

ABSTRACT
CcrM is an orphan DNA methyltransferase nearly universally conserved in a vast group of Alphaproteobacteria. In Caulobacter crescentus, it controls the expression of key genes involved in the regulation of the cell cycle and cell division. Here, we demonstrate, using an experimental evolution approach, that C. crescentus can significantly compensate, through easily accessible genetic changes like point mutations, the severe loss in fitness due to the absence of CcrM, quickly improving its growth rate and cell morphology in rich medium. By analyzing the compensatory mutations genome-wide in 12 clones sampled from independent ΔccrM populations evolved for ~300 generations, we demonstrated that each of the twelve clones carried at least one mutation that potentially stimulated ftsZ expression, suggesting that the low intracellular levels of FtsZ are the major burden of ΔccrM mutants. In addition, we demonstrate that the phosphoenolpyruvate-carbohydrate phosphotransfer system (PTS) actually modulates ftsZ and mipZ transcription, uncovering a previously unsuspected link between metabolic regulation and cell division in Alphaproteobacteria. We present evidence that point mutations found in genes encoding proteins of the PTS provide the strongest fitness advantage to ΔccrM cells cultivated in rich medium despite being disadvantageous in minimal medium. This environmental sign epistasis might prevent such mutations from getting fixed under changing natural conditions, adding a plausible explanation for the broad conservation of CcrM.
IMPORTANCE
In bacteria, DNA methylation has a variety of functions, including the control of DNA replication and/or gene expression. The cell cycle-regulated DNA methyltransferase CcrM modulates the transcription of many genes and is critical for fitness in Caulobacter crescentus. Here, we used an original experimental evolution approach to determine which of its many targets make CcrM so important physiologically. We show that populations lacking CcrM evolve quickly, accumulating an excess of mutations affecting, directly or indirectly, the expression of the ftsZ cell division gene. This finding suggests that the most critical function of CcrM in C. crescentus is to promote cell division by enhancing FtsZ intracellular levels. During this work, we also discovered an unexpected link between metabolic regulation and cell division that might extend to other Alphaproteobacteria.
INTRODUCTION
Homologs of the solitary DNA adenine methyltransferase CcrM are almost universally conserved in the ancient and ecologically very diverse clade of non-Rickettsiales Alphaproteobacteria (1). This extensive conservation supports the idea that CcrM homologs have been essential for fitness (2, 3) in these Alphaproteobacteria over hundreds of millions of years of evolution and suggests that virtually no strain or species in which the ccrM gene was inactivated by accident would have been able to compete efficiently with its ccrM-encoding relatives. Accordingly, CcrM homologs show major signs of phylogenetic persistence, being very conserved in sequence, highly expressed in tested models, and located in the vicinity of other essential or persistent genes (1, 4–7). Moreover, the nearly universal conservation of CcrM suggests that the function of DNA methylation by CcrM in Alphaproteobacteria is difficult to compensate genetically under common evolutionary scenarios in natural environments. It has indeed been shown that essential genes whose function can be replaced through easily accessible genetic changes, like a small number of single nucleotide substitutions or a change in expression of a paralogous gene, tend to be less conserved than genes whose function cannot be so easily compensated (8, 9). Our objective was to shed some light on the striking persistence of CcrM DNA methyltransferases by using experimental evolution, whole-genome sequencing and classical genetics to study the compensability of the CcrM homolog in the alphaproteobacterial model Caulobacter crescentus.
CcrM plays a central role in the regulation of the cell cycle of Caulobacter crescentus, methylating the adenine in nearly all GANTC sequences along the chromosome in a cell cycle-dependent way (5, 10, 11). Since CcrM is active during only a short time window before cell division and after the replication of the chromosome, the methylation state of GANTC motifs alternates periodically during the cell cycle between a fully methylated and a hemimethylated form (after the replication of each GANTC motif until the predivisional stage, when CcrM is expressed again) (11, 12). The methylation state of GANTC motifs influences among others the transcription rate of the genes coding for two global regulators of the cell cycle: DnaA, which initiates DNA replication and regulates the transcription of about 40 genes, and CtrA, which represses DNA replication and regulates the transcription of about 95 genes (13–16). In addition, the activity of a third key regulator of the cell cycle, the transcription factor GcrA, seems to be influenced by the methylation state of GANTC motifs located at or close to its binding sites on the promoter of at least some of its many target genes (17, 18). On its own, a regulatory cascade connecting DnaA, GcrA, and CtrA might control the cell cycle-dependent transcription of nearly 200 genes (13). An estimate based on a transcriptomic study suggests that the transcription of at least 40 genes could be influenced directly by GANTC methylation; among them, essential functions related to cell division and DNA metabolism are overrepresented (1). In particular, the transcription of mipZ and ftsZ, whose products are involved in the early steps of the divisome assembly, is strongly activated by the methylation of GANTC motifs located in their promoter region (19). Overall, these results indicate that CcrM is an important element in the architecture of the network regulating the cell cycle and, especially, in the control of cell division in C. crescentus.
Despite its key regulatory role, the ccrM gene is not essential for viability in C. crescentus: ΔccrM mutants can easily be constructed and cultured in minimal medium (M2G [M2 minimal salts plus 0.2% glucose]), where they have a moderate growth disadvantage relative to wild-type cells (19). However, when they are transferred to rich medium (peptone yeast extract [PYE]), ΔccrM mutants lose the capacity to divide, forming long filaments that quickly end up lysing. Complementation with an increased expression of the FtsZ protein can partially restore cell division and viability of ΔccrM mutants, suggesting that low levels of FtsZ are responsible for the most severe phenotypes of the mutant or that other missing functions can be compensated through higher expression of FtsZ. Overall, it appears that, in the absence of CcrM, C. crescentus cannot cope with elevated growth rates under conditions where organic matter (amino acids) is abundant, where it probably fails to coordinate growth and cell division (19). Such a limitation would be extremely disadvantageous in a natural environment where changes in nutrient abundance and type might be quick and radical (20). Naturally arising ΔccrM mutants could simply not be able to survive sudden “feast” conditions, missing most opportunities to increase in population size. This might apply to other Alphaproteobacteria as well, given that the ccrM gene seems to be essential in rich medium in several of them (4, 21, 22).
As the starting point of our research, we asked whether and to what extent the fitness disadvantage associated with the disruption of ccrM could be compensated through mutations in C. crescentus cells cultivated under very selective conditions (rich medium). We took advantage of the fact that CcrM is not essential in minimal medium and that cell death is not immediate upon transfer to rich medium (19). Indeed, for potential compensatory mutations to appear and spread in the population, cells have to be able to survive and, ideally, divide, even slowly, in the restrictive conditions. Since fitter mutants appeared quickly and reproducibly in our experimental setup, we prolonged the experiment for a total of 300 generations to get a more extensive view of the mutational landscape of evolving ΔccrM mutants. Through the analysis of the genetic basis of the adaptation, we were able to learn more about the fitness determinants of C. crescentus and to get a better understanding of CcrM interactions within the complex regulatory network that underlies the bacterial cell cycle.
RESULTS
Phenotypic adaptation of ΔccrM populations growing in rich medium.
We let 12 independent populations of a C. crescentus strain lacking CcrM (NA1000 ΔccrM) evolve in rich medium (PYE) at 28°C for about 300 doublings in optical density (corresponding to doublings in cell mass; referred to here as “generations”), rediluting the cultures every day and collecting/freezing samples every 50 generations. We started the cultures from three independently obtained ΔccrM clones; the evolving populations were numbered 1.1 to 1.4 (ancestral strain, JC1149), 2.1 to 2.4 (ancestral strain, JC1150), and 3.1 to 3.4 (ancestral strain, JC1151). As controls, 10 independent populations of wild-type NA1000 C. crescentus were cultured under the same conditions for the same number of generations. During the course of the experiment, we monitored the growth rate and the morphology of the cells in the evolving populations. In each of the 12 ΔccrM populations, but in none of the NA1000 populations, we observed significant changes in growth and morphology over the 300 generations. In particular, we distinguished two phases during the evolution of the ΔccrM populations: a phase of rapid adaptation, between the start of the experiment and the ~40th generation, involving rapid changes in cell shape and growth rates, and a phase of slower evolution, between the ~40th and the 300th generation (Fig. 1A).
FIG 1 .

Changes in growth rate and morphology during the evolution of ΔccrM populations in rich medium. Twelve independent ΔccrM populations and 10 independent wild-type (NA1000) populations were transferred from minimal medium (M2G) to rich medium (PYE) at an OD660 of 0.01 at the beginning of the experiment. Each second day for the first 40 doublings and each day for the following doublings, the culture was rediluted to an OD660 of 0.001. (A) Changes in average overnight generation time during the course of the experiment for the 12 ΔccrM populations and for the 10 NA1000 control populations. The overnight generation time was measured, taking the initial and final OD660 and time interval into account. The error bars show standard deviations from the means for the 10 or 12 populations of each starting genotype. (B) Morphology of representative cells in a ΔccrM population during the initial phase of evolution: before transfer to rich medium (M2G) and 6, 15, and 32 generations (gen.) after transfer. After 6 generations in rich medium, cells formed 10- to 40-µm-long filaments that did not show signs of lysis; after 15 generations, more than 50% of the filamentous cells seemed to lyse (light grey cells); after 32 generations, viable shorter cells invaded the population. (C) Between generation 50 and generation 300, the clones sampled from the ΔccrM populations gained in fitness (growth rate). Two clones were isolated from each of the 12 ΔccrM populations at generation 50 and generation 300 and grown independently in rich medium. The doubling times were measured in exponential phase. The thick line represents the median, the limits of the box show the 25th and 75th quartiles, and the whiskers show the 5th and 95th quantiles.
