

Abstract
Neurofibromatosis type 1 (NF1) is a common autosomal dominant neurologic condition characterized by significant clinical heterogeneity, ranging from malignant cancers to cognitive deficits. Recent studies have begun to reveal rare genotype–phenotype correlations, suggesting that the specific germline NF1 gene mutation may be one factor underlying disease heterogeneity. The purpose of this study was to define the impact of the germline NF1 gene mutation on brain neurofibromin function relevant to learning. Herein, we employ human NF1-patient primary skin fibroblasts, induced pluripotent stem cells and derivative neural progenitor cells (NPCs) to demonstrate that NF1 germline mutations have dramatic effects on neurofibromin expression. Moreover, while all NF1-patient NPCs exhibit increased RAS activation and reduced cyclic AMP generation, there was a neurofibromin dose-dependent reduction in dopamine (DA) levels. Additionally, we leveraged two complementary Nf1 genetically-engineered mouse strains in which hippocampal-based learning and memory is DA-dependent to establish that neuronal DA levels and signaling as well as mouse spatial learning are controlled in an Nf1 gene dose-dependent manner. Collectively, this is the first demonstration that different germline NF1 gene mutations differentially dictate neurofibromin function in the brain.
Introduction
Neurofibromatosis type 1 (NF1) is a monogenic neurodevelopmental disorder affecting ∼1 in 2500 individuals worldwide (1). While NF1 is typically regarded as a cancer predisposition syndrome, affected individuals can manifest a wide variety of clinical features involving the central and peripheral nervous systems, ranging from benign cutaneous neurofibromas and malignant nerve sheath tumors to benign and malignant gliomas (1). Importantly, >50% of individuals with NF1 exhibit cognitive deficits, severely affecting their scholastic abilities and impacting on their overall quality of life (2). These cognitive impairments include specific learning disabilities, attention deficits, and visuospatial learning/memory problems (2,3).
Despite their frequency, the specific cognitive symptoms as well as their severity vary greatly among individuals with NF1. Some children with NF1 may have significant problems with reading or mathematic achievement (4), while others exhibit deficits in visual perception or response inhibition (5). In addition, there may be multiple cellular and molecular etiologies for these cognitive delays, which may partly explain why a small number of children respond to targeted therapeutic interventions. In this regard, neurofibromin functions as a negative regulator of RAS as well as a positive regulator of DA homeostasis. In the brains of Nf1 genetically-engineered mice, high levels of RAS activation and low levels of DA have been reported (6,7). Correction of these abnormalities using Lovastatin (RAS inactivation) (8) or DA uptake blockers (methylphenidate) (7) ameliorates the spatial learning and memory deficits in these mouse strains. However, clinical trials using these agents have had limited effectiveness when treating potentially heterogeneous cohorts of children with NF1-associated cognitive problems (9–11).
A second challenge inherent to the management of children with NF1 is the lack of predictive markers available to identify those individuals at higher risk for specific morbidities. Whereas tumor (neurofibroma, glioma) development requires bi-allelic NF1 gene inactivation, reflecting the combination of germline and somatic NF1 gene mutations (12,13), a single germline NF1 gene mutation is sufficient for neuronal dysfunction (11,14,15). In this regard, the germline NF1 gene mutation would be predicted to impact most significantly on neural cell types relevant to cognitive and behavioral function. Emerging evidence from NF1 mutation studies has revealed that some specific types of germline NF1 gene mutations may be associated with particular clinical features. For example, individuals from different families harboring the c.2970-2972_delAAT germline NF1 gene mutation do not develop dermal neurofibromas, despite having many other features of NF1 (16). Similarly, individuals with germline c.5425C>T NF1 gene mutations exhibit mild symptoms of NF1 without neurofibromas or optic gliomas (17). In addition, individuals with NF1 genomic microdeletions are prone to early neurofibroma formation, mental retardation and increased cancer incidence (18,19), while those with 5′ end NF1 gene frame shift and premature truncation mutations have an increased incidence of optic gliomas (20). These intriguing population-based studies prompted us to critically examine the molecular basis for potential genotype–phenotype correlations.
Results
We first established a collection of primary fibroblast lines from unrelated male (n = 5) and female (n = 8) patients diagnosed with NF1 using NIH Consensus Development Conference diagnostic criteria (21) as well as from four sex- and age-matched control individuals with no known neurological problems. Analysis of neurofibromin levels revealed two distinct subgroups (Fig. 1A): those with minor reductions (Group 1; <25% following normalization to α-tubulin) and those with >70% reductions (Group 2) in neurofibromin expression. This differential neurofibromin expression pattern was observed using both carboxyl- and amino-terminal neurofibromin antibodies (Supplementary Material, Fig. S1A) and was not related to NF1 RNA expression levels (Supplementary Material, Fig. S1B) nor to patient sex, age or the in vitro growth conditions employed (with or without serum; data not shown). In all NF1-patient fibroblasts, there was increased RAS activity (Fig. 1B) relative to control patient fibroblasts.
Figure 1.

