

Abstract
We aimed to investigate the molecular characteristics of Korean breast cancer. A cDNA microarray study (>42k clones) was performed on 69 breast cancers and three normal breast tissues. The subjects had a high percentage of HER-2 expression, hormone receptor negativity, and young onset. Molecular subtypes according to gene expression profiles were determined and their correlations to the clinicopathologic characteristics and patients outcome were analyzed. The tumors were subdivided into luminal-, normal breast-like, ERBB2+, and basal-like subtypes according to the correlations to the previously described intrinsic genes and five centroids. Only a few tumors were highly correlated to the luminal B and normal-like centroids. The high grade tumors with high p53 and Ki-67 were found more commonly in non-luminal tumors. Distant recurrence-free survival was worse in ERBB2+ and basal-like subgroups than luminal tumors. In an unsupervised clustering with 864 genes, many interesting gene clusters were observed, some of which had not been previously described. Although the Korean breast cancers showed generally similar molecular phenotypes as Western studies, some distinct gene expression patterns and their association to clinical outcomes were observed.
Keywords: breast neoplasms, gene expression profiling, Asian, race, prognosis
Introduction
It is known that race/ethnicity can impact molecular pathways of various cancers in human and as a result, make difference in clinical and pathological features (1). Breast cancer in Korean women has distinct characteristics different from Caucasian breast cancer, possibly as a result of genetic differences between the races. The incidence of breast cancer is still low in Korea, similar to most other Asian countries, while breast cancer is the most frequent cancer in women, and its incidence is increasing rapidly (2,3). The age distribution is also different than in western countries: the median age at diagnosis in Korean women is 45 years, ~15 years younger than American women; 9.5–12% of Korean breast cancers develop before the age of 35, which is much higher than in Western countries (4,5). In histologic subtype, lobular carcinoma is relatively rare in Korea. Based on a study from two major hospitals in Korea, almost 90% of all breast cancers are ductal carcinoma, with lobular histology comprising <3% (3). In a molecular marker study, Choi et al (6) reported a higher percentage of Korean breast cancers that overexpress HER-2 by immunohistochemistry (IHC) and fluorescence in situ hybridization (FISH) than Caucausian breast cancers. In another genetic study, the prevalence of BRCA1/2 mutation in Korean breast cancer is comparable with that of Western patients. However, the penetrance appears to be lower than in Caucasians, suggesting the effect of a different genetic factor (7).
Previous microarray studies of mainly Caucasian patients have shown that breast cancers can be classified into molecular subtypes by their gene expression profiles and each subtype shows characteristic clinicopathological features and different outcomes (8–11). Sorlie et al (10) showed the universality of the distinction between basal-like and luminal-like subtypes in two independent data sets comprising different patient populations whose gene expression profiles had been determined using different microarray technology platforms.
In the present study, we analyzed 72 samples of breast cancer and normal breast tissue from ethnically homogeneous Korean patients using cDNA microarrays containing >42,000 clones. We intended to determine whether: i) molecular subtype patterns previously defined in Western studies would be observed in this racially different tumor set, ii) new race-specific tumor subtypes or gene clusters would be found, and iii) subtypes based on gene expression profiles would correlate with clinicopathological phenotypes and disease outcomes.
To address these questions, previously described ‘intrinsic gene’ list and five centroids (10) were used and unsupervised hierarchical clustering was performed. We also analyzed any associations between observed gene expression patterns and clinicopathological data.
Materials and methods
Patients and samples
A total of 69 primary invasive breast cancer, and 3 normal breast tissues from different individuals were studied. They were randomly selected from the tissue archives in Cancer Research Institute, Seoul National University. All tumors were excised between 1996 and 2002 and were histopathologically confirmed. Informed consents, approved by an Institutional Review Board of Seoul National University Hospital (H-0205/091-007), were obtained from all participants before operation. Most patients received adjuvant treatment after surgery, consisting of chemotherapy (84.1%), radiotherapy (46.4%), and endocrine therapy (46.4%). Chemotherapy regimens used were doxorubicin-based regimen ± taxane in 58.6% and six cycles of CMF (cyclophosphamide, methotrexate, and 5-fluorouracil) in 41.4%. 93.8% of ER+ or PR+ patients received tamoxifen. Radiotherapy was done for all breast-conserving cases and for 78.6% of locally advanced breast cancers after mastectomy. Median follow-up time for survival analysis was 69 months. Patient age at diagnosis, histologic subtypes, tumor size, lymph node status, histological grade (Scarff-Bloom-Richardson classification), and nuclear grade (Black’s nuclear grade) were reviewed. IHC study was performed to determine expression of the following tumor markers: estrogen receptor (ER), progesterone receptor (PR), HER-2, p53 and Ki-67. The primary antibodies used, staining and scoring methods, and cut-off values were previously described (12).