Right after the transfer from minimal medium (M2G) to rich medium at the beginning of the experiment, the C. crescentus cells lacking CcrM started to elongate and to form filaments more than 10 times longer than the wild-type cells, as previously reported (19) (Fig. 1B). The filamentous cells eventually started to lyse: the populations sampled at generations ~15 and ~23 were composed of more than 50% “ghost” cells either lacking DNA (no 4′,6-diamidino-2-phenylindole [DAPI] coloration) or permeable to the dead stain propidium iodide (data not shown), which is a known characteristic of the ΔccrM strain cultivated in rich medium (19); by that time, most live cells were filamentous or very elongated (Fig. 1B). However, between generations 30 and 40, the average length of the live cells decreased suddenly in all the populations simultaneously, while the frequency of dying and highly filamentous cells dropped (Fig. 1B). The morphological evolution correlated with changes in growth rate: while the average doubling time remained between 3 and 4 h during the 5 to 7 first generations in rich medium, it increased up to more than 6 h during the next 20 generations, as dead cells were accumulating in the populations (Fig. 1A; also, see Fig. S1A in the supplemental material). After that, the doubling time dropped back to about 3 h, as the average cell size and the frequency of dead cells decreased.
The changes observed during the rapid evolution phase could have been caused either by a physiological adaptation (epigenetic or regulatory) or by a change in the genetic composition of the population (selective sweep). To test this second alternative, we isolated colonies at generation 50 on rich medium agar plates, grew them in minimal medium and then transferred them back to rich medium. No obvious decrease in growth rate was observed upon transfer (data not shown), indicating that the adaptation of the cells to rich medium was stable and, thus, most likely had a genetic rather than epigenetic or regulatory basis.
A further slower decrease in doubling time was observed after generation 40 for most evolving ΔccrM populations on the basis of their generation time (see Fig. S1A and B in the supplemental material). To confirm this result, we calculated the generation time in rich medium by a more traditional method using pure cultures of clones isolated from the 12 ΔccrM evolving populations at generations 50 and 300. The median difference in generation time between the first and the second set of clones was 24 min and statistically significant (Student’s t test P value, <0.01) (Fig. 1C). We could not demonstrate significant differences in cell morphology or size between two sets of clones sampled from the 12 evolving populations at the 50th and 300th generations by either microscopy or flow cytometry (data not shown). The existence of consistent and stable differences in growth rate between the two sets of clones suggested that the adaptation during the second phase of evolution might also have a genetic origin.
We concluded that, over time, the growth rate and morphology of ΔccrM populations evolving in rich medium improved greatly, most probably as a consequence of the appearance of fitter mutants that could invade and outcompete the ancestral ΔccrM strain.
Genetic basis of the adaptation.
To determine which mutations led to an increase in fitness in these ΔccrM strains, we sequenced the chromosome of one clone isolated from each of the 12 ΔccrM populations frozen after 300 generations and one clone isolated from 2 different bacterial populations frozen after 50 generations; as controls, we also sequenced the three independent ancestral ΔccrM strains that we used to start the evolution experiment and our NA1000 laboratory strain.
A total of 55 point mutations (single nucleotide substitutions, insertions, or deletions) was found in the genomes of the 14 evolved ΔccrM strains, each of which contained 2 to 7 individual point mutations (Table 1). The 55 point mutations were not distributed randomly along the genome of C. crescentus (Fig. 2). Ten point mutations were found in at least 2 of the 14 evolved strains (Table 1). Among the 35 different point mutations detected, 25 were nonsynonymous mutations affecting coding sequences, 3 were synonymous mutations, and 7 were found in intergenic regions. The ratio of nonsynonymous point mutations per nonsynonymous site to synonymous point mutations per synonymous site (Ka/Ks) was 5.24; the ratio for intergenic sites (Ki/Ks) was 7.71; the expected Ka/Ks and Ki/Ks ratios would have been 1 in a neutral evolution scenario. The 35 different point mutations were distributed in a total of 25 functional regions (coding sequences or intergenic regions). The recurrence of almost 40% of the point mutations (Fig. 2; Table 1), their concentration in a limited number of functional regions and the biased Ka/Ks and Ki/Ks ratios strongly suggest that the fixation of the point mutations in the different populations was promoted primarily by selection rather than drift.
Table 1 .
Point mutations identified in the 14 evolved ΔccrM strains
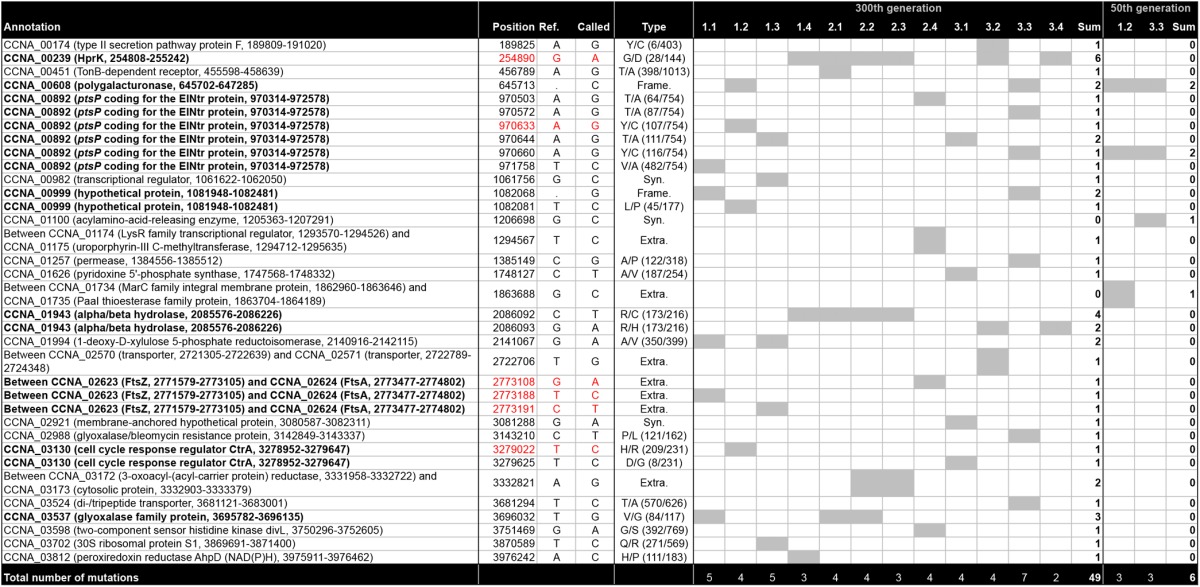
Bold text in “Annotation” indicates recurrent mutations. In “Type”: Syn., synonymous; Extra., extragenic; Frame., frameshift; for nonsynonymous mutations, amino acid in reference strain/amino acid in mutant strain (position/total amino acid). Grey cells, presence of the mutation in the strain. Red color, mutations confirmed as sufficient to induce ftsZ expression in this study.
FIG 2 .
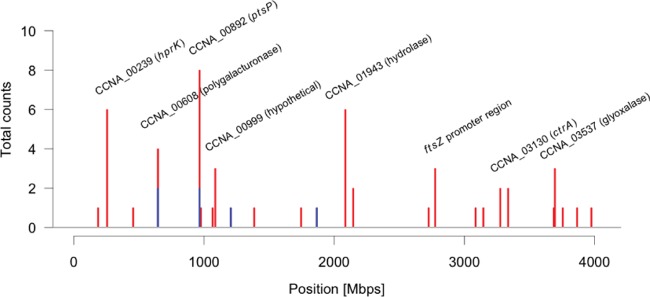
Distribution of the 55 identified point mutations along the chromosomes of a total of 14 evolved ΔccrM clones. One clone was isolated from 2 ΔccrM populations at generation 50 and from all 12 ΔccrM populations at generation 300. The graph shows the point mutation counts in 10-kb windows for the clones isolated at generation 50 (blue) or 300 (red).
Point mutations in PTS or PTS-associated genes.
All 14 evolved ΔccrM strains that we sequenced contained at least one point mutation affecting the phosphoenolpyruvate-carbohydrate phosphotransferase system (PTS), whereas none of the 10 NA1000 strains evolved in rich medium for 300 generations presented mutations in the same regions (see Table S1 in the supplemental material). The PTS is a pathway that mediates the transfer of a phosphate group from phosphoenolpyruvate to a sugar via several protein intermediates (Fig. 3A); it often controls the import of carbohydrates into the cytoplasm and regulates the metabolism in response to nutrient availability (23). In the genomes of the evolved ΔccrM strains, point mutations were found in sequences encoding either EINtr, which is often the only EI enzyme in Alphaproteobacteria (24), or the HPr serine kinase HPrK (Fig. 3B and C). One particular mutation was found in 6 different strains (Table 1): it changed into an aspartate a very conserved glycine 28 residue in the ATP-binding motif of the HprK serine kinase (encoded by hprK, also called CCNA_00239) (Fig. 3C). All the strains carrying the hprK mutation also carried one of two mutations in the CCNA_01943 gene, which changed an arginine into a histidine or a cysteine (Table 1). The CCNA_01943 gene codes for a protein of unknown function that is highly conserved among Alphaproteobacteria (including members of divergent orders, like Rickettsiales) and beyond. The co-occurrence of hprK and CCNA_01943 mutations suggests that there is a genetic interaction between the two proteins. The 8 strains that did not carry the hprK mutation harbored at least one mutation in the gene coding for the PTS EINtr (ptsP, also called CCNA_00892) (Fig. 3B; Table 1). In 7 of these strains, the mutation affected different residues in the N-terminal regulatory GAF domain of the protein.
FIG 3 .