Germline NF1 gene mutations result in different levels of neurofibromin expression in NF1-patient fibroblasts. (A) Neurofibromin immunoblot of NF1-patient fibroblasts reveals two groups; Group 1 patients have <25% neurofibromin expression (n = 4) and Group 2 patients have >70% reductions in neurofibromin expression (n = 7 shown) compared with sex- and age-matched controls. Tubulin was used as an internal protein loading control. (B) RAS activity is significantly elevated in all NF1-patient fibroblasts. (C) Schematic representation of the NF1 gene with the identified NF1-patient mutations and summary of NF1-patient germline NF1 gene mutations, demographic information and genotyping results. The number of the exon mutated is indicated above the mutation location. Light and dark boxes indicate alternate exons. Horizontal bars represent the length of the predicted transcribed mRNA. TGD, total genomic deletion; FS, frame shift mutation. All data are represented as means ± SEM (*P < 0.0001, one-way ANOVA with Bonferroni post-test).
To determine whether these differences in neurofibromin protein expression reflected the underlying germline NF1 gene mutation, all NF1-patient fibroblast samples were analyzed (Fig. 1C). In these individuals, we conclusively identified 10 distinct mutations. The mutations in Group 1 causing mild reductions in neurofibromin expression were frameshift (NF1-1, NF1-5) or nonsense mutations (NF1-8, NF1-10) and the mutations in Group 2 with more dramatic reductions in neurofibromin expression included nonsense mutations (NF1-4, NF1-7, NF1-9, NF1-11) and large microdeletions of the NF1 locus (NF1-6, NF1-12). Together, these data suggest that there is no apparent correlation between the location or nature of the germline NF1 mutation and the level of neurofibromin expression. Therefore, for further analysis we chose to include data only from two control- and six of the NF1-lines; three from Group 1 and three from Group 2 (Supplementary Material, Fig. S1A).
Leveraging induced pluripotent stem cell (iPSC) technology and integration-free Sendai virus infection, we reprogrammed primary skin fibroblasts into iPSCs (Fig. 2A). Following immunoblot analysis, we demonstrated that the same differential pattern of neurofibromin expression in fibroblasts was observed in the derivative iPSCs (Fig. 2C). Consistent with its established role as negative RAS regulator (22), the levels of active RAS (RAS-GTP) were elevated in all NF1-iPSCs, irrespective of neurofibromin levels (Fig. 2D), similar to our findings in NF1-patient fibroblasts.
Figure 2.

Germline NF1 gene mutations promote differential neurofibromin expression in NF1-patient-derived iPSCs. (A) Immunostaining of the iPSC cultures with stem cell-specific antibodies confirmed the pluripotent nature of the cultured iPSCs. Scale bar, 50 µm. (B) Bright-field imaging of representative iPSCs. Scale bar, 100 µm. (C) Neurofibromin expression is only significantly reduced in group 2 NF1-patient-derived iPSCs. (D) All NF1-patient-derived iPSCs have elevated RAS activity levels (RAS-GTP) relative to control iPSC lines. All data are represented as means ± SEM (*P < 0.0001, one-way ANOVA with Bonferroni post-test).
In order to examine the effect of the germline NF1 gene mutation on central nervous system lineage cells, we directed iPSC differentiation into neural progenitor cells (NPCs) (Fig. 3A). We found that the neurofibromin expression observed similarly matched the patterns found in the parental fibroblasts and iPSCs (Fig. 3B). Previous work from our laboratory revealed that neurofibromin positively controls cyclic AMP (cAMP) in a RAS-dependent manner in brain neurons (15). Consistent with this mechanism, all NF1-NPCs exhibited 2–3-fold elevated RAS-GTP levels (Fig. 3C), irrespective of the germline NF1 gene mutation. Similarly all NF1-NPCs showed significantly reduced cAMP (>50%) levels relative to control NPCs (Fig. 3D).
Figure 3.

Germline NF1 gene mutations promote differential neurofibromin expression in NF1-patient-derived NPCs. (A) Bright field imaging and immunostaining of the NPC cultures with the SMI-312 pan-axonal (neuronal) antibody. Scale bar left panel, 100 µm; Scale bar right panel, 50 µm. (B) Neurofibromin expression is only significantly different in group 2 NF1-patient samples compared with control NPCs. (C) RAS activity is significantly elevated in all NF1-patient NPCs. (D) cAMP is decreased in all NF1-NPCs compared with control NPCs.
In contrast, neurofibromin control of DA homeostasis in the striatum and hippocampus is RAS-independent (7,11). As such, Group 1 NF1-NPCs exhibited <25% reductions in DA levels, while Group 2 NF1-NPCs had ∼75% reductions (Fig. 4A). Importantly, phosphorylation of the DA downstream effector, DA and cAMP-regulated phosphoprotein of 32 kDa (DARPP-32), was only significantly reduced in Group 2 NF1-NPCs (Fig. 4C). Together, these findings establish a strong correlation between neurofibromin expression and DA homeostasis/signaling (Fig. 4B and D).
Figure 4.

Germline NF1 gene mutations regulate DA signaling in a gene dose-dependent manner in NF1-patient NPCs. (A) DA levels are reduced in group 2 NF1-patient samples compared with control NPCs. (B) Correlation of relative neurofibromin expression and DA levels. (C) DARPP-32 activity (phosphorylation) is only reduced in group 2 NF1-patient samples compared with control NPCs. (D) Correlation of relative neurofibromin expression and DARPP-32 phosphorylation. All data are represented as means ± SEM (*P < 0.0001, one-way ANOVA with Bonferroni post-test).