Tissue samples were frozen in liquid nitrogen within 20 min following surgical devascularization and stored at −80°C. All tumor specimens contained >50% tumor cells.
RNA preparation, amplification, labeling, hybridization, and imaging
Total RNA was isolated from primary tumor tissue using TRIzol solution (Invitrogen, Carlsbad, CA). Briefly, the RNA pellet was dissolved in diethylpyrocarbonate (DEPC)-treated H2O to give a concentration in the range 0.5–1.0 μg/μl and stored at −70°C. The quantity and quality of the RNA preparations were determined by absorbance at 260 and 280 nm. Sample preparation and RNA extraction was done in the Cancer Research Institute, Seoul National University College of Medicine and transferred to Stanford University for expression profiling. Total RNA concentration was determined using a GeneSpec I spectrophotometer (Hitachi, Yokohama, Japan), and RNA integrity was assessed using a 2100 Bio-analyzer (Agilent Technologies, Palo Alto, CA). Amplification of total RNA was performed using an optimized protocol described previously (13). Amplified tumor RNA was labeled by Cy5 and amplified RNA from Universal Human Reference total RNA (Stratagene, La Jolla, CA) was labeled by Cy3. The labeling and hybridization of amplified RNA to cDNA microarrays containing >42,000 elements, was performed as described previously (13). Complete experimental protocols can be found at http://www.stanford.edu/group/sjeffreylab/. Details of the normalization of the intensity levels can be found at http://genomewww5.stanford.edu/help/results_normalization.shtml.
The arrays with hybridized probes were scanned using an Axon scanner. The scanned images were analyzed first using GenePix Pro3.0 software (Axon Instruments, Foster City, CA), and spots of poor quality determined by visual inspection were removed from further analysis. The resulting data collected from each array was submitted to the Stanford Microarray Database (SMD; http://genome-www5.stanford.edu/microarray/SMD) (14,15).
Microarray data analysis
Microarray data quality filters and data transformations
All expression data were retrieved from the Stanford Microarray Database as log ratio data with the same stringency filter: signal intensity 1.5 over background in both Cy5 and Cy3 channels, or spot regression correlation >0.6. Two datasets were retrieved and analyzed: the tumor samples under study, and the public expression data that generated the intrinsic list of genes and the five tumor subtype centroids for breast cancer in the study of Sorlie et al (10). Clones comprising the intrinsic list of genes were retained if they had data on 70% of the arrays used for computing the five centroids and missing values in this smaller data file were imputed using a k-nearest neighbors algorithm (16,17). Microarray print batches partitioned the data and an ANOVA batch correction was performed on each batch. The entire batch-corrected data was then centered by subtracting from each gene (row) the median value for the gene. The arrays that contributed to the construction of centroids, as obtained in Sorlie et al (10) were then grouped together according to their centroids, and the five centroids were reconstructed by taking the average gene data for the arrays in each centroids group. Microarray data for the new tumor samples under study was retrieved, and individual clones were retained if they had data for at least 80% of the arrays. Missing values in the remaining data file were imputed using the k-nearest neighbors algorithm. The data for each gene was then corrected by subtracting the mean of the data for the gene. This ANOVA correction was introduced to diminish the protocol batch distinction between the new data and the centroids data.
Computing the molecular subtype assignment
The Pearson’s correlations of samples to each of the five tumor subtype centroids was computed along the genes in the intrinsic list, and used to assign the sample to one of the five tumor subtypes. For each sample, the difference between the highest and second highest correlations to centroids was also computed. Highest correlations to these centroids were also computed for virtual tumor sample arrays obtained by random permutations of the data for each gene. The 95th percentile of all the second best correlations of samples to centroids, and the 95th percentile of the highest correlations to centroids of the virtual samples obtained by random permutations are seen in Fig. 1.
Figure 1.

Correlation to five centroids and clustering with intrinsic genes. Top, dendrogram of hierarchical clustering analysis of the 69 primary tumors and three normal tissues by using 275 intrinsic clone set. Sixty-five of 72 samples were categorized into one of the five subtypes of breast carcinomas identified previously based on their Pearson’s r. The branches are colored as basal-like subtype in red, ERBB2+ subtype in pink, normal-like subtype in green, luminal A subtype in dark blue, and luminal B subtype in light blue. Seven samples colored in gray showed correlation below threshold. The purple colored sample IDs are correspond to low confidence tumors of which difference between 1st and 2nd high correlation (r) with centroids is <0.1. Bottom, Pearson’s correlation coefficients between each of the 72 samples and the centroid with highest correlation to each sample of the five sets of centroids derived from 122 breast samples published previously (10). The line for 95 percentile of second highest correlation coefficient to centroids of all samples (gray dots) and threshold line estimated by random permutations (black dots) are shown. *This figure was published as a Supplementary figure in Han W, et al: DNA copy number alterations and expression of relevant genes in triple-negative breast cancer. Genes Chromosomes Cancer 47: 490–499, 2008. The figure is used here with permission of Wiley-Liss, Inc., a subsidiary of John Wiley & Sons, Inc.
