

Abstract
Recent research has established clear connections between G-quadruplexes and human disease. Features of quadruplex structures that promote genomic instability have been determined. Quadruplexes have been identified as transcriptional, translational and epigenetic regulatory targets of factors associated with human genetic disease. An expandable GGGGCC motif that can adopt a G4 structure, located in the previously obscure C9ORF72 locus, has been shown to contribute to two well-recognized neurodegenerative diseases, amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). This review focuses on these advances, which further dispel the view that genomic biology is limited to the confines of the canonical B-form DNA duplex, and show how quadruplexes contribute spatial and temporal dimensionalities to linear sequence information. This recent progress also has clear practical ramifications, as prevention, diagnosis, and treatment of disease depend on understanding the underlying mechanisms.
Keywords: ALS, FTD, G4, helicase, quadruplex
Introduction
G-quadruplex or “G4” structures form in G-rich regions of single-stranded DNA or RNA 1,2. Quadruplexes are distinct in structure from the canonical B-form duplex, and they are recognized by a distinct subset of nuclear factors. Formation of quadruplexes can be regulated, since it occurs only in single-stranded regions, which are generated during replication and transcription as a result of high density supercoiling. This review discusses some recent advances in our understanding of the many ways in which genomic biology exploits the special properties of quadruplexes, and how quadruplexes are involved in human health and disease.
G4 motifs and quadruplex structures
A G4 motif is a sequence that can form a quadruplex structure (Sidebar A). The G4 signature motif is comprised of four adjacent runs of guanines separated by other nucleotides: G≥3NxG≥3NxG≥3NxG≥3. The recurrent unit in a quadruplex is a G-quartet 3, a planar array of four hydrogen-bonded guanines (Fig1A). The guanines in the G-runs in the G4 sequence motif typically participate in G-quartet formation. Strands in parallel or antiparallel orientation may contribute guanines to a G-quartet (Fig1B). The planar quartets are connected by “loops” formed by the nucleotides between the G-runs, denoted as “N” in the G4 sequence motif.
Sidebar A: Terminology: G4, G4 motifs, and G-quadruplexes.
G4: The shorthand term “G4” is currently used to refer both to sequence motifs that have the potential to form quadruplex structures and to actual quadruplex structures. This does not need to be a source of confusion, but it can be. It is critical to distinguish between sequences with the potential to form stable structures containing G-quartets, and the structures themselves. Unfortunately, this distinction is not always clearly drawn in the literature. Calling both sequences and structures “G4” can give the misimpression that a sequence referred to as a “G4” is in a quadruplex conformation in a living cell, when that has not been demonstrated. More precise terminology that observes this important distinction is shown below and will be used in this review.
G4 sequence motif or G4 motif: a nucleic acid sequence that either conforms to the algorithm G≥3NxG≥3NxG≥3NxG≥3, or that has been shown to form a stable G-quadruplex structure in vitro.
G-quadruplex structure, G-quadruplex, quadruplex, G4 DNA, or G4 RNA: a structure stabilized by G-quartets, which may be made of DNA or RNA.”
Figure 1.

G-quartets, G-quadruplex structures, and formation of G4 DNA during replication or transcription
(A) G-quartet, a planar array of four guanines stabilized by pairwise hydrogen-bonding and coordination by a monovalent cation. (B) Antiparallel and parallel G-quadruplexes formed by (GGGGCC)4. The parallel conformation is observed in RNA formed by this sequence 66. Underlined G’s participate in G-quartets. G-quartets, green; loops connecting quartets, dotted lines; arrows, 3′ DNA ends. (C) Left, parallel G-quadruplex formed by the human telomeric RNA. Right, structure formed by the long TERRA transcript, containing quadruplexes stacked 5′-5′. Reprinted (adapted) with permission from: Structure of long human telomeric RNA (TERRA): G-quadruplexes formed by four and eight UUAGGG repeats are stable building blocks. Biochemistry 50: 6455–6461. Copyright 2011 American Chemical Society 12. Notations as in (B). (D) Left, parallel G-quadruplex formed by the core quadruplex of the CEB25 VNTR, d(AAGGGTGGGTGTAAGTGTGGGTGGGT). Right, a “pearl necklace” composed of quadruplexes strung along a chain of unstructured DNA. Reprinted (adapted) with permission from: Formation of pearl-necklace monomorphic G-quadruplexes in the human CEB25 minisatellite. J Am Chem Soc 134: 5807–5816. Copyright 2012 American Chemical Society 13.
The G≥3NxG≥3NxG≥3NxG≥3 sequence motif constitutes a loose rather than strict consensus. Some sequences that do not strictly conform to the consensus do form quadruplexes—CGG repeats are one example. Conversely, about 10% of oligonucleotides bearing sequences that conform to that motif do not readily form quadruplex structures in vitro, although it remains unclear whether the failure of some G4 motifs to form quadruplexes reflects flaws in the algorithm or in the conditions used to test structure formation.
Quadruplex structures are diverse, and they may be quite intricate 4,5. Quadruplex conformation is determined by the orientation of the glycosidic bonds of the guanines in the quartets, the parallel, anti-parallel, or mixed orientation of the strands contributing guanine bases to the quartets, and the lengths and sequences of the loops connecting the guanine runs (Fig1B). Quadruplexes exhibit considerable thermodynamic stability, largely derived from stacking of the hydrophobic G-quartets, and modulated by the length and sequence of the loops. It is not yet possible to predict details of a quadruplex conformation based on sequence alone, but that should become possible in the future.
Evidence that distinct G4 motifs adopt distinctive quadruplex structures fueled efforts to develop small molecule ligands that would bind to a postulated quadruplex and affect expression of a nearby gene 6. This strategy has not yet generated any drugs that can be used in the clinic, but G4 ligands have proved useful for other purposes. Notably, the G4 ligand PhenDC3 has been used to exacerbate instability at G4 motifs in S. cerevisiae 7, and a different compound, pyridostatin, has been shown to cause instability at G4 motifs in human cells 8. The interest in developing G4 ligands remains high. One target of continued therapeutic interest is the telomere, with its clear connection to cell proliferation and aging, and where abundant G4 motifs form well-defined structures that should in principle be good targets for G4 ligands 9.
Quadruplexes formed by sequences bearing repetitive G4 motifs can fold to produce regular structures. The best-studied example is the human TTAGGG telomeric repeat, which forms characteristic parallel structures as DNA or RNA 10,11. In TERRA RNAs transcribed from telomeric repeats, quadruplexes composed of r(UUAGGG)4 repeats readily stack on each other at their 5′ ends (Fig1C; 12). The 52 nt human CEB25 VNTR, located on chromosome 10, contains a 26 nt core sequence, d(AAGGGTGGGTGTAAGTGTGGGTGGGT), which forms a parallel quadruplex in which a 9 nt loop connects the top of one G-quartet to the bottom of another; and in long repeats, a 26 nt chain of relatively unstructured DNA connects these quadruplexes to create a “pearl necklace” conformation (13; Fig1D). The quartets, the loops within them, and the chains that connect them are all potential targets for protein binding.
A number of factors have been shown to bind quadruplexes in human cells, and some recurrent polypeptide domains have been shown to promote binding to quadruplexes. These include the RRM/RBD domains and RGG motifs found in many nuclear RNA binding proteins 14, the RSM domain found at the N-terminus of RHAU RNA helicase 15, and the RQC domain found in most RECQ family helicases 16. Loops within quadruplexes may confer specificity to protein binding. Some of the factors that interact with quadruplexes will be discussed later in this review.
G4 motifs in the genome
We have proposed that G4 motifs and quadruplex structures that participate in key processes comprise the G4 genome, analogous to the transcriptome, proteome, or metabolome 17. This poses the challenge of identifying those G4 motifs that do form quadruplexes in living cells, and determining their specific functions.
G4 motifs in a genome can be tallied and mapped by algorithms that search for matches to the G4 sequence motif, G≥3NxG≥3NxG≥3NxG≥3. Search algorithms typically allow loop length to be specified by the user. As discussed later in this review, searches have been published that allow very long loop lengths, and this can yield results that are misleading or simply incorrect. Rigorous searches, specifying loops of 7 nt, identify 360,000 G4 motifs in the human genome 18, but only 27 in the S. cerevisiae genome 19. Thus, the S. cerevisiae genome, which is about 0.4% the size of the human genome (1.2 × 107 vs. 3.2 × 109 bp), has only 0.01% as many G4 motifs. This suggests that that the functions of G4 motifs have changed over the course of evolution.