DA signaling is particularly relevant to Nf1 mouse learning and memory. In this regard, we have previously shown that Nf1 mutant mice have exploratory (attention) and learning/memory deficits that are corrected by pharmacologic elevation of brain DA bioavailability (11). While reduced neurofibromin function in heterozygous Nf1 null mice (Nf1+/− mice) maintained on a mixed genetic background is associated with hippocampal-based learning and memory defects (6,23,24), we were unable to convincingly demonstrate any spatial learning or memory deficits in C57BL/6J Nf1+/− mice with reduced neurofibromin expression (Fig. 5A). Motor skills, swimming speeds and exploratory behaviors were also unimpaired in these Nf1+/− mice (Supplementary Material, Fig. S2). This is in striking contrast to our experience with Nf1+/− mice that also harbor Nf1 loss in neuroglial progenitor cells (Nf1flox/−; GFAP-Cre mice, Nf1FMC): Nf1FMC mice exhibit highly reproducible spatial memory and learning defects (7,11).
Figure 5.

Mouse learning is regulated in an Nf1 gene dose-dependent manner. (A) Both Nf1+/− and Nf1+/− (WT) mice preferentially occupy the target quadrant and exhibit equivalent occupancies in the target quadrant during probe trial 2 of the Morris water maze test. Dashed line depicts chance occupancy of a quadrant in the water maze. (B) Neurofibromin expression is reduced by 54% in Nf1+/− mice and by 85% in Nf1FMC mice. (C) Both Nf1FFC and Nf1FF (control) mice preferentially occupy the target quadrant. Null Nf1FFC mice spent significantly less time in the target quadrant relative to controls. (D) Neurofibromin expression is reduced by 50% in Nf1FWC mice and by 80% in Nf1FFC mice compared with controls. (E) Extensive recombination of GFAP-Cre (green) in NeuN+ neurons (red) is depicted by immunofluorescent staining within the CA1, CA2, CA3 and dentate gyrus (DG) of the hippocampus of RosaGREENGFAP-Cre mice. Scale bars, 50 µm. All data are represented as means ± SEM (***P < 0.0001; **P < 0.001; *P < 0.01, one-way/two-way ANOVA with Bonferroni post-test).
Based on the differential effects of NF1-patient germline NF1 gene mutations on neurofibromin expression and DA homeostasis, we hypothesized that differences in neuronal neurofibromin expression may explain the discrepancies observed between the two Nf1 mouse model results. As such, we reasoned that Nf1+/− mice have reduced neurofibromin expression, similar to Group 1 individuals with NF1, whereas Nf1FMC mice have more dramatically decreased neurofibromin expression, sufficient to impair DA levels and signaling, similar to Group 2 individuals with NF1. Consistent with this model, Nf1+/− mice exhibited ∼50% reductions in neurofibromin expression, while Nf1FMC mice had >80% reductions (Fig. 5B).
To establish a clear mechanistic connection between neurofibromin expression and mouse learning and memory, we employed the original GFAP-Cre driver line (25) to generate mice with normal neurofibromin (Nf1flox/flox mice, Nf1F/F), reduced (Nf1flox/wt; GFAP-Cre mice, Nf1FWC), and absent (Nf1flox/flox; GFAP-Cre mice, Nf1FFC) neurofibromin expression in GFAP+ neuroglial progenitors. As predicted, only Nf1FFC mice exhibited impaired performance in probe trial 2 (Fig. 5C). Similar to Nf1FMC mice, there was a gene dose-dependent reduction in neurofibromin expression, with Nf1FMC mice exhibiting >70% reductions (Fig. 5D). To explain the magnitude of decrease in neurofibromin expression in Nf1FMC and Nf1FFC mice, we crossed the GFAP-Cre mice with the RosaGREEN (26) reporter strain (Fig. 5E). Analysis of these RosaGREENGFAP-Cre mice revealed extensive Cre-mediated recombination in neurons throughout the hippocampus, suggesting that the majority of Nf1FMC and Nf1FFC hippocampal neurons exhibit bi-allelic inactivation of the Nf1 gene and harbor markedly reduced neurofibromin expression levels.
Next, we examined DA levels in the hippocampi of all Nf1 mutant mice (Fig. 6A and B). Heterozygous (Nf1+/− and Nf1FWC) and homozygous (Nf1FMC and Nf1FFC) mice exhibited 50 and 65% reductions in DA levels, respectively, correlating with neurofibromin expression (R2 = 0.863) (Fig. 6C). Similarly, DARPP-32 phosphorylation was also reduced in a gene dose-dependent manner in mouse hippocampi (Fig. 6D and E) and showed an even more robust correlation with neurofibromin expression (R2 = 0.966) (Fig. 6F).
Figure 6.

