xpa (−/−) mice
We have reported that Xpa (−/−) mice are deficient in the repair of UV-damage and show a higher incidence of UVB-induced skin tumorigenesis and mutation rate in UVB-irradiated skin than Xpa (+/+) mice (2, 4, 5). In the present work, we examined whether spontaneous tumorigenesis is increased in Xpa (−/−) mice. No distinct difference in longevity between the Xpa (+/+) and Xpa (−/−) mice was observed in Kaplan-Meier format (data not shown). After 3, 6, 12, 18 and 24 months, the mice were necropsied by the same examiner (H.N.) at each time point. We examined 54 Xpa (+/+) mice and 49 Xpa (−/−) mice at the age of 3 months, 55 Xpa (+/+) mice and 55 Xpa (−/−) mice at the age of 6 months, 49 Xpa (+/+) mice and 55 Xpa (−/−) mice at the age of 12 months, and 52 Xpa (+/+) mice and 51 Xpa (−/−) mice at the age of 18–24 months. In the 3 and 6-month-old mice, we did not detect any spontaneous tumors in Xpa (+/+) or Xpa (−/−) mice. In the 12-month-old mice, four Xpa (−/−) mice had tumors (4/55; 7.3%) while no Xpa (+/+) mice had tumors. Thus, the Xpa (−/−) mice developed spontaneous tumors earlier than the Xpa (+/+) mice. In the 18- to 24-month-old mice, eighteen Xpa (−/−) mice (18/51; 35.3%) and ten Xpa (+/+) mice (10/52; 19.2%) had tumors (Fig. 4). The total number of tumor-bearing mice was 22 out of 106 (21%) in the Xpa (−/−) mice and 10 out of 101 (10%) in the Xpa (+/+) mice. These results indicated that the Xpa (−/−) mice developed more spontaneous tumors than the Xpa (+/+) mice (p=0.0352<0.05 by chi-square test).
Fig. 4. Spontaneous malignant tumor yield during 24 months.

The time course of tumor formation and tumor yield in the Xpa (−/−) mice (closed circles) and Xpa (+/+) mice (closed triangles) are shown. Tumor numbers of 3, 6, 12, 24-month-old Xpa (−/−) and Xpa (+/+) mice were counted and divided by the number of mice examined in each group. Xpa (−/−) mice had more tumors than Xpa (+/+) mice through 24 months. We examined 54 Xpa (+/+) mice and 49 Xpa (−/−) mice at 3 months of age, 55 Xpa (+/+) mice and 55 Xpa (−/−) mice at 6 months, 49 Xpa (+/+) mice and 55 Xpa (−/−) mice at 12 months, and 52 Xpa (+/+) mice and 51 Xpa (−/−) mice at 24 months (24 month old mice contained 18,21 month old mice.). When a statistical analysis was carried out comparing the total number of mice bearing tumors between the 12-, 24-month-old-Xpa (−/−) and Xpa (+/+) groups, a significant difference was found in these two groups: 22 of 106 (21%) Xpa (−/−) mice compared with 10 of 101 (10%) Xpa (+/+) mice (**P=0.0352<0.05:chi-square test).
The results of histological examinations of all the spontaneous tumors in the Xpa (−/−) and Xpa (+/+) mice are summarized in Tables 1 and 2. Some of them are shown in Fig. 5. Four of the 21- to 24-month-old Xpa (−/−) mice (mouse 7, 8, 9, and 12) had multiple tumors, but no such tumors were detected in the 24 month-old Xpa (+/+) mice. Hemangiosarcoma (mouse 5), renal cell carcinoma (mouse 8), and squamous cell carcinoma (mouse 11) were detected only in the Xpa (−/−) mice. Taken together, these results indicate that the Xpa (−/−) mice show a higher incidence of spontaneous tumorigenesis than the Xpa (+/+) mice, as well as a different spectrum of tumors.
Table 1.
Histopathological findings of spontaneous tumors in the Xpa (−/−) and Xpa (+/+) mice.