Filtering and unsupervised clustering
In parallel, we computed the unsupervised hierarchical clustering of the cohort using a multi-step filter for identifying clones which distinguish tumors from one another. For each clone, the mean expression level was computed; clones passed a stringent and less-stringent filter, respectively, if their expression deviated from the mean by at least log24 and log23 respectively, in at least 3 arrays. Clones (2659) passed the stringent filter, and 6510 clones were identified by the less stringent filter. To identify clones that were able to distinguish well among distinct tumors, as well as occurring in highly correlated clusters, the correlation coefficients of each of the 6510 clones to each of the 2659 clones were computed, as well as the top and bottom 0.1 percentiles of these correlation coefficients for each clone. Only clones for which the top or bottom 0.1 percentiles of these correlation coefficients were >0.7 were retained for hierarchichal clustering tumors. There were 864 clones representing 677 distinct UniGene cluster IDs passed the filter. For clustering, fractional weights were assigned to clones associated to same unique UniGene cluster ID.
Survival analysis
Survival estimates were computed using the Kaplan-Meier method and differences between survival times were assessed using the log-rank test. SPSS version 17.0 (Chicago, IL, USA) was used for statistical analysis.
Results
Patient characteristics
Compared with the consecutive patients in SNUH during the same period, the study subjects had higher T and N stage, higher grade, more HER-2 expression (IHC), more hormone receptor negativity, and higher proliferation index (Ki-67) and were younger (Table I). Because of technical details related to tumor harvesting, the tumors tended to be larger and of a more advanced stage.
Table I.
Characteristics of 69 breast cancer subjects compared with total breast cancer cases in Seoul National University Hospital during the same period.
| No. of subjects (%) | No. (%) in SNUH (n=2428)a | |
|---|---|---|
| Age distribution | ||
| <30 | 3 (4.3) | 67 (2.8) |
| 30–39 | 22 (31.9) | 473 (19.5) |
| 40–49 | 24 (34.8) | 990 (40.8) |
| 50–59 | 12 (17.4) | 630 (25.9) |
| ≥60 | 8 (11.6) | 268 (11.0) |
| Median age at diagnosis, years (range) | 43 (25–86) | 46 (20–89) |
| Histological subtype | ||
| Ductal | 65 (94.2) | 2169 (89.3) |
| Medullary | 4 (5.8) | 31 (1.3) |
| Histological grade | ||
| 1 or 2 | 30 (43.5) | 1126 (46.4) |
| 3 | 33 (47.8) | 749 (30.8) |
| Nuclear grade | ||
| 1 or 2 | 28 (40.6) | 1289 (53.1) |
| 3 | 39 (56.5) | 768 (31.6) |
| T stage | ||
| T1 | 16 (23.2) | 1163 (47.9) |
| T2 | 46 (66.7) | 1119 (46.1) |
| T3 | 7 (10.1) | 118 (4.9) |
| T4 | 0 (0) | 28 (1.2) |
| Lymph node status | ||
| N0 | 24 (34.8) | 1463 (60.3) |
| N1 | 20 (29.0) | 571 (23.5) |
| N2 | 12 (17.4) | 230 (9.5) |
| N3 | 13 (18.8) | 164 (6.8) |
| 2 (2.9) | 86 (3.5) | |
| ER and PR status | ||
| ER(+) or PR(+) | 32 (47.1) | 1404 (61.2) |
| ER(−) and PR(−) | 36 (52.9) | 890 (38.8) |
| HER-2 (IHC) | ||
| Negative (0 or 1+) | 32 (47.1) | 1051 (51.7) |
| Positive (2+ or 3+) | 36 (52.9) | 983 (48.3) |
| P53 | ||
| <10% | 38 (56.8) | |
| ≥10 | 29 (43.2) | |
| Ki-67 | ||
| <10% | 26 (46.4) | |
| ≥10, <50% | 25 (44.6) | |
| ≥50% | 5 (8.9) | |
Number of consecutive patients operated on primary invasive breast cancer from January 1996 to June 2003.
Hierarchical clustering and correlation to the five centroids
We performed Pearson’s correlation by using the five sets of centroids defined by Sorlie et al (10). These sets of centroids consist of the average expression of the 500 intrinsic genes corresponding to each of the five subtypes. The Pearson’s correlation coefficients between the expression ratio of intrinsic genes in our 69 carcinomas and three non-malignant breast samples, and the five sets of centroids were calculated. Sixty-five of 72 samples were assigned to a subtype by the highest r, confirming the existence of the five centroids also in this set of tumors. The seven tumors that could not be classified using an r threshold of 0.15 (determined by random permutation of gene expression values) were located near the normal cluster or ERBB2 overexpressing cluster. The basal subtype had the highest correlation with the centroid compared with other subtypes, suggesting a highly consistent gene expression pattern associated with basal subtype tumors also in this Korean data set. All the four medullary carcinomas belonged to this basal subtype.
