

Significance
Defining a core functional proteome supporting the living process has importance for both developing fundamental understanding of cell functions and for synthetic biology applications. Comparative genomics has been the primary approach to achieve such a definition. Here, we use genome-scale models to define a core proteome that computationally supports basic cellular function. This core proteome for metabolism and protein expression, defined through systems biology methods, is validated and characterized by using multiple disparate data types.
Keywords: constraint-based modeling, metabolism, gene expression, minimal genome, core proteome
Abstract
Finding the minimal set of gene functions needed to sustain life is of both fundamental and practical importance. Minimal gene lists have been proposed by using comparative genomics-based core proteome definitions. A definition of a core proteome that is supported by empirical data, is understood at the systems-level, and provides a basis for computing essential cell functions is lacking. Here, we use a systems biology-based genome-scale model of metabolism and expression to define a functional core proteome consisting of 356 gene products, accounting for 44% of the Escherichia coli proteome by mass based on proteomics data. This systems biology core proteome includes 212 genes not found in previous comparative genomics-based core proteome definitions, accounts for 65% of known essential genes in E. coli, and has 78% gene function overlap with minimal genomes (Buchnera aphidicola and Mycoplasma genitalium). Based on transcriptomics data across environmental and genetic backgrounds, the systems biology core proteome is significantly enriched in nondifferentially expressed genes and depleted in differentially expressed genes. Compared with the noncore, core gene expression levels are also similar across genetic backgrounds (two times higher Spearman rank correlation) and exhibit significantly more complex transcriptional and posttranscriptional regulatory features (40% more transcription start sites per gene, 22% longer 5′UTR). Thus, genome-scale systems biology approaches rigorously identify a functional core proteome needed to support growth. This framework, validated by using high-throughput datasets, facilitates a mechanistic understanding of systems-level core proteome function through in silico models; it de facto defines a paleome.
In 1995 the full genome sequence of the prokaryote Haemophilus influenzae was published (1), giving researchers their first glimpse into the entire gene complement of an organism. Improved sequencing technologies have led to the full genome sequences of more thn 30,000 organisms now available in the National Center for Biotechnology Information (NCBI) Reference Sequence database (RefSeq) (2). In contrast to individual gene sequences or viral genomes that had been sequenced before 1995, the genome sequence of a living cell contains a comprehensive list of genes capable of sustaining life. The availability of the complete set of genes for many organisms leads to the question: Which subset of these genes is fundamental to supporting cellular life?
Less than a year after the publication of the first two prokaryotic genomes, Mushegian and Koonin (3) proposed the genetic content of a theoretical minimal cell, defined as the minimal number of genes required to sustain cellular life in a nutrient-rich optimal environment. Using sequence and functional homology between H. influenzae and Mycoplasma genitalium, the authors identified 254 genes that they considered sufficient to support life (3). As more bacterial genomes were sequenced, other definitions of minimal cell genomes soon followed (3–7). Of particular interest were insect endosymbionts: bacteria that had coexisted mutualistically in the gut of various insects for thousands of years. These species, such as Buchnera and Ricketsiella, are characterized by extremely small genomes that are still capable of self-replication, making them a good starting point for defining the minimal cell genome (4). Another approach by Antoine Danchin (8) made use of rational categorization of the genome and persistence of genes across species to define what he termed the paleome. This definition differed slightly from the minimal cell genome in that it did not require genes to be present in all of the organisms it compared, but instead, only persistent (found in a quorum number of those organisms) (9). Separate paleomes could also be defined for each species, or across species living within an environmental niche, and the number of genes could differ greatly from 500 to more than 1,500 (8).