Mouse DA signaling and learning is regulated in an Nf1 gene dose-dependent manner. (A and B) DA is reduced in gene dose-dependent manner in heterozygous (Nf1+/− and Nf1FWC) and null (Nf1FMC and Nf1FFC) mice compared with controls. (C) Correlation between neurofibromin expression and DA levels. (D and E) DARPP-32 phosphorylation is reduced in a neurofibromin dose-dependent manner. (F) Neurofibromin expression highly correlates with pDARPP32 levels in mouse hippocampi. All data are represented as means ± SEM (***P < 0.0001; **P < 0.001; *P < 0.01, one-way ANOVA with Bonferroni post-test).
Finally, to confirm the dependency of Nf1 gene dose on dopaminergic signaling, we selectively targeted dopaminergic cells. To accomplish this, we employed the tyrosine hydroxylase (TH)-Cre driver strain (27) (Fig. 7A). As above, mice were generated with reduced (Nf1TH+/−) or absent (Nf1TH−/−) neurofibromin expression in TH+ neurons. In these experiments, Nf1TH−/−, but not Nf1TH+/− or WT mice demonstrated a defect in spatial learning (Fig. 7B). Consistent with these behavioral results, there was a dose-dependent reduction in neurofibromin expression (Fig. 7C) that correlated with comparable decreases in DA and pDARPP-32 levels (Fig. 7D and E).
Figure 7.
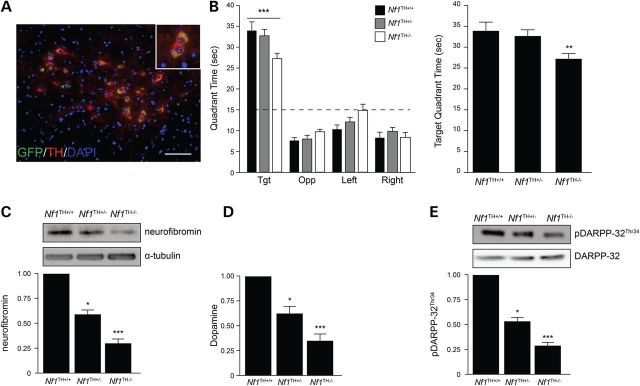
Neurofibromin regulates dopaminergic neurons in a gene dose-dependent manner. (A) TH-Cre (green) recombination occurs exclusively in TH+ dopaminergic neurons (red) in the VTA (top) of Rosa-GREENTH-Cre mice. Scale bar, 50 µm. (B) All Nf1TH mice exhibited preferential occupancy for the target quadrant. Nf1TH−/− mice exhibited reduced quadrant occupancy in the target quadrant relative to Nf1TH+/− and Nf1TH+/+ mice during probe trial 2 of the Morris water maze test. Dashed line depicts chance occupancy of a quadrant in the water maze. (C) Neurofibromin expression is reduced by 42% in Nf1TH+/− mice and by 72% in Nf1TH−/− mice compared with controls. (D) Hippocampus DA levels are reduced by 38% in Nf1TH+/− mice and by 69% in Nf1TH−/− mice compared with controls. (E) Western blots (left) and quantification (right) of DARPP-32 Thr34 phosphorylation (n = 5 mice per genotype). All data are represented as means ± SEM (***P < 0.0001; **P < 0.05; *P < 0.01, one-way/ two-way ANOVA with Bonferroni post-test).
Discussion
In the current study, we employed a combination of NF1-patient primary fibroblasts as well as derivative iPSCs and NPCs to explore the mechanistic relationship between the germline NF1 gene mutation and neurofibromin expression/function. The deployment of these unique NF1-patient biospecimens along with novel Nf1 genetically-engineered mouse (GEM) strains provided complementary evidence relevant to disease heterogeneity, biomarker implementation and risk assessment in this common neurogenetic condition.
First, we demonstrate that germline NF1 gene mutations result in dramatically different effects on neurofibromin expression in primary cells from individuals with NF1. In this regard, we found that one group of individuals with NF1 harbor >70% reductions in neurofibromin expression after a single germline mutation regardless of the cell lineage. This is likely due to post-translational modifications causing allelic imbalance, rather than the germline mutation affecting transcriptional regulation. As such, previous studies have shown that 30% of control individuals (28) and 75–80% of people with NF1, and not exclusively those with premature stop mutations, exhibit allelic variation in NF1 RNA expression (29,30), resulting in dramatic differences in neurofibromin expression similar in magnitude to our findings (30). However, this differential neurofibromin expression is not accurately recapitulated in the current Nf1 GEM models. Herein, we show that the standard Nf1 knockout (Nf1+/−) mice, which harbor an inactivating allele created by the insertion of a neomycin targeting cassette in exon 31 (24), express 50% reduced neurofibromin levels compared with wild-type littermates. Although this model permits step-wise genetic manipulation of neurofibromin levels to methodically study neurofibromin function in the hippocampus, it is not representative of either of the two identified NF1-patient groups. The generation and culture of human NF1-patient-derived iPSCs provide unprecedented opportunities to explore the molecular function of unique germline NF1 gene mutations on cell signaling and biology.