In a hierarchical clustering with 275 intrinsic genes, the major distinction seen was between the tumors showing high expression of luminal epithelial specific genes including ESR1 and all other tumors showing low or no expression of these genes. Samples tended to cluster based on their correlation to the centroids of the subtypes (Fig. 1).
Of the five subtypes, luminal A, basal-like, and ERBB2+ subtype clusters were well-defined. However, the three tumors showing highest correlation with luminal B centroids were scattered in luminal A and ERBB2+ clusters. The typically expressed genes in luminal B cluster in the study of Sorlie et al (10), GGH, LAPTM4B, PRDX4 and SQLE, did not produce any cluster at all in this study. While significant number of tumors was clustered with normal tissues, only one tumor was highly correlated with normal centroid. Two tumors that did not have high correlation with any of five centroids and three luminal A tumors were clustered with this normal centroid tumor and normal tissues.
Association with clinicopathologic data and patient prognosis
The clinicopathologic data of tumors are shown in Fig. 2. The lymph node status (positive or negative) was not different between molecular subtypes (p>0.05), while luminal/normal showed higher ER/PR positivity than ERBB2+/basal subtypes (85.3 and 8.8%, respectively, p<0.001). The proportion of nuclear grade 3 tumors were significantly higher in ERBB2/basal compared to luminal/normal subtypes (76.5 and 37.1%, respectively, p<0.001). Tumors with high p53 (≥10%) and high proliferating tumors with Ki-67 ≥10% were also more in ERBB2+/basal than luminal/normal tumors (55.9 vs. 30.3% for p53, p=0.035; 76.9 vs. 33.3% for Ki-67, p=0.001). The percentage of tumors with very high Ki-67 expression (≥50%) and p53 mutation score (≥50%) were higher in basal subtype than ERBB2+ tumors (25 vs. 10% for Ki-67; 61.1 vs. 30.8% for p53). HER-2 positivity with IHC was found frequently in luminal (50%) as well as in ERBB2+ (87.5%) subtypes, but rarely in basal tumors (27.8%).
Figure 2.
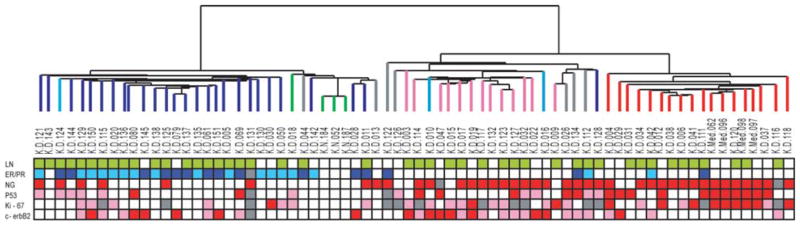
Dendrogram of 69 breast cancer specimens and three normal tissues analyzed by hierarchical clustering using intrinsic gene set (top), and pathological and IHC parameters of each tumor samples (bottom). The color scheme of dendrogram is identical to that of Fig. 1. LN, lymph node status of each tumor. Green squares are for positive lymph node metastasis and blanks for negative lymph node; ER/PR, dark blue squares are for tumors with both ER and PR positive by IHC, light blue squares for tumors positive for only one of ER or PR, and blanks for tumors negative for both ER and PR; NG, nuclear grade. Red squares are for grade 3 tumors and blanks for grade 1 or 2; P53 and Ki-67, red squares are for tumors with percentage of positively stained cells in IHC ≥50%, pink squares for ≥10% and <50%, and blanks for <10%; HER-2, red squares are for HER-2 score of 3+ by IHC, pink squares are for 2+, and blank for 1+ or 0. All the gray squares shown in this figures are for missing data of each variable.
In a univariate Kaplan-Meier analysis for the distant metastasis-free survival, patients with luminal A type tumors showed considerably better outcome, whereas the basal and ERBB2+ subgroups had worse prognosis (p=0.01) (Fig. 3A). This is consistent with the results of previous studies (9–11).
Figure 3.

Kaplan-Meier analysis of disease outcome. (A) Time to development of distant metastasis of the Korean patients stratified according to the molecular subtypes defined by correlation to the five centroids as shown in Fig. 1. Curves for luminal B and normal-like subtypes were not shown here because of the small number of cases assigned to those subtypes. (B) Distant recurrence-free survival curves showing the excellent outcome of the tumors which were low-confidence and clustered with normal tissues in the intrinsic gene analysis, and the worst outcome of the tumors which were low-confidence but not clustered with normal tissues. Patients with distant metastasis at the time of diagnosis (operation) were excluded from all the survival analysis.