G4 motifs exhibit a distinct distribution within the human genome and among human genes 17. As G4 motifs can confer or correlate with specific properties and functions, it is useful to distinguish regions that are significantly enriched or depleted for G4 motifs relative to the genomic average as “G4hi” and “G4lo”, respectively. G4 motifs are enriched in three G4hi chromosomal domains: the telomeres, composed of 3–20 kb TTAGGG repeats; the rDNA, where G4 motifs are abundant in sequences transcribed into rRNA as well as spacer DNA; and the immunoglobulin heavy chain switch regions, 1–10 kb degenerate repetitive regions that are sites of class switch recombination junctions. Like CEB25 (see above), many minisatellites or VNTRs contain G4 motifs.
Among the approximately 30,000 human RefSeq genes, promoters are typically G4hi, especially in the 1 kb region flanking the TSS; coding regions are G4lo; and G4 motifs are greatly enriched at the very 5′ end of the first intron at a subset of genes. At some genes, G4 motifs are significantly enriched or depleted relative to the genome-wide average, and this correlates with gene function 20. G4hi genes include oncogenes, transcriptional activators, and regulators of development, and they are enriched for G4 motifs in both exons and introns. Conversely, tumor suppressor genes are typically G4lo.
As discussed below, G4 motifs may influence genomic stability, transcriptional and translational regulation, and chromatin structure, so enrichment or depletion of G4 motifs may confer special properties or regulation.
Quadruplexes add a temporal dimension to genomic structure and function
The human genome contains many G4 motifs: over 360,000 when searched using an algorithm that allows up to 7 nt in the loops between G-runs, and approximately twice that number allowing loops up to 12 nt in length. The fraction of genomic G4 motifs that actually form quadruplex structures in a living human cell is not known.
Quadruplexes can form in regions of B-form duplex DNA that are denatured by replication or transcription, or in which local superhelicity has been altered (Fig2). Quadruplex density within DNA molecules displays striking dependence on negative superhelicity 21, which increases in the wake of the advancing transcription apparatus, and can also increase in response to topoisomerase activity or protein binding. Conversely, a quadruplex can stall replication or transcription, or alter DNA structure to inhibit recognition of the duplex by a sequence-specific DNA binding protein (Fig2). Thus, quadruplex formation is regulated and can respond to temporal signals associated with the cell cycle and development.
Figure 2.

Induction of quadruplex structures and potential impact on essential processes
(A) Replication, transcription, or local increases in negative superhelicity may expose a single-stranded region bearing a G4 motif, and lead to quadruplex formation. [G4] identifies one or more G4 motifs. G4 RNA may form in the transcript of a gene carrying a G4 motif on the non-transcribed strand (center). (B) A quadruplex may stall replication or transcription, or prevent a sequence-specific transcription factor from recognizing or binding its site in duplex DNA.
Tools that detect, enumerate and map quadruplexes would be very useful for identifying those G4 motifs that do indeed form quadruplexes, and establishing the conditions that promote or prevent quadruplex formation. Antibodies have been reported that bind quadruplexes specifically in vitro, and that stain living cells (reviewed by 22). If those antibodies were able to recognize all quadruplex structures, then it would in principle be possible to generate a catalog of quadruplexes by ChIP-Seq. However, no anti-G4 antibody has yet been shown to recover G4 motifs as a specific or even predominant target by genome-wide approaches. Some efforts are directed toward the development of ligands that light up upon quadruplex binding 23, but these have not reached the stage where they could be used to enumerate quadruplex structures.
A more fruitful approach to identification of quadruplexes in a living cell is to map sites of association of endogenous quadruplex binding proteins. ChIP-Seq analysis has shown that G4 motifs are greatly overrepresented (> 40%; P = 0) among sites of association of two endogenous G4 helicases, XPB and XPD, and that 2% of G4 motifs associated with these factors (P = 0; 24). Those experiments depended on the availability of highly specific antibodies to XPB and XPD, which thus far are the only quadruplex binding proteins for which genome-wide ChIP-Seq has been carried out. It has become possible to use CRISPR/Cas9 to add an epitope tag to essentially any gene, circumventing the need for specific antibodies for ChIP-Seq. This means that it is now possible in principle to catalog the nuclear DNA targets of all endogenous quadruplex binding proteins and provide a more complete picture of the “G4 Genome”.
Quadruplexes pose challenges for maintaining genomic stability
A novel role for Pol θ in promoting G4 motif stability in C. elegans
A G-quadruplex structure on the template strand for replication may cause the replication apparatus to stall (Fig2A). This may in turn promote formation of one-ended breaks in the chromosome which can initiate genomic instability if not repaired faithfully 25. Our understanding of replication-associated instability at G4 motifs has benefited from experiments carried out in model organisms, especially the nematode C. elegans and the yeast S. cerevisiae.
Replicative instability at G4 motifs was first demonstrated in experiments by Tijsterman and collaborators that analyzed DNA deletions in nematodes deficient in dog-1, an ortholog of human FANCJ 26 (reviewed by 27). Deletion endpoints mapped to G4 motifs, and only to G4 motifs. Unexpectedly, repair at G4-induced breaks in the nematode depends on a novel repair mechanism, in which the A-family polymerase, Pol θ, tethers the end of the break and carries out error-prone repair synthesis to generate occasional insertions at the break sites 28. The importance of Pol θ is especially evident in backgrounds lacking low fidelity polymerases Pol η and Pol κ 29 (reviewed in 30).
Replication-associated genomic instability at the G4L1 motif in S. cerevisiae
Many DNA repair pathways are highly conserved, and S. cerevisiae has proved to be an excellent model for eukaryotic DNA repair in many contexts. Nicolas and collaborators used a human G4hi VNTR, CEB1 (GGGGGGAGGGAGGGTGGCCTGCGGAGGTCCCTGGGCTGA), as a reporter for replicative instability at G4 motifs, and identified Pif1 helicase as necessary for G4 motif stability 7. Instability exhibited a clear strand bias and occurred only if the G-rich strand was the template for leading strand replication 31 (reviewed by 32). Analysis of another G4hi human VNTR, CEB25 33, suggested that G4 motifs differ in their ability to cause replicative instability. This has now been validated by a systematic analysis from the Nicolas laboratory, showing that G4 motifs that contain single nucleotide loops (“G4L1 motifs”) are most detrimental to genomic stability 19.
G4L1 motifs are depleted in most genomes, including those of S. cerevisiae, S. pombe, nematode, and human. Depletion particularly affects G4 motifs with loops containing pyrimidines rather than purines, which exhibit the greatest genetic instability and the greatest thermodynamic stability. The entire yeast genome contains only four G4 motifs with short loops (≤ 3 nt), and even these are not likely sites of replication-associated genomic instability, as they are purine rather than pyrimidine loops. If the same rules of genomic maintenance that have been documented in S. cerevisiae extend to human cells, the encouraging take home message is that only 5% of the G4 motifs in the human genome (18,000 of 360,000) are G4L1 motifs, and thus likely to be at risk of maximal genomic instability.
The recent evidence that potentially unstable G4 motifs are dramatically depleted from the S. cerevisiae genome 19 has raised questions about a report claiming that the S. cerevisiae Pif1 helicase is enriched at G4 motifs and suppresses G4-induced genomic instability at those sites 34,35. There were initially questions about how G4 motifs were identified in that report, as it employed a search algorithm that allowed a maximal loop length of 25 nt 36, which will considerably inflate the numbers of G4 motifs scored. A search algorithm that allows loops up to 25 nt in length tallies 552 G4 motifs in the S. cerevisiae genome, while the more standard algorithm that allows loops up to 7 nt in length tallies only 27. If the endogenous G4 motifs in S. cerevisiae are not sites of replicative instability, as suggested 19, then enrichment of Pif1 at or near a G4 motif is unlikely to reflect a role for this factor in suppressing G4-induced genomic instability.
Analysis of sites of genomic instability in nematodes and human cells provides further support for the notion that G4L1 motifs—and not G4 motifs in general—are sequences at risk 19. The genome of the nematode C. elegans contains unusual mono-G runs, which account for essentially all of the G4L1 motifs in this organism. DNA deletions in nematodes deficient in dog-1, an ortholog of human FANCJ, map almost exclusively to mono-G runs 26. A corresponding effect is evident in human cells treated with the G4-ligand pyridostatin, which induces genomic instability accompanied by induction of the alternate histone, γ-H2AX 8. Sites of γ-H2AX accumulation proved to be enriched for pyrimidine-containing but not purine-containing G4L1 motifs 19.
Further experiments are necessary to better define the G4 motifs that do in fact contribute to genomic instability in human cells, and to understand which factors normally enable smooth replication of quadruplexes. A significant enrichment of G4 motifs has been documented at chromosomal breakpoints in human tumor genomes 37, and more detailed examination of loop length and composition of those motifs may provide useful rules for identifying sites of instability. In addition, as increasing numbers of normal and cancer genomes are systematically sequenced, it should be possible to make statistically compelling correlations between mutations and polymorphisms in repair and replication factors and instability at G4 motifs genome-wide. These same factors may contribute to preventing expansion of G4 motifs associated with neurodegenerative diseases, as discussed below.