Point mutations in genes encoding components of the phosphoenolpyruvate-carbohydrate phosphotransfer system (PTS) are the most frequent in evolved ΔccrM clones. (A) A simplified model for the PTS in C. crescentus based on homology searches (24, 47) and on the experimental work done on the Rhizobiales members Brucella melitensis and Sinorhizobium meliloti (36, 41, 48). The PTS allows a phosphate group to be transferred from phosphoenolpyruvate (PEP) to specific carbohydrates via several protein intermediates—EINtr, HPr, and EII—to regulate their import into the cytoplasm. EINtr and HPr may have additional regulatory functions via unidentified factors in Alphaproteobacteria. (B) Amino acid changes (counts) caused by point mutations found in the coding sequence of ptsP (EINtr) in 12 clones isolated at generation 300 (blue, whole-genome sequencing) or at generation 50 (red, targeted PCR and Sanger sequencing). (C) Amino acid changes (counts) caused by point mutations found in the coding sequence of hprK (HprK) in 12 clones isolated at generation 300 (blue, whole-genome sequencing) or at generation 50 (red, targeted PCR and Sanger sequencing).
The fact that the two clones isolated from two independent populations at generation 50 already carried a mutation in genes encoding a PTS component suggested that the initial increase in fitness of the ΔccrM strain cultivated in PYE was specifically linked to PTS mutations (Table 1). To confirm this, we sequenced the hprK and ptsP loci in 12 clones isolated from the 12 evolving ΔccrM populations at generation 50. We found that all but one (2.1) had a nonsynonymous mutation in the ptsP gene (8 strains) or the hprK gene (3 strains) (see Table S2 in the supplemental material). None of these strains carried a mutation in the CCNA_01943 gene, suggesting that mutations in this gene appear secondarily in strains already carrying a mutation in the hprK gene (see Table S2 in the supplemental material).
To characterize the effect of point mutations affecting the PTS in the absence of other mutations, we constructed wild-type and ΔccrM strains carrying the individual point mutations found in the evolved strains 1.2 (ptsP 1.2, an A-to-G change at position 970633 [970633 A/G]) and 1.4 (hprK 1.4, 254890 G/A). The growth of wild-type strains carrying these point mutations was heavily impaired in rich medium: the doubling times of the mutants were 270 and 210 min, respectively, compared to about 110 min for the control isogenic strains under the same conditions (Fig. 4A). In a ΔccrM genetic background, in contrast, the point mutations affecting the PTS were very advantageous in rich medium, as these strains had growth rates on average 1.7 times higher than that of the control ΔccrM strain; in minimal medium, the same mutations were only slightly disadvantageous (300 min of doubling time compared to 250 min for the control) (Fig. 4B and C). Phase-contrast microscopy confirmed that the morphology of the ΔccrM strains carrying the PTS mutations did not obviously change upon transfer from minimal to rich medium (Fig. 5; also, see Fig. S2 in the supplemental material), while control ΔccrM cells became highly filamentous and lysed (Fig. 1). All together, these results indicate that either mutation is sufficient to compensate for the most deleterious phenotypes of the ΔccrM strain in rich medium. Interestingly, one additional characteristic of these ΔccrM cells carrying a mutation in PTS-encoding genes cultivated in both rich and minimal media was a long-stalk phenotype (Fig. 5; also, see Fig. S2 in the supplemental material) similar to the one seen in wild-type C. crescentus under phosphate limitation (25, 26); this suggests a possible cross talk between the PTS and phosphate sensing in C. crescentus.
FIG 4 .
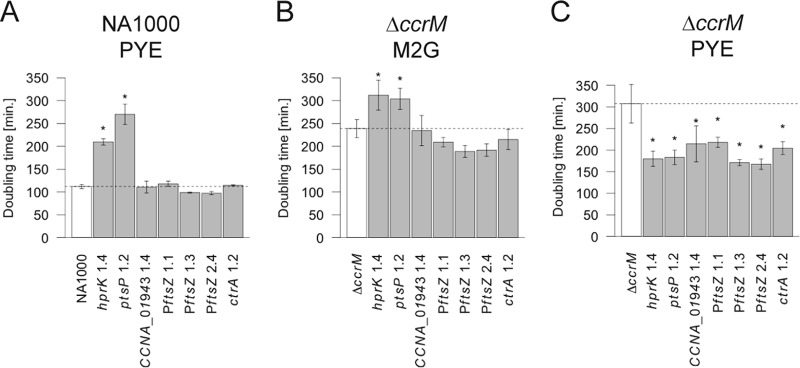
Growth rates of NA1000 and ΔccrM strains carrying a single point mutation in hprK, ptsP, CCNA_01943, the promoter region of ftsZ (PftsZ) (three different mutations), or ctrA. (A) Average growth rate in exponential phase of NA1000 mutants (hprK 1.4, JC1323; ptsP 1.2, JC1336; CCNA_01943 1.4, JC1327; PftsZ 1.1, JC1325; PftsZ 1.3, JC1328; PftsZ 2.4, JC1338; ctrA 1.2, JC1343) and control strains (NA1000, JC1322) in rich medium based on three replicates. (B) Average growth rate in exponential phase of ΔccrM mutant (hprK 1.4, JC1339; ptsP 1.2, JC1340; CCNA_01943 1.4, JC1344; PftsZ 1.1, JC1345; PftsZ 1.3, JC1341; PftsZ 2.4, JC1342; ctrA 1.2, JC1346) and control (ΔccrM, JC1347) strains in minimal medium based on three replicates. (C) Average growth rate of ΔccrM mutant and control strains during the 23 generations after transfer from minimal medium to rich medium based on two replicates. Error bars show standard deviations from the means; the dotted line represents the average for the control strain.
FIG 5 .
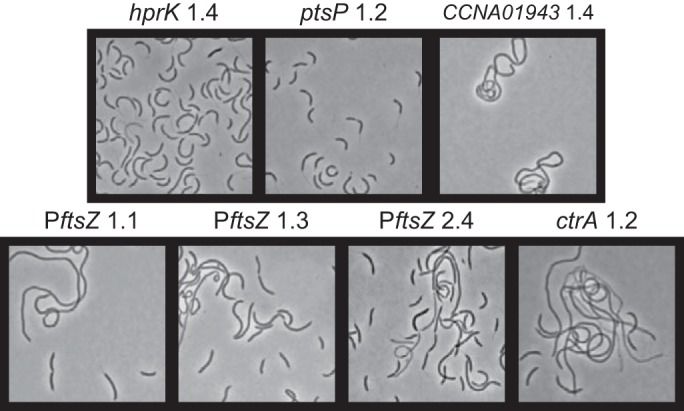
Phase-contrast micrographs of ΔccrM strains with single point mutations 7 generations after transfer from minimal to rich medium. Cells from strains JC1323, JC1325, JC1327, JC1328, JC1336, JC1338, and JC1343 are shown. Compare these images with the micrographs of a ΔccrM population after ~6 generations in rich medium (PYE) (Fig. 1B). The morphology of the same mutants in minimal medium (M2G) and at later generations in rich medium is presented in Fig. S2 in the supplemental material.
Since we had previously shown that the main phenotypes of the ΔccrM strain could be complemented by enhanced ftsZ transcription (19), we conjectured that the PTS mutations that we identified might increase the intracellular concentration of FtsZ and thereby promote cell division (Fig. 5). To determine whether the transcription of ftsZ might be promoted by the point mutations affecting the PTS, we quantified the activity of the ftsZ promoter in the wild-type and ΔccrM strains carrying the hprK 1.4 or ptsP 1.2 point mutation. We found that, in both PTS mutants, the ftsZ promoter was ~1.6 times more active than in the wild-type strain, suggesting that the PTS actually regulates ftsZ transcription in C. crescentus (Fig. 6A). In a ΔccrM background, the effect was similar: ftsZ transcription increased ~1.5 times in both PTS mutants, thereby compensating for 50% of the decrease in expression of ftsZ due to the absence of CcrM (Fig. 6A and B). This suggests that part of the fitness advantage that the ΔccrM PTS mutants have over their ΔccrM ancestors could be a consequence of partially restored levels of FtsZ.
FIG 6 .
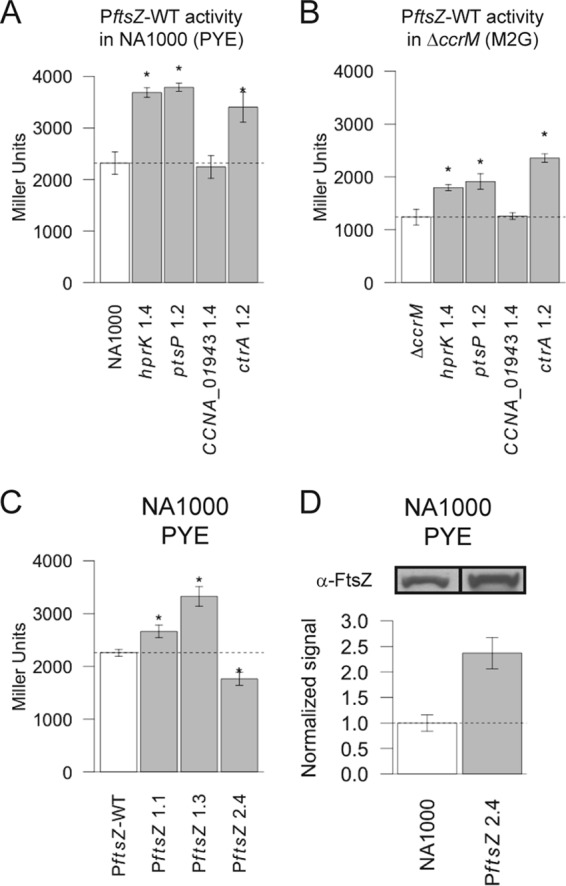
Effect of selected point mutations on ftsZ transcription and/or on the intracellular levels of the FtsZ protein. (A) Average activity of the wild-type ftsZ promoter in selected NA1000 mutant strains (NA1000, JC1353; hprK 1.4, JC1354; ptsP 1.2, JC1369; CCNA_01943 1.4, JC1368; ctrA 1.2, JC1370) as measured by β-galactosidase assays using cells cultivated in rich medium (PYE). (B) Average activity of the wild-type ftsZ promoter in selected ΔccrM mutant strains (ΔccrM, JC1371; hprK 1.4, JC1372; ptsP 1.2, JC1386; CCNA_01943 1.4, JC1387; ctrA 1.2, JC1388) as measured by β-galactosidase assays using cells cultivated in minimal medium (M2G). (C) Effect of the three different point mutations found in the ftsZ promoter region of evolved ΔccrM strains on ftsZ transcription in NA1000, as measured by β-galactosidase assays using cells cultivated in PYE (NA1000, JC1353; PftsZ 1.1, JC1383; PftsZ 1.3, JC1384; PftsZ 2.4, JC1385). (D) Relative quantification of FtsZ levels, from immunoblots, in an NA1000 derivative and in an isogenic strain carrying the 2.4 point mutation in the ftsZ promoter region (strains JC1347 and JC1342). In panels A through C, the averages are based on 3 or 4 replicates and error bars indicate standard deviations from the means; the dotted line represents the average for the control strain. In panel D, the averages are based on duplicates, and error bars indicate standard deviations from the means.