| Mouse No. (Genotype) | Sex | Age (Months) | Histological type | Anatomic site |
|---|---|---|---|---|
| 1(−/−) | M | 12 | malignant lymphoma | spleen, LN |
| 2(−/−) | M | 12 | well-differentiated adenocarcinoma (papillary) | lung |
| 3(−/−) | F | 12 | malignant lymphoma | spleen |
| 4(−/−) | F | 12 | malignant lymphoma | spleen, liver, kidney |
| 5(−/−) | F | 18 | hemangiosarcoma (haemoangiopericytoma) | right leg |
| 6(−/−) | F | 18 | malignant lymphoma | spleen |
| 7(−/−) | M | 21 | HCC alveolar tumor | liver lung |
| 8(−/−) | M | 24 | HCC renal cell carcinoma | liver kidney |
| 9(−/−) | M | 24 | well-differentiated adenocarcinoma (papillary) malignant lymphoma | lung LN |
| 10(−/−) | M | 24 | malignant lymphoma | kidney |
| 11(−/−) | M | 24 | moderately differentiated squamous cell carcinoma | left ear |
| 12(−/−) | M | 24 | malignant lymphoma well-differentiated adenocarcinoma (papillary) | spleen, liver, kidney lung |
| 13(−/−) | M | 24 | HCC | liver |
| 14(−/−) | F | 24 | malignant lymphoma | lung, spleen, liver, kidney |
| 15(−/−) | F | 24 | malignant lymphoma | LN |
| 16(−/−) | F | 24 | malignant lymphoma | LN |
| 17(−/−) | F | 24 | malignant lymphoma | LN |
| 18(−/−) | F | 24 | HCC | liver |
| 19(+/+) | M | 24 | well-differentiated adenocarcinoma (papillary) | lung |
| 20(+/+) | M | 24 | adenocarcinoma | lung |
| 21(+/+) | M | 24 | HCC | liver |
| 22(+/+) | M | 24 | malignant lymphoma | spleen |
| 23(+/+) | M | 24 | adenocarcinoma | lung |
| 24(+/+) | F | 24 | maliganat lymphoma | LN |
| 25(+/+) | F | 24 | malignant lymphoma | spleen, LN |
| 26(+/+) | F | 24 | malignant lymphoma | spleen, LN |
| 27(+/+) | F | 24 | malignant lymphoma | spleen, lung |
| 28(+/+) | F | 24 | malignant lymphoma | spleen, liver, LN, lung |
LN: lymph node, HCC: hepatocellular carcinoma
Table 2.
Tumor spectrum in Xpa (−/−) and Xpa (+/+) mice
| Xpa (−/−) | Xpa (+/+) | |||
|---|---|---|---|---|
| 12mo. | 24* mo. | 12mo. | 24* mo. | |
| Type of tumor | No.(% of tumor incidence) | No.(% of tumor incidence) | No.(% of tumor incidence) | No.(% of tumor incidence) |
| Lung cancer (adenocarcinoma, alveolar tumor) | 1 (1.8) | 3 (5.9)a, b, c | 0 | 3 (5.8) |
| Hepatocellular carcinoma(HCC) | 0 | 4 (7.8)a, d | 0 | 1 (1.9) |
| Malignant lymphoma | 3 (5.5) | 8 (15.7)b, c | 0 | 6 (11.5) |
| Other cancer | ||||
| hemangiosarcoma | 0 | 1 (2) | 0 | 0 |
| renal cell carcinoma | 0 | 1 (2)d | 0 | 0 |
| squamous cell carcinoma | 0 | 1 (2) | 0 | 0 |
| Total | 4 (7.3) | 18 (35.3) | 0 | 10 (19.2) |
| Total No. of mice examined | 55 | 51 | 49 | 52 |
Xpa (−/−) mice bearing multiple tumors
lung cancer, hepatocellular carcinoma
24 month old mice contained 18, 21 month old mice.
lung cancer, malignant lymphoma
(): % of tumor incidence
hepatocellular carcinoma, renal cell carcinoma
Fig. 5. Spontaneous tumorigenesis in the Xpa (−/−) mice and Xpa (+/+) littermates.
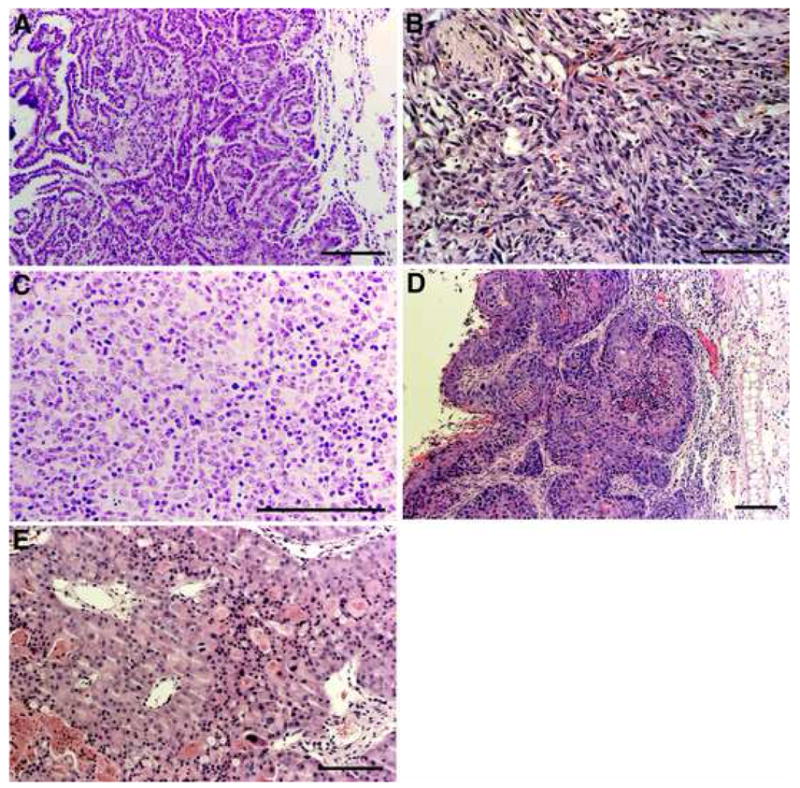
(A) Histologic section of well-differentiated (papillary) lung adenocarcinoma developed in a 12-month-old Xpa (−/−) mouse (mouse 2 in Table 1). (B) Histologioc section of a hemangiosarcoma developed in the right leg of a 18-month-old Xpa (−/−) mouse (mouse 5 in Table 1). (C) Histologic section of a malignant lymphoma invading into the kidney of a 24-month-old Xpa (−/−) mouse (mouse 10 in Table 1). (D) Histologic section of a moderately differentiated squamous cell cabrcinoma in the left ear of a 24-month-old Xpa (−/−) mouse (mouse 11 in Table 1). (E) Histologic section of a renal cell carcinoma developed in a 24-month-old Xpa (−/−) mouse (mouse 8 in Table 1). Bars correspond to 100 μm. Paraffin-embedded sections were stained with hematoxylin and eosin.