Another interesting finding is that ‘low-confidence tumors’ showed distinct survival patterns. Low-confidence tumors were those that had no exclusive correlation with a specific centroid. We arbitrarily defined low-confidence as when the difference between 1st and 2nd high correlation with centroids is <0.1. There were 18 such tumors in this data set. Five of them were clustered with normal tissues and showed excellent outcome without recurrence during follow-up. On the other hand, 13 non-normal clustered low-confidence tumors had the worst prognoses (p<0.01) (Fig. 3B).
Unsupervised hierarchical clustering
With the 864 clones that passed a gene filtering based on analysis of variance for highly correlated groups of genes, the 69 carcinomas and three normal breast tissues were analyzed by hierarchical clustering (Fig. 4). We identified distinct gene clusters consisting of highly correlated clones. The uppermost gene cluster in Fig. 4B-a showed high expression in normal tissues and the tumors clustered with them but low expression in most of the other tumors. The genes included in this cluster were early B-cell factor, PDGF receptor β, LIM domain binding 2, IGF1, and fibulin 1. Caveolin 1 and 2 in this cluster (Fig. 4B-a) have been suggested to act as a tumor suppressor in breast cancer (18–20). On the other hand, Sotiriou et al (11) found caveolins in their ‘basal 2 subgroup’. The second gene cluster is of histones (Fig. 4B-b). Histones were mainly overexpressed in luminal tumors (blue colored in dendrogram) but not in normal breast-like tumors. Some tumors in ERBB2+ cluster also expressed histones. The cluster next to this is the famous large cluster involving luminal-enriched genes (Fig. 4B-c). It includes ESR1 (ERα), GATA3, BCMP11, SCUBE2, RERG, IGF-1 receptor, and STATIP1. The representative genes that Perou et al (8) described to be in this cluster, X-box binding protein 1, trefoil factor 3, hepatocyte nuclear factor 3 and LIV-1 were filtered. The well-known luminal epithelial markers, keratins 8 and 18 were generally expressed in luminal-and normal-like tumors but not in normal tissues (Fig. 4B-d). They also expressed in about a half of the ERBB2 over-expressing tumors. The RERG low-expressing luminal-like tumors were found to make separate cluster in which the three ERBB2+ luminal tumors were included (Fig. 4B-c′). Finlin et al (21) reported that RERG is an inhibitor of growth of MCF-7 cells and tumorigenesis in nude mice, and suggested that loss of RERG expression may contribute to tumor growth. The next gene cluster is ‘stromal genes’, like SPARC, lumican, various collagens, and fibrillin 1 (Fig. 4B-e). Their expression was very low in normal tissues and basal-like tumors, especially in the medullary tumors. The next is the cluster of genes amplified with ERBB2 on chromosome 17q11–21 (Fig. 4B-f).
Figure 4.

Unsupervised hierarchical clustering of 69 tumors and 3 non-malignant tissues using the clones that passed the filtering described in Materials and methods. (A) A scaled-down representation of the entire cluster of 864 clones and 72 tissue samples based on similarities in gene expression. The colors of the representative gene clusters in the left are correspond to the colors of the lowercase letters shown in the left of B. (B) Top, dendrogram representing similarities in the expression patterns between experimental samples. The dendrogram further branched into smaller subgroups based on their basal and luminal characteristics, clustering with normal tissues and overexpression of ERBB2 amplicon: normal-like subgroup is shown in green; luminal in dark blue; ERBB2+ in pink; and basal in red. Bottom, (a) genes mainly expressed in normal-like subgroup; (b) histones; (c) luminal cluster including ESR1; (c′) RERG; (d) keratin 8; (e) stromal cluster; (f) ERBB2 amplicon; (g) cell cycle and check-point control (proliferation cluster); (h) interferon-related genes; (i) genes derived from immune cells; (j) TNFSF10 (TRAIL); (k) keratin 5/6 and 17; l, EGFR; (m) genes up-regulated in normal tissues and basal tumors, not in ERBB2+ subgroup; (n) genes exclusively up-regulated in basal tumors.
A large gene cluster composed of even and highly correlated genes mainly involved in cell cycle and check-point control was observed (Fig. 4B-g). They were highest in basal-like tumors and lowest in normal tissues and normal-like tumors. CCNB2, CDCs, and some serine/threonine kinases (STK6, AURKB, PLK, NEK2, MELK, and BUB1) were included in this category.