We also reconstructed the CCNA_01943 (2086092 C/T) mutation, which is systematically associated with hprK mutations, in wild-type and ΔccrM backgrounds and did not observe major effects on growth rate or ftsZ expression (Fig. 4A and B, 5, and 6A; also, see Fig. S2 in the supplemental material). The only significant difference relative to the control strain was found upon transfer of the ΔccrM CCNA_01943 single mutant to rich medium: cells grew slightly faster than ΔccrM cells (Fig. 4C). These results suggest that CCNA_01943 point mutations do not promote ftsZ transcription or cell division per se; they might rather modulate potential negative effects that the mutations found in hprK have on growth, consistent with their late appearance during our evolution experiment.
Taking all these data together, we concluded that mutants carrying specific point mutations in hprK or ptsP quickly invaded the ancestral ΔccrM populations cultivated in rich medium. We show that, on their own, these mutations are sufficient to suppress cell filamentation and lysis and to give a strong selective advantage to their carriers during adaptation to rich medium. Finally, we provide evidence that this selective advantage could be a consequence of enhanced transcription of the ftsZ gene, uncovering an interesting connection between the PTS and cell division.
Point mutations affecting cell cycle regulators and cell division.
Among the 28 mutations that did not affect the PTS or CCNA_01943, 6 (21%) were connected with the control of the cell cycle and, especially, of cell division (Table 1).
A point mutation was found in the promoter region of the ftsZ gene (PftsZ) in three different strains (Table 1), while none of the 10 wild-type control strains sampled from populations that evolved under the same conditions carried any mutation in this genetic context (see Table S1 in the supplemental material). The fact that 3 out of 8 point mutations found in intergenic regions were located in a narrow 100-bp window suggests that this region is a strong determinant of the adaptation of ΔccrM to rich medium. To determine whether any point mutation was present in PftsZ after the first selection sweep, we analyzed the ftsZ promoter region in one clone isolated from each of the 12 populations sampled at the 50th generation. No point mutation was detected in this region in any of the strains, suggesting that the mutations located in PftsZ became predominant in the population only during the phase of slower evolution, after the 50th generation (see Table S2 in the supplemental material).
One of the PftsZ mutations (2773108 G/A, strain 2.4) was located 3 bp upstream of the translational start codon of ftsZ. The two other mutations (2773188 T/C, strain 1.1; 2773191 C/T, strain 1.3) were located 2 and 5 nucleotides upstream of the transcription start site of ftsZ (27). To evaluate the effect of the PftsZ point mutations on growth and morphology, we constructed wild-type and ΔccrM strains carrying one of each of the three PftsZ mutations. While no significant effect on growth was observed in the wild-type genetic background (Fig. 4A), we found that each ΔccrM derivative had a higher growth rate in minimal medium than either the parental ΔccrM strain or the ΔccrM strains carrying the mutations affecting the PTS (Fig. 4B). In rich medium, the growth rates of the ΔccrM derivatives were much higher than that of the ΔccrM strain and similar to or slightly higher than those of the ΔccrM PTS mutants (Fig. 4C). However, the PftsZ point mutations had a much milder suppressive effect than PTS mutations on ΔccrM cell morphology: the PftsZ mutant cells were on average longer than the PTS mutant ones, and the frequency of filamentous cells was higher in the population (Fig. 5; also, see Fig. S2 in the supplemental material).
Considering the position of the point mutations found in PftsZ, we hypothesized that these could lead to increased ftsZ expression. We constructed transcriptional reporters to compare the activity of the three mutant promoters with the wild-type promoter using β-galactosidase assays. We found that the mutated ftsZ promoters found in the evolved ΔccrM strains 1.1 and 1.3 were 1.2 and 1.5 times more active than the wild-type promoter (Fig. 6C). In contrast, the mutated ftsZ promoter found in the evolved ΔccrM strain 2.4 was less active than the wild type (Fig. 6C). Since the point mutation present in the latter promoter region was located close to the translational start site, we conjectured that translation efficiency might be enhanced rather than transcription. We therefore evaluated the impact of the PftsZ 2.4 mutation on the intracellular levels of the FtsZ protein using immunoblots; we found that FtsZ levels were 2.4 times higher in the mutant strain than in the wild-type strain, suggesting that the PftsZ 2.4 mutation enhances ftsZ translation (Fig. 6D). In conclusion, all three point mutations found in the ftsZ promoter region in our evolved ΔccrM strains enhance ftsZ expression, either by increasing transcription or by a posttranscriptional mechanism.
Two other point mutations were found in the sequence encoding the cell cycle regulator CtrA, which represses ftsZ transcription (27). One of them (ctrA3.2; 8 D/G) affects the predicted active site (“acidic pocket”) of the response regulator (28). The other one (ctrA 1.2; 209 H/R) is located in the C-terminal part of the protein very close to a determinant of CtrA degradation (29). These two mutations could potentially lower the repressing activity of CtrA on the ftsZ promoter by affecting either the phosphorylation state or the proteolysis of CtrA. Another point mutation (divL2.4) was found in the sequence encoding the DivL histidine kinase. DivL affects the phosphorylation and the degradation of CtrA, and although this mutation (392 G/S) should not affect the phosphorylation of DivL at Y550, it might disturb its regulation or possible phosphorylation-independent functions and modulate CtrA activity (30–32).
We reconstructed the ctrA 1.2 mutation in the wild-type and ΔccrM genetic backgrounds, to test whether it had an effect on growth or ftsZ transcription on its own. We found that the ctrA 1.2 mutation had a negligible effect on growth in the wild-type strain cultivated in rich medium or in the ΔccrM strain cultivated in minimal medium (Fig. 4A and B). Upon transfer to rich medium, in contrast, the ΔccrM ctrA 1.2 mutant grew much faster than the ΔccrM strain (Fig. 4C); in contrast with the ΔccrM control (Fig. 1B), shorter cells were present in the ΔccrM ctrA 1.2 mutant population, although the morphology of a significant proportion of the cells was still filamentous (Fig. 5; also, see Fig. S2 in the supplemental material). These observations indicated that this mutation enhances growth rate but that it is not sufficient to improve cell division as much as PTS mutations. To test whether it might derepress ftsZ transcription in cells cultivated in rich medium, we compared the transcriptional activity of the ftsZ promoter in a wild-type strain carrying or lacking the ctrA 1.2 mutation. We observed a significant increase (1.6-fold) in ftsZ transcription in the presence of the ctrA 1.2 mutation (Fig. 6A); in a ΔccrM background, the effect was even stronger (1.9-fold increase) (Fig. 6B). We therefore suppose that the aberrant morphology of many cells seen in the ΔccrM background despite the presence of the ctrA 1.2 mutation might be due to a pleiotropic effect associated with a dysfunctional CtrA protein (28), rather than to a strong deficiency in ftsZ expression.
The occurrence of three independent mutations in the ftsZ promoter region supports our previous hypothesis that a low FtsZ concentration is a major burden on the fitness of the ΔccrM strain cultivated in rich medium (19). The presence of three additional mutations in sequences encoding two essential regulators of the cell cycle, CtrA and DivL, more generally confirms the genetic connection between DNA methylation by CcrM and the cell cycle control circuit (1).
Other point mutations of interest.
In addition to the point mutations affecting PTS and PTS-associated genes or converging on the regulation of ftsZ expression, 28 additional mutations were found in the evolved ΔccrM strains (Table 1). Some of them were recurrent, like a frameshift mutation found in four strains in the CCNA_00608 gene (predicted to code for a polygalacturonase [pectin hydrolase]), the same 84 V/G substitution found in three strains in the CCNA_03537 gene (encoding a glyoxalase family protein), and two different point mutations found in CCNA_00999 (encoding a conserved hypothetical protein). Several independent nonsynonymous mutations could affect membrane receptors, transporters, or permeases (in the CCNA_00451, CCNA_03524 or CCNA_01257 coding sequences or in the intergenic region between CCNA_02570 and CCNA_02571). Some of these mutations might modulate adverse effects that PTS mutations have on growth rate and thus enhance the fitness of the strains rather than having a direct impact on the ΔccrM phenotype.
Other mutations.