4. Discussion
We found that the Xpa (−/−) mice showed several of the pathologies observed in human XP-A patients, including smaller organ, major abnormalities of the testes, and increased spontaneous carcinogenesis. Below we discuss possible mechanistic explanations for these observations.
4.1. The weight of some organs is reduced in Xpa (−/−) mice
Human XP-A patients are smaller and show reduced size of some organs when compared with normal individuals. Consistent with this, our Xpa (−/−) mice were also slightly smaller than Xpa (+/+) controls, and had significantly reduced weights of some organs. While the smaller brain weight in both 12-months-old male and female Xpa (−/−) mice is intriguing in view of the neurologic disease observed in human XP-A patients, it should be noted that the human patients develop a progressive atrophy of the brain, due to the degeneration of neuronal cells (7). It appears that the brain of Xpa (−/−) mice does not undergo progressive atrophy, in contrast to the human XP-A patients.
4.2. Impaired spermatogenesis in the testis of Xpa (−/−) mice
Our most striking finding is the dramatic reduction in the weight and degenerative seminiferous tubules of the testis of Xpa (−/−) mice. In contrast to the other organs analyzed, the testis clearly undergoes atrophy over time (Fig. 1G), and the atrophy is accompanied by striking changes in the cellular composition of the testis. Specifically, by the age of 24 months, almost all the spermatogenic cells had degenerated, while Leydig cells proliferated. In contrast, essentially all the seminiferous tubules were normal in the Xpa (+/+) mice even at the age of 24 months (Fig. 2Ag and Fig. 2Cb).
Importantly, abnormalities of the testis have also been consistently observed in human XP patients. It was reported that 12% of XP patients with neurological abnormalities show delayed secondary sexual development (1, 8). DeSanctis and Cacchione reported 3 brothers with XP having immature testicular development (8, 21), and Yano noted a complete lack of spermatozoa in the testicles of Japanese XP patient (22). We also found that adult XP-A patients had abnormalities of spermatogenesis in their testis (our unpublished data).
4.3. Mechanisms of testicular abnormalities in Xpa (−/−) mice
Since the main known function of the Xpa protein is in nucleotide excision repair, the most likely explanation for our observations is a failure to repair some type of endogenous DNA damage by NER. However, this explanation is complicated by the cell-specific pattern of pathology we observe. Specifically we observed an age-dependent loss of spermatogenic cells, maintenance of Sertoli cells, and proliferation of Leydig cells in the testis of Xpa (−/−) mice. While several genes involved in NER or a related mechanism, including ERCC1, XPF, XAB1, XPB, XPC, HR23A and HR23B, are highly expressed in the testis (23, 24, 25, 26, 27, 28), one possible explanation for these results would be that in the testis, NER is only active in spermatogenic cell types, and therefore these cells are specifically affected by NER deficiency. However, the results from two studies indicate that NER in spermatogenic cells is either the same or lower than in somatic cells, although the NER activity varied with cell type-specificity during spermatogenesis (29, 30). Although neither Leydig or Sertoli cells were directly assayed in these experiments, our Northern blotting data show that Xpa mRNA is expressed in both somatic and germ cells of the mouse. Thus the available data do not support the idea that NER is restricted to germ cells in the testis.
Another way in which an NER defect could explain the spermatogenic cell defects we observe in Xpa (−/−) testis could be a depletion in stem cells. An inability to maintain tissue homeostasis, which is regulated by stem cells, is a major characteristic of aging. It has been shown that DNA repair deficiencies, that is, the accumulation of DNA damage, severely affected hematopoietic stem cell functions to maintain homeostasis after exposure to acute stress or injury (31, 32, 33, 34). Spermatogenic stem cell function of aged Xpa (−/−) mice may be affected by the accumulation of 8,5′-cyclopurine deoxynucleosides (cPu) or other endogenous DNA lesions, leading to a loss of proliferative potential and diminished self-renewal of spermatogenic stem cells. The relatively slow time course of spermatogenic cell failure would seem to be consistent with this mechanism.