G4 helicases promote stability at G4 motifs in human cells
Connections between quadruplexes and human disease (summarized in Table1) first emerged from studies of the roles of G4 helicases at regions of the genome significantly enriched for G4 motifs. Genome-wide analyses have recently established the importance of smooth replication for the maintenance of genomic stability at G4 motifs.
Table 1.
Human disease-associated factors that bind quadruplexes
| Helicase | Disease | Process |
|---|---|---|
| XPB/XPD | Xeroderma pigmentosum (UV sensitivity, cancer); trichothiodystrophy and Cockayne syndrome (development) | TFIIH components, with functions in nucleotide excision repair |
| BLM | Primordial dwarfism, genomic instability, cancer predisposition | DNA replication; immunoglobulin class switch recombination |
| WRN | Premature aging, cancer predisposition, cell senescence | Telomere maintenance |
| RTEL1 | Hoyeraal-Hreidarsson syndrome (cf. dyskeratosis congenita), cell senescence | Telomere maintenance |
| DNA2 | Cell senescence | Telomere maintenance; helicase and G4 nuclease |
| FANCJ | Fanconi anemia (bone marrow failure) | Replication with REV1 polymerase (incorporates only C) |
| CHL1 | Warsaw break syndrome (cohesinopathy) | Chromosome segregation |
| ATRX | Alpha thalassemia with mental retardation | Remodels chromatin at G4hi regions (telomeres, VNTRs, etc.) |
| eIF4A | T cell leukemia (overexpression) | Translation: unwinds mRNA to scan for 5′-AUG initiator codon |
BLM helicase at the immunoglobulin switch regions
BLM helicase is one of five human RecQ family helicases 38. BLM is deficient in the genetic disease Bloom syndrome, a rare autosomal recessive disease characterized by primordial dwarfism and predisposition to cancer. Affected individuals frequently present with immunodeficiency due to impaired immunoglobulin heavy chain class switch recombination. In class switch recombination, regulated DNA deletion joins the expressed variable region to a new constant region, conferring new effector functions on the encoded antibody without changing its specificity for antigen. Recombination junctions map to intronic “switch” regions, 1–10 kb degenerative repeats that are enriched for G4 motifs. Recombination to a particular switch region is preceded by its active transcription, which promotes quadruplex formation. BLM is a G4 helicase, which preferentially unwinds quadruplex relative to duplex DNA 39, and can thereby remove quadruplexes from the transcribed switch regions to enable replication of switched B cells and a robust immune response.
Telomeric activities of WRN, RTEL1, and DNA2
WRN helicase is a member of the conserved RecQ helicase family, and a close relative of BLM 38. WRN is deficient in the human genetic disease Werner syndrome, characterized by premature aging and cancer predisposition 40. As young adults, affected individuals exhibit signs of aging including greying and thinning hair, bilateral cataract formation, and an elevated risk of atherosclerosis, but without increased risk for Alzheimer’s disease or other dementias associated with normal aging. WRN is a G4 helicase, and in WRN-deficient cells, telomeric sequence is lost specifically due to impaired replication of the G-rich strand, an adverse consequence of persistence of G4 DNA during replication 41,42.
Telomere maintenance also depends on at least two other proteins that are active on G4 substrates, RTEL1 helicase and DNA2 helicase/nuclease. RTEL1 is an iron–sulfur domain protein, and one of four human paralogs of the highly conserved XPD helicase 43. RTEL1 maintains telomere length by resolving D-loops, telomeric T loops, and G4 DNA 44. Deficiencies in RTEL1 cause Hoyeraal–Hreidarsson syndrome, characterized by some symptoms that overlap those of dyskeratosis congenita, caused by deficiency of telomerase RNA. These include nail dysplasia, abnormal skin pigmentation, and oral leukoplakia, along with more severe manifestations such as microcephaly, cerebellar hypoplasia, bone marrow failure, and severe immunodeficiency and intrauterine growth retardation 45—47.
Another iron–sulfur domain protein, the highly conserved helicase/nuclease DNA2, promotes telomere stability by cleaving G4 structures 48. DNA2 deficiencies have recently been associated with Seckel syndrome, characterized by primordial dwarfism (like BLM deficiency), and also caused by mutations in other genes, including ATR, which encodes a key member of the DNA damage response pathway 49. Cellular senescence is accelerated in WRN-deficient cells 50, in cells in which the interaction between RTEL1 and PCNA is disrupted by mutation 51 and in cells that bear DNA2 mutations 49.
DNA replication at G4 motifs genome-wide: FANCJ and REV1
FANCJ is an iron–sulfur domain helicase, related to RTEL1 43. FANCJ is one of 16 genes that are found mutated in Fanconi anemia, a genetic disease characterized by bone marrow failure, predisposition to cancer, and cellular sensitivity to agents that cause inter-strand DNA cross-links. FANCJ unwinds G4 DNA 52,53, and FANCJ enables replication through quadruplex structures without stalling and causing genomic instability 54.
The Y family polymerase, REV1, may cooperate with FANCJ to replicate past quadruplex structures. REV1 is a polymerase that incorporates only C, an activity that would be very useful in restarting replication opposite a G at a stalled replication fork 55. It is also an ATP-independent resolvase, which can bind to and disrupt quadruplexes and prevent their refolding to promote replication 56.
Expandable G4 motifs in neurological diseases
At least thirty neurological diseases are associated with expansions at DNA repeats, including G4 motifs and trinucleotide repeats (TNRs) 57.
Fragile X syndrome
The best recognized of the human G4 expansion diseases is Fragile X syndrome, the most common cause of inherited mental retardation, with symptoms that frequently include behavioral problems. Fragile X syndrome is a neurodevelopmental disease caused by expansions of a CGG repeat in the FMR1 gene, with disease-associated repeat numbers of more than 200 58. This repeat does not strictly conform to the G4 motif, as the G-runs are only two nucleotides in length, but it does form quadruplex structures. Fragile X syndrome is characterized by anticipation: disease severity increases and age of onset decreases from one generation to the next, reflecting increased repeat lengths between generations. However, CGG repeat lengths are identical among tissues of an individual affected with Fragile X syndrome, as the CGG repeat expands during cell divisions that occur in germ cells and early embryonic development, but not in fully differentiated somatic tissues. The developmental regulation of CGG repeat expansion reflects a programmed switch in replication origin use. Expansion occurs when the expandable sequence is the template for leading strand replication, reflecting structure formation within Okazaki fragments replicating the lagging strand 59,60.
Shorter CGG repeat expansions, on the order of 55–200, can cause a distinct, late onset neurodegenerative disease, Fragile X-associated tremor/ataxia syndrome, or FXTAS 61. This is referred to as the “premutation state”, and predisposes to repeat expansion that may occur in the course of transmission of the affected chromosome from the mother to the child. FXTAS symptoms include tremor, ataxia, and cognitive decline. The disease mechanism is not well understood.
GGGGCC repeat expansions in ALS and FTD
Expansion of a GGGGCC repeat in a gene at the C9ORF72 locus on chromosome 9 is associated with more than 10% of familial cases and some sporadic cases of two different neurodegenerative diseases, amyotrophic lateral sclerosis (ALS), and frontotemporal dementia (FTD) 62—65. This sequence motif conforms to the G4 consensus and has been shown to form quadruplexes as either RNA 66 or DNA 67. In ALS, the death of motor neurons leads to stiff muscles and muscle wasting accompanied by difficulty in speaking, swallowing, breathing, and eventual death from respiratory failure. In FTD, neurons are lost from the frontal and/or temporal lobes, causing dementia that begins in late adulthood and is characterized by compulsive behaviors but—in contrast to Alzheimer’s disease—not by loss of memory. C9ORF72 GGGGCC repeat expansions have also been documented in other neurodegenerative diseases, including primary progressive aphasia 68 and depressive pseudodementia 69. It is not understood why apparently identical molecular pathology gives rise to distinct symptoms.
Most individuals carry only two or three GGGGCC repeats at the C9ORF72 locus. Pathology is typically associated with at least 30 repeats, and in some cases, expansions may be much longer. Exact lengths can be difficult to determine, as GGGGCC repeats are not readily amplified by PCR. For diagnostic purposes, the standard assay is Southern blotting, which is more labor-intensive and requires more tissue, limiting the numbers of samples that can be analyzed and the accuracy of length measurements 70. Like CGG repeat expansion diseases, GGGGCC repeat expansion exhibits anticipation, with age of presentation decreasing in successive affected generations 71. However, in contrast to CGG expansions, GGGGCC expansions are variable in length among tissues of affected individuals. Thus, expansion must occur in somatic cells. In particular, long expansions of GGGGCC repeats have been reported in neural tissues, where they exert their pathological effects. Neural cells are non-dividing, so sequence expansion must occur in the absence of DNA replication.