The clinical importance of differential neurofibromin expression caused by a single germline mutation is especially germane to the interpretation of Nf1 GEM studies that focus on phenotypes dictated by NF1 gene heterozygosity, such as behavior and learning. Previous studies have shown that 129T2/SvEmsJ or 129T2/SvEmsJ x C57BL/6 F1 hybrid mice display impaired spatial learning and memory on the Morris water maze test (6,24). In contrast, when these Nf1+/− mice are maintained on a pure C57BL/6J background, these abnormalities are transient (probe trial 2 only) and subtle (11). Similarly, we demonstrated that hemizygous Nf1+/− C57BL/6J mutant mice exhibit normal learning and memory. This discrepancy between the two strains may be due to differential basal levels of neurofibromin between 129T2 and C57BL/6 mice (31). In this regard, we found that >70% reductions in neurofibromin expression in C57BL/6J mice were required to reliably detect impaired learning and memory in the Morris Water Maze. Based on these observations, there is a need for a second generation of Nf1 mutant mouse strains harboring specific germline NF1 gene mutations to more accurately recapitulate the allelic imbalance observed in individuals with NF1 relevant to learning and memory. These ‘personalized’ mouse models will facilitate the identification of targeted approaches to predicting disease pathogenesis and designing effective treatments.
Second, we found that all NF1-patient fibroblasts, iPSCs and NPCs exhibited high levels of RAS activity, regardless of the level of neurofibromin expression. This intriguing finding suggests that the RAS-GAP activity of neurofibromin is it is comparably impaired in all individuals with NF1, irrespective of the nature or location of the germline NF1 gene mutation, and is thus highly sensitive to NF1 gene mutation. Importantly, it argues against the predictive value of the germline mutation or neurofibromin levels in phenotypes primarily driven by neurofibromin RAS regulation, such as leukemia or tibial dysplasia. However, other tumors types, like plexiform neurofibromas and optic gliomas (13), which are dependent on stromal cell types harboring only a single germline NF1 gene mutation, may be heavily influenced by the effect of neurofibromin function in mast cells (32) and microglia (33), respectively. Future investigation will be required to develop more reliable biomarkers for RAS pathway dysfunction in non-neuronal cells.
Third, we established a clear gene dose dependency for neurofibromin regulation of DA signaling in two independent Nf1 mouse model systems. We show that low hippocampal DA levels and downstream signaling, as measured by DARPP-32 phosphorylation, are associated with impaired spatial learning in mice. Since cognitive problems in neurological disorders can be difficult to identify, particularly in non-verbal or young children, predictive molecular biomarkers would be especially useful for the early diagnosis of these problems and the appropriate treatment of affected individuals. Initial studies have begun to identify blood serum potential biomarker for Alzheimer's disease (34), schizophrenia (35) and Asperger's syndrome (36). In this respect, the translation of the findings reported herein will require future research focused on defining the association between neurofibromin expression, DA signaling, and spatial learning deficits in NF1 patients.
Materials and Methods
Human subjects
Thirteen adults with an established diagnosis of NF1 (21) who receive their medical care in the Washington University/St. Louis Children's Hospital Neurofibromatosis Clinical Program were randomly selected and underwent a skin punch biopsy under an approved Human Studies Protocol at the Washington University School of Medicine.
Mice
Nf1+/− (37), RosaGREENGFAP (RosaGREEN (26); GFAP-Cre (25)) and RosaGREENTH (RosaGREEN; TH-Cre (27)), Nf1GFAP flox/+ (Nf1flox/+ (13,37); GFAP-Cre, Nf1FWC), Nf1GFAPflox/− (Nf1flox/−; GFAP-Cre, Nf1FMC), Nf1GFAPflox/flox (Nf1flox/flox; GFAP-Cre, Nf1FFC) (13), Nf1TH flox/+ (Nf1flox/+; TH-Cre, Nf1TH+/−), Nf1TH flox/flox (Nf1flox/flox; TH-Cre, Nf1TH−/−) and littermate control Nf1flox/flox (Nf1FF) mice were all maintained on an inbred C57BL/6 background and used in accordance with an approved Animal Studies protocol at the Washington University School of Medicine. All mice had ad libitum access to food and water. Unless otherwise stated, all experiments were performed on 3-month-old mice.
Comprehensive NF1 mutational analysis
The specific germline NF1 gene mutation was identified following comprehensive mutation analysis of DNA and RNA extracted from primary skin fibroblasts using an RNA-core assay complemented with dose analysis by multiplex ligation-dependent probe amplification (MLPA) as previously described (38) at the University of Alabama at Birmingham Medical Genomics Laboratory under the direction of Dr. Ludwine Messiaen. NF1 gene nucleotide numbering is based on GenBank reference sequence NM_000267.3 and protein numbering based on NP_000258.1. Exon numbering is assigned according to the NCB1 reference sequence along with the widely known legacy numbering in parenthesis. Nomenclature of the mutations follows the recommendations of the Human Genome Variation Society (HGVS).
Human induced pluripotent stem cell generation and neural progenitor cell differentiation
Primary fibroblasts were isolated and cultured from plated skin biopsies collected from 13 individuals with NF1 and four age- and sex-matched control individuals for ∼3 weeks. Established lines were reprogrammed into iPSCs as previously described (39). Briefly, using Cyto-Tune technology (Invitrogen), confluent fibroblasts were infected with a Sendai virus carrying four stem cell reprogramming factors (OCT4, KLF4, SOX2, C-MYC). Six weeks later, iPSC colonies were isolated, and their pluripotency confirmed by morphological assessment and the expression of stem cell markers (Nanog, SOX2, OCT4, SSEA-4, TRA-1-60, TRA-1-81). Chromosomal analysis demonstrated normal karyotypes in all lines (data not shown). Two separate clones from each iPSC line were cultured in Neural Induction Medium (NIM; STEMCELL Technologies) for 5 days. Embryoid body aggregates were plated in NIM on adhesive plates pre-coated with poly-ornithine/laminin. Once neural rosettes formed, gentle dissociation and re-plating facilitated their differentiation into NPCs. A portion of NPCs were differentiated into dopaminergic neurons (40).