The advent of genome-scale models of metabolism in bacteria opened a new facet to the study of core proteomes and minimal gene sets, allowing a mechanistic understanding of the metabolic network underlying cell physiology (10). Pál et al. (11) showed that they could model the evolution of several of these endosymbionts such as Buchnera aphidicola from genome-scale metabolic models of Escherichia coli through a random gene deletion approach (11). More recently, genome-scale models that account for metabolism and expression (ME models) have been developed (12, 13). In addition to metabolism, these models account for transcription and translation at the genome scale, providing an opportunity to investigate the importance of these processes for cellular viability. Although it only accounts for 1,554 of the 4,500+ genes in E. coli, the iOL1554-ME (ME model) has been shown to be highly accurate and representative of the physiological state and capabilities of E. coli, representing close to 80% (g/g) of the expressed proteome (13).
Here, we propose a method for determining the minimal set of genes fundamental to cellular life: genes that are used consistently across numerous and varied environmental conditions. Such varied conditions can be simulated by using the iOL1554-ME model, predicting the genes used for optimal growth across 333 different environmental conditions. We have labeled this gene set the core proteome, because these genes code for the portion of the proteome that is consistent across all of the 333 environments. We show here that the so-defined core proteome genes can be distinguished from noncore genes at the transcriptomic, proteomic, and transcriptional regulatory level.
Results
The model-based core proteome was defined to be the list of 356 genes (Dataset S1) that are required for growth across all 333 ME model simulations for E. coli K-12 MG1655. In each simulation, the main carbon, nitrogen, phosphorus, or sulfur source in the media was changed, using the base glucose/M9-minimal media conditions for the other three nutrient sources. In total, 180 different carbon sources, 49 phosphorus sources, 93 nitrogen sources, and 11 sulfur sources were examined. The ME model provides a mechanistic representation of metabolism and expression in the cell, making it possible to determine computationally the steady-state gene expression and translation flux values for each gene in the simulations (Dataset S2). Making use of glucose M9-minimal media as the reference condition and varying one nutrient source at a time forces the cell to use a wide array of its biochemical pathways, in both anabolic and catabolic capacities. Thus, the intersection of model-predicted cellular function in these simulations defines a scope of metabolic and expression capabilities encompassing growth, even in rich media. By defining a core proteome as those genes expressed across all simulation conditions, we select those that are used regardless of nutrient availability.
Consistency with Comparative Genomics-Based Minimal Genome Definitions.
We compared our iOL1554-ME model-based core proteome definition against two minimal gene sets previously identified in literature (Fig. 1A) (3, 4). Both gene sets (3, 4) were derived by using a combination of sequence-based and functional homology, followed by a deeper analysis of functions that the authors deemed necessary for life in a minimal organism growing in rich optimal media. However, because the criteria for defining all three gene sets are slightly different, we aimed to classify the content of each based on the different approaches. Although there is a significant overlap (60–77%) between the Mushegian and Gil gene sets (Fig. 1A, blue and red circles, respectively), at least 25% are exclusive to each, illustrating the difficulty researchers had in defining a true cross-species minimal gene set (Dataset S1).
Fig. 1.

Comparison of computationally derived core proteome and hypothesized minimal gene sets from literature (3, 4). Area enclosed in the dashed line in A refers to genes in the core proteome that are excluded from the minimal genome sets. Area enclosed in the dotted line in A refers to genes identified in ref. 3 that were excluded from the computationally derived core proteome. Area enclosed in the bolded line represents genes identified in ref. 4 that were excluded from the computationally derived core proteome. COG categories are shown in B or each of these three sets of genes. Hashed bars refer to genes not currently implemented in the ME model.
Clusters of Orthologous Groups (COG) (14) was used to classify the functions of genes in each minimal genome set. The core proteome was consistent with 55% (135) and 63% (128) of the genes proposed by Mushegian and Gil, respectively (Fig. 1 C and D). Of the 254 M. genitalium genes in the minimal cell genome described by Mushegian, 14 had no E. coli functional homologs, whereas others mapped to 244 functionally equivalent E. coli genes in a one-to-many or many-to-one fashion, resulting in a total of 258 gene IDs. However, 60% of the computationally derived core (212 genes) had not been previously identified in either of the minimal cell genomes (Fig. 1B).