An alternative and non-mutually exclusive possibility is that the testis abnormalities we observed are due in part to the loss of a non-NER function of XPA. It is known that Ser196 of XPA is phosphorylated by ATR (ataxia telangiectasia-mutated and Rad3-related) in UV-irradiated cells, increasing cell survival following UV irradiation, and that the nuclear accumulation of XPA and UV-induced nuclear focus formation of XPA are dependent on ATR (35, 36). Therefore, NER and ATR-dependent cell cycle checkpoint activation processes are coordinated via XPA. Moreover, UV-induced ATR signaling is compromised in XP-A cells during S-phase, but not in XP-C, CS-B, XP-F, and XP-G cells. These observations indicate that XPA has functions other than NER, suggesting that the impaired spermatogenesis in Xpa (−/−) mice is not only due to NER-deficiency but also due to a defect in a DNA damage dependent ATR signaling pathway (35,36). Notably, antibodies against ATR strongly stain meiotic chromosomes in spermatocytes (37,38).
To address these various hypotheses, it will be necessary to monitor DNA lesion levels in specific cell types over time in the Xpa (−/−) testis, particularly at early ages prior to the onset of cell loss. In view of the changes in the cellular composition of the testis in Xpa (−/−) versus Xpa (+/+) mice over time, the measurement of DNA lesion levels in total testis DNA samples would be very difficult to interpret and therefore of limited value.
4.4. Testicular abnormalities in other NER mouse models
It has been reported that mice deficient in a number of DNA repair genes such as ERCC1, HR23B, PMS2, and MLH1 were infertile (23, 39, 40, 41). Of these, the two that are of most potential relevance to our work are HR23B and Ercc1, because of their relationship to NER. While HR23B (−/−) mice are severely affected, a few surviving adult HR23B (−/−) mice showed a reduction in the size of testes with no spermatogenesis. However, it is unlikely that this defect was due to defective NER, because cells from these mice were shown to be proficient in both GG-NER and TC-NER. Also, HR23B functions not only as the main damage detector and initiator of NER but also as a regulator of protein stability. Thus the phenotypes observed in HR23B (−/−) mice may be due to a defective regulation of protein stability via HR23B-dependent ubiquitin/proteasome pathway that relates to a development of testis, in contrast to the fertility problem in Xpa (−/−) mice (39).
Melton and colleagues reported that the testis weights of Ercc1 (−/−) mice were 50 % of those of control mice at 3 – 6 weeks of age. On days 3 – 22, Ercc1 (−/−) mice contained smaller and more variable numbers of germ cells within the seminiferous epithelium than control littermates. Some tubules containing only Sertoli cells and germ cells with an abnormal morphology were seen at all ages. While an increased level of DNA strand breaks and oxidative DNA damage was found in the Ercc1 (−/−) testis, as the authors noted, strand breaks could have arisen from stalled replication forks due to unrepaired DNA lesions, as well as from a failure of ERCC1 to function in the single-strand annealing pathway for double-strand break repair and homologous recombination (23). Since XPA is not required for these functions, this could explain the earlier onset of testicular failure in the Ercc1 (−/−) mice than the Xpa (−/−) mice. In addition, the XPF/ERCC1 complex plays a role in the repair of intrastrand crosslinks that is independent of NER (42), and this could also contribute to the earlier onset of testicular failure in Ercc1 (−/−) mice. Regarding 7,8-dihydro-8-oxoguanine (8-oxoG), it is possible that the increased levels of this lesion were the result of oxidative stress due to the apoptosis and phagocytosis which were also observed in the testes of Ercc1 (−/−) mice (23).
It was recently reported that Xpe (−/−) mice had similar phenotypes to Xpa (−/−) mice including decreased body weight and a higher incidence of spontaneous tumors. In contrast to Xpa (−/−) mice testis, the Xpe (−/−) mice have larger testes, and the testicular germ cells show decreased apoptosis with reduction of p53 and its serine 15 phosphorylation. This distinction may be related to various aspects of the DNA damage response in testis (43).
4.5. High incidence of spontaneous tumorigenesis in Xpa (−/−) mice
Kraemer et al. reported that XP patients under 20 years of age showed an estimated 2000-fold increase in the frequency of cancers in sun-exposed skin in comparison to the general population, and that they also showed an estimated 12-fold increase in the occurrence of neoplasms at sites not exposed to sunlight. Among XP patients under 40 years of age with internal cancer, a higher incidence of brain sarcomas and oral cavity cancer was found compared with the general population of same aged US whites (9). Consistent with these findings, the present findings indicated that the Xpa (−/−) mice showed a higher incidence of spontaneous tumorigenesis than the Xpa (+/+) mice. A histological diagnosis of all the spontaneous tumors in the Xpa (−/−) and Xpa (+/+) mice is summarized in Tables 1 and 2. Both Xpa (−/−) and Xpa (+/+) mice developed malignant tumors such as malignant lymphoma, hepatocellular carcinoma, and lung adenocarcinoma. On the other hand, other Xpa (−/−) mice, established by a different gene targeting strategy (replacement of exons 3–4 by neo gene) and containing the Ola129 and C57BL/6 genetic background (44), developed only spontaneous benign tumors such as lung adenoma and hepatocellular adenoma at the age of 12–20 months, in spite of a high mutation frequency in liver (45, 46). The difference in spontaneous tumorigenesis between these two Xpa (−/−) mice is probably due to different genetic backgrounds, although our Xpa (−/−) mice with the C3H/HeN background also showed a high incidence of spontaneous malignant liver tumors (47).
