

SUMMARY
The signaling output of protein kinase C (PKC) is exquisitely controlled, with its disruption resulting in pathophysiologies. Identifying the structural basis for autoinhibition is central to developing effective therapies for cancer, where PKC activity needs to be enhanced, or neurodegenerative diseases, where PKC activity should be inhibited.
Here, we reinterpret a previously reported crystal structure of PKCβII and use docking and functional analysis to propose an alternative structure that is consistent with previous literature on PKC regulation.
Mutagenesis of predicted contact residues establishes that the Ca2+-sensing C2 domain interacts intramolecularly with the kinase domain and the carboxyl-terminal tail, locking PKC in an inactive conformation. Ca2+-dependent bridging of the C2 domain to membranes provides the first step in activating PKC via conformational selection. Although the placement of the C1 domains remains to be determined, elucidation of the structural basis for autoinhibition of PKCβII unveils a unique direction for therapeutically targeting PKC.
INTRODUCTION
Protein kinase C isozymes transduce a myriad of signals that result in phospholipid hydrolysis. As such, they play key roles in a multitude of cellular processes, including controlling the balance between cell survival and death, and their dysregulation has been implicated in numerous diseases. Mounting evidence suggests that PKC activity suppresses survival signaling (Reyland, 2007); thus, it functions as a tumor suppressor and cancer-associated PKC mutations are generally loss-of-function (Antal et al., 2015). In marked contrast, its activity is elevated in neurodegenerative diseases such as spinocerebellar ataxia 14 (Ji et al., 2014; Verbeek et al., 2005), ischemic neurodegeneration (Sieber et al., 1998), and in heart disease (Belin et al., 2007; Bowling et al., 1999; Takeishi et al., 2000). From a therapeutic standpoint, identifying intramolecular interactions between the different PKC domains is essential to design small molecules or peptides that can either disrupt these contacts to open up and activate PKC or clamp the domains closed to prevent PKC activation.
The PKC family consists of 9 genes that are grouped according to their regulatory domains and thus the second messengers that regulate them (Parker and Murray-Rust, 2004). Conventional PKC isozymes (α, β, γ) contain tandem C1 domains, C1A and C1B, that bind diacylglycerol (DAG) and a C2 domain that binds anionic phospholipids in a Ca2+-dependent manner (Figure 1A); the C2 domain also contains phosphatidylinositol-4,5-bisphosphate (PIP2)-binding determinants that direct conventional PKC isozymes to the plasma membrane. Novel PKC (δ, ε, η, θ) isozymes lack a functional C2 domain, and thus are activated solely by DAG binding to the C1 domain, whereas atypical PKC (ι, ζ) isozymes bind neither of these second messengers. Conventional and novel PKC isozymes are constitutively phosphorylated at three priming sites (activation loop, turn motif, and hydrophobic motif), that trigger a series of conformational changes that allow PKC to adopt an autoinhibited conformation that is catalytically competent but unable to signal in the absence of agonists (Antal et al., 2015; Feng et al., 2000; Stensman et al., 2004). Specifically, the C1 domains become masked to prevent basal recognition of DAG, and the pseudosubstrate binds the substrate-binding cavity to prevent substrate phosphorylation. Signals that result in phospholipid hydrolysis activate conventional PKC isozymes by a two-step mechanism: generation of Ca2+ recruits PKC to the plasma membrane where it binds its membrane-embedded ligand, DAG. This latter event releases the pseudosubstrate, thus activating PKC.
Figure 1. The C2 Domain of PKCβII Interacts with the Kinase Domain and C-Terminal Tail.

(A) Schematic of the primary structure of PKCβII showing domain composition, priming phosphorylation sites (activation loop in pink, turn motif in orange, and hydrophobic motif in green), and the proteolytically-labile hinge that separates the regulatory and catalytic moieties.
(B) Crystal structure of PKCβII (PDBID:3PFQ) showing the original interpretation of the location of the C2 domain that traces polypeptide from the C1B to the C2 (mode i) and the alternative interpretation in which the C2 domain binds the kinase domain by an intramolecular interaction (mode ii). Both modes are present in the crystal packing.
(C) C2 domain (yellow) docked onto a complex of the PKCβII kinase domain (cyan) and the modeled pseudosubstrate (red).
(D) Complex of the PKCβII kinase domain (cyan) and the mode ii C2 domain (yellow) from the crystal packing with the modeled pseudosubstrate (red), superimposed with the docked C2 domain (brown).
(E) Structure of the kinase domain:C2 domain:pseudosubstrate complex showing the opening between the kinase domain (cyan) and the C2 domain (yellow), through which the pseudosubstrate:C1A linker (red) can be accommodated.
See also Figure S2.
Elucidation of the structure of PKC has been challenging, given that it is a highly dynamic, multi-module protein that undergoes large conformational changes. The structures of the isolated C1, C2, and kinase domains of conventional PKC isozymes have been previously solved (Grodsky et al., 2006; Guerrero-Valero et al., 2009; Hommel et al., 1994). The most complete PKC crystal structure to date is that of PKCβII, in which electron density is clearly evident for the C1B, C2, and kinase domains, and the carboxyl-terminal (C-term) tail (Leonard et al., 2011). However, because the structure lacks adequate electron density for the pseudosubstrate, the C1A domain, or any of the regions connecting the domains to one another, the assignment of which domains belong to a particular polypeptide, as opposed to other symmetry mates, was challenging. The crystal lattice revealed two possible contacts between the C2 and catalytic domains: mode i involving intermolecular contacts with a distal surface of the kinase domain C-term lobe and mode ii involving intramolecular contacts with the catalytic cleft of the kinase (Figure 1B). The authors hypothesized that binding of the pseudosubstrate in the catalytic site of the kinase would cause a steric clash with the C2 domain and thus dismissed mode ii as being functionally irrelevant. However, this latter pose is supported by extensive literature establishing intramolecular contacts between the C2 domain and C-term tail of conventional PKC isozymes (Banci et al., 2002; Conrad et al., 1994; Corbalan-Garcia et al., 2003; Edwards and Newton, 1997a, b; Feng et al., 2000; Kheifets and Mochly-Rosen, 2007). This raises the question as to whether the mode ii intradomain contacts might represent the biologically relevant structure.