Quadruplex RNA: targets of pathology in G4 expansion diseases
Most motifs that form G-quadruplex structures as DNA also do so as RNA, and the expansion-associated G4 motifs are not an exception. Cellular pathology in G4 expansion diseases can be ascribed in part to protein binding to quadruplexes in the pre-mRNA or mRNA transcript. G4 RNA in the FMR1 gene transcript titrates key factors that promote normal nuclear mRNA processing, contributing to some of the pathology resulting from CGG repeat expansions 72. The GGGGCC motif forms parallel quadruplexes as RNA 66. It is claimed that one source of pathology caused by GGGGCC expansion may be binding to RNA quadruplexes by the major nucleolar protein, nucleolin, which is claimed to deform nucleoli and disrupt normal nucleolar functions 67. Altered nucleolar function would affect ribosome biogenesis and protein synthesis, and could cause many kinds of cellular pathology, though none specifically associated with ALS or FTD thus far.
Are there other expandable G4 motifs?
Currently, two additional unstable repeats are known that bear sequence motifs predictive of formation of G4 DNA within the gene itself and G4 RNA in the encoded transcript: the GGGCCT repeat in intron 1 of the NOP56 gene, on chromosome 20, associated with SCA36, an adult-onset disease of spinocerebellar ataxia and the CCCCATGGTGGTGGCTGGGGACAG repeat in the PRNP gene, which encodes the prion protein.
Normal alleles of the NOP56 gene carry 5–14 GGGCCT repeats, and expansions typically associated with disease are very long, in the range of 800–2,000 73,74. The expandable GGGCCT repeat is always found within a shared haplotype rather than occurring as a result of direct expansion. While relatively few patients have been analyzed, disease severity does not seem to vary with the length of the expansion, in contrast to GGGCCC and GGGGCC expansion diseases. The intronic position of the expandable GGGCCT repeat raises the possibility that G4 RNA contributes to pathology, as in CGG and GGGGCC repeat expansion diseases.
The normal prion gene contains 5 tandem G-rich repeats that encode a repetitive sequence within the larger prion protein, and are known as oligopeptide repeats (OPRs). Expansions of these repeats were one of the first genetic polymorphisms associated with CJD, a human transmissible spongiform encephalopathy 75,76. CJD presents as dementia and results from destruction of neural cells in the brain due to aggregation by the prion protein. Expansions, typically from 5 to 10–14 OPRs, cause a dominant form of disease exhibiting early onset and slow progression. Those features led to initial identification of the disease caused by OPR expansion as Huntington’s disease phenocopy prior to definitive molecular diagnosis and reclassification as familial CJD 77. OPR expansion results in protein misfolding, evidenced by plaque formation by the prion protein in vivo and formation of insoluble protein aggregates in vitro 78,79. It is an interesting possibility that G4 motifs are critical to disease-associated OPR expansions, but a role for them has not yet been demonstrated.
G-rich regions are notoriously difficult to amplify by PCR, so polymorphisms and instability at G4 motifs may go unscored by standard genomic analyses. The very recent identification of GGGGCC motif expansion as a cause of ALS and FTD 62—65 suggests that expandable G4 motifs may contribute to additional sporadic or familial neurodegenerative diseases.
Quadruplex targets for antigenic variation in microbial pathogens
Many pathogenic microbes evade the immune response by expressing a variety of different surface antigens. Switching surface antigen expression can be carried out by regulated recombination, which moves a genetic cassette into an expression site. In the obligate pathogen Neisseria gonorrhoeae, which causes the venereal disease gonorrhea, antigenic variation depends on recombination. Variation is regulated by a recombination activator element that functions in cis to promote gene conversion of the active pilin expression locus, pilE, using a reservoir of silent pilS loci as sequence donors. The activator element is a G4 motif, (G3TG3TTG3TG3), and it must form a transcription-induced quadruplex structure to promote variation 80,81. The conserved RecA recombination protein recognizes this specific quadruplex structure to stimulate strand exchange, and other G4 motifs do not support regulated recombination at the pilE locus 82.
G4-regulated antigen variation may also occur in other pathogens. A recent review 83 has compiled tantalizing examples of microbial pathogens which carry G4 motifs at sites of antigenic variation, which may occur by mechanisms related to those defined in N. gonorrhea. Candidates include N. meningitides, which causes meningitis and other meningococcal diseases; the Borrelia spirochetes, among them B. burgdorferi, which causes Lyme disease; and Trypanosoma brucei, which causes sleeping sickness.
Quadruplex targets of transcriptional, translational, and epigenetic regulation
The sections below focus on recent genome-wide analyses that have identified quadruplexes as regulatory targets (summarized in Table1). Numerous reports connecting specific G4 motifs with regulation of specific genes complement these genome-wide analyses, but the genome-wide studies are especially powerful because they provide new and unanticipated insights into how factors that act at G4 DNA or G4 RNA may contribute to cancer and genetic disease.
Quadruplex targets of transcriptional regulation
G4 DNA targets of XPD/XPB are enriched at the promoter
XPD and XPB are the helicase components of the general transcription complex TFIIH. They are essential for nucleotide excision repair; and specific mutant alleles are associated with three genetic diseases: Xeroderma pigmentosum, characterized by defective repair of damage from UV radiation, greatly increased predisposition to cancer, especially skin cancer and melanoma, trichothiodystrophy, characterized by distinctive developmental defects, especially brittle hair, but no cancer predisposition and Cockayne syndrome, characterized by developmental defects and progressive neurological degeneration, again without cancer predisposition 84,85. We showed by biochemical analysis that XPD is a robust G4 helicase and that XPB binds but does not unwind G4 DNA, and by complementary ChIP-Seq mapping that XPB/XPD is greatly enriched at the TSS (Fig3A), where 60% of binding sites overlap with G4 motifs (P = 0; 24). Systems analysis shows that XPB/XPD is bound to G4 motifs in genes and pathways that are dysregulated in the diseases associated with deficiencies in these factors. Strikingly, highly significant enrichment of XPB/XPD was evident at G4 motifs near genes that escape the genome-wide shutdown of transcription that typically follows UV irradiation of human cells, and instead exhibit transcriptional upregulation in response to UV (P < 1e-12). That observation raises the intriguing possibility that not only failed UV repair but also dysregulated transcription may contribute to the predisposition to skin cancer and melanoma associated with XPB or XPD deficiencies.
Figure 3.
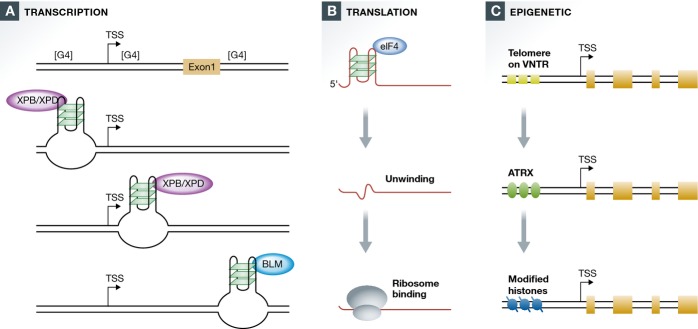
Quadruplex targets of regulation
(A) Three regions of human genes are G4hi ([G4]) and thus prone to quadruplex formation: upstream and downstream of the TSS and the 5′ end of intron 1 (right). Quadruplexes upstream and downstream of the TSS are predominant among targets of XPB/XPD, while quadruplexes at the 5′ end of intron 1 are predominant among targets of BLM. (B) Translation of mRNAs containing a quadruplex in the 5′ UTR requires unwinding by the G4 RNA helicase, eIF4, which exposes the ribosome binding site to allow translation. (C) ATRX promotes epigenetic modifications at quadruplexes within G4hi telomeres and VNTRs.
XPB/XPD bind approximately 2% of all G4 motifs in the human genome. The features that distinguish XPB/XPD targets from other quadruplexes have yet to be defined. Quadruplex conformation is likely to be one of them. Transcriptional status and local DNA supercoiling (Fig2) are also likely to be important, and interactions with other chromatin bound proteins may be necessary to promote stable associations.
Quadruplex targets of BLM at the 5′ end of intron 1
In order to determine whether BLM helicase might regulate transcription, we compared gene expression in fibroblasts from individuals with Bloom syndrome and normal individuals 86. In BLM-deficient cells, mRNA expression of genes connected with cellular growth, proliferation, and cell death/survival was reduced. Altered expression of genes in these pathways may contribute to the primordial dwarfism that is a defining characteristic of Bloom syndrome. Genes downregulated in the absence of BLM were especially enriched in G4 motifs on the non-transcribed strand at the 5′ end of the first intron (FDR < 0.001), where approximately 20% of human genes carry a G4hi region that correlates with transcriptional pausing 87,88. This suggests that BLM may normally unwind quadruplexes formed within the 5′ end of the first intron, thereby enabling transcription (Fig3A).