A cluster of genes regulated by the interferon pathway was also seen in this analysis showing substantial variation in expression among the tumors, as was observed in a smaller set of breast tumors (22) (Fig. 4B-h). Close neighbor of interferon cluster is of the genes from lymphocytes and macrophages (Fig. 4B-i). These genes possibly from immune cells were highest in basal-like tumors, moderate in ERBB2+ tumors, and lowest in normal-like and luminal-like tumors. Although STAT1 was included in this cluster, the expression pattern was similar with interferon-related genes, which is consistent with previous result (8). Two basal-like tumors, K-D-111 and 118 showed very low expression of genes in this big cluster, probably making them apart from the main basal-like tumors. Small cluster of genes including TNFSF10 (TRAIL) showed distinct expression pattern (Fig. 4B-j). They seemed to be a part of genes from immune cells.
Basal-like tumors in this analysis were quite identical to the tumors identified by the correlation to the five centroids and intrinsic genes except for one tumor (K-D-032). The remarkable finding in this data set is that the well-known basal keratins 5, 6, and 17 showed high expression in some of ERBB2+ tumors as well (Fig. 4B-k). EGFR, another candidate for the marker of basal-like tumors (23), also showed non-specific expression pattern within ER-negative tumors (Fig. 4B-l). Meanwhile, a gene cluster involving SFRP1 was only highly expressed in basal-like tumors (Fig. 4B-m) and normal tissues. We found a novel gene cluster increased exclusively in basal-like tumors. It includes SKP2, lipin 1, forkhead box C1, and FTHFSDC1 (C-1-tetrahydrofolate synthase).
Discussion
This is one of the few studies on molecular phenotype of ethnically homogeneous breast cancer other than in Caucasians, and the first and only study of Korean breast cancer. It is noteworthy in this study that the proportion of ER-negative tumors, especially the ERBB2+ tumors were much higher than in previous Western studies. As shown in Table I, the proportion of ER negative and HER-2 positive tumors with IHC in this study subjects was even higher than the average incidence of Seoul National University Hospital. This case selection could reveal the characteristic gene expression pattern of Korean breast cancer, and also could disclose the hidden gene expression profiles of ER-negative and ERBB2+ tumors that had not been observed in Western data, although it is possible that they may not be the intrinsic feature of Korean breast cancer.
The major factor discriminating molecular profile, clinicopathological parameters, and the disease outcome was estrogen receptor positivity. It has been observed in many previous studies (9,23–25), and means that ER/PR-positive cancers are biologically quite different from ER/PR negative cancers across races.
In general, the gene expression pattern of this Korean tumor set was not different from previous Western studies in that the distinction of at least three major molecular subtypes, such as luminal, basal, and ERBB2+ were also evident in this study. However, there were some different features from Western data in our study. The luminal B tumors were very few and did not make a cluster and the genes relevant to them were either filtered or did not show any significant difference among the luminal tumors. Sorlie et al (10) found substantial numbers of luminal B tumors in the Stanford-Norway patient data set and even in the van’t Veer et al data set (24) performed on a different array platform. Sotiriou et al (11) also found a subtype of luminal tumors with worse outcome in the unsupervised clustering. Chang et al (26) showed (in the supporting information of their report) that number of tumors classified as Luminal A:B = 47:45 with the centroid analysis similar to ours.
A small number of normal-like subtype in centroid analysis is another different feature from previous data. Considering that a significant number of samples clustered with normal tissue in cluster analysis using intrinsic genes and unsupervised clustering, it is possible that the genetic profile of normal-like tumor is different from Western study and the normal-like centroid may not be applicable across races.
Basal-like subtype breast cancer has been recognized as most homogeneous and discrete molecular category and is characterized by aggressive biologic behavior (9–11). In these Korean tumors, basal-like subtype with such characteristics was evident.
The well known basal keratins 5, 6, and 17 were highly expressed in a considerable number of ERBB2 overexpressing tumors as well as in basal subtype tumors. This finding is contradictory to the previous idea that basal keratins are the specific marker for basal subtype tumors never expressing ERBB2 (27,28). As shown in Fig. 4, ERBB2+ tumors could be divided into basal keratin expressing and luminal keratin expressing tumors.
A most remarkable finding in this study is the presence of the low-confidence tumors with the worst or the best outcome. The low-confidence tumors would not have dominant expression of genes of any one subtype exclusively. We hypothesized that the low-confidence tumors not clustered with normal tissues might be most undifferentiated pluripotent tumors in terms of gene expression. They showed relatively even distribution across the dendrogram in the unsupervised clustering. In centroid analysis for their 1st correlation, two of them were luminal A, three luminal B, two basal, and six were ERBB2+ subtype, showing that they did not converge into a specific subtype. With IHC, five of the 13 tumors were ER+/PR+. On the other hand, the five low-confidence tumors clustered with normal tissues have their 1st correlation to luminal A centroid in four, or normal centroid in one. In our hypothesis, the undifferentiated pluripotent tumors express mixtures of marker genes of different subtypes at the same time, and they differentiate into tumors with dominant marker genes, such as either luminal, basal, or ERBB2+ genes. In further differentiated tumors they express both luminal and non-luminal genes while the luminal profile is dominant. The aggressiveness of tumor cells diminishes along with the differentiation process. It is clinically very important and helpful if we could distinguish the most violent tumor group using their gene expression profile. Prediction of good prognosis tumor is also useful in making treatment decision. Kun et al (29) first described low-confidence ER+ tumors exhibiting significantly worse survival compared with ‘high-confidence’ ER+ counterparts. They indicated that ERBB2 may contribute to the aggressive behavior of the low-confidence subtype. Their results partly support our theory, although they did not consider ERBB2+ tumors as independent subtype. Our hypothesis is also consistent with the recent concept of cancer stem cell in breast carcinogenesis. Dontu et al (30) exhibited that type 1 cells originated from stem cells, and are mainly ER-negative and histologically undifferentiated. These cells are expressing markers of both luminal epithelial and myoepithelial cells and are more aggressive.