Here we use structure/function analysis to test whether the C2 domain clamps over the kinase domain to provide a previously undescribed mechanism of autoinhibition in which not only is the pseudosubstrate in the substrate-binding cavity, but the C2 domain clamps this autoinhibited conformation. From the crystal packing, we identify key ion pairs between the C2 domain and kinase domain or C-term tail and show that reversal of one charge unfolds PKC and that reversal of both charges re-clamps PKC in a closed conformation. Using this information with subsequent structural modeling, we propose a model for the two-step activation of PKC. In this model 1] Ca2+ binding to the C2 domain pushes the equilibrium towards the open conformation (C2 removed from kinase domain) because the C2 domain is now retained at the plasma membrane via Ca2+-bridging to anionic phospholipids and 2] binding of DAG to the C1B domain repositions the pseudosubstrate-C1A moiety to relieve autoinhibition. Our findings suggest that PKC is activated via conformational selection of the open state of the enzyme. Additionally, they open new avenues for therapeutically targeting PKC with small molecules or peptides that could disrupt or strengthen intramolecular contacts in order to modulate PKC activity.
RESULTS
PKCβII Crystal Structure Packing Reveals that the C2 Domain Interfaces with the Kinase Domain
Given the differences between the PKCβII structure and model of activation (Leonard et al., 2011) with previous biochemical analyses of PKC, we examined the reported crystal packing to determine whether any other conformations could support the known biology of PKC. Docking of the C2 domain onto a complex of the kinase domain with a modeled pseudosubstrate (Figure 1C) showed that the C2 domain can be bound to the kinase domain with the pseudosubstrate present in its active site in a way that is very similar to the mode ii with the RMSD being 13Å (1095 atoms) (Figure 1D). In this model, an opening between the catalytic and C2 domains would readily accommodate the presence of the linker between the pseudosubstrate and C1A domain (Figures 1E). If this interaction were biologically relevant, it would unveil yet another mechanism of autoinhibition, with the C2 domain clamping over the kinase domain to maintain the pseudosubstrate in the substrate-binding cavity.
Mutational Analysis Corroborates a C2:Kinase Domain Interface
To test whether the C2 domain of PKCβII interfaces with the kinase domain and the C-term tail in the closed, autoinhibited conformation, we determined which residues are involved in this interaction. Based on the crystal packing, Asp382 within the kinase domain and Lys209 within the C2 domain were predicted to form hydrogen bonds (Figure 2A). To test this potential interaction, we mutated the negatively charged Asp382 to a positively charged Lys and assessed whether this induced an open conformation of the enzyme by displacing regulatory moieties from the kinase domain. Note, we refer to an open conformation of PKC as one in which the C1 and/or C2 domains of PKC are displaced from the kinase domain and the pseudosubstrate is out of the substrate-binding site, and to a closed conformation as one in which PKC is autoinhibited through intramolecular interactions with its pseudosubstrate and regulatory domains. We have previously shown that unphosphorylated PKCβII adopts an open conformation that, because of unmasked C1A and C1B domains, translocates more rapidly to the plasma membrane upon treatment with the C1 domain ligand, phorbol dibutyrate (PDBu), compared with matured (phosphorylated) PKC that has undergone conformational transitions to mask its C1 domains (Antal et al., 2014). Indeed, when the Asp382-Lys209 interaction was disrupted by a D382K mutation, the protein translocated more rapidly (Figure 2B; t1/2= 1.7 ± 0.1 min versus 3.5 ± 0.2 min), indicating that it was in a more open conformation with its C1 domains exposed. Simultaneously inverting the charges of both Asp382 and Lys209 (D382K/K209D) rescued the translocation kinetics (t1/2=3.4 min ± 0.2 min), corroborating the interpretation that these residues interact with each other. Glu655 and Lys205 are also positioned in proximity such that they could interact electrostatically (Figure 2A). Similar to the Asp382-Lys209 pair, mutating Glu655 to a Lys also increased the translocation rate (t1/2=1.6 min ± 0.1 min) induced by PDBu, and simultaneously reversing the charges of both Glu655 and Lys205 rescued the translocation kinetics (Figure 2C; t1/2=2.9 min ± 0.2 min). Lys K209 is part of the C2 domain lysine-rich cluster that binds PIP2 (Corbalan-Garcia et al., 2003) and Lys205 interacts with the PS head groups (Verdaguer et al., 1999). Because these mutations disrupted the plasma membrane-sensing role of the C2 domain (Scott et al., 2013), they augmented basal and PDBu-dependent PKC association with the DAG-rich Golgi through the now dominant C1 domain binding (data not shown). Thus, we could not assess the plasma membrane translocation kinetics of these mutants. However, our results are consistent with these C2 domain mutants adopting an open conformation in which the C1 domain is able to bind basal DAG at the Golgi. As mentioned above, the plasma membrane translocation kinetics of either charge reversal double mutant (K209D/D382K and K205E/E655K) was rescued, and so was their localization (data not shown), because they were able to adopt a closed conformation. As a negative control, mutating the nearby Glu657 within the C-term tail to a Lys had no effect on the translocation kinetics (Figure 2C; t1/2=4.0 min ± 0.2 min) because this residue does not interface with the C2 domain. Furthermore, mutating both residues involved in the interaction with the C2 domain (D382K and E655K) resulted in a more open conformation, as it further increased the rate of translocation (Figure 2D; t1/2=1.1 min ± 0.1 min) induced by phorbol dibutyrate (PDBu). However, this conformation was not as open as that of unprocessed, kinase-dead PKCβII (Antal et al., 2014), suggesting that there are other contributing points of contact within the regulatory domain. A quadruple mutant in which charges in both ion pairs were reversed (i.e. K205E/E655K/K209D/D382K) displayed only partial recovery (t1/2=1.7 min ± 0.1 min), likely because mutating all four residues significantly reduced processing phosphorylations (data not shown). Note that all other mutants were effectively processed by phosphorylation (Figure S1).