Quadruplex targets of translational regulation
G4 RNA targets of eIF4A-dependent translation
Just as BLM helicase may smoothen the path for transcription through regions containing G4 DNA, the RNA helicase eIF4A may unwind G4 RNA to promote translation (Fig3B). One well-documented function of this helicase is to scan the 5′ end of mRNA to reveal the site for translational initiation. Overexpression of eIF4A can drive development of T-cell acute lymphoblastic leukemia by inducing overexpression of other potent oncogenes, such as MYC 89. Ribosome footprinting showed that many genes that are translational regulatory targets of eIF4A bear a G4 motif in the 5′ UTR, and the (CGG)4 motif appears to be a hallmark of mRNAs that depend on eIF4A for translation. These include oncogenes and transcription factors, classes of genes that were previously shown to be G4hi (20; see above). Dependence of these genes on eIF4A for efficient translation may provide an extra level of regulation that normally prevents overexpression of G4hi genes.
Quadruplex targets of epigenetic regulation
ATRX
The possibility that G4 motifs function as targets of epigenetic regulation was first raised by analyses of the SWI/SNF chromatin remodeler family member, ATRX 90. ATRX deficiency is associated with a genetic disease, X-linked alpha thalassemia/mental retardation syndrome 90,91. Somatic mutations in ATRX are common in pancreatic neuroendocrine tumors and tumors of the central nervous system, where they correlate with telomere maintenance by the telomerase-independent ALT pathway 92. ATRX has robust G4 DNA binding activity, and it is enriched at telomeres, rDNA, and other sites. One of the targets of ATRX is a G-rich VNTR at the α-globin gene (CGCGGGGCGGGGG)n, which is predicted to form quadruplexes. In the absence of ATRX, this α-globin VNTR undergoes silencing, downregulating α-globin expression to cause the characteristic α-thalassemia. ATRX enables deposition of modified histones compatible with gene expression (Fig3C). The G-rich α-globin VNTR reaches lengths of nearly 500 bp in some individuals, though it is much shorter in others, and the severity of α-thalassemia resulting from ATRX deficiency correlates with VNTR length 90.
Perturbed replication promotes epigenetic instability at G4 motifs
Epigenetic dysregulation is a well-documented feature of many tumors, and the mechanisms that cause this are not clearly understood. An intriguing series of experiments suggests that slowed replication at G4 motifs may be one source of epigenetic instability 93—95.
Epigenetic modifications at or near G4 motifs are altered in cells deficient in repair factors that promote smooth replication through quadruplexes, including FANCJ, BLM, or WRN helicases or the repair polymerase REV1 93,94. This causes epigenetic dysregulation evident either as increased expression of silenced genes or reduced expression of active genes. Conversely, an individual G4 motif can function as an epigenetic regulatory element that directs histone modifications at an adjacent promoter 95. One model for how that may occur is as follows:
Histone deposition immediately after replication ensures correct propagation of specific histone marks. The normally rapid tempo of replication may be interrupted when the fork encounters a quadruplex structure, leading to diffusion of modified histones away from the stalled fork, and coating of the new daughter DNAs with histones bearing default marks that cannot exert appropriate regulation. This raises the possibility that some or all G4hi genes may be epigenetically labile, with chromatin structures tending toward the default setting with each round of replication. This would provide a level of control ensuring that G4hi oncogenes and transcription factors 20 could be expressed at high levels only if appropriate factors continued to be available following cell division.
Do other disease-associated G4 helicases regulate gene expression?
Further genome-wide analyses are necessary to map the quadruplex targets of other factors, an important step toward functional understanding of the G4 genome. Candidate regulators among the disease-associated human helicases include WRN and another RECQ family helicase, REQ4, deficient in Rothmund–Thomson syndrome, DNA2 and the XPD paralogs FANCJ, RTEL1, and CHL1 (CHLR1/DDX11), deficient in Warsaw breakage syndrome, a cohesinopathy 96. The iron–sulfur domain proteins (XPD, FANCJ, RTEL1, CHL1, and DNA2) are particularly interesting, as proteins in this family readily transport charge and they have been suggested to thereby relay intracellular signals 97.
G-quadruplex DNAs can themselves transport charge, over distances sufficiently long (100 nM) to be of potential utility for nano-devices 98. This property could contribute a novel dimension to the functions of a quadruplex regulatory target in vivo. For example, iron–sulfur domain proteins may bind to intracellular quadruplexes to stimulate charge transport between them, or along a region of DNA that is enriched for quadruplexes. This could in principle enable coordination of transcription or repair among physically distant nodes of a G4 DNA-based intracellular regulatory network.
Conclusions and future directions
We have begun to understand the connections between G-quadruplexes and human disease, at the levels of genomic stability, transcriptional, and translational regulation and chromatin structure. Detailed mechanistic understanding will enable rapid diagnosis of G4-associated disease, and development of new and better drugs with appropriate specificity. Recent progress now points to important questions for the immediate future (Sidebar B).
Sidebar B: In need of answers.
Which G4 motifs form quadruplexes in living human cells? A catalog of interactions of all endogenous quadruplex binding proteins with genomic DNA will provide a picture of this functional G4 genome.
Which human G4 motifs are at risk of genomic instability? Do the same rules of G4 motif maintenance extend from yeast to human cells? If so, sites of instability in tumors are predicted to be enriched for G4L1 motifs.
What is the mechanism of expansion of CGG motifs in germ cells or early embryonic development? How does it differ from the mechanism of expansion of GGGGCC motifs, which seems to occur in somatic cells?
Does the prion gene G4 motif form a quadruplex structure in vivo, and does this promote sequence expansion in neurodegenerative disease?
Which human G4 motifs are targets of transcriptional, translational, and epigenetic regulation?
Do all the enzymes identified as G4 helicases also function as transcriptional regulators?
Acknowledgments
I thank the US National Institutes of Health and National Cancer Institute for supporting our research related to this review (P01 CA77852).
Glossary
- ALS
amyotrophic lateral sclerosis
- ATR
ataxia telangiectasia and Rad3 related
- ATRX
alpha thalassemia/mental retardation syndrome X-linked
- BLM
Bloom syndrome, RecQ helicase-like
- C9ORF27
chromosome 9, open reading frame 27
- CEB25
Centre d’étude du polymorphisme humain, probe 25
- CJD
Creutzfeldt–Jacob disease
- DDX11/CHL1/CHLR1
DEAD/H (Asp-Glu-Ala-Asp/His) box helicase 11
- DNA2
DNA replication helicase/nuclease 2
- dog-1
deletion of guanine-rich DNA
- eIF4A
eukaryotic translation initiation factor 4A
- FANCJ/BACH1/BRIP1
Fanconi anemia, complementation group J
- FMR1
fragile X mental retardation 1
- FDR
false discovery rate
- FTD
frontotemporal dementia
- FXTAS
Fragile X-associated tremor/ataxia syndrome
- G4L1
G4 motif with a one nucleotide loop
- MYC
v-myc avian myelocytomatosis viral oncogene homolog
- NOP56
NOP56 ribonucleoprotein
- OPR
oligopeptide repeat
- PCNA
proliferating cell nuclear antigen
- PIF1
PIF1 5′-to-3′ DNA helicase
- PRNP
prion protein
- RECQ
Gene family related to E. coli RecQ helicase
- REV1
REV1, polymerase (DNA directed)
- RGG
Arg-Gly-Gly protein sequence motifs
- RHAU
RNA helicase associated with AU-rich element
- RQC
RECQ-conserved domain
- RRM/RBD
RNA recognition motif/RNA binding domain
- RSM
RHAU-specific motif
- RTEL1
regulator of telomere elongation helicase 1
- SCA36
spinocerebellar ataxia 36
- SWI/SNF
Switch/sucrose non-fermentable
- TFIIH
transcription factor II H
- TNR
trinucleotide repeat
- TSS
transcription start site
- VNTR
variable number tandem repeat
Conflict of interest
The author declares that she has no conflict of interest.