It has been reported that Xpc (−/−) mice (16–17 months old) developed multiple spontaneous lung tumors including non-small cell lung adenocarcinoma (48), and Xpe (−/−) mice developed various types of spontaneous tumors in the later stages of life (20–25 months) (49). Although XPC has been shown to play a role in the repair of cPu and 8-hydroxyguanine (8-OH-Gua) following exposure to ionizing radiation, the basal levels of these lesions did not differ between XP-C and wild type human cells (50).
Finally, in view of our previous observation that Xpa (−/−) mice had an increased frequency of UVB-induced CC-TT mutations (4, 5), it is interesting to note that Reid and Loeb (51) showed that CC->TT mutations are a signature for oxidative stress, perhaps resulting from hydroxyl radical induced cytosine crosslinks. Consistent with this idea, it has been shown that exposure of DNA to oxygen radicals generates DNA lesions at tandem cytosines, as well as tandem purines, that are substrates for the bacterial NER system (52), and presumably for mammalian NER as well. Clearly, a greater knowledge of the mutation spectrum observed in internal tissues of our Xpa (−/−) mice and tumors, as well as a fuller appreciation of endogenous DNA lesions that are substrates for repair by NER, will shed light on the carcinogenic mechanisms due to NER deficiency.
In summary, we have discovered age-dependent phenotypes of Xpa (−/−) mice including small organ weight, impaired spermatogenesis and a higher incidence of spontaneous malignant tumorigenesis. Thus these Xpa (−/−) mice are a valuable animal model with which to study some of the pathological processes that affect human XP patients.
Acknowledgments
This work was supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (Grant numbers: 18591782, 20591898 to HN and 17109006 to KT), and by a Grant for Solution Oriented Research for Science and Technology (SORST) of Japan Science and Technology Agency (JST) to KT. We thank Dr. Junji Tsuchida for helpful advice, and Akinori Fukuyama, Kenji Morihara and Toshio Kameie for excellent technical support.
Footnotes
Conflict of Interest statement
The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. DNA Repair and Mutagenesis. ASM Press; Washington, DC: 2006. [Google Scholar]
- 2.Nakane H, Takeuchi S, Yuba S, Saijo M, Nakatsu Y, Murai H, Nakatsuru Y, Ishikawa T, Hirota S, Kitamura Y, Kato Y, Tsunoda Y, Miyauchi H, Horio T, Tokunaga T, Matsunaga T, Nikaido O, Nishimune Y, Okada Y, Tanaka K. High incidence of ultraviolet-B- or chemical-carcinogen-induced skin tumours in mice lacking the xeroderma pigmentosum group A gene. Nature. 1995;377:165–8. doi: 10.1038/377165a0. [DOI] [PubMed] [Google Scholar]
- 3.Ichikawa M, Nakane H, Marra G, Corti C, Jiricny J, Fitch M, Ford JM, Ikejima M, Shimada T, Yoshino M, Takeuchi S, Nakatsu Y, Tanaka K. Decreased UV sensitivity, mismatch repair activity and abnormal cell cycle checkpoints in skin cancer cell lines derived from UVB-irradiated XPA-deficient mice. Mutat Res. 2000;459:285–98. doi: 10.1016/s0921-8777(00)00005-7. [DOI] [PubMed] [Google Scholar]
- 4.Ikehata H, Yanase F, Mori T, Nikaido O, Tanaka K, Ono T. Mutation spectrum in UVB-exposed skin epidermis of Xpa-knockout mice: frequent recovery of triplet mutations. Environ Mol Mutagen. 2007;48:1–13. doi: 10.1002/em.20262. [DOI] [PubMed] [Google Scholar]
- 5.Murai H, Takeuchi S, Nakatsu Y, Ichikawa M, Yoshino M, Gondo Y, Katsuki M, Tanaka K. Studies of in vivo mutations in rpsL transgene in UVB-irradiated epidermis of XPA-deficient mice. Mutat Res. 2000;450:181–92. doi: 10.1016/s0027-5107(00)00024-5. [DOI] [PubMed] [Google Scholar]
- 6.Tanaka K, Kamiuchi S, Ren Y, Yonemasu R, Ichikawa M, Murai H, Yoshino M, Takeuchi S, Saijo M, Nakatsu Y, Miyauchi-Hashimoto H, Horio T. UV-induced skin carcinogenesis in xeroderma pigmentosum group A (XPA) gene-knockout mice with nucleotide excision repair-deficiency. Mutat Res. 2001;477:31–40. doi: 10.1016/s0027-5107(01)00093-8. [DOI] [PubMed] [Google Scholar]