Several large groups of genes were involved in this discrepancy. Many genes involved with replication, which has not yet been implemented in the ME model, were not included in the core proteome (24% and 25% of the discrepancies in the Mushegian and Gil gene sets, respectively) (Fig. 1B). Other differences lie in the COG categories of translation, amino acid, carbohydrate, and nucleotide metabolism. Because of the detailed mechanistic representation of translation in the ME model, many genes involved in rRNA and tRNA modification that were not included in either of the minimal genome sets are included in the core proteome. Some genes, such as several ribosomal proteins, are essential for growth in E. coli, whereas others involved with rRNA and tRNA modifications might not be essential but play important roles to ensure replication stability and high growth rates. At the same time, by using a minimal medium in our simulations, we require the cell to synthesize all other precursors essential for growth from a single nutrient source. To determine the robustness of this gene set, we ran ME model simulations of E. coli on rich media, which more closely match the environmental conditions of the Gil and Mushegian paleomes. This set of 386 genes had a large overlap with our core proteome (273 genes). Differences between the two gene sets were largely metabolic genes (SI Appendix, Fig. S9A), and DAVID functional annotation clustering (15) found that the 113 genes not in the core proteome are enriched for cell membrane synthesis pathways (enrichment score 16.98), oxidative phosphorylation (16.80) and amino acid transport (8.11), whereas the 83 core proteome genes which were unaccounted for in the rich media simulations are enriched for amino acid biosynthesis (33.99) (Dataset S3). Additionally, the rich media simulations only marginally improved consistency with the Mushegian or Gil paleomes (nine and four genes, respectively) (SI Appendix, Fig. S9 B and C). Hence, by selecting only biosynthetic pathways that are consistently used across all conditions, we have identified a robust core that is used in all minimal media environments and better represents the core genes expressed by a cell growing in varied media conditions. The validity of this systems biology-based definition can be examined based on disparate data types.
The Core Proteome Can Be Distinguished from Noncore at Both the Transcriptomic and Proteomic Levels.
We investigated whether the expression of core genes is variable or remains constant across growth conditions and genetic backgrounds. First, we compared RNA-Seq data for E. coli MG1655 across three growth conditions: glucose, fructose, and acetate (sbrg.ucsd.edu/Downloads/SupplementalData). We identified 763 and 480 genes that were differentially expressed (|log2(fold-change)|>1; P < 0.05) in acetate and fructose, respectively, relative to glucose. Core proteome genes were significantly depleted for differentially expressed genes (hypergeometric test, P < 0.05), whereas the homology-based minimal genomes were not (Fig. 2). Therefore, the ME model provided enhanced discriminatory power, beyond homology-based methods, distinguishing genes with core functionality vs. those without.
Fig. 2.

Depletion analysis of genes differentially expressed in E. coli when grown on fructose and acetate, relative to growth on glucose. (A) Proportion of core genes that are differentially and nondifferentially expressed. (B) A hypergeometric P value cutoff of 0.05 (below the dashed lines) was used to determine significant depletion: The ME model-based core was statistically significantly depleted for differentially expressed genes, whereas the homology-based minimal gene sets were not.
To compare genetic backgrounds, we used RNA-Seq data from eight adaptively evolved strains grown on glucose minimal media (16). These strains show a number of mutations leading to fitness increases up to 1.6-fold over the wild type. From these strains, a set of genes was consistently differentially expressed across all of the evolved strains. The core and minimal proteomes were significantly enriched for commonly up-regulated genes, and significantly depleted for commonly down-regulated genes (hypergeometric test, P < 0.05) (SI Appendix, Fig. S1 and Dataset S4). In turn, the commonly up-regulated genes were involved in translation, protein folding, and amino acid metabolism (16). Therefore, the core proteome is comprised of a gene set enriched for rapid growth and depleted for genes that do not contribute to growth.