References
- Sen D, Gilbert W. Formation of parallel four-stranded complexes by guanine rich motifs in DNA and its implications for meiosis. Nature. 1988;334:364–366. doi: 10.1038/334364a0. [DOI] [PubMed] [Google Scholar]
- Kim J, Cheong C, Moore PB. Tetramerization of an RNA oligonucleotide containing a GGGG sequence. Nature. 1991;351:331–332. doi: 10.1038/351331a0. [DOI] [PubMed] [Google Scholar]
- Gellert M, Lipsett MN, Davies DR. Helix formation by guanylic acid. Proc Natl Acad Sci USA. 1962;48:2014–2018. doi: 10.1073/pnas.48.12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan AT, Kuryavyi V, Patel DJ. DNA architecture: from G to Z. Curr Opin Struct Biol. 2006;16:288–298. doi: 10.1016/j.sbi.2006.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel DJ, Phan AT, Kuryavyi V. Human telomere, oncogenic promoter and 5′-UTR G-quadruplexes: diverse higher order DNA and RNA targets for cancer therapeutics. Nucleic Acids Res. 2007;35:7429–7455. doi: 10.1093/nar/gkm711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramanian S, Hurley LH, Neidle S. Targeting G-quadruplexes in gene promoters: a novel anticancer strategy? Nat Rev Drug Discov. 2011;10:261–275. doi: 10.1038/nrd3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piazza A, Boulé JB, Lopes J, Mingo K, Largy E, Teulade-Fichou MP, Nicolas A. Genetic instability triggered by G-quadruplex interacting Phen-DC compounds in Saccharomyces cerevisiae. Nucleic Acids Res. 2010;38:4337–4348. doi: 10.1093/nar/gkq136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez R, Miller KM, Forment JV, Bradshaw CR, Nikan M, Britton S, Oelschlaegel T, Xhemalce B, Balasubramanian S, Jackson SP. Small-molecule-induced DNA damage identifies alternative DNA structures in human genes. Nat Chem Biol. 2012;8:301–310. doi: 10.1038/nchembio.780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagano B, Amato J, Iaccarino N, Cingolani C, Zizza P, Biroccio A, Novellino E, Randazzo A. Looking for efficient G-quadruplex ligands: evidence for selective stabilizing properties and telomere damage by drug-like molecules. ChemMedChem. 2015;10:640–649. doi: 10.1002/cmdc.201402552. [DOI] [PubMed] [Google Scholar]
- Phan AT. Human telomeric G-quadruplex: structures of DNA and RNA sequences. FEBS J. 2010;277:1107–1117. doi: 10.1111/j.1742-4658.2009.07464.x. [DOI] [PubMed] [Google Scholar]
- Lim KW, Ng VC, Martin-Pintado N, Heddi B, Phan AT. Structure of the human telomere in Na+ solution: an antiparallel (2+2) G-quadruplex scaffold reveals additional diversity. Nucleic Acids Res. 2013;41:10556–10562. doi: 10.1093/nar/gkt771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martadinata H, Heddi B, Lim KW, Phan AT. Structure of long human telomeric RNA (TERRA): G-quadruplexes formed by four and eight UUAGGG repeats are stable building blocks. Biochemistry. 2011;50:6455–6461. doi: 10.1021/bi200569f. [DOI] [PubMed] [Google Scholar]
- Amrane S, Adrian M, Heddi B, Serero A, Nicolas A, Mergny JL, Phan AT. Formation of pearl-necklace monomorphic G-quadruplexes in the human CEB25 minisatellite. J Am Chem Soc. 2012;134:5807–5816. doi: 10.1021/ja208993r. [DOI] [PubMed] [Google Scholar]
- Hanakahi LA, Sun H, Maizels N. High affinity interactions of nucleolin with G-G-paired rDNA. J Biol Chem. 1999;274:15908–15912. doi: 10.1074/jbc.274.22.15908. [DOI] [PubMed] [Google Scholar]
- Lattmann S, Giri B, Vaughn JP, Akman SA, Nagamine Y. Role of the amino terminal RHAU-specific motif in the recognition and resolution of guanine quadruplex-RNA by the DEAH-box RNA helicase RHAU. Nucleic Acids Res. 2010;38:6219–6233. doi: 10.1093/nar/gkq372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber MD, Duquette ML, Shiels JC, Maizels N. A conserved G4 DNA binding domain in RecQ family helicases. J Mol Biol. 2006;358:1071–1080. doi: 10.1016/j.jmb.2006.01.077. [DOI] [PubMed] [Google Scholar]
- Maizels N, Gray LT. The G4 genome. PLoS Genet. 2013;9:e1003468. doi: 10.1371/journal.pgen.1003468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huppert JL, Balasubramanian S. Prevalence of quadruplexes in the human genome. Nucleic Acids Res. 2005;33:2908–2916. doi: 10.1093/nar/gki609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piazza A, Adrian M, Samazan F, Heddi B, Hamon F, Serero A, Lopes J, Teulade-Fichou MP, Phan AT, Nicolas A. Short loop length and high thermal stability determine genomic instability induced by G-quadruplex-forming minisatellites. EMBO J. 2015;34:1718–1734. doi: 10.15252/embj.201490702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy J, Maizels N. Gene function correlates with potential for G4 DNA formation in the human genome. Nucleic Acids Res. 2006;34:3887–3896. doi: 10.1093/nar/gkl529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvam S, Koirala D, Yu Z, Mao H. Quantification of topological coupling between DNA superhelicity and G-quadruplex formation. J Am Chem Soc. 2014;136:13967–13970. doi: 10.1021/ja5064394. [DOI] [PubMed] [Google Scholar]
- Murat P, Balasubramanian S. Existence and consequences of G-quadruplex structures in DNA. Curr Opin Genet Dev. 2014;25:22–29. doi: 10.1016/j.gde.2013.10.012. [DOI] [PubMed] [Google Scholar]
- Largy E, Granzhan A, Hamon F, Verga D, Teulade-Fichou MP. Visualizing the quadruplex: from fluorescent ligands to light-up probes. Top Curr Chem. 2013;330:111–177. doi: 10.1007/128_2012_346. [DOI] [PubMed] [Google Scholar]
- Gray LT, Vallur AC, Eddy J, Maizels N. G quadruplexes are genomewide targets of transcriptional helicases XPB and XPD. Nat Chem Biol. 2014;10:313–318. doi: 10.1038/nchembio.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta A, Haber JE. Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harb Perspect Biol. 2014;6:a016428. doi: 10.1101/cshperspect.a016428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruisselbrink E, Guryev V, Brouwer K, Pontier DB, Cuppen E, Tijsterman M. Mutagenic capacity of endogenous G4 DNA underlies genome instability in FANCJ-defective C. elegans. Curr Biol. 2008;18:900–905. doi: 10.1016/j.cub.2008.05.013. [DOI] [PubMed] [Google Scholar]
- Maizels N. Genomic Stability: FANCJ-Dependent G4 DNA Repair. Curr Biol. 2008;18:R613–614. doi: 10.1016/j.cub.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koole W, van Schendel R, Karambelas AE, van Heteren JT, Okihara KL, Tijsterman M. A Polymerase Theta-dependent repair pathway suppresses extensive genomic instability at endogenous G4 DNA sites. Nat Commun. 2014;5:3216. doi: 10.1038/ncomms4216. [DOI] [PubMed] [Google Scholar]
- Roerink SF, van Schendel R, Tijsterman M. Polymerase theta-mediated end joining of replication-associated DNA breaks in C. elegans. Genome Res. 2014;24:954–962. doi: 10.1101/gr.170431.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Kregten M, Tijsterman M. The repair of G-quadruplex-induced DNA damage. Exp Cell Res. 2014;329:178–183. doi: 10.1016/j.yexcr.2014.08.038. [DOI] [PubMed] [Google Scholar]
- Lopes J, Piazza A, Bermejo R, Kriegsman B, Colosio A, Teulade-Fichou MP, Foiani M, Nicolas A. G-quadruplex-induced instability during leading-strand replication. EMBO J. 2011;30:4033–4046. doi: 10.1038/emboj.2011.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis L, Maizels N. G4 DNA: at risk in the genome. EMBO J. 2011;30:3878–3879. doi: 10.1038/emboj.2011.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piazza A, Serero A, Boulé JB, Legoix-Né P, Lopes J, Nicolas A. Stimulation of gross chromosomal rearrangements by the human CEB1 and CEB25 minisatellites in Saccharomyces cerevisiae depends on G-quadruplexes or Cdc13. PLoS Genet. 2012;8:e1003033. doi: 10.1371/journal.pgen.1003033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paeschke K, Capra JA, Zakian VA. DNA Replication through G-Quadruplex motifs Is promoted by the Saccharomyces cerevisiae Pif1 DNA helicase. Cell. 2011;145:678–691. doi: 10.1016/j.cell.2011.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabouri N, Capra JA, Zakian VA. The essential Schizosaccharomyces pombe Pfh1 DNA helicase promotes fork movement past G-quadruplex motifs to prevent DNA damage. BMC Biol. 2014;12:101. doi: 10.1186/s12915-014-0101-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capra JA, Paeschke K, Singh M, Zakian VA. G-quadruplex DNA sequences are evolutionarily conserved and associated with distinct genomic features in Saccharomyces cerevisiae. PLoS Comput Biol. 2010;6:e1000861. doi: 10.1371/journal.pcbi.1000861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De S, Michor F. DNA replication timing and long-range DNA interactions predict mutational landscapes of cancer genomes. Nat Biotechnol. 2011;29:1103–1108. doi: 10.1038/nbt.2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croteau DL, Popuri V, Opresko PL, Bohr VA. Human RecQ helicases in DNA repair, recombination, and replication. Annu Rev Biochem. 2014;83:519–552. doi: 10.1146/annurev-biochem-060713-035428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Karow JK, Hickson ID, Maizels N. The Bloom’s syndrome helicase unwinds G4 DNA. J Biol Chem. 1998;273:27587–27592. doi: 10.1074/jbc.273.42.27587. [DOI] [PubMed] [Google Scholar]