Figure 2. Mutational Analysis Corroborates a C2:Kinase Domain Interface.

(A) Crystal structure of PKCβII (PDBID:3PFQ) showing predicted ion pairs between the kinase domain (cyan) or the C-terminal tail (gray) and the C2 domain (yellow).
(B–D) Normalized FRET ratio changes (mean ± SEM) representing PDBu- (200 nM) induced PKC translocation in COS7 cells co-expressing YFP-tagged PKCβII WT or mutants and plasma membrane-targeted CFP.
(E) FRET-ratio changes (mean ± SEM) representing thapsigargin- (5μM) followed by PDBu- (200 nM) induced PKC translocation in COS7 cells co-expressing YFP-tagged PKCβII WT or mutant and plasma membrane-targeted CFP.
See also Figure S1.
To ensure that the mutations that disrupted the C2:kinase domain interface indeed disengaged the C2 domain to allow it to favor Ca2+-dependent lipid binding, we monitored the steady-state levels of binding of the PKCβII-D382K/E655K mutant enzyme to the plasma membrane upon elevation of intracellular Ca2+ with thapsigargin, a sarco/endoplasmic reticulum Ca2+-ATPase inhibitor (Rogers et al., 1995). Elevation of Ca2+ resulted in an approximately 2-fold increase in the steady-state binding of the PKCβII-D382K/E655K to the plasma membrane (Figure 2E), consistent with disruption of these ion pairs favoring an open conformation with an exposed C2 domain. Moreover, this mutant translocated to membranes with much faster kinetics upon subsequent stimulation with PDBu, corroborating the open conformation. Kinase-dead PKCβII-D466N, which has fully exposed C1A and C1B domains (Antal et al., 2014; Gould et al., 2011; Shi et al., 2010), translocated rapidly and more completely to the membrane upon elevation of Ca2+(Figure 2E), revealing that the C2 domain, similar to the C1A and C1B domains, is highly exposed in unprimed PKC. Thus, the C2 domain of the PKCβII-D382K/E655K favors a more open conformation than that of wild-type, but not as open as that of the kinase-dead PKC, likely because of additional points of contact between the C2 and kinase domains.
The C2:Kinase Domain Interaction Is Intramolecular
To exclude the possibility that the C2:kinase domain interaction is intermolecular, as opposed to intramolecular, we examined whether an intermolecular interaction between a YFP-tagged PKCβII E655K mutant and a RFP-tagged PKCβII K205E mutant could rescue the fast translocation kinetics of the E655K mutant (Figure 3A). In contrast to the rescue induced by introducing a complementary C2 domain mutation into the same polypeptide as the kinase domain mutation (see Figure 2C; t1/2=1.0 min ± 0.1 min versus 2.9 min ± 0.2 min), the presence of a C2 domain mutation on another PKCβII molecule did not rescue the fast translocation kinetics for the C-term tail mutant (Figure 3B). These data are consistent with an intramolecular, and not intermolecular, C2:kinase domain interaction and support biophysical studies showing that cellular PKCβII translocates to plasma membranes as a monomer (L. Kaestner and P. Lipp, personal communication) and biochemical studies showing that pure PKC is fully active as a monomer (Hannun and Bell, 1986).
Figure 3. The PKCβII Kinase Domain Binds the C2 Domain through an Intramolecular Interaction.
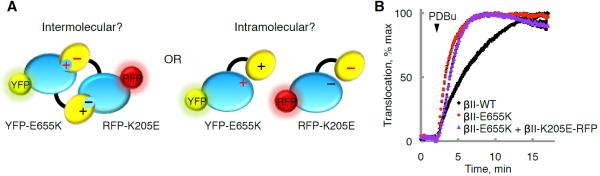
(A) Charge reversal of the ion pair partner in the C2 domain (K205E) would rescue the fast translocation kinetics of the E655K C-terminal tail mutation in the case of an intermolecular (left) but not intramolecular (right) C2:kinase interaction.
(B) Normalized FRET ratio changes (mean ± SEM) representing PDBu- (200 nM) induced PKC translocation in COS7 cells co-expressing plasma membrane-targeted CFP and either YFP-PKCβII-WT, YFP-PKCβII-E655K, or both YFP-PKCβII-E655K and RFP-PKCβII-K205E.
PKC Activation is Insensitive to Phe629
We next investigated the physiological significance of the previously suggested allosteric activation model, which postulates that Phe629 of the NFD helix (residues 628–630) controls the activity of PKC (Leonard et al., 2011). Specifically, the C1B was proposed to clamp the NFD in a low activity conformation in which the Phe is displaced from the active site, with binding of the C1B domain to membranes releasing the Phe to interact with the adenine ring of ATP. However, mutation of Phe629 to Ala resulted in a kinase whose cellular activation kinetics and magnitude were indistinguishable from that of the wild-type enzyme, both in response to natural agonists and phorbol esters (Figure 4C). This suggests that Phe629 is not a key regulator of the physiological activation of PKC.
Figure 4. Mutation of Phe629 Does Not Affect PKC Activation.
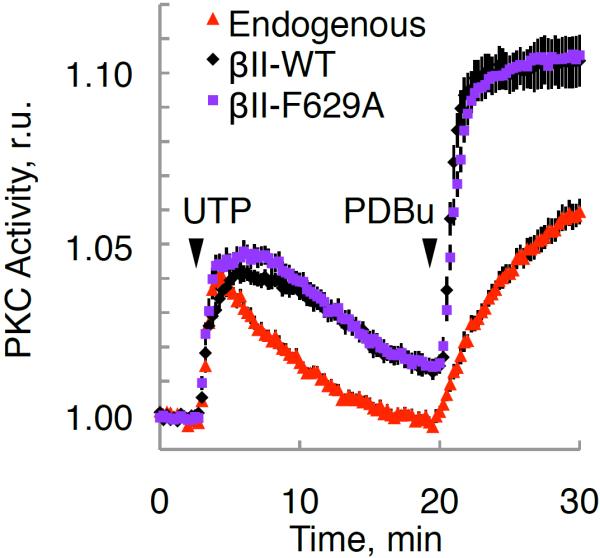
Normalized FRET ratio changes (mean ± SEM) showing agonist-dependent PKC activity of the indicated RFP-tagged PKCβII constructs or RFP control (endogenous) in COS7 cells co-expressing CKAR.