A few larger mutational events specific to the evolved ΔccrM strains were identified during the analysis. A deletion of nearly 50 kbp encompassing 56 predicted coding sequences (bp 2932736 to 2982516 in the genome) was detected as missing coverage in 11 out of the 12 sequenced strains collected after 300 generations but not in the wild-type or in the ancestral ΔccrM strains (see Fig. S3A in the supplemental material). The presence of the deletion was confirmed by PCR using primers annealing on the flanking regions of the deletion. Two 100% identical sequences of 927 bp coding for a transposase (CCNA_02772 and CCNA_02828) belonging to an IS511 transposable element (33, 34) were found on each side of the deleted region, suggesting that the ~50 kbp was excised through homologous recombination between these two sequences. The deletion was not present in any of 10 wild-type strains that evolved for the same number of generations under the same conditions, indicating that this deletion is not an adaptation to long-term culture in rich medium but might confer a specific advantage in the ΔccrM or ΔccrM/PTS background. Surprisingly, this large deletion was already present in 3 out of 12 ΔccrM clones sampled after the 50th generation (see Fig. S4B in the supplemental material). This early presence suggests that the advantage provided by this mutation is strong enough for its carriers to quickly outcompete ΔccrM/PTS mutants. Another possibility is that homologous recombination is more efficient in ΔccrM cells than wild-type cells, promoting the excision of this region.
One of four IS511 insertion sequences present in the C. crescentus genome (33) had been transposed in two of the ΔccrM evolved strains sampled at the 300th generation to position 1054365 (CCNA_00974) in population 1.3 and to position 1111235 (CCNA_01023) in population 3.1. No point mutation was found in these two genes in any of the sequenced strains, suggesting that the insertion position might be neutral from a selective viewpoint. It is possible that IS511 transposition frequency could be enhanced in the ΔccrM strain, considering that three transposases were found to be overexpressed 2-fold or more in that strain (1).
DISCUSSION
We previously showed that DNA methylation by CcrM is critical for the viability of C. crescentus in rich medium and for the regulation of its cell cycle (1, 19). In this study, using an original experimental evolution approach, we demonstrated that the most important function of CcrM is to stimulate ftsZ transcription and found an unexpected connection between the regulation of cell division and the central metabolism (through the PTS).
The lack of FtsZ is a major fitness burden for ΔccrM cells.
Twelve independent ΔccrM populations were cultivated for ~300 generations in rich medium. All ancestral ΔccrM populations were quickly invaded by mutants with improved cell division, able to partially compensate for the loss of fitness due to the absence of CcrM (Fig. 1). Whole-genome sequencing revealed that each evolved strain carried two to seven different mutations in addition to the ΔccrM deletion (Table 1) and that two thirds of these mutations (32/55) clustered in only eight chromosomal regions (Fig. 2). More detailed analysis of several of these mutations demonstrated that nearly 40% (21/55) of all detected point mutations could be connected with the regulation of ftsZ expression and that each of the twelve evolved strains carried at least one mutation promoting ftsZ transcription (Fig. 6; Table 1).
Three of these mutations were located upstream of the ftsZ coding sequence (Fig. 7) and were sufficient to strongly ameliorate the growth and division of ΔccrM cells in rich medium (resulting in a doubling time nearly two times shorter) (Fig. 4). Since these mutations promote ftsZ transcription or translation in cis (Fig. 6), they are unlikely to affect cellular factors other than FtsZ. Consistently, we previously showed that an artificial induction of ftsZ transcription was sufficient to restore the viability of ΔccrM cells cultivated in rich medium (19). Taken together, these findings demonstrate that the lack of FtsZ is a major burden slowing the growth of ΔccrM cells.
FIG 7 .
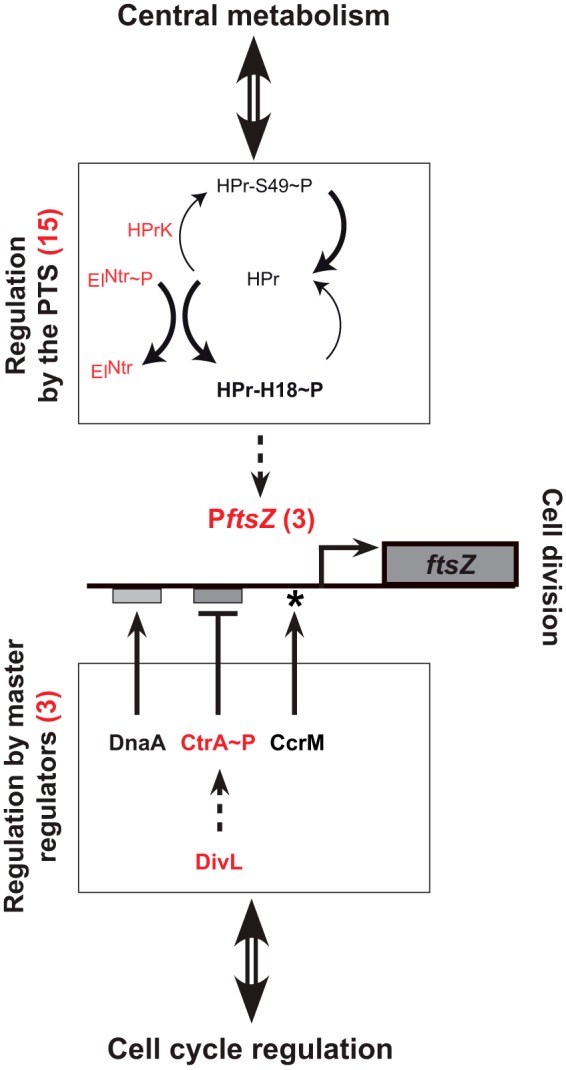
Model for the network connecting ftsZ transcription with cell cycle progression (through master cell cycle regulators) and central metabolism (through PTS proteins). About 40% (21/55) of the point mutations identified in the 14 evolved ΔccrM strains converge on promoting ftsZ expression. The red numbers in parentheses are the actual mutation counts identified in the 14 evolved ΔccrM strains. Fifteen mutations were found in sequences encoding the EINtr and HprK PTS proteins; these mutations probably promote the accumulation of Hpr-H18~P (bold arrows), leading to an activation of the transcription of ftsZ through unknown intermediates. Three mutations were found in the ftsZ promoter region (two close to the transcription start site and one near the ribosome binding site). Two mutations were found in the sequence encoding CtrA, a repressor of ftsZ transcription, and one in the sequence encoding DivL, which activates the phosphorylation and the activation of CtrA. Dashed arrows indicate potentially indirect effects.
Three other mutations were identified in the ctrA and divL genes (Table 1), encoding the CtrA master regulator and the DivL regulator of CtrA phosphorylation and degradation (Fig. 7) (30). Consistent with the fact that CtrA is a direct inhibitor of ftsZ transcription (27), we demonstrated that one of these mutations (ctrA 1.2) promoted ftsZ transcription (Fig. 6) and the growth of ΔccrM cells cultivated in rich medium (Fig. 4).
Finally, at least one mutation in the hprK or ptsP genes, encoding PTS components, was identified on the chromosome of each of the twelve strains evolved for ~300 generations (Table 1; Fig. 7). We showed that cells carrying one of these mutations were already frequent in 11 ΔccrM populations after only ~50 generations (Table 1; also, see Table S2 in the supplemental material) and that mutations in hprK or ptsP improved the division of ΔccrM cells cultivated in rich medium very efficiently compared to other mutations (Fig. 5; also, see Fig. S2 in the supplemental material). Altogether, these findings indicate that PTS mutations might provide the easiest and most efficient way to improve the fitness of ΔccrM cells cultivated in rich medium. Supporting our proposal that the lack of FtsZ is one of the main factors limiting the growth of ΔccrM cells, we showed that mutations in hprK or ptsP alone are sufficient to increase ftsZ transcription (Fig. 6).
Control of ftsZ transcription by the PTS.
The components of the PTS have not been characterized so far in C. crescentus, but their orthologs in other Alphaproteobacteria have been studied (23, 35). The C. crescentus HprK protein is the homolog of the Sinorhizobium meliloti HprK serine kinase, which phosphorylates the Ser53 residue of the S. meliloti HPr protein (36). By analogy, it is very likely that C. crescentus HprK phosphorylates the conserved Ser49 residue of the C. crescentus HPr (Fig. 3A). The mutation that we identified six times in hprK (Table 1) and that stimulates ftsZ transcription (Fig. 6) changed the conserved Gly28 residue of HprK into an aspartate. This residue is located in the putative ATP-binding motif of the kinase (Fig. 3C), suggesting that the mutation leads to a loss of function that blocks the phosphorylation of the conserved Ser49 residue of HPr by HprK (Fig. 7). This mutation may at the same time promote the phosphorylation of the His18 residue of HPr, leading to an accumulation of HPr with His18 phosphorylated (HPr-H18~P) (36). Similarly, the C. crescentus EINtr protein (encoded by ptsP) is the homolog of the S. meliloti EINtr protein (Fig. 3A). This protein phosphorylates the His22 residue of the S. meliloti HPr protein when its activity is not inhibited by glutamine binding to its GAF domain (36, 37). By analogy, it is likely that the five different mutations that we identified in the GAF domain of the C. crescentus EINtr protein (Fig. 3B), including the one that was confirmed to stimulate ftsZ transcription (ptsP 1.2) (Fig. 6), promote the phosphorylation of the conserved His18 residue of the C. crescentus HPr by the mutant EINtr, leading again to an increase in HPr-H18~P levels (Fig. 7). Taken together, our results suggest that ftsZ transcription might be directly or indirectly affected by the phosphorylation state of the HPr protein, with HPr-H18~P appearing to be, potentially, the most efficient at stimulating ftsZ transcription (Fig. 7). HPr has no obvious DNA binding domain, but it was shown that it binds to transcription factors or to anti-sigma factors in other bacterial species and that it sometimes phosphorylates transcriptional regulators, to control gene expression indirectly in a manner that is often dependent on its phosphorylation state (23, 38, 39). None of these known HPr-interacting proteins (CcpA, BglG, Rsd, etc.) seems to be conserved in the C. crescentus proteome, suggesting that the C. crescentus HPr might regulate ftsZ transcription through novel intermediates.