- Goto M, Ishikawa Y, Sugimoto M, Furuichi Y. Werner syndrome: a changing pattern of clinical manifestations in Japan (1917∼2008) Biosci Trends. 2013;7:13–22. [PubMed] [Google Scholar]
- Rossi ML, Ghosh AK, Bohr VA. Roles of Werner syndrome protein in protection of genome integrity. DNA Repair (Amst) 2010;9:331–344. doi: 10.1016/j.dnarep.2009.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damerla RR, Knickelbein KE, Strutt S, Liu FJ, Wang H, Opresko PL. Werner syndrome protein suppresses the formation of large deletions during the replication of human telomeric sequences. Cell Cycle. 2012;11:3036–3044. doi: 10.4161/cc.21399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White MF. Structure, function and evolution of the XPD family of iron-sulfur-containing 5′-3′ DNA helicases. Biochem Soc Trans. 2009;37:547–551. doi: 10.1042/BST0370547. [DOI] [PubMed] [Google Scholar]
- Vannier JB, Pavicic-Kaltenbrunner V, Petalcorin MI, Ding H, Boulton SJ. RTEL1 dismantles T loops and counteracts telomeric G4-DNA to maintain telomere integrity. Cell. 2012;149:795–806. doi: 10.1016/j.cell.2012.03.030. [DOI] [PubMed] [Google Scholar]
- Deng Z, Glousker G, Molczan A, Fox AJ, Lamm N, Dheekollu J, Weizman OE, Schertzer M, Wang Z, Vladimirova O, et al. Inherited mutations in the helicase RTEL1 cause telomere dysfunction and Hoyeraal-Hreidarsson syndrome. Proc Natl Acad Sci USA. 2013;110:E3408–3416. doi: 10.1073/pnas.1300600110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Guen T, Jullien L, Touzot F, Schertzer M, Gaillard L, Perderiset M, Carpentier W, Nitschke P, Picard C, Couillault G, et al. Human RTEL1 deficiency causes Hoyeraal-Hreidarsson syndrome with short telomeres and genome instability. Hum Mol Genet. 2013;22:3239–3249. doi: 10.1093/hmg/ddt178. [DOI] [PubMed] [Google Scholar]
- Ballew BJ, Joseph V, De S, Sarek G, Vannier JB, Stracker T, Schrader KA, Small TN, O’Reilly R, Manschreck C, et al. A recessive founder mutation in regulator of telomere elongation helicase 1, RTEL1, underlies severe immunodeficiency and features of Hoyeraal Hreidarsson syndrome. PLoS Genet. 2013;9:e1003695. doi: 10.1371/journal.pgen.1003695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Sampathi S, Dai H, Liu C, Zhou M, Hu J, Huang Q, Campbell J, Shin-Ya K, Zheng L, et al. Mammalian DNA2 helicase/nuclease cleaves G-quadruplex DNA and is required for telomere integrity. EMBO J. 2013;32:1425–1439. doi: 10.1038/emboj.2013.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaheen R, Faqeih E, Ansari S, Abdel-Salam G, Al-Hassnan ZN, Al-Shidi T, Alomar R, Sogaty S, Alkuraya FS. Genomic analysis of primordial dwarfism reveals novel disease genes. Genome Res. 2014;24:291–299. doi: 10.1101/gr.160572.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Y, Murnane JP. Telomere instability in a human tumor cell line expressing a dominant-negative WRN protein. Hum Genet. 2003;113:337–347. doi: 10.1007/s00439-003-0972-y. [DOI] [PubMed] [Google Scholar]
- Vannier JB, Sandhu S, Petalcorin MI, Wu X, Nabi Z, Ding H, Boulton SJ. RTEL1 is a replisome-associated helicase that promotes telomere and genome-wide replication. Science. 2013;342:239–242. doi: 10.1126/science.1241779. [DOI] [PubMed] [Google Scholar]
- London TB, Barber LJ, Mosedale G, Kelly GP, Balasubramanian S, Hickson ID, Boulton SJ, Hiom K. FANCJ is a structure-specific DNA helicase associated with the maintenance of genomic G/C tracts. J Biol Chem. 2008;283:36132–36139. doi: 10.1074/jbc.M808152200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Shin-Ya K, Brosh RM., Jr FANCJ helicase defective in Fanconi anemia and breast cancer unwinds G-quadruplex DNA to defend genomic stability. Mol Cell Biol. 2008;28:4116–4128. doi: 10.1128/MCB.02210-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo Bosch P, Segura-Bayona S, Koole W, van Heteren JT, Dewar JM, Tijsterman M, Knipscheer P. FANCJ promotes DNA synthesis through G-quadruplex structures. EMBO J. 2014;33:2521–2533. doi: 10.15252/embj.201488663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickramasinghe CM, Arzouk H, Frey A, Maiter A, Sale JE. Contributions of the specialised DNA polymerases to replication of structured DNA. DNA Repair (Amst) 2015;29:83–90. doi: 10.1016/j.dnarep.2015.01.004. [DOI] [PubMed] [Google Scholar]
- Eddy S, Ketkar A, Zafar MK, Maddukuri L, Choi JY, Eoff RL. Human Rev1 polymerase disrupts G-quadruplex DNA. Nucleic Acids Res. 2014;42:3272–3285. doi: 10.1093/nar/gkt1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirkin SM. Expandable DNA repeats and human disease. Nature. 2007;447:932–940. doi: 10.1038/nature05977. [DOI] [PubMed] [Google Scholar]
- Santoro MR, Bray SM, Warren ST. Molecular Mechanisms of Fragile X Syndrome: A Twenty-Year Perspective. Annu Rev Pathol. 2011;7:219–245. doi: 10.1146/annurev-pathol-011811-132457. [DOI] [PubMed] [Google Scholar]
- Gerhardt J, Tomishima MJ, Zaninovic N, Colak D, Yan Z, Zhan Q, Rosenwaks Z, Jaffrey SR, Schildkraut CL. The DNA replication program is altered at the FMR1 locus in fragile X embryonic stem cells. Mol Cell. 2014;53:19–31. doi: 10.1016/j.molcel.2013.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirkin EV, Mirkin SM. To switch or not to switch: at the origin of repeat expansion disease. Mol Cell. 2014;53:1–3. doi: 10.1016/j.molcel.2013.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loesch D, Hagerman R. Unstable mutations in the FMR1 gene and the phenotypes. Adv Exp Med Biol. 2012;769:78–114. doi: 10.1007/978-1-4614-5434-2_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renton AE, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rademakers R. C9orf72 repeat expansions in patients with ALS and FTD. Lancet Neurol. 2012;11:297–298. doi: 10.1016/S1474-4422(12)70046-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majounie E, Abramzon Y, Renton AE, Perry R, Bassett SS, Pletnikova O, Troncoso JC, Hardy J, Singleton AB, Traynor BJ. Repeat expansion in C9ORF72 in Alzheimer’s disease. N Engl J Med. 2012;366:283–284. doi: 10.1056/NEJMc1113592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fratta P, Mizielinska S, Nicoll AJ, Zloh M, Fisher EM, Parkinson G, Isaacs AM. C9orf72 hexanucleotide repeat associated with amyotrophic lateral sclerosis and frontotemporal dementia forms RNA G-quadruplexes. Sci Rep. 2012;2:1016. doi: 10.1038/srep01016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeusler AR, Donnelly CJ, Periz G, Simko EA, Shaw PG, Kim MS, Maragakis NJ, Troncoso JC, Pandey A, Sattler R, et al. C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature. 2014;507:195–200. doi: 10.1038/nature13124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simón-Sánchez J, Dopper EG, Cohn-Hokke PE, Hukema RK, Nicolaou N, Seelaar H, de Graaf JR, de Koning I, van Schoor NM, Deeg DJ, et al. The clinical and pathological phenotype of C9ORF72 hexanucleotide repeat expansions. Brain. 2012;135:723–735. doi: 10.1093/brain/awr353. [DOI] [PubMed] [Google Scholar]