DISCUSSION
We propose an alternate structure for PKCβII, based on re-analysis of the crystal packing of PKCβII (Leonard et al., 2011), that reveals how the C2 domain provides an additional layer of autoinhibition to ensure minimal signaling in the absence of activators. This structure (mode ii in Figure 1B), in which the C2 domain clamps over the kinase domain, is validated by mutagenesis of interacting surfaces. Specifically, disruption of ion pairs at the C2:kinase domain/C-term tail interface unclamps the C2 domain, and reversal of charges in this ion pair maintains the clamped conformation. Additionally, we show that disruption of an ion pair is not rescued by introducing the opposite charge into a separate PKC molecule, establishing that monomeric PKC binds its own C2 domain. These data support a model in which the C2 domain forms an intramolecular clamp with the kinase domain and C-term tail. Taken together with the extensive biochemical analyses on PKCβII regulation, we propose a model (Figure 5) for the activation mechanism of PKC that reveals how conformational regulation results in minimal signaling in the absence of activators, thus optimizing the signaling output of the enzyme.
Figure 5. Model of PKCβII Activation.

(A) Unprimed PKCβII is in a membrane-associated, open conformation in which its C1A, C1B, and C2 domains are fully exposed and the pseudosubstrate and C-terminal tail are unmasked.
(B) Upon priming phosphorylation at its activation loop (T500, magenta) by PDK-1, followed by autophosphorylation at the turn motif (T641, orange) and the hydrophobic motif (S660, green), PKCβII matures into a closed conformation in which the C2 domain interfaces with the kinase domain and traps the pseudosubstrate into the substrate-binding site, both C1 domains become masked, and the primed enzyme localizes to the cytosol.
(C) In response to agonists that promote PIP2 hydrolysis, Ca2+ binds cytosolic PKCβII via a low affinity interaction such that upon the next diffusion-controlled membrane encounter, the Ca2+-bound C2 domain is retained at the plasma membrane via Ca2+-bridging to anionic lipids and binding to PIP2.
(D) Pre-targeted PKC binds the membrane-embedded ligand, DAG, predominantly via the C1B domain, resulting in release of the pseudosubstrate from the substrate-binding cavity, thereby activating PKC. Only one of the C1 domains binds DAG in the membrane at a time.
Our structure and model are supported by prior biochemical studies indicating that the C2 domain of PKCβII interfaces with the C-term tail to maintain PKC in a closed, inactive conformation. Specifically, the C-term tail of PKCβII has been previously suggested to interface with the C2 domain because the C2 domain-mediated Ca2+ affinity is sensitive to the composition of the C-term tail (Edwards and Newton, 1997b) and to the phosphorylation state of the hydrophobic motif within the C-term tail (Edwards and Newton, 1997a); additionally, constructs with an Ala at this phosphorylation site adopt an open conformation that associates with the plasma membrane, unless Ca2+ is chelated (Feng et al., 2000). Yet another study established that PKC is in a closed conformation in which neither PIP2 nor DAG can bind in the absence of Ca2+ (Corbalan-Garcia et al., 2003). Further evidence for intramolecular autoinhibitory contacts comes from studies by Mochly-Rosen and colleagues that revealed that the Receptor for Activated C-kinase (RACK)-binding site within the C-term tail of PKCβII interacts with a sequence in the C2 domain that mimics the PKC binding site on RACK (pseudo-RACK), maintaining PKC in an inactive conformation (Banci et al., 2002). These studies are consistent with the C2 domain of PKCβII interacting with the C-term tail to maintain the enzyme unresponsive to basal levels of agonists.
The C2 domain of the related PKCα has also been reported to contribute to its autoinhibition (Parissenti et al., 1998; Riedel et al., 1993). For example, deletion of 20 amino acids within its C2 domain induced constitutive activity of PKCα (Rotenberg et al., 1998). This autoinhibitory interaction was mediated by negatively charged residues in the C-term tail and a lysine-rich cluster in the C2 domain (Stensman and Larsson, 2007). Consistent with our work on PKCβII, reversing the charge of either of these interacting residues resulted in a higher sensitivity to DAG and thus enhanced membrane translocation, whereas simultaneously reversing both charges rescued the enhanced translocation phenotype (Stensman and Larsson, 2007). Taken together, these findings suggest that the C2 domain of PKCα has a similar placement to that of PKCβII, with conservation of charge at critical residues within the interfacing surfaces.
Of the two observed modes in the crystal lattice (Leonard et al., 2011), several arguments were presented against the intramolecularly clamped mode ii that our biochemical data support. First, Hurley and coworkers hypothesized that the pseudosubstrate would be excluded from the substrate-binding cavity in this conformation. However, molecular modeling of the pseudosubstrate in the substrate-binding cavity reveals that there is minimal steric hindrance, especially given the highly flexible nature of the Ca2+-binding loops (Figure 1C). Indeed, a clear opening is present and would accommodate threading of the segment following the pseudosubstrate to allow it to connect to the C1A domain that begins 10 residues past the pseudosubstrate (Figures 1E). Thus, the pseudosubstrate could occupy the active site in mode ii without significant structural changes. Although protein-protein docking is not a flawless method as it cannot directly address flexibility of protein main chains, our mutagenesis data confirm this mode ii conformation. Second, the authors reasoned that Ca2+ would not bind the C2 domain in mode ii. However, the affinity of the C2 domain for Ca2+ is over three-orders of magnitude lower in the absence of anionic lipids (Nalefski and Newton, 2001), because anionic lipids are required to stabilize Ca2+ binding (Nalefski and Falke, 1996). Stopped flow kinetic experiments suggest that PKC collides with membranes at the diffusion-controlled limit but rapidly dissociates because of unfavorable electrostatic interactions. Elevation of intracellular Ca2+ allows anionic lipids to retain PKC on membranes, increasing the lifetime of the membrane-bound complex by several orders of magnitude. This induces a conformational change that results in a two orders of magnitude increase in proteolytic sensitivity of the hinge connecting the C2 domain and kinase domain (Keranen and Newton, 1997; Kishimoto et al., 1983; Young et al., 1988). In fact, no cleavage was observed between the C1B and C2 domains under any condition tested (Keranen and Newton, 1997), as the model proposed by Leonard et al. (2011) suggests based on the presence of a large flexible linker between these domains. Rather, Ca2+-dependent binding to membranes results in unmasking of the hinge separating the C2 domain from the kinase domain (Keranen and Newton, 1997), with subsequent activation resulting in exposure of the pseudosubstrate segment (Orr et al., 1992). This supports a model in which Ca2+ provides conformational selection by favoring an equilibrium in which the C2 domain is pulled away from the kinase domain via bridging to the membrane.