- 7.Mimaki T, Itoh N, Abe J, Tagawa T, Sato K, Yabuuchi H, Takebe H. Neurological manifestations in xeroderma pigmentosum. Ann Neurol. 1986;20:70–5. doi: 10.1002/ana.410200112. [DOI] [PubMed] [Google Scholar]
- 8.Kraemer KH, Lee MM, Scotto J. Xeroderma pigmentosum. Cutaneous, ocular, and neurologic abnormalities in 830 published cases. Arch Dermatol. 1987;123:241–50. doi: 10.1001/archderm.123.2.241. [DOI] [PubMed] [Google Scholar]
- 9.Kraemer KH, Lee MM, Scotto J. DNA repair protects against cutaneous and internal neoplasia: evidence from xeroderma pigmentosum. Carcinogenesis. 1984;5:511–4. doi: 10.1093/carcin/5.4.511. [DOI] [PubMed] [Google Scholar]
- 10.Kuraoka I, Bender C, Romieu A, Cadet J, Wood RD, Lindahl T. Removal of oxygen free-radical-induced 5′,8-purine cyclodeoxynucleosides from DNA by the nucleotide excision-repair pathway in human cells. Proc Natl Acad Sci US A. 2000;97:3832–7. doi: 10.1073/pnas.070471597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brooks PJ, Wise DS, Berry DA, Kosmoski JV, Smerdon MJ, Somers RL, Mackie H, Spoonde AY, Ackerman EJ, Coleman K, Tarone RE, Robbins JH. The oxidative DNA lesion 8,5′-(S)-cyclo-2′-deoxyadenosine is repaired by the nucleotide excision repair pathway and blocks gene expression in mammalian cells. J Biol Chem. 2000;275:22355–62. doi: 10.1074/jbc.M002259200. [DOI] [PubMed] [Google Scholar]
- 12.Johnson KA, Fink SP, Marnett LJ. Repair of propanodeoxyguanosine by nucleotide excision repair in vivo and in vitro. J Biol Chem. 1997;272:11434–8. doi: 10.1074/jbc.272.17.11434. [DOI] [PubMed] [Google Scholar]
- 13.Marnett LJ. Oxyradicals and DNA damage. Carcinogenesis. 2000;21:361–70. doi: 10.1093/carcin/21.3.361. [DOI] [PubMed] [Google Scholar]
- 14.Niedernhofer LJ, Daniels JS, Rouzer CA, Greene RE, Marnett LJ. Malondialdehyde, a product of lipid peroxidation, is mutagenic in human cells. J Biol Chem. 2003;278:31426–33. doi: 10.1074/jbc.M212549200. [DOI] [PubMed] [Google Scholar]
- 15.Wang Y. Bulky DNA lesions induced by reactive oxygen species. Chem Res Toxicol. 2008;21:276–81. doi: 10.1021/tx700411g. [DOI] [PubMed] [Google Scholar]
- 16.Russell ES. Hereditary anemias of the mouse: a review for geneticists. Adv Genet. 1979;20:357–459. [PubMed] [Google Scholar]
- 17.Coulombre JL, Russell ES. Analysis of the pleiotropism at the W locus in the mouse. The effects of W and Wv substitutions upon post natal development of germ cells. J Exp Zool. 1954;126:277–96. [Google Scholar]
- 18.Ohta H, Tohda A, Nishimune Y. Proliferation and differentiation of spermatogonial stem cells in the W/Wv mutant mouse testis. Biol Reprod. 2003;69:1815–21. doi: 10.1095/biolreprod.103.019323. [DOI] [PubMed] [Google Scholar]
- 19.Leblond CP, Clermont Y. Definition of the stages of the cycle of the seminiferous epithelium in the rat. Ann NY Acad Sci. 1952;55:548–573. doi: 10.1111/j.1749-6632.1952.tb26576.x. [DOI] [PubMed] [Google Scholar]
- 20.Oakberg EF. A description of spermiogenesis in the mouse and its use in analysis of the cycle of the seminiferous epithelium and germ cell renewal. Am J Anat. 1956;99:391–413. doi: 10.1002/aja.1000990303. [DOI] [PubMed] [Google Scholar]
- 21.DeSanctis C, Cacchione A. L’idiozia xerodermica. Riv Sper Freniatr. 1932;56:269–92. [Google Scholar]
- 22.Yano K. Xeroderma pigmentosum with disorders of the central nervous system; a histopathological study. Folia Psychiatr Neurol Jpn. 1950;4:143–75. [PubMed] [Google Scholar]
- 23.Hsia KT, Millar MR, King S, Selfridge J, Redhead NJ, Melton DW, Saunders PT. DNA repair gene Ercc1 is essential for normal spermatogenesis and oogenesis and for functional integrity of germ cell DNA in the mouse. Development. 2003;130:369–78. doi: 10.1242/dev.00221. [DOI] [PubMed] [Google Scholar]
- 24.Shannon M, Lamerdin JE, Richardson L, McCutchen-Maloney SL, Hwang MH, Handel MA, Stubbs L, Thelen MP. Characterization of the mouse Xpf DNA repair gene and differential expression during spermatogenesis. Genomics. 1999;62:427–35. doi: 10.1006/geno.1999.6016. [DOI] [PubMed] [Google Scholar]