Western blotting, immunocytochemistry and immunohistochemistry
Western blotting was performed as previously described (14) using appropriate primary antibodies (Supplementary Material, Table S1), secondary horseradish peroxidase-conjugated antibodies (Sigma) and ECL (Fisher) chemiluminescence. Neurofibromin (C) antibody (sc-67) was used instead of neurofibromin (N) (sc-68) unless otherwise specified (Fig. 1A). Immunocytochemistry was performed as previously described (15) on 4% paraformaldehyde (PFA) fixed cultured cells. Immunohistochemistry was performed on mice transcardially perfused with 4% PFA (Sigma) in 0.1 M sodium phosphate buffer (pH 7.4) and post-fixed in 4% PFA prior to paraffin embedding, as previously described (7). Appropriate primary antibodies (Supplementary Material, Table S1) and secondary antibodies were used.
cAMP, DA and RAS activity assays
All assays were performed on dissected embryonic hippocampi immediately snap frozen in liquid nitrogen following isolation. cAMP levels were quantitated from tissue homogenized in 0.1 M HCl, using a cAMP ELISA immunoassay kit (Enzo Life Sciences) following the manufacturer's instructions. DA levels were determined following tissue homogenization in 0.01 M HCl following the manufacturer's instructions (Rocky Mountain Diagnostics). Active Ras (Ras-GTP) was detected by Raf1-RBD immunoprecipitation using the RAS activation kit (Millipore) according to the manufacturer's instructions.
Behavioral testing
Morris Water Maze testing was conducted as previously described (7). Mice were trained for two consecutive days in cued water maze trials where the platform is visibly marked but its location is varied, followed by place water maze trials for 5 consecutive days where the submerged platform position is static. The probe trial (platform removed) was performed 1 h after the end of the last place trial on the third and fifth days of the trials. The escape path lengths (distances traveled to platform) and latencies (swimming times to platform) were recorded for all training trials. The time spent in the water maze quadrant of the former location of the platform (target quadrant) and the spatial bias for the target quadrant (time in target versus other quadrants) were used as readouts for the probe trials.
Statistical analyses
All statistical analyses were performed using GraphPad Prism 5 software (GraphPad Software). Unpaired two-tailed Student's T-tests were used for experiments analyzing data between two groups. One-way or two-way ANOVA with Bonferroni post-test correction analyses were employed for multiple comparisons. Some behavioral data were analyzed by repeated measures (rm) ANOVA models containing one between-subjects variable (genotype) and one within-subjects (repeated measures) variable (e.g. blocks of trials or time blocks). The Huynh-Feldt adjustment of alpha levels was utilized for all within-subjects effects containing more than two levels to protect against violations of sphericity/compound symmetry assumptions underlying rmANOVA models.
Supplementary Material
Supplementary Material is available at HMG online.
Conflict of Interest statement. The authors declare no competing financial interests.
Funding
This work was partially funded by generous support from the Walk Family. Control patient fibroblast lines were obtained from Matthew B. Harms, MD and were generated with support from the P30 Neuroscience Blueprint Interdisciplinary Center Core award to Washington University (P30 NS057105).
Supplementary Material
References
- 1.Jett K., Friedman J.M. (2010) Clinical and genetic aspects of neurofibromatosis 1. Genet. Med., 12, 1–11. [DOI] [PubMed] [Google Scholar]
- 2.Hyman S.L., Shores A., North K.N. (2005) The nature and frequency of cognitive deficits in children with neurofibromatosis type 1. Neurology, 65, 1037–1044. [DOI] [PubMed] [Google Scholar]
- 3.Isenberg J.C., Templer A., Gao F., Titus J.B., Gutmann D.H. (2013) Attention skills in children with neurofibromatosis type 1. J. Child Neurol., 28, 45–49. [DOI] [PubMed] [Google Scholar]
- 4.Orraca-Castillo M., Estevez-Perez N., Reigosa-Crespo V. (2014) Neurocognitive profiles of learning disabled children with neurofibromatosis type 1. Front. Hum. Neurosci., 8, 386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lorenzo J., Barton B., Arnold S.S., North K.N. (2013) Cognitive features that distinguish preschool-age children with neurofibromatosis type 1 from their peers: a matched case-control study. J. Pediatr., 163, 1479–1483 e1471. [DOI] [PubMed] [Google Scholar]