SAXS data revealed an elongated shape of PKCβII (Leonard et al., 2011), which the authors considered at odds with the intramolecularly clamped mode ii. The program AutoGNOM was used by Leonard et al. (2011) to determine that the maximum linear dimension (Dmax) of the complex was 100 Å. Our model of the kinase domain and C2 domain in mode ii is ~ 83Å (Figure 1E), but excludes the C1A and C1B domains, which would increase the Dmax, because their exact placement within the full-length structure is yet to be determined. Additional experiments such as sedimentation studies or electron microscopy would be required for the independent estimate of Dmax (Moore, 1980). Another limitation of SAXS analysis is that it considers proteins as rigid bodies; however, our model suggests that the C2 domain is highly dynamic and alternates between various conformations until Ca2+-binding drives the equilibrium towards the membrane-bound, open conformation. It is known that interdomain dynamics can significantly alter SAXS profiles and lead to misinterpretation of SAXS data (Bernado, 2010). Therefore, the existing SAXS data (Leonard et al., 2011) cannot distinguish between the two models in question. A complete crystal structure, in which all domains are present and with high enough resolution to resolve the linker regions, corroborated biochemically by mutagenesis, would be necessary to determine the exact conformation of the C1 domains in the inactive conformation of PKCβII. However, our mutagenesis data clearly support our model of the C2 domain interfacing with the kinase domain and C-term tail.
Our data suggest that the NFD helix does not serve as the linchpin for activation of PKC, as proposed by Hurley and colleagues. suggested that binding of the NFD helix to the C1B domain positions Phe629 away from the ATP binding site to reduce catalysis. However, a Phe to Ala mutation results in a PKC whose activity in cells is indistinguishable from that of wild-type enzyme. Mutation of the corresponding residue in PKA (Phe327) to Ala resulted in only a modest decrease in kcat (from 26 sec−1 to 20 sec−1), and although the Km for ATP was increased approximately 10-fold (from 20 μM to 249 μM) (Yang et al., 2009), this is 20-fold lower than the intracellular concentration of ATP, suggesting limited biological relevance. Based on this mechanism, the authors proposed an intermediate step in the activation of PKC in which the pseudosubstrate is released from the active site but the enzyme is inactive because the C1B is still bound to the kinase domain, positioning Phe629 away from the ATP binding site. However, release of the pseudosubstrate correlates with the activation of PKC under all conditions examined (Orr et al., 1992; Orr and Newton, 1994). Thus, the NFD helix of PKCβII is not a key regulator of kinase activity; rather release of the pseudosubstrate is the key determinant for activation.
Figure 5 presents a model for the regulation of PKC that takes into account the conformational sensing by the C2 domain. We have previously shown that newly-synthesized PKC (Figure 5A) is in an open conformation (Antal et al., 2014). Upon maturation by phosphorylation, conformational rearrangements mask PKC's C1 domains, thus reducing its apparent affinity for DAG so there is no binding to basal DAG. We now build on this model of autoinhibition to show that the C2 domain clamps over the kinase domain, tethering the pseudosubstrate in place (Figure 5B) for even more effective autoinhibition than previously proposed. This explains why the activation loop is inaccessible to PDK-1 or to phosphatases in the autoinhibited conformation (Dutil and Newton, 2000). Upon elevation of intracellular Ca2+, conformational selection allows the Ca2+-bound C2 domain to engage on the membrane, inducing a large hinge motion that renders the C2:kinase domain linker 100-fold more sensitive to limited proteolysis (Figure 5C) (Keranen and Newton, 1997). This membrane-bound species is now able to bind DAG, via the C1B domain, an event that pulls the pseudosubstrate out of the substrate-binding cavity to allow full activation of PKC (Figure 5D). Note that only one C1 domain binds ligand at a time, as determined by seminal studies by Nishizuka and Blumberg establishing that the stoichiometry of ligand binding of full-length PKC is one mole DAG or one mole phorbol ester per mole of PKC (Kikkawa et al., 1983; Konig et al., 1985b), a finding that has been confirmed many times (Giorgione et al., 2003; Hannun et al., 1985; Quest and Bell, 1994; Solodukhin et al., 2007).
Two striking features of the PKC structure are now apparent. First, the C2 domain provides an additional layer of autoinhibition to ensure no basal signaling of PKC in the absence of agonists. Thus, activation requires the release of the C2 domain from the kinase domain, followed by the release of the pseudosubstrate from the substrate-binding cavity. Second, intramolecular interactions can now be targeted in therapies. For example, in cancer therapies, where PKC activity should be enhanced (Antal et al., 2015), small molecules or peptides that destabilize the clamped conformation of PKCβII will allow it to be more responsive to second messengers. Conversely, in therapies for neurodegenerative or heart diseases where PKC activity should be reduced, small molecules or peptides can be designed to stabilize the clamped conformation. Because the C-term tails of the PKC isozymes are highly variable, this method could provide a unique isozyme-specific method of regulating the activity of individual isozymes.