- 25.Nitta M, Saijo M, Kodo N, Matsuda T, Nakatsu Y, Tamai H, Tanaka K. A novel cytoplasmic GTPase XAB1 interacts with DNA repair protein XPA. Nucl Acid Res. 2000;28:4212–18. doi: 10.1093/nar/28.21.4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weeda G, Ma L, van Ham RC, Bootsma D, van der Eb AJ, Hoeijmakers JH. Characterization of the mouse homolog of the XPBC/ERCC-3 gene implicated in xeroderma pigmentosum and Cockayne’s syndrome. Carcinogenesis. 1991;12:2361–8. doi: 10.1093/carcin/12.12.2361. [DOI] [PubMed] [Google Scholar]
- 27.Li L, Peterson C, Legerski R. Sequence of the mouse XPC cDNA and genomic structure of the human XPC gene. Nucl Acids Res. 1996;24:1026–8. doi: 10.1093/nar/24.6.1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van der Spek PJ, Visser CE, Hanaoka F, Smit B, Hagemeijer A, Bootsma D, Hoeijmakers JH. Cloning, comparative mapping, and RNA expression of the mouse homologues of the Saccharomyces cerevisiae nucleotide excision repair gene RAD23. Genomics. 1996;31:20–7. doi: 10.1006/geno.1996.0004. [DOI] [PubMed] [Google Scholar]
- 29.Xu G, Spivak G, Mitchell DL, Mori T, McCarrey JR, McMahan CA, Walter RB, Hanawalt PC, Walter CA. Nucleotide excision repair activity varies among murine spermatogenic cell types. Biol Reprod. 2005;73:123–30. doi: 10.1095/biolreprod.104.039123. [DOI] [PubMed] [Google Scholar]
- 30.Jansen J, Olsen AK, Wiger R, Naegeli H, de Boer P, van Der Hoeven F, Holme JA, Brunborg G, Mullenders L. Nucleotide excision repair in rat male germ cells: low level of repair in intact cells contrasts with high dual incision activity in vitro. Nucleic Acids Res. 2001;29:1791–800. doi: 10.1093/nar/29.8.1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Prasher JM, Lalai AS, Heijmans-Antonissen C, Ploemacher RE, Hoeijmakers JH, Touw IP, Niedernhofer LJ. Reduced hematopoietic reserves in DNA interstrand crosslink repair-deficient Ercc1−/− mice. EMBO J. 2005;24:861–71. doi: 10.1038/sj.emboj.7600542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nijnik A, Woodbine L, Marchetti C, Dawson S, Lambe T, Liu C, Rodrigues NP, Crockford TL, Cabuy E, Vindigni A, Enver T, Bell JI, Slijepcevic P, Goodnow CC, Jeggo PA, Cornall RJ. DNA repair is limiting for haematopoietic stem cells during ageing. Nature. 2007;447:686–90. doi: 10.1038/nature05875. [DOI] [PubMed] [Google Scholar]
- 33.Rossi DJ, Bryder D, Seita J, Nussenzweig A, Hoeijmakers J, Weissman IL. Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature. 2007;447:725–9. doi: 10.1038/nature05862. [DOI] [PubMed] [Google Scholar]
- 34.Niedernhofer LJ. DNA repair is crucial for maintaining hematopoietic stem cell function. DNA Repair. 2008;7:523–9. doi: 10.1016/j.dnarep.2007.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu X, Shell SM, Yang Z, Zou Y. Phosphorylation of nucleotide excision repair factor xeroderma pigmentosum group A by ataxia telangiectasia mutated and Rad3-related-dependent checkpoint pathway promotes cell survival in response to UV irradiation. Cancer Res. 2006;66:2997–3005. doi: 10.1158/0008-5472.CAN-05-3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bomgarden RD, Lupardus PJ, Soni DV, Yee MC, Ford JM, Cimprich KA. Opposing effects of the UV lesion repair protein XPA and UV bypass polymerase eta on ATR checkpoint signaling. EMBO J. 2006;25:2605–14. doi: 10.1038/sj.emboj.7601123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Keegan KS, Holtzman DA, Plug AW, Christenson ER, Brainerd EE, Flaggs G, Bentley NJ, Taylor EM, Meyn MS, Moss SB, Carr AM, Ashley T, Hoekstra MF. The Atr and Atm protein kinases associate with different sites along meiotically pairing chromosomes. Genes Dev. 1996;10:2423–37. doi: 10.1101/gad.10.19.2423. [DOI] [PubMed] [Google Scholar]
- 38.Moens PB, Tarsounas M, Morita T, Habu T, Rottinghaus ST, Freire R, Jackson SP, Barlow C, Wynshaw-Boris A. The association of ATR protein with mouse meiotic chromosome cores. Chromosoma. 1999;108:95–102. doi: 10.1007/s004120050356. [DOI] [PubMed] [Google Scholar]