It was previously shown that ftsZ transcription is inhibited or activated by the direct binding of CtrA or DnaA, respectively, to the ftsZ promoter (Fig. 7) (15, 27, 40), raising the possibility that the PTS might affect the regulation of the ftsZ promoter by these regulators. To test this hypothesis, we measured the effect of the ptsP 1.2 mutation on the activity of mutant ftsZ promoters that cannot bind to DnaA or CtrA. We found that these DnaA- or CtrA-independent mutant ftsZ promoters were still more active in the ptsP 1.2 mutant than in the wild-type strain (see Fig. S4 in the supplemental material), suggesting that the binding of DnaA or CtrA to the ftsZ promoter is not required for the activation of ftsZ transcription by PTS components (Fig. 7). We also found that the integrity of the GANTC motif methylated by CcrM in the ftsZ promoter (19) is unnecessary (see Fig. S4 in the supplemental material), consistent with our previous finding that PTS mutations promote the activity of the methylated (in wild-type cells) or nonmethylated (in ΔccrM cells) ftsZ promoter (Fig. 6). Taken together, our observations suggest that two independent modules might regulate ftsZ transcription (Fig. 7). One module would include the DnaA, CtrA, and CcrM master cell cycle regulators, which bind or methylate the ftsZ promoter and which coordinate cell division with other events of the cell cycle, such as DNA replication (15, 19, 27). The other module would be connected with the activity of PTS components, maybe to coordinate cell division with central metabolism. Understanding how PTS components such as HPr influence ftsZ transcription in a manner that seems independent of known direct regulators of ftsZ transcription will require further investigations.
The PTS might connect cell division with central metabolism.
PTS play a great variety of biological roles in bacteria, including detecting available nutrients or metabolites and controlling central metabolism, chemotaxis, or virulence (for example, see reference 23). Still, despite sparse and very indirect evidence in Escherichia coli (24), no clear connection between the PTS and cell division in bacteria had been described.
Canonical PTS from Enterobacteriaceae, Vibrionales, and Firmicutes regulate the assimilation of alternative carbon sources through the uptake and phosphorylation of carbohydrates (23, 35). The presence or absence of specific sugars is translated into relative levels of phosphorylated versus nonphosphorylated forms of enzyme II (EII) permeases and HPr, causing diverse effects, including carbon catabolite repression. As many Alphaproteobacteria lack EII permeases, it has been proposed that Alphaproteobacteria could sense carbon and nitrogen availability through their PTS (24, 35). Consistent with this hypothesis, the PTS of Brucella melitensis has been connected with central metabolism through interactions of EINtr and EIIA with the α-ketoglutarate dehydrogenase and its substrates (41); in S. meliloti, EINtr binds to glutamine to control its activity in response to carbon and nitrogen levels in the cell (37). S. meliloti hprK mutants grow slowly on all types of carbon sources, suggesting that the PTS is connected with central metabolic pathways in this bacterium (36). Considering that wild-type C. crescentus strains carrying hprK and ptsP 1.2 mutations showed severe growth defects in rich medium (Fig. 4A), it is tempting to assume that the C. crescentus PTS may communicate the metabolic state of the cell to the ftsZ promoter to coordinate cell division with cell growth (Fig. 7).
Interestingly, we observed that the activity of a second promoter controlling the transcription of another important cell division gene, mipZ, was also stimulated in hprK and ptsP 1.2 mutants (see Fig. S5 in the supplemental material). This gene encodes the MipZ protein, which controls the subcellular localization of the FtsZ ring before cell division in C. crescentus (42). It is also regulated by the DnaA, CtrA, and CcrM regulators, like ftsZ (15, 16, 19). The coregulation of ftsZ and mipZ by a PTS-dependent mechanism might explain why ΔccrM mutants carrying PTS mutations divided more efficiently than ΔccrM mutants carrying suppressor mutations in the ftsZ promoter (Fig. 5). This finding suggests that master regulators of the C. crescentus cell cycle and the PTS share common targets to coordinate cell division with other cell cycle events (through master regulators) and with the central metabolism of the cell (through the PTS) (Fig. 7). This study also illustrates the power of genetic screens using CcrM-deficient strains to identify novel pathways controlling the cell cycles of Alphaproteobacteria.
Concluding remarks on the conservation of CcrM.
During the evolution experiment described in this study, C. crescentus ΔccrM cells quickly gained fitness through point mutations, suggesting that the ccrM gene might be lost if compensating PTS mutations arise during natural evolution. This appears to be in contradiction with the high degree of conservation of the ccrM gene in Alphaproteobacteria (1). However, PTS mutations improved the fitness of ΔccrM cells in rich medium, but not in minimal medium (Fig. 4), providing an example of environmental sign epistasis (43). Since most Alphaproteobacteria are frequently exposed to changing environments in their natural habitats, including nutrient-poor environments, PTS mutations that could have compensated for or allowed the loss of ccrM would be strongly counterselected. Furthermore, even in rich medium, ΔccrM cells with PTS mutations, although fitter than ΔccrM cells (Fig. 4C), were still much less fit and competitive than wild-type C. crescentus cells. Taken together, these observations add a possible explanation for the phylogenetic conservation of CcrM in so many Alphaproteobacteria.
MATERIALS AND METHODS
Culture conditions and strains.
C. crescentus strains were cultivated in peptone yeast extract (PYE) rich medium or in M2 minimal salts plus 0.2% glucose (M2G) minimal medium at 28°C (44), except when indicated otherwise. Agar (1.5%) was added to make solid media (M2GA or PYEA). Strains used in this study are listed and described in Table S4 in the supplemental material.
Experimental evolution.
Overnight cultures in M2G of three independently obtained ΔccrM strains (JC1149, JC1150, and JC1151 [19]) were diluted in 3 ml of PYE (rich medium) to an optical density at 660 nm (OD660) of 0.01, each in four replicates numbered 1.1 to 1.4 (ancestral culture, JC1149), 2.1 to 2.4 (ancestral culture, JC1150) and 3.1 to 3.4 (ancestral culture, JC1151). After that, for a total of 300 doublings in OD660 (37 days), the 12 populations were rediluted to an OD660 of 0.001 in 3 ml of PYE as soon as the cultures reached an OD660 of 0.5 to 1.0 (late exponential phase). The doubling times of each population were estimated on the basis of the initial and final OD660 and the time interval at every dilution. Aliquots of each population were frozen every ~50 doublings. As a control, a similar experiment was carried out with 10 wild-type NA1000 populations; we used a total volume of PYE of 5 ml and daily dilutions to an OD660 of 0.0001 for these populations.
Whole-genome sequencing and assembly.
One clone was reisolated from the 12 ΔccrM populations sampled at generation 300 and from 2 populations (1.2 and 3.3) sampled at generation 50 for sequencing. As controls, one colony from our NA1000 C. crescentus strain and one colony from each of the three ΔccrM strains JC1149, JC1150, and JC1151 were also included. Illumina libraries of 400 to 500 bp were constructed from the extracted genomic DNA using barcoded adaptors optimized for high GC content and sequenced (single reads) on a HiSeq 2500 system in a single lane (18-plex sequencing). The filtering, alignment of the reads to the reference NA1000 genome (NC_011916), point mutations and new junction (rearrangements) calling was done using breseq 0.22 software (45, 46). The mean coverage depth was >180 for all strains.
Other methods, including those used to calculate generation times and to construct strains and transcriptional reporters, are described in Text S1 in the supplemental material.
SUPPLEMENTAL MATERIAL
Supplemental materials and methods. Download
Changes in growth rate during the evolution of ΔccrM populations in rich medium. (A) Changes in the doubling time in PYE medium of the 12 individual evolved ΔccrM populations over the course of experimental evolution. (B) Linear regression of the doubling time in PYE medium of individual ΔccrM populations as a function of time between generation ~75 and generation ~300. A trend toward a decrease in doubling time is noticeable for all populations. For additional information, see the legend to Fig. 1A. Download
Morphology of ΔccrM point mutants in minimal and rich medium. Phase-contrast micrographs of ΔccrM point mutant strains in minimal medium (M2G) and about 7 and 15 generations after transfer from minimal to rich medium (PYE) are shown. Compare these with the micrographs of ΔccrM strains (Fig. 1B). Download
A 50-kbp deletion occurs frequently during ΔccrM evolution. (A) Mean coverage over 50 bp along the entire chromosome of C. crescentus for the genome of a clone from population 1.2 sampled at generation 50 and 300. In the clone sampled at generation 300, coverage is missing in a region framed in red corresponding to a 50 kbp deletion (bp 2932736 to 2982516). (B) Detection by PCR of an ~1,500-bp fragment bridging the flanks of a recurrent 50-kbp deletion in one clone sampled from each of the 12 evolved ΔccrM populations at generation 50. The amplification product is present in strains 1.1, 1.3, and 1.4; a faint nonspecific product of a slightly higher molecular weight is present in most lanes. Download
The effect of the ptsP 1.2 point mutation on the activity of the ftsZ promoter is independent of its conserved GANTC site, of its CtrA binding site, and of its DnaA binding site. The graph shows the average activity of wild-type or mutant ftsZ promoters in NA1000 and ptsP 1.2 strains, as measured by β-galactosidase assays using cells cultivated in PYE. PftsZ corresponds to the wild-type ftsZ promoter. PftsZ-GANTC corresponds to a mutant ftsZ promoter that lacks its conserved GANTC motif methylated by CcrM. PftsZ-ctrA and PftsZ-dnaA correspond to mutant ftsZ promoters that lack their CtrA-binding or DnaA-binding motifs, respectively. Each wild-type or mutant ftsZ promoter is more active in the ptsP 1.2 mutant strain than in the wild-type strain, indicating that the PTS can modulate ftsZ transcription in a manner that is neither dependent on the binding of CtrA or DnaA to the ftsZ promoter nor dependent on the methylation of the ftsZ promoter by CcrM (Fig. 7). The transcriptional activity of each promoter was normalized so that its value equaled 1 in the NA1000 strain to make all four pairs of assays comparable. The averages are based on 3 replicates, and error bars indicate standard deviations from the means. Strains in the NA1000 background include JC133 (PftsZ), JC1398 (PftsZ-GANTC), JC1401 (PftsZ-ctrA), and JC1399 (PftsZ-dnaA). Strains in the ptsP 1.2 background include JC1369 (PftsZ), JC1402 (PftsZ-GANTC), JC1405 (PftsZ-ctrA), and JC1403 (PftsZ-dnaA). Download
The transcription of mipZ is also stimulated by PTS mutations promoting ftsZ transcription. The graph shows the average activity of the mipZ promoter in NA1000 (JC1378), hprK 1.4 (JC1379) and ptsP 1.2 (JC1381) strains, as measured by β-galactosidase assays using cells cultivated in PYE. The averages are based on 3 replicates, and error bars indicate standard deviations from the means. Download
Identification of mutations in targeted regions of the chromosomes of 10 NA1000 strains that evolved in PYE for 300 generations.