- 6.Costa R.M., Federov N.B., Kogan J.H., Murphy G.G., Stern J., Ohno M., Kucherlapati R., Jacks T., Silva A.J. (2002) Mechanism for the learning deficits in a mouse model of neurofibromatosis type 1. Nature, 415, 526–530. [DOI] [PubMed] [Google Scholar]
- 7.Brown J.A., Emnett R.J., White C.R., Yuede C.M., Conyers S.B., O'Malley K.L., Wozniak D.F., Gutmann D.H. (2010) Reduced striatal dopamine underlies the attention system dysfunction in neurofibromatosis-1 mutant mice. Hum. Mol. Genet., 19, 4515–4528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li W., Cui Y., Kushner S.A., Brown R.A., Jentsch J.D., Frankland P.W., Cannon T.D., Silva A.J. (2005) The HMG-CoA reductase inhibitor lovastatin reverses the learning and attention deficits in a mouse model of neurofibromatosis type 1. Curr. Biol., 15, 1961–1967. [DOI] [PubMed] [Google Scholar]
- 9.Krab L.C., de Goede-Bolder A., Aarsen F.K., Pluijm S.M., Bouman M.J., van der Geest J.N., Lequin M., Catsman C.E., Arts W.F., Kushner S.A., et al. (2008) Effect of simvastatin on cognitive functioning in children with neurofibromatosis type 1: a randomized controlled trial. JAMA, 300, 287–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mautner V.F., Kluwe L., Thakker S.D., Leark R.A. (2002) Treatment of ADHD in neurofibromatosis type 1. Dev. Med. Child Neurol., 44, 164–170. [DOI] [PubMed] [Google Scholar]
- 11.Diggs-Andrews K.A., Tokuda K., Izumi Y., Zorumski C.F., Wozniak D.F., Gutmann D.H. (2013) Dopamine deficiency underlies learning deficits in neurofibromatosis-1 mice. Ann. Neurol., 73, 309–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jouhilahti E.M., Peltonen S., Heape A.M., Peltonen J. (2011) The pathoetiology of neurofibromatosis 1. Am. J. Pathol., 178, 1932–1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bajenaru M.L., Hernandez M.R., Perry A., Zhu Y., Parada L.F., Garbow J.R., Gutmann D.H. (2003) Optic nerve glioma in mice requires astrocyte Nf1 gene inactivation and Nf1 brain heterozygosity. Cancer Res., 63, 8573–8577. [PubMed] [Google Scholar]
- 14.Brown J.A., Gianino S.M., Gutmann D.H. (2010) Defective cAMP generation underlies the sensitivity of CNS neurons to neurofibromatosis-1 heterozygosity. J. Neurosci., 30, 5579–5589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anastasaki C., Gutmann D.H. (2014) Neuronal NF1/RAS regulation of cyclic AMP requires atypical PKC activation. Hum. Mol. Genet., 23, 6712–6721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Upadhyaya M., Huson S.M., Davies M., Thomas N., Chuzhanova N., Giovannini S., Evans D.G., Howard E., Kerr B., Griffiths S., et al. (2007) An absence of cutaneous neurofibromas associated with a 3-bp inframe deletion in exon 17 of the NF1 gene (c.2970–2972 delAAT): evidence of a clinically significant NF1 genotype-phenotype correlation. Am. J. Hum. Genet., 80, 140–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pinna V., Lanari V., Daniele P., Consoli F., Agolini E., Margiotti K., Bottillo I., Torrente I., Bruselles A., Fusilli C., et al. (2014) p.Arg1809Cys substitution in neurofibromin is associated with a distinctive NF1 phenotype without neurofibromas. Eur. J. Hum. Genet., doi:10.1038/ejhg.2014.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kehrer-Sawatzki H., Vogt J., Mussotter T., Kluwe L., Cooper D.N., Mautner V.F. (2012) Dissecting the clinical phenotype associated with mosaic type-2 NF1 microdeletions. Neurogenetics, 13, 229–236. [DOI] [PubMed] [Google Scholar]
- 19.De Raedt T., Brems H., Wolkenstein P., Vidaud D., Pilotti S., Perrone F., Mautner V., Frahm S., Sciot R., Legius E. (2003) Elevated risk for MPNST in NF1 microdeletion patients. Am. J. Hum. Genet., 72, 1288–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sharif S., Upadhyaya M., Ferner R., Majounie E., Shenton A., Baser M., Thakker N., Evans D.G. (2011) A molecular analysis of individuals with neurofibromatosis type 1 (NF1) and optic pathway gliomas (OPGs), and an assessment of genotype-phenotype correlations. J. Med. Genet., 48, 256–260. [DOI] [PubMed] [Google Scholar]
- 21.Neurofibromatosis. (1988) Conference statement, National Institutes of Health Consensus Development Conference. Arch. Neurol., 45, 575–578. [PubMed] [Google Scholar]
- 22.Basu T.N., Gutmann D.H., Fletcher J.A., Glover T.W., Collins F.S., Downward J. (1992) Aberrant regulation of ras proteins in malignant tumour cells from type 1 neurofibromatosis patients. Nature, 356, 713–715. [DOI] [PubMed] [Google Scholar]