EXPERIMENTAL PROCEDURES
Plasmid Constructs, Antibodies, and Reagents
C-terminally tagged rat PKCβII-YFP (Dries et al., 2007) and the membrane-targeted CFP (Violin et al., 2003) have been previously described. Rat PKCβII was RFP-tagged at the C-terminus. All mutants were generated by QuikChange site-directed mutagenesis (Stratagene). The pan anti-phospho-PKC activation loop antibody was previously described (Dutil et al., 1998) and the anti-PKCβ (610128) antibody was from BD Transduction Laboratories. Phorbol 12,13-dibutyrate (PDBu) and thapsigargin were purchased from Calbiochem. All other materials were reagent grade.
Cell Culture, Transfection, and Immunoblotting
COS7 cells were cultured in DMEM (Cellgro) containing 10% fetal bovine serum (Atlanta Biologicals) and 1% penicillin/streptomycin (Gibco) at 37 °C in 5% CO2. Transient transfection was carried out using jetPRIME (PolyPlus Transfection) or FuGENE 6 transfection reagents (Roche Applied Science) for ~24h. Cells were lysed in 50 mM Tris, pH 7.4, 1% Triton X-100, 50 mM NaF, 10 mM Na4P2O7, 100 mM NaCl, 5 mM EDTA, 1 mM Na3VO4, 1 mM PMSF, 40 μg/ml leupeptin,, and 1μM microcystin. Whole cell lysates were analyzed by SDS-PAGE and Western blotting via chemiluminescence on a FluorChem Q imaging system (ProteinSimple).
FRET Imaging and Analysis
Cells were imaged as described previously (Gallegos et al., 2006). COS7 cells were co-transfected with the indicated YFP-tagged PKC construct and plasma membrane-targeted. Base-line images were acquired every 7 or 15 sec for ≥ 2 min before ligand addition. FRET ratios represent mean ± SEM from at least 3 independent experiments. Data were normalized to the baseline FRET ratios and because the maximal amplitude of translocation of the mutants varied, possibly because changes in the orientation or distance of the fluorophores caused by differential folding of the kinase, data were also normalized to the maximal amplitude of translocation for each cell as assessed following PDBu addition, as previously described (Antal et al., 2015). Statistical significance was determined via a Student's t-test performed in Graph Pad Prism 6.0a (GraphPad Software). The half-time of translocation was calculated by fitting the data to a non-linear regression using a one-phase exponential association equation, with Graph Pad Prism 6.0a (GraphPad Software).
Supplementary Material
ACKNOWLEDGMENTS
We thank Emily Kang for experimental assistance and the Newton and Taylor labs for helpful discussions. This work was supported by NIH GM43154 to A.C.N. and DK54441 to S.S.T. C.E.A. was supported in part by the UCSD Graduate Training Program in Cellular and Molecular Pharmacology (T32 GM007752) and the NSF Graduate Research Fellowship (DGE1144086).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
SUPPLEMENTAL INFORMATION Supplemental Information includes 2 Figures and can be found with this article online.
AUTHOR CONTRIBUTIONS C.E.A and J.A.C. performed the experiments. C.E.A and A.C.N wrote the manuscript. A.P.K. performed the modeling and docking. S.S.T. and A.C.N. conceived the project.
REFERENCES
- Antal CE, Hudson AM, Kang E, Zanca C, Wirth C, Stephenson NL, Trotter EW, Gallegos LL, Miller CJ, Furnari FB, et al. Cancer-Associated Protein Kinase C Mutations Reveal Kinase's Role as Tumor Suppressor. Cell. 2015;160:489–502. doi: 10.1016/j.cell.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antal CE, Violin JD, Kunkel MT, Skovso S, Newton AC. Intramolecular conformational changes optimize protein kinase C signaling. Chemistry & biology. 2014;21:459–469. doi: 10.1016/j.chembiol.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banci L, Cavallaro G, Kheifets V, Mochly-Rosen D. Molecular dynamics characterization of the C2 domain of protein kinase Cbeta. The Journal of biological chemistry. 2002;277:12988–12997. doi: 10.1074/jbc.M106875200. [DOI] [PubMed] [Google Scholar]
- Belin RJ, Sumandea MP, Allen EJ, Schoenfelt K, Wang H, Solaro RJ, de Tombe PP. Augmented protein kinase C-alpha-induced myofilament protein phosphorylation contributes to myofilament dysfunction in experimental congestive heart failure. Circulation research. 2007;101:195–204. doi: 10.1161/CIRCRESAHA.107.148288. [DOI] [PubMed] [Google Scholar]
- Bernado P. Effect of interdomain dynamics on the structure determination of modular proteins by small-angle scattering. Eur Biophys J. 2010;39:769–780. doi: 10.1007/s00249-009-0549-3. [DOI] [PubMed] [Google Scholar]
- Bowling N, Walsh RA, Song G, Estridge T, Sandusky GE, Fouts RL, Mintze K, Pickard T, Roden R, Bristow MR, et al. Increased protein kinase C activity and expression of Ca2+-sensitive isoforms in the failing human heart. Circulation. 1999;99:384–391. doi: 10.1161/01.cir.99.3.384. [DOI] [PubMed] [Google Scholar]
- Conrad R, Keranen LM, Ellington AD, Newton AC. Isozyme-specific inhibition of protein kinase C by RNA aptamers. The Journal of biological chemistry. 1994;269:32051–32054. [PubMed] [Google Scholar]
- Corbalan-Garcia S, Garcia-Garcia J, Rodriguez-Alfaro JA, Gomez-Fernandez JC. A new phosphatidylinositol 4,5-bisphosphate-binding site located in the C2 domain of protein kinase Calpha. The Journal of biological chemistry. 2003;278:4972–4980. doi: 10.1074/jbc.M209385200. [DOI] [PubMed] [Google Scholar]
- Dries DR, Gallegos LL, Newton AC. A single residue in the C1 domain sensitizes novel protein kinase C isoforms to cellular diacylglycerol production. The Journal of biological chemistry. 2007;282:826–830. doi: 10.1074/jbc.C600268200. [DOI] [PubMed] [Google Scholar]