Identification of mutations in targeted regions of the chromosomes of 12 ΔccrM strains that evolved in PYE for 50 generations.
(A) Single nucleotide mutations found in NA1000. (B) Point mutations found in the ancestral ΔccrM strains.
Bacterial strains, plasmids, and oligonucleotides used in this study.
ACKNOWLEDGMENTS
The Illumina sequencing could not have been carried out without the help of Keith Harshman, Johann Weber and their collaborators at the Lausanne Genomic Technologies Facility. We thank Noémie Matthey and Guillaume Michon for their help with the evolution experiment and growth curves and Carmen Fernández-Fernández, Katharina Eich, and Céline Terrettaz for stimulating discussions.
This work was supported by the Swiss National Science Foundation (SNSF) fellowship 31003A_140758 and the SNSF R’Equip grant 316030_121391.
Footnotes
Citation Gonzalez D, Collier J. 2015. Genomic adaptations to the loss of a conserved bacterial DNA methyltransferase. mBio 6(4):e00952-15. doi:10.1128/mBio.00952-15.
REFERENCES
- 1.Gonzalez D, Kozdon JB, McAdams HH, Shapiro L, Collier J. 2014. The functions of DNA methylation by CcrM in Caulobacter crescentus: a global approach. Nucleic Acids Res 42:3720–3735. doi: 10.1093/nar/gkt1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gerdes S, Edwards R, Kubal M, Fonstein M, Stevens R, Osterman A. 2006. Essential genes on metabolic maps. Curr Opin Biotechnol 17:448–456. doi: 10.1016/j.copbio.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 3.Khatiwara A, Jiang T, Sung SS, Dawoud T, Kim JN, Bhattacharya D, Kim HB, Ricke SC, Kwon YM. 2012. Genome scanning for conditionally essential genes in Salmonella enterica serotype Typhimurium. Appl Environ Microbiol 78:3098–3107. doi: 10.1128/AEM.06865-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kahng LS, Shapiro L. 2001. The CcrM DNA methyltransferase of Agrobacterium tumefaciens is essential, and its activity is cell cycle regulated. J Bacteriol 183:3065–3075. doi: 10.1128/JB.183.10.3065-3075.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stephens CM, Zweiger G, Shapiro L. 1995. Coordinate cell cycle control of a Caulobacter DNA methyltransferase and the flagellar genetic hierarchy. J Bacteriol 177:1662–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fang G, Rocha E, Danchin A. 2005. How essential are nonessential genes? Mol Biol Evol 22:2147–2156. doi: 10.1093/molbev/msi211. [DOI] [PubMed] [Google Scholar]
- 7.Acevedo-Rocha CG, Fang G, Schmidt M, Ussery DW, Danchin A. 2013. From essential to persistent genes: a functional approach to constructing synthetic life. Trends Genet 29:273–279. doi: 10.1016/j.tig.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Geissler B, Elraheb D, Margolin W. 2003. A gain-of-function mutation in ftsA bypasses the requirement for the essential cell division gene zipA in Escherichia coli. Proc Natl Acad Sci U S A 100:4197–4202. doi: 10.1073/pnas.0635003100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bergmiller T, Ackermann M, Silander OK. 2012. Patterns of evolutionary conservation of essential genes correlate with their compensability. PLoS Genet 8:e1002803. doi: 10.1371/journal.pgen.1002803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zweiger G, Marczynski G, Shapiro L. 1994. A Caulobacter DNA methyltransferase that functions only in the predivisional cell. J Mol Biol 235:472–485. doi: 10.1006/jmbi.1994.1007. [DOI] [PubMed] [Google Scholar]
- 11.Kozdon JB, Melfi MD, Luong K, Clark TA, Boitano M, Wang S, Zhou B, Gonzalez D, Collier J, Turner SW, Korlach J, Shapiro L, McAdams HH. 2013. Global methylation state at base-pair resolution of the Caulobacter genome throughout the cell cycle. Proc Natl Acad Sci U S A 110:E4658–E4667. doi: 10.1073/pnas.1319315110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marczynski GT. 1999. Chromosome methylation and measurement of faithful, once and only once per cell cycle chromosome replication in Caulobacter crescentus. J Bacteriol 181:1984–1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Collier J, McAdams HH, Shapiro L. 2007. A DNA methylation ratchet governs progression through a bacterial cell cycle. Proc Natl Acad Sci U S A 104:17111–17116. doi: 10.1073/pnas.0708112104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reisenauer A, Shapiro L. 2002. DNA methylation affects the cell cycle transcription of the CtrA global regulator in Caulobacter. EMBO J 21:4969–4977. doi: 10.1093/emboj/cdf490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hottes AK, Shapiro L, McAdams HH. 2005. DnaA coordinates replication initiation and cell cycle transcription in Caulobacter crescentus. Mol Microbiol 58:1340–1353. doi: 10.1111/j.1365-2958.2005.04912.x. [DOI] [PubMed] [Google Scholar]
- 16.Laub MT, Chen SL, Shapiro L, McAdams HH. 2002. Genes directly controlled by CtrA, a master regulator of the Caulobacter cell cycle. Proc Natl Acad Sci U S A 99:4632–4637. doi: 10.1073/pnas.062065699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fioravanti A, Fumeaux C, Mohapatra SS, Bompard C, Brilli M, Frandi A, Castric V, Villeret V, Viollier PH, Biondi EG. 2013. DNA binding of the cell cycle transcriptional regulator GcrA depends on N6-adenosine methylation in Caulobacter crescentus and other Alphaproteobacteria. PLoS Genet 9:e1003541. doi: 10.1371/journal.pgen.1003541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murray SM, Panis G, Fumeaux C, Viollier PH, Howard M. 2013. Computational and genetic reduction of a cell cycle to its simplest, primordial components. PLoS Biol 11:e1001749. doi: 10.1371/journal.pbio.1001749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gonzalez D, Collier J. 2013. DNA methylation by CcrM activates the transcription of two genes required for the division of Caulobacter crescentus. Mol Microbiol 88:203–218. doi: 10.1111/mmi.12180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Poindexter JS. 1981. Oligotrophy: fast and famine existence. Adv Microb Ecol 5:63–89. [Google Scholar]
- 21.Curtis PD, Brun YV. 2014. Identification of essential alphaproteobacterial genes reveals operational variability in conserved developmental and cell cycle systems. Mol Microbiol 93:713–735. doi: 10.1111/mmi.12686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Robertson GT, Reisenauer A, Wright R, Jensen RB, Jensen A, Shapiro L, Roop RM. 2000. The Brucella abortus CcrM DNA methyltransferase is essential for viability, and its overexpression attenuates intracellular replication in murine macrophages. J Bacteriol 182:3482–3489. doi: 10.1128/JB.182.12.3482-3489.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deutscher J, Francke C, Postma PW. 2006. How phosphotransferase system-related protein phosphorylation regulates carbohydrate metabolism in bacteria. Microbiol Mol Biol Rev 70:939–1031. doi: 10.1128/MMBR.00024-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barabote RD, Saier MH Jr. 2005. Comparative genomic analyses of the bacterial phosphotransferase system. Microbiol Mol Biol Rev 69:608–634. doi: 10.1128/MMBR.69.4.608-634.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Klein EA, Schlimpert S, Hughes V, Brun YV, Thanbichler M, Gitai Z. 2013. Physiological role of stalk lengthening in Caulobacter crescentus. Commun Integr Biol 6:e24561. doi: 10.4161/cib.24561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wagner JK, Setayeshgar S, Sharon LA, Reilly JP, Brun YV. 2006. A nutrient uptake role for bacterial cell envelope extensions. Proc Natl Acad Sci U S A 103:11772–11777. doi: 10.1073/pnas.0602047103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kelly AJ, Sackett MJ, Din N, Quardokus E, Brun YV. 1998. Cell cycle-dependent transcriptional and proteolytic regulation of FtsZ in Caulobacter. Genes Dev 12:880–893. doi: 10.1101/gad.12.6.880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quon KC, Marczynski GT, Shapiro L. 1996. Cell cycle control by an essential bacterial two-component signal transduction protein. Cell 84:83–93. doi: 10.1016/S0092-8674(00)80995-2. [DOI] [PubMed] [Google Scholar]
- 29.Ryan KR, Judd EM, Shapiro L. 2002. The CtrA response regulator essential for Caulobacter crescentus cell-cycle progression requires a bipartite degradation signal for temporally controlled proteolysis. J Mol Biol 324:443–455. doi: 10.1016/S0022-2836(02)01042-2. [DOI] [PubMed] [Google Scholar]