- Bieniek KF, van Blitterswijk M, Baker MC, Petrucelli L, Rademakers R, Dickson DW. Expanded C9ORF72 Hexanucleotide Repeat in Depressive Pseudodementia. JAMA Neurol. 2014;71:775–781. doi: 10.1001/jamaneurol.2013.6368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akimoto C, Volk AE, van Blitterswijk M, Van den Broeck M, Leblond CS, Lumbroso S, Camu W, Neitzel B, Onodera O, van Rheenen W. A blinded international study on the reliability of genetic testing for GGGGCC-repeat expansions in C9orf72 reveals marked differences in results among 14 laboratories. J Med Genet. 2014;51:419–424. doi: 10.1136/jmedgenet-2014-102360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper-Knock J, Shaw PJ, Kirby J. The widening spectrum of C9ORF72-related disease; genotype/phenotype correlations and potential modifiers of clinical phenotype. Acta Neuropathol. 2014;127:333–345. doi: 10.1007/s00401-014-1251-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qurashi A, Li W, Zhou JY, Peng J, Jin P. Nuclear accumulation of stress response mRNAs contributes to the neurodegeneration caused by Fragile X premutation rCGG repeats. PLoS Genet. 2011;7:e1002102. doi: 10.1371/journal.pgen.1002102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi H, Abe K, Matsuura T, Ikeda Y, Hitomi T, Akechi Y, Habu T, Liu W, Okuda H, Koizumi A. Expansion of intronic GGCCTG hexanucleotide repeat in NOP56 causes SCA36, a type of spinocerebellar ataxia accompanied by motor neuron involvement. Am J Hum Genet. 2011;89:121–130. doi: 10.1016/j.ajhg.2011.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obayashi M, Stevanin G, Synofzik M, Monin ML, Duyckaerts C, Sato N, Streichenberger N, Vighetto A, Desestret V, Tesson C, et al. Spinocerebellar ataxia type 36 exists in diverse populations and can be caused by a short hexanucleotide GGCCTG repeat expansion. J Neurol Neurosurg Psychiatry. 2014 doi: 10.1136/jnnp-2014-309153. doi: 10.1136/jnnp-2014-309153. [DOI] [PubMed] [Google Scholar]
- Owen F, Poulter M, Lofthouse R, Collinge J, Crow TJ, Risby D, Baker HF, Ridley RM, Hsiao K, Prusiner SB. Insertion in prion protein gene in familial Creutzfeldt-Jakob disease. Lancet. 1989;1:51–52. doi: 10.1016/s0140-6736(89)91713-3. [DOI] [PubMed] [Google Scholar]
- Mead S, Webb TE, Campbell TA, Beck J, Linehan JM, Rutherfoord S, Joiner S, Wadsworth JD, Heckmann J, Wroe S. Inherited prion disease with 5-OPRI: phenotype modification by repeat length and codon 129. Neurology. 2007;69:730–738. doi: 10.1212/01.wnl.0000267642.41594.9d. [DOI] [PubMed] [Google Scholar]
- Moore RC, Xiang F, Monaghan J, Han D, Zhang Z, Edström L, Anvret M, Prusiner SB. Huntington disease phenocopy is a familial prion disease. Am J Hum Genet. 2001;69:1385–1388. doi: 10.1086/324414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfarb LG, Brown P, McCombie WR, Goldgaber D, Swergold GD, Wills PR, Cervenakova L, Baron H, Gibbs CJ, Jr, Gajdusek DC. Transmissible familial Creutzfeldt-Jakob disease associated with five, seven, and eight extra octapeptide coding repeats in the PRNP gene. Proc Natl Acad Sci USA. 1991;88:10926–10930. doi: 10.1073/pnas.88.23.10926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann S, Harris DA. Two mutant prion proteins expressed in cultured cells acquire biochemical properties reminiscent of the scrapie isoform. Proc Natl Acad Sci USA. 1996;93:5610–5614. doi: 10.1073/pnas.93.11.5610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahoon LA, Seifert HS. An alternative DNA structure is necessary for pilin antigenic variation in Neisseria gonorrhoeae. Science. 2009;325:764–767. doi: 10.1126/science.1175653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahoon LA, Seifert HS. Transcription of a cis-acting, noncoding, small RNA Is required for pilin antigenic variation in Neisseria gonorrhoeae. PLoS Pathog. 2013;9:e1003074. doi: 10.1371/journal.ppat.1003074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuryavyi V, Cahoon LA, Seifert HS, Patel DJ. RecA-binding pilE G4 sequence essential for pilin antigenic variation forms monomeric and 5′ end-stacked dimeric parallel G-quadruplexes. Structure. 2012;20:2090–2102. doi: 10.1016/j.str.2012.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris LM, Merrick CJ. G-quadruplexes in pathogens: a common route to virulence control? PLoS Pathog. 2015;11:e1004562. doi: 10.1371/journal.ppat.1004562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egly JM, Coin F. A history of TFIIH: two decades of molecular biology on a pivotal transcription/repair factor. DNA Repair (Amst) 2011;10:714–721. doi: 10.1016/j.dnarep.2011.04.021. [DOI] [PubMed] [Google Scholar]
- Kamileri I, Karakasilioti I, Garinis GA. Nucleotide excision repair: new tricks with old bricks. Trends Genet. 2012;28:566–573. doi: 10.1016/j.tig.2012.06.004. [DOI] [PubMed] [Google Scholar]
- Nguyen GH, Tang W, Robles AI, Beyer RP, Gray LT, Welsh JA, Schetter AJ, Kumamoto K, Wang XW, Hickson ID, et al. Regulation of gene expression by the BLM helicase correlates with the presence of G-quadruplex DNA motifs. Proc Natl Acad Sci USA. 2014;111:9905–9910. doi: 10.1073/pnas.1404807111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy J, Maizels N. Conserved elements with potential to form polymorphic G-quadruplex structures in the first intron of human genes. Nucleic Acids Res. 2008;36:1321–1333. doi: 10.1093/nar/gkm1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy J, Vallur AC, Varma S, Liu H, Reinhold WC, Pommier Y, Maizels N. G4 motifs correlate with promoter-proximal transcriptional pausing in human genes. Nucleic Acids Res. 2011;39:4975–4983. doi: 10.1093/nar/gkr079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe AL, Singh K, Zhong Y, Drewe P, Rajasekhar VK, Sanghvi VR, Mavrakis KJ, Jiang M, Roderick JE, Van der Meulen J, et al. RNA G-quadruplexes cause eIF4A-dependent oncogene translation in cancer. Nature. 2014;513:65–70. doi: 10.1038/nature13485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law MJ, Lower KM, Voon HP, Hughes JR, Garrick D, Viprakasit V, Mitson M, De Gobbi M, Marra M, Morris A, et al. ATR-X syndrome protein targets tandem repeats and influences allele-specific expression in a size-dependent manner. Cell. 2010;143:367–378. doi: 10.1016/j.cell.2010.09.023. [DOI] [PubMed] [Google Scholar]
- Wong LH, McGhie JD, Sim M, Anderson MA, Ahn S, Hannan RD, George AJ, Morgan KA, Mann JR, Choo KH. ATRX interacts with H3.3 in maintaining telomere structural integrity in pluripotent embryonic stem cells. Genome Res. 2010;20:351–360. doi: 10.1101/gr.101477.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaphy CM, de Wilde RF, Jiao Y, Klein AP, Edil BH, Shi C, Bettegowda C, Rodriguez FJ, Eberhart CG, Hebbar S, et al. Altered telomeres in tumors with ATRX and DAXX mutations. Science. 2011;333:425. doi: 10.1126/science.1207313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkies P, Reams C, Simpson LJ, Sale JE. Epigenetic instability due to defective replication of structured DNA. Mol Cell. 2010;40:703–713. doi: 10.1016/j.molcel.2010.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkies P, Murat P, Phillips LG, Patel KJ, Balasubramanian S, Sale JE. FANCJ coordinates two pathways that maintain epigenetic stability at G-quadruplex DNA. Nucleic Acids Res. 2011;40:1485–1498. doi: 10.1093/nar/gkr868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiavone D, Guilbaud G, Murat P, Papadopoulou C, Sarkies P, Prioleau MN, Balasubramanian S, Sale JE. Determinants of G quadruplex-induced epigenetic instability in REV1-deficient cells. EMBO J. 2014;33:2507–2520. doi: 10.15252/embj.201488398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Lelij P, Chrzanowska KH, Godthelp BC, Rooimans MA, Oostra AB, Stumm M, Zdzienicka MZ, Joenje H, de Winter JP. Warsaw breakage syndrome, a cohesinopathy associated with mutations in the XPD helicase family member DDX11/ChlR1. Am J Hum Genet. 2010;86:262–266. doi: 10.1016/j.ajhg.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grodick MA, Muren NB, Barton JK. DNA charge transport within the cell. Biochemistry. 2015;54:962–973. doi: 10.1021/bi501520w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livshits GI, Stern A, Rotem D, Borovok N, Eidelshtein G, Migliore A, Penzo E, Wind SJ, Di Felice R, Skourtis SS, et al. Long-range charge transport in single G-quadruplex DNA molecules. Nat Nanotechnol. 2014;9:1040–1046. doi: 10.1038/nnano.2014.246. [DOI] [PubMed] [Google Scholar]