- Dutil EM, Newton AC. Dual role of pseudosubstrate in the coordinated regulation of protein kinase C by phosphorylation and diacylglycerol. The Journal of biological chemistry. 2000;275:10697–10701. doi: 10.1074/jbc.275.14.10697. [DOI] [PubMed] [Google Scholar]
- Dutil EM, Toker A, Newton AC. Regulation of conventional protein kinase C isozymes by phosphoinositide-dependent kinase 1 (PDK-1) Current biology : CB. 1998;8:1366–1375. doi: 10.1016/s0960-9822(98)00017-7. [DOI] [PubMed] [Google Scholar]
- Edwards AS, Newton AC. Phosphorylation at conserved carboxyl-terminal hydrophobic motif regulates the catalytic and regulatory domains of protein kinase C. The Journal of biological chemistry. 1997a;272:18382–18390. doi: 10.1074/jbc.272.29.18382. [DOI] [PubMed] [Google Scholar]
- Edwards AS, Newton AC. Regulation of protein kinase C betaII by its C2 domain. Biochemistry. 1997b;36:15615–15623. doi: 10.1021/bi9718752. [DOI] [PubMed] [Google Scholar]
- Feng X, Becker KP, Stribling SD, Peters KG, Hannun YA. Regulation of receptor-mediated protein kinase C membrane trafficking by autophosphorylation. The Journal of biological chemistry. 2000;275:17024–17034. doi: 10.1074/jbc.275.22.17024. [DOI] [PubMed] [Google Scholar]
- Gallegos LL, Kunkel MT, Newton AC. Targeting protein kinase C activity reporter to discrete intracellular regions reveals spatiotemporal differences in agonist-dependent signaling. The Journal of biological chemistry. 2006;281:30947–30956. doi: 10.1074/jbc.M603741200. [DOI] [PubMed] [Google Scholar]
- Giorgione J, Hysell M, Harvey DF, Newton AC. Contribution of the C1A and C1B domains to the membrane interaction of protein kinase C. Biochemistry. 2003;42:11194–11202. doi: 10.1021/bi0350046. [DOI] [PubMed] [Google Scholar]
- Gould CM, Antal CE, Reyes G, Kunkel MT, Adams RA, Ziyar A, Riveros T, Newton AC. Active site inhibitors protect protein kinase C from dephosphorylation and stabilize its mature form. The Journal of biological chemistry. 2011;286:28922–28930. doi: 10.1074/jbc.M111.272526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grodsky N, Li Y, Bouzida D, Love R, Jensen J, Nodes B, Nonomiya J, Grant S. Structure of the catalytic domain of human protein kinase C beta II complexed with a bisindolylmaleimide inhibitor. Biochemistry. 2006;45:13970–13981. doi: 10.1021/bi061128h. [DOI] [PubMed] [Google Scholar]
- Guerrero-Valero M, Ferrer-Orta C, Querol-Audi J, Marin-Vicente C, Fita I, Gomez-Fernandez JC, Verdaguer N, Corbalan-Garcia S. Structural and mechanistic insights into the association of PKCalpha-C2 domain to PtdIns(4,5)P2. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:6603–6607. doi: 10.1073/pnas.0813099106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannun YA, Bell RM. Phorbol ester binding and activation of protein kinase C on triton X-100 mixed micelles containing phosphatidylserine. The Journal of biological chemistry. 1986;261:9341–9347. [PubMed] [Google Scholar]
- Hannun YA, Loomis CR, Bell RM. Activation of protein kinase C by Triton X-100 mixed micelles containing diacylglycerol and phosphatidylserine. The Journal of biological chemistry. 1985;260:10039–10043. [PubMed] [Google Scholar]
- Hommel U, Zurini M, Luyten M. Solution structure of a cysteine rich domain of rat protein kinase C. Nature structural biology. 1994;1:383–387. doi: 10.1038/nsb0694-383. [DOI] [PubMed] [Google Scholar]
- Ji J, Hassler ML, Shimobayashi E, Paka N, Streit R, Kapfhammer JP. Increased protein kinase C gamma activity induces Purkinje cell pathology in a mouse model of spinocerebellar ataxia 14. Neurobiology of disease. 2014;70:1–11. doi: 10.1016/j.nbd.2014.06.002. [DOI] [PubMed] [Google Scholar]
- Keranen LM, Newton AC. Ca2+ differentially regulates conventional protein kinase Cs' membrane interaction and activation. The Journal of biological chemistry. 1997;272:25959–25967. doi: 10.1074/jbc.272.41.25959. [DOI] [PubMed] [Google Scholar]
- Kheifets V, Mochly-Rosen D. Insight into intra- and inter-molecular interactions of PKC: design of specific modulators of kinase function. Pharmacological research : the official journal of the Italian Pharmacological Society. 2007;55:467–476. doi: 10.1016/j.phrs.2007.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishimoto A, Kajikawa N, Shiota M, Nishizuka Y. Proteolytic activation of calcium-activated, phospholipid-dependent protein kinase by calcium-dependent neutral protease. The Journal of biological chemistry. 1983;258:1156–1164. [PubMed] [Google Scholar]
- Leonard TA, Rozycki B, Saidi LF, Hummer G, Hurley JH. Crystal structure and allosteric activation of protein kinase C betaII. Cell. 2011;144:55–66. doi: 10.1016/j.cell.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore PB. Small-Angle Scattering - Information-Content and Error Analysis. J Appl Crystallogr. 1980;13:168–175. [Google Scholar]
- Nalefski EA, Falke JJ. The C2 domain calcium-binding motif: structural and functional diversity. Protein science : a publication of the Protein Society. 1996;5:2375–2390. doi: 10.1002/pro.5560051201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalefski EA, Newton AC. Membrane binding kinetics of protein kinase C betaII mediated by the C2 domain. Biochemistry. 2001;40:13216–13229. doi: 10.1021/bi010761u. [DOI] [PubMed] [Google Scholar]