- 39.Jessica MYNg, Vrieling H, Sugasawa K, Ooms MP, Grootegoed JA, Vreeburg JT, Visser P, Beems RB, Gorgels TG, Hanaoka F, Hoeijmakers JH, van der Horst GT. Developmental defects and male sterility in mice lacking the ubiquitin-like DNA repair gene mHR23B. Mol Cell Biol. 2002;22:1233–45. doi: 10.1128/MCB.22.4.1233-1245.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Baker SM, Bronner CE, Zhang L, Plug AW, Robatzek M, Warren G, Elliott EA, Yu J, Ashley T, Arnheim N, Flavell RA, Liskay RM. Male mice defective in the DNA mismatch repair gene PMS2 exhibit abnormal chromosome synapsis in meiosis. Cell. 1995;82:309–19. doi: 10.1016/0092-8674(95)90318-6. [DOI] [PubMed] [Google Scholar]
- 41.Edelmann W, Cohen PE, Kane M, Lau K, Morrow B, Bennett S, Umar A, Kunkel T, Cattoretti G, Chaganti R, Pollard JW, Kolodner RD, Kucherlapati R. Meiotic pachytene arrest in MLH1-deficient mice. Cell. 1996;85:1125–34. doi: 10.1016/s0092-8674(00)81312-4. [DOI] [PubMed] [Google Scholar]
- 42.Niedernhofer LJ, Odijk H, Budzowska M, van Drunen E, Maas A, Theil AF, de Wit J, Jaspers NG, Beverloo HB, Hoeijmakers JH, Kanaar R. The structure-specific endonuclease Ercc1-Xpf is required to resolve DNA interstrand cross-link-induced double-strand breaks. Mol Cell Biol. 2004;24:5776–87. doi: 10.1128/MCB.24.13.5776-5787.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Itoh T, Iwashita S, Cohen MB, Meyerholz DK, Linn S. Ddb2 is a haploinsufficient tumor suppressor and controls spontaneous germ cell apoptosis. Hum Mol Genet. 2007;16:1578–86. doi: 10.1093/hmg/ddm107. [DOI] [PubMed] [Google Scholar]
- 44.de Vries A, van Oostrom CT, Hofhuis FM, Dortant PM, Berg RJ, de Gruijl FR, Wester PW, van Kreijl CF, Capel PJ, van Steeg H. Increased susceptibility to ultraviolet-B and carcinogens of mice lacking the DNA excision repair gene XPA. Nature. 1995;377:169–73. doi: 10.1038/377169a0. [DOI] [PubMed] [Google Scholar]
- 45.de Vries A, van Oostrom CT, Dortant PM, Beems RB, van Kreijl CF, Capel PJ, van Steeg H. Spontaneous liver tumors and benzo[a]pyrene-induced lymphomas in XPA-deficient mice. Mol Carcinog. 1997;19:46–53. [PubMed] [Google Scholar]
- 46.Giese H, Dolle ME, Hezel A, van Steeg H, Vijg J. Accelerated accumulation of somatic mutations in mice deficient in the nucleotide excision repair gene XPA. Oncogene. 1999;18:1257–60. doi: 10.1038/sj.onc.1202404. [DOI] [PubMed] [Google Scholar]
- 47.Takahashi Y, Nakatsuru Y, Zhang S, Shimizu Y, Kume H, Tanaka K, Ide F, Ishikawa T. Enhanced spontaneous and aflatoxin-induced liver tumorigenesis in xeroderma pigmentosum group A gene-deficient mice. Carcinogenesis. 2002;23:627–33. doi: 10.1093/carcin/23.4.627. [DOI] [PubMed] [Google Scholar]
- 48.Hollander MC, Philburn RT, Patterson AD, Velasco-Miguel S, Friedberg EC, Linnoila RI, Fornace AJ., Jr Deletion of XPC leads to lung tumors in mice and is associated with early events in human lung carcinogenesis. Proc Natl Acad Sci US A. 2005;102:13200–5. doi: 10.1073/pnas.0503133102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yoon T, Chakrabortty A, Franks R, Valli T, Kiyokawa H, Raychaudhuri P. Tumor-prone phenotype of the DDB2-deficient mice. Oncogene. 2005;24:469–78. doi: 10.1038/sj.onc.1208211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.D’Errico M, Parlanti E, Teson M, de Jesus BM, Degan P, Calcagnile A, Jaruga P, Bjørås M, Crescenzi M, Pedrini AM, Egly JM, Zambruno G, Stefanini M, Dizdaroglu M, Dogliotti E. New functions of XPC in the protection of human skin cells from oxidative damage. EMBO J. 2006;25:4305–15. doi: 10.1038/sj.emboj.7601277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reid TM, Loeb LA. Tandem double CC-->TT mutations are produced by reactive oxygen species. Proc Natl Acad Sci U S A. 1993;90:3904–7. doi: 10.1073/pnas.90.9.3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Roldán-Arjona T, Sedgwick B. DNA base damage induced by ionizing radiation recognized by Escherichia coli UvrABC nuclease but not Nth or Fpg proteins. Mol Carcinog. 1996;16:188–96. doi: 10.1002/(SICI)1098-2744(199608)16:4<188::AID-MC2>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]