- 23.Cui Y., Costa R.M., Murphy G.G., Elgersma Y., Zhu Y., Gutmann D.H., Parada L.F., Mody I., Silva A.J. (2008) Neurofibromin regulation of ERK signaling modulates GABA release and learning. Cell, 135, 549–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Silva A.J., Frankland P.W., Marowitz Z., Friedman E., Laszlo G.S., Cioffi D., Jacks T., Bourtchuladze R. (1997) A mouse model for the learning and memory deficits associated with neurofibromatosis type I. Nat. Genet., 15, 281–284. [DOI] [PubMed] [Google Scholar]
- 25.Bajenaru M.L., Zhu Y., Hedrick N.M., Donahoe J., Parada L.F., Gutmann D.H. (2002) Astrocyte-specific inactivation of the neurofibromatosis 1 gene (NF1) is insufficient for astrocytoma formation. Mol. Cell. Biol., 22, 5100–5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mao X., Fujiwara Y., Chapdelaine A., Yang H., Orkin S.H. (2001) Activation of EGFP expression by Cre-mediated excision in a new ROSA26 reporter mouse strain. Blood, 97, 324–326. [DOI] [PubMed] [Google Scholar]
- 27.Lindeberg J., Usoskin D., Bengtsson H., Gustafsson A., Kylberg A., Soderstrom S., Ebendal T. (2004) Transgenic expression of Cre recombinase from the tyrosine hydroxylase locus. Genesis, 40, 67–73. [DOI] [PubMed] [Google Scholar]
- 28.Jentarra G.M., Rice S.G., Olfers S., Rajan C., Saffen D.M., Narayanan V. (2012) Skewed allele-specific expression of the NF1 gene in normal subjects: a possible mechanism for phenotypic variability in neurofibromatosis type 1. J. Child Neurol., 27, 695–702. [DOI] [PubMed] [Google Scholar]
- 29.Hoffmeyer S., Assum G. (1994) An RsaI polymorphism in the transcribed region of the neurofibromatosis (NF1)-gene. Hum. Genet., 93, 481–482. [DOI] [PubMed] [Google Scholar]
- 30.Hoffmeyer S., Assum G., Griesser J., Kaufmann D., Nurnberg P., Krone W. (1995) On unequal allelic expression of the neurofibromin gene in neurofibromatosis type 1. Hum. Mol. Genet., 4, 1267–1272. [DOI] [PubMed] [Google Scholar]
- 31.Hawes J.J., Tuskan R.G., Reilly K.M. (2007) Nf1 expression is dependent on strain background: implications for tumor suppressor haploinsufficiency studies. Neurogenetics, 8, 121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perry A., Roth K.A., Banerjee R., Fuller C.E., Gutmann D.H. (2001) NF1 deletions in S-100 protein-positive and negative cells of sporadic and neurofibromatosis 1 (NF1)-associated plexiform neurofibromas and malignant peripheral nerve sheath tumors. Am. J. Pathol., 159, 57–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang F.C., Ingram D.A., Chen S., Zhu Y., Yuan J., Li X., Yang X., Knowles S., Horn W., Li Y., et al. (2008) Nf1-dependent tumors require a microenvironment containing Nf1+/−- and c-kit-dependent bone marrow. Cell, 135, 437–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ray S., Britschgi M., Herbert C., Takeda-Uchimura Y., Boxer A., Blennow K., Friedman L.F., Galasko D.R., Jutel M., Karydas A., et al. (2007) Classification and prediction of clinical Alzheimer's diagnosis based on plasma signaling proteins. Nat. Med., 13, 1359–1362. [DOI] [PubMed] [Google Scholar]
- 35.Carlino D., Leone E., Di Cola F., Baj G., Marin R., Dinelli G., Tongiorgi E., De Vanna M. (2011) Low serum truncated-BDNF isoform correlates with higher cognitive impairment in schizophrenia. J. Psychiatr. Res., 45, 273–279. [DOI] [PubMed] [Google Scholar]
- 36.Schwarz E., Guest P.C., Rahmoune H., Wang L., Levin Y., Ingudomnukul E., Ruta L., Kent L., Spain M., Baron-Cohen S., et al. (2011) Sex-specific serum biomarker patterns in adults with Asperger's syndrome. Mol. Psychiatry, 16, 1213–1220. [DOI] [PubMed] [Google Scholar]
- 37.Brannan C.I., Perkins A.S., Vogel K.S., Ratner N., Nordlund M.L., Reid S.W., Buchberg A.M., Jenkins N.A., Parada L.F., Copeland N.G. (1994) Targeted disruption of the neurofibromatosis type-1 gene leads to developmental abnormalities in heart and various neural crest-derived tissues. Genes Dev., 8, 1019–1029. [DOI] [PubMed] [Google Scholar]
- 38.Messiaen L.M.W. (2008) D, K. (ed.), In Monogr. Hum. Genet. Karger, Basel, Vol. 16, pp. 63–77.
- 39.Yagi T., Ito D., Okada Y., Akamatsu W., Nihei Y., Yoshizaki T., Yamanaka S., Okano H., Suzuki N. (2011) Modeling familial Alzheimer's disease with induced pluripotent stem cells. Hum. Mol. Genet., 20, 4530–4539. [DOI] [PubMed] [Google Scholar]
- 40.Devine M.J., Ryten M., Vodicka P., Thomson A.J., Burdon T., Houlden H., Cavaleri F., Nagano M., Drummond N.J., Taanman J.W., et al. (2011) Parkinson's disease induced pluripotent stem cells with triplication of the alpha-synuclein locus. Nat. Commun., 2, 440. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.