The major limitation of this study is that the number of cases was not large enough to show clearly the small subgroups which we proposed as the novel finding and characteristics in Korean tumor set. The patients’ composition also can be a bias causing factor. It is possible that the significant findings in this study might be simply a result of eccentric clinicopathologic characteristics of the study subjects.
In conclusion, our cDNA microarray data showed that the general molecular profiles and their correlation to clinicopathological phenotypes are not different across races. However, we found some significant novel gene expression patterns and association with clinical outcomes in these ethnically homogeneous Korean breast cancers. Much of the remarkable findings are suspected to be secondary to the characteristic composition of samples such as, high ER(−) and high HER-2(+). Further study will follow to elucidate whether these findings were ethnicity-specific molecular phenotype pattern or they resulted from different clinicopathology that can be generalized to all breast cancer.
Acknowledgments
This work was supported by a grant from the Seoul National University Hospital Research Fund (09-2006-006-0), and a grant from the Korea Health 21 R&D Project, Ministry of Health & Welfare, R.O.K (01-PJ3-PG6-01GN07-0004); by grants NIH K01 HG00030 (MN), NIH U01 CA85129 (SSJ), by funds from the California Breast Cancer Research Program of the University of California, grant no. 10 EB-1086 (SSJ), and by funds from the John and Marva Warnock Cancer Research Fund, and the Rachleff family fund.
References
- 1.Wiencke JK. Impact of race/ethnicity on molecular pathways in human cancer. Nat Rev Cancer. 2004;4:79–84. doi: 10.1038/nrc1257. [DOI] [PubMed] [Google Scholar]
- 2.Ahn SH. Clinical characteristics of breast cancer patients in Korea in 2000. Arch Surg. 2004;139:27–31. doi: 10.1001/archsurg.139.1.27. [DOI] [PubMed] [Google Scholar]
- 3.Yoo KY, Kang D, Park SK, et al. Epidemiology of breast cancer in Korea: occurrence, high-risk groups, and prevention. J Korean Med Sci. 2002;17:1–6. doi: 10.3346/jkms.2002.17.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Han W, Kim SW, Park IA, et al. Young age: an independent risk factor for disease-free survival in women with operable breast cancer. BMC Cancer. 2004;4:82. doi: 10.1186/1471-2407-4-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ahn SH, Son BH, Kim SW, et al. Poor outcome of hormone receptor-positive breast cancer at very young age is due to tamoxifen resistance: nationwide survival data in Korea - a report from the korean breast cancer society. J Clin Oncol. 2007;25:2360–2368. doi: 10.1200/JCO.2006.10.3754. [DOI] [PubMed] [Google Scholar]
- 6.Choi DH, Shin DB, Lee MH, et al. A comparison of five immunohistochemical biomarkers and her-2/neu gene amplification by fluorescence in situ hybridization in white and Korean patients with early-onset breast carcinoma. Cancer. 2003;98:1587–1595. doi: 10.1002/cncr.11703. [DOI] [PubMed] [Google Scholar]
- 7.Choi DH, Lee MH, Bale AE, Carter D, Haffty BG. Incidence of BRCA1 and BRCA2 mutations in young Korean breast cancer patients. J Clin Oncol. 2004;22:1638–1645. doi: 10.1200/JCO.2004.04.179. [DOI] [PubMed] [Google Scholar]
- 8.Perou CM, Sorlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature. 2000;406:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 9.Sorlie T, Perou CM, Tibshirani R, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA. 2001;98:10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sorlie T, Tibshirani R, Parker J, et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci USA. 2003;100:8418–8423. doi: 10.1073/pnas.0932692100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sotiriou C, Neo SY, McShane LM, et al. Breast cancer classification and prognosis based on gene expression profiles from a population-based study. Proc Natl Acad Sci USA. 2003;100:10393–10398. doi: 10.1073/pnas.1732912100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jung SY, Han W, Lee JW, et al. Ki-67 expression gives additional prognostic information on St. Gallen 2007 and Adjuvant! Online risk categories in early breast cancer. Ann Surg Oncol. 2009;16:1112–1121. doi: 10.1245/s10434-009-0334-7. [DOI] [PubMed] [Google Scholar]