- 30.Reisinger SJ, Huntwork S, Viollier PH, Ryan KR. 2007. DivL performs critical cell cycle functions in Caulobacter crescentus independent of kinase activity. J Bacteriol 189:8308–8320. doi: 10.1128/JB.00868-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brown PJ, Hardy GG, Trimble MJ, Brun YV. 2009. Complex regulatory pathways coordinate cell-cycle progression and development in Caulobacter crescentus. Adv Microb Physiol 54:1–101. doi: 10.1016/S0065-2911(08)00001-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Childers WS, Xu Q, Mann TH, Mathews II, Blair JA, Deacon AM, Shapiro L. 2014. Cell fate regulation governed by a repurposed bacterial histidine kinase. PLoS Biol 12:e1001979. doi: 10.1371/journal.pbio.1001979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mullin DA, Zies DL, Mullin AH, Caballera N, Ely B. 1997. Genetic organization and transposition properties of IS511. Mol Gen Genet 254:456–463. PubMed. [DOI] [PubMed] [Google Scholar]
- 34.Nierman WC, Feldblyum TV, Laub MT, Paulsen IT, Nelson KE, Eisen JA, Heidelberg JF, Alley MR, Ohta N, Maddock JR, Potocka I, Nelson WC, Newton A, Stephens C, Phadke ND, Ely B, DeBoy RT, Dodson RJ, Durkin AS, Gwinn ML. 2001. Complete genome sequence of Caulobacter crescentus. Proc Natl Acad Sci U S A 98:4136–4141. doi: 10.1073/pnas.061029298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cases I, Velázquez F, de Lorenzo V. 2007. The ancestral role of the phosphoenolpyruvate-carbohydrate phosphotransferase system (PTS) as exposed by comparative genomics. Res Microbiol 158:666–670. doi: 10.1016/j.resmic.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 36.Pinedo CA, Gage DJ. 2009. HPrK regulates succinate-mediated catabolite repression in the gram-negative symbiont Sinorhizobium meliloti. J Bacteriol 191:298–309. doi: 10.1128/JB.01115-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goodwin RA, Gage DJ. 2014. Biochemical characterization of a nitrogen-type phosphotransferase system reveals that enzyme EI(Ntr) integrates carbon and nitrogen signaling in Sinorhizobium meliloti. J Bacteriol 196:1901–1907. doi: 10.1128/JB.01489-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Görke B, Stülke J. 2008. Carbon catabolite repression in bacteria: many ways to make the most out of nutrients. Nat Rev Microbiol 6:613–624. doi: 10.1038/nrmicro1932. [DOI] [PubMed] [Google Scholar]
- 39.Park YH, Lee CR, Choe M, Seok YJ. 2013. HPr antagonizes the anti-sigma70 activity of Rsd in Escherichia coli. Proc Natl Acad Sci U S A 110:21142–21147. doi: 10.1073/pnas.1316629111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fernandez-Fernandez C, Gonzalez D, Collier J. 2011. Regulation of the activity of the dual-function DnaA protein in Caulobacter crescentus. PLoS One 6:e26028. doi: 10.1371/journal.pone.0026028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dozot M, Poncet S, Nicolas C, Copin R, Bouraoui H, Mazé A, Deutscher J, De Bolle X, Letesson JJ. 2010. Functional characterization of the incomplete phosphotransferase system (PTS) of the intracellular pathogen Brucella melitensis. PLoS One 5:e12679. doi: 10.1371/journal.pone.0012679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thanbichler M, Shapiro L, Mip Z. 2006. MipZ, a spatial regulator coordinating chromosome segregation with cell division in Caulobacter. Cell 126:147–162. doi: 10.1016/j.cell.2006.05.038. [DOI] [PubMed] [Google Scholar]
- 43.Lindsey HA, Gallie J, Taylor S, Kerr B. 2013. Evolutionary rescue from extinction is contingent on a lower rate of environmental change. Nature 494:463–467. doi: 10.1038/nature11879. [DOI] [PubMed] [Google Scholar]
- 44.Ely B. 1991. Genetics of Caulobacter crescentus. Methods Enzymol 204:372–384. [DOI] [PubMed] [Google Scholar]
- 45.Barrick JE, Lenski RE. 2009. Genome-wide mutational diversity in an evolving population of Escherichia coli. Cold Spring Harb Symp Quant Biol 74:119–129. doi: 10.1101/sqb.2009.74.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barrick JE, Yu DS, Yoon SH, Jeong H, Oh TK, Schneider D, Lenski RE, Kim JF. 2009. Genome evolution and adaptation in a long-term experiment with Escherichia coli. Nature 461:1243–1247. doi: 10.1038/nature08480. [DOI] [PubMed] [Google Scholar]
- 47.Ren Q, Paulsen IT. 2007. Large-scale comparative genomic analyses of cytoplasmic membrane transport systems in prokaryotes. J Mol Microbiol Biotechnol 12:165–179. doi: 10.1159/000099639. [DOI] [PubMed] [Google Scholar]
- 48.Pinedo CA, Bringhurst RM, Gage DJ. 2008. Sinorhizobium meliloti mutants lacking phosphotransferase system enzyme HPr or EIIA are altered in diverse processes, including carbon metabolism, cobalt requirements, and succinoglycan production. J Bacteriol 190:2947–2956. doi: 10.1128/JB.01917-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental materials and methods. Download
Changes in growth rate during the evolution of ΔccrM populations in rich medium. (A) Changes in the doubling time in PYE medium of the 12 individual evolved ΔccrM populations over the course of experimental evolution. (B) Linear regression of the doubling time in PYE medium of individual ΔccrM populations as a function of time between generation ~75 and generation ~300. A trend toward a decrease in doubling time is noticeable for all populations. For additional information, see the legend to Fig. 1A. Download
Morphology of ΔccrM point mutants in minimal and rich medium. Phase-contrast micrographs of ΔccrM point mutant strains in minimal medium (M2G) and about 7 and 15 generations after transfer from minimal to rich medium (PYE) are shown. Compare these with the micrographs of ΔccrM strains (Fig. 1B). Download
A 50-kbp deletion occurs frequently during ΔccrM evolution. (A) Mean coverage over 50 bp along the entire chromosome of C. crescentus for the genome of a clone from population 1.2 sampled at generation 50 and 300. In the clone sampled at generation 300, coverage is missing in a region framed in red corresponding to a 50 kbp deletion (bp 2932736 to 2982516). (B) Detection by PCR of an ~1,500-bp fragment bridging the flanks of a recurrent 50-kbp deletion in one clone sampled from each of the 12 evolved ΔccrM populations at generation 50. The amplification product is present in strains 1.1, 1.3, and 1.4; a faint nonspecific product of a slightly higher molecular weight is present in most lanes. Download
The effect of the ptsP 1.2 point mutation on the activity of the ftsZ promoter is independent of its conserved GANTC site, of its CtrA binding site, and of its DnaA binding site. The graph shows the average activity of wild-type or mutant ftsZ promoters in NA1000 and ptsP 1.2 strains, as measured by β-galactosidase assays using cells cultivated in PYE. PftsZ corresponds to the wild-type ftsZ promoter. PftsZ-GANTC corresponds to a mutant ftsZ promoter that lacks its conserved GANTC motif methylated by CcrM. PftsZ-ctrA and PftsZ-dnaA correspond to mutant ftsZ promoters that lack their CtrA-binding or DnaA-binding motifs, respectively. Each wild-type or mutant ftsZ promoter is more active in the ptsP 1.2 mutant strain than in the wild-type strain, indicating that the PTS can modulate ftsZ transcription in a manner that is neither dependent on the binding of CtrA or DnaA to the ftsZ promoter nor dependent on the methylation of the ftsZ promoter by CcrM (Fig. 7). The transcriptional activity of each promoter was normalized so that its value equaled 1 in the NA1000 strain to make all four pairs of assays comparable. The averages are based on 3 replicates, and error bars indicate standard deviations from the means. Strains in the NA1000 background include JC133 (PftsZ), JC1398 (PftsZ-GANTC), JC1401 (PftsZ-ctrA), and JC1399 (PftsZ-dnaA). Strains in the ptsP 1.2 background include JC1369 (PftsZ), JC1402 (PftsZ-GANTC), JC1405 (PftsZ-ctrA), and JC1403 (PftsZ-dnaA). Download
The transcription of mipZ is also stimulated by PTS mutations promoting ftsZ transcription. The graph shows the average activity of the mipZ promoter in NA1000 (JC1378), hprK 1.4 (JC1379) and ptsP 1.2 (JC1381) strains, as measured by β-galactosidase assays using cells cultivated in PYE. The averages are based on 3 replicates, and error bars indicate standard deviations from the means. Download
Identification of mutations in targeted regions of the chromosomes of 10 NA1000 strains that evolved in PYE for 300 generations.
Identification of mutations in targeted regions of the chromosomes of 12 ΔccrM strains that evolved in PYE for 50 generations.
(A) Single nucleotide mutations found in NA1000. (B) Point mutations found in the ancestral ΔccrM strains.
Bacterial strains, plasmids, and oligonucleotides used in this study.