- Orr JW, Keranen LM, Newton AC. Reversible exposure of the pseudosubstrate domain of protein kinase C by phosphatidylserine and diacylglycerol. The Journal of biological chemistry. 1992;267:15263–15266. [PubMed] [Google Scholar]
- Orr JW, Newton AC. Intrapeptide regulation of protein kinase C. The Journal of biological chemistry. 1994;269:8383–8387. [PubMed] [Google Scholar]
- Parissenti AM, Kirwan AF, Kim SA, Colantonio CM, Schimmer BP. Inhibitory properties of the regulatory domains of human protein kinase Calpha and mouse protein kinase Cepsilon. The Journal of biological chemistry. 1998;273:8940–8945. doi: 10.1074/jbc.273.15.8940. [DOI] [PubMed] [Google Scholar]
- Parker PJ, Murray-Rust J. PKC at a glance. Journal of cell science. 2004;117:131–132. doi: 10.1242/jcs.00982. [DOI] [PubMed] [Google Scholar]
- Quest AF, Bell RM. The regulatory region of protein kinase C gamma. Studies of phorbol ester binding to individual and combined functional segments expressed as glutathione S-transferase fusion proteins indicate a complex mechanism of regulation by phospholipids, phorbol esters, and divalent cations. The Journal of biological chemistry. 1994;269:20000–20012. [PubMed] [Google Scholar]
- Reyland ME. Protein kinase Cdelta and apoptosis. Biochemical Society transactions. 2007;35:1001–1004. doi: 10.1042/BST0351001. [DOI] [PubMed] [Google Scholar]
- Riedel H, Su L, Hansen H. Yeast phenotype classifies mammalian protein kinase C cDNA mutants. Molecular and cellular biology. 1993;13:4728–4735. doi: 10.1128/mcb.13.8.4728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers TB, Inesi G, Wade R, Lederer WJ. Use of thapsigargin to study Ca2+ homeostasis in cardiac cells. Bioscience reports. 1995;15:341–349. doi: 10.1007/BF01788366. [DOI] [PubMed] [Google Scholar]
- Rotenberg SA, Zhu J, Hansen H, Li XD, Sun XG, Michels CA, Riedel H. Deletion analysis of protein kinase Calpha reveals a novel regulatory segment. Journal of biochemistry. 1998;124:756–763. doi: 10.1093/oxfordjournals.jbchem.a022176. [DOI] [PubMed] [Google Scholar]
- Scott AM, Antal CE, Newton AC. Electrostatic and hydrophobic interactions differentially tune membrane binding kinetics of the C2 domain of protein kinase Calpha. The Journal of biological chemistry. 2013;288:16905–16915. doi: 10.1074/jbc.M113.467456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi F, Telesco SE, Liu Y, Radhakrishnan R, Lemmon MA. ErbB3/HER3 intracellular domain is competent to bind ATP and catalyze autophosphorylation. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:7692–7697. doi: 10.1073/pnas.1002753107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieber FE, Traystman RJ, Brown PR, Martin LJ. Protein kinase C expression and activity after global incomplete cerebral ischemia in dogs. Stroke; a journal of cerebral circulation. 1998;29:1445–1452. doi: 10.1161/01.str.29.7.1445. discussion 1452–1443. [DOI] [PubMed] [Google Scholar]
- Solodukhin AS, Kretsinger RH, Sando JJ. Initial three-dimensional reconstructions of protein kinase C delta from two-dimensional crystals on lipid monolayers. Cellular signalling. 2007;19:2035–2045. doi: 10.1016/j.cellsig.2007.05.010. [DOI] [PubMed] [Google Scholar]
- Stensman H, Larsson C. Identification of acidic amino acid residues in the protein kinase C alpha V5 domain that contribute to its insensitivity to diacylglycerol. The Journal of biological chemistry. 2007;282:28627–28638. doi: 10.1074/jbc.M702248200. [DOI] [PubMed] [Google Scholar]
- Stensman H, Raghunath A, Larsson C. Autophosphorylation suppresses whereas kinase inhibition augments the translocation of protein kinase Calpha in response to diacylglycerol. The Journal of biological chemistry. 2004;279:40576–40583. doi: 10.1074/jbc.M405560200. [DOI] [PubMed] [Google Scholar]
- Takeishi Y, Ping P, Bolli R, Kirkpatrick DL, Hoit BD, Walsh RA. Transgenic overexpression of constitutively active protein kinase C epsilon causes concentric cardiac hypertrophy. Circulation research. 2000;86:1218–1223. doi: 10.1161/01.res.86.12.1218. [DOI] [PubMed] [Google Scholar]
- Verbeek DS, Knight MA, Harmison GG, Fischbeck KH, Howell BW. Protein kinase C gamma mutations in spinocerebellar ataxia 14 increase kinase activity and alter membrane targeting. Brain : a journal of neurology. 2005;128:436–442. doi: 10.1093/brain/awh378. [DOI] [PubMed] [Google Scholar]
- Verdaguer N, Corbalan-Garcia S, Ochoa WF, Fita I, Gomez-Fernandez JC. Ca(2+) bridges the C2 membrane-binding domain of protein kinase Calpha directly to phosphatidylserine. The EMBO journal. 1999;18:6329–6338. doi: 10.1093/emboj/18.22.6329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Violin JD, Zhang J, Tsien RY, Newton AC. A genetically encoded fluorescent reporter reveals oscillatory phosphorylation by protein kinase C. The Journal of cell biology. 2003;161:899–909. doi: 10.1083/jcb.200302125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Kennedy EJ, Wu J, Deal MS, Pennypacker J, Ghosh G, Taylor SS. Contribution of non-catalytic core residues to activity and regulation in protein kinase A. The Journal of biological chemistry. 2009;284:6241–6248. doi: 10.1074/jbc.M805862200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young S, Rothbard J, Parker PJ. A monoclonal antibody recognising the site of limited proteolysis of protein kinase C. Inhibition of down-regulation in vivo. European journal of biochemistry / FEBS. 1988;173:247–252. doi: 10.1111/j.1432-1033.1988.tb13991.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.