- 13.Zhao H, Hastie T, Whitfield ML, Borresen-Dale AL, Jeffrey SS. Optimization and evaluation of T7 based RNA linear amplification protocols for cDNA microarray analysis. BMC Genomics. 2002;3:31. doi: 10.1186/1471-2164-3-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gollub J, Ball CA, Binkley G, et al. The Stanford Microarray Database: data access and quality assessment tools. Nucleic Acids Res. 2003;31:94–96. doi: 10.1093/nar/gkg078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sherlock G, Hernandez-Boussard T, Kasarskis A, et al. The Stanford Microarray Database. Nucleic Acids Res. 2001;29:152–155. doi: 10.1093/nar/29.1.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tibshirani R, Hastie T, Narasimhan B, Chu G. Diagnosis of multiple cancer types by shrunken centroids of gene expression. Proc Natl Acad Sci USA. 2002;99:6567–6572. doi: 10.1073/pnas.082099299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Troyanskaya O, Cantor M, Sherlock G, et al. Missing value estimation methods for DNA microarrays. Bioinformatics. 2001;17:520–525. doi: 10.1093/bioinformatics/17.6.520. [DOI] [PubMed] [Google Scholar]
- 18.Sagara Y, Mimori K, Yoshinaga K, et al. Clinical significance of caveolin-1, caveolin-2 and her2/neu mRNA expression in human breast cancer. Br J Cancer. 2004;91:959–965. doi: 10.1038/sj.bjc.6602029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sloan EK, Stanley KL, Anderson RL. Caveolin-1 inhibits breast cancer growth and metastasis. Oncogene. 2004;23:7893–7897. doi: 10.1038/sj.onc.1208062. [DOI] [PubMed] [Google Scholar]
- 20.Williams TM, Cheung MW, Park DS, et al. Loss of caveolin-1 gene expression accelerates the development of dysplastic mammary lesions in tumor-prone transgenic mice. Mol Biol Cell. 2003;14:1027–1042. doi: 10.1091/mbc.E02-08-0503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Finlin BS, Gau CL, Murphy GA, et al. Rerg is a novel ras-related, estrogen-regulated and growth-inhibitory gene in breast cancer. J Biol Chem. 2001;276:42259–42267. doi: 10.1074/jbc.M105888200. [DOI] [PubMed] [Google Scholar]
- 22.Perou CM, Jeffrey SS, van de Rijn M, et al. Distinctive gene expression patterns in human mammary epithelial cells and breast cancers. Proc Natl Acad Sci USA. 1999;96:9212–9217. doi: 10.1073/pnas.96.16.9212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gruvberger S, Ringner M, Chen Y, et al. Estrogen receptor status in breast cancer is associated with remarkably distinct gene expression patterns. Cancer Res. 2001;61:5979–5984. [PubMed] [Google Scholar]
- 24.Van’t Veer LJ, Dai H, van de Vijver MJ, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–536. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- 25.West M, Blanchette C, Dressman H, et al. Predicting the clinical status of human breast cancer by using gene expression profiles. Proc Natl Acad Sci USA. 2001;98:11462–11467. doi: 10.1073/pnas.201162998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chang HY, Nuyten DS, Sneddon JB, et al. Robustness, scalability, and integration of a wound-response gene expression signature in predicting breast cancer survival. Proc Natl Acad Sci USA. 2005;102:3738–3743. doi: 10.1073/pnas.0409462102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nielsen TO, Hsu FD, Jensen K, et al. Immunohistochemical and clinical characterization of the basal-like subtype of invasive breast carcinoma. Clin Cancer Res. 2004;10:5367–5374. doi: 10.1158/1078-0432.CCR-04-0220. [DOI] [PubMed] [Google Scholar]
- 28.Van de Rijn M, Perou CM, Tibshirani R, et al. Expression of cytokeratins 17 and 5 identifies a group of breast carcinomas with poor clinical outcome. Am J Pathol. 2002;161:1991–1996. doi: 10.1016/S0002-9440(10)64476-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kun Y, How LC, Hoon TP, et al. Classifying the estrogen receptor status of breast cancers by expression profiles reveals a poor prognosis subpopulation exhibiting high expression of the ERBB2 receptor. Hum Mol Genet. 2003;12:3245–3258. doi: 10.1093/hmg/ddg347. [DOI] [PubMed] [Google Scholar]
- 30.Dontu G, El-Ashry D, Wicha MS. Breast cancer, stem/progenitor cells and the estrogen receptor. Trends Endocrinol Metab. 2004;15:193–197. doi: 10.1016/j.tem.2004.05.011. [DOI] [PubMed] [Google Scholar]