As an additional characterization of the core proteome under varying genetic backgrounds, we calculated the correlation of proteomics data between MG1655 (17) and four strains of BW25113 (18) (SI Appendix, Fig. S2). Core genes showed statistically significantly higher correlation over noncore genes, in terms of both Pearson (3.6–5.5-fold higher, Fisher’s Z procedure P < 1 × 10−5) and Spearman rank (1.7- to twofold higher, permutation test P < 1 × 10−3). Therefore, the expression of core genes appears to be more tightly regulated across both growth conditions and genetic backgrounds than noncore genes. We additionally tested this hypothesis by computing each gene’s coefficient of variation (19) across a large-scale microarray compendium (20). The core proteome had a higher fraction of low-variation genes, and lower fraction of high-variation genes, than noncore genes; both results were statistically significant (permutation test, P < 1 × 10−4) (SI Appendix, Fig. S10).
We then investigated whether the tight regulation observed at the transcriptome level was also reflected at the sequence level, specifically in the organization of regulatory regions. To this end, we used genome-wide profiling of transcription start sites (TSSs) (21) and estimated the number of TSSs upstream of each gene, for core and noncore genes. This high-throughput, experimental TSS profiling method enables assessment of regulatory complexity of genes that is unbiased by the extent to which a gene has been studied. A greater percentage of core genes was annotated with two or more TSSs (41.7% core vs. 24.4% noncore; SI Appendix, Fig. S3). Additionally, core genes were annotated with 40% more TSSs per gene compared with noncore genes (average 1.89 vs. 1.35; Wilcoxon rank-sum test P = 2.0 × 10−8), indicating that core genes involve more complex transcriptional regulation. In addition, core genes had significantly longer 5′ untranslated regions (5′UTRs) than noncore genes (Wilcoxon rank-sum test P = 0.005; SI Appendix, Fig. S3), implying differences in posttranscriptional regulation between core and noncore genes. We found similar regulatory feature differences between core and noncore genes with orthologs in Klebsiella pneumoniae (SI Appendix, Fig. S4).
To further characterize regulatory feature differences, we analyzed the use of regulatory elements upstream of orthologs between E. coli and K. pneumoniae. As expected, calculation of sequence conservation showed more conservation in the Shine-Dalgarno region, high conservation of the first (ATG) and second codons, and lowest conservation of the third codon (SI Appendix, Fig. S5). Core genes had 8.4% higher median conservation (Wilcoxon rank-sum test P = 3.8 × 10−52; SI Appendix, Fig. S5), suggesting that core genes have higher protein similarity, at least in the genomic regions surrounding TSSs. The level of conservation between the first, second, and third nucleotides in codons of coding regions showed that the second nucleotide had the lowest difference, whereas the third nucleotide had the greatest difference (all 3 groups statistically significantly different by rank-sum tests with P < 1 × 10−7). Collectively, these results indicated that the E. coli core proteome and the orthologous core in K. pneuomoniae were both characterized by significantly different organization of regulatory regions from the noncore proteome.
We next extended our analysis to two large-scale microarray compendia. First, using a compendium containing negative control probes (22, 23), we found that 86% of the core proteome was always expressed across 69 experiments, which was significant (hypergeometric test, P = 8.0 × 10−28). In contrast, only five core genes were never expressed (Dataset S5). A sensitivity analysis (SI Appendix, SI Methods and Fig. S12) showed that the ME model selected these genes over alternatives (e.g., isozymes) based on the predicted gene product efficiency, a function of effective rate constant parameters. Thus, such inconsistencies are expected to be reduced as enzyme rate constants are better determined for future ME models.
We next analyzed 444 microarray experiments (20) in the context of functional gene sets (COG and KEGG pathways) and the core proteome. First, core genes showed statistically significantly higher expression for nearly every COG (Wilcoxon rank-sum P < 0.01), with the sole exception of COG N (cell motility) (SI Appendix, Fig. S8). This high percentage of COGs containing significantly higher expression of core genes was statistically significantly higher than an equivalently defined percentage based on randomly chosen genes from the ME model (permutation test, P = 0.048). We then identified 48, 24, and 11 biclusters (Dataset S6 and SI Appendix, Figs. S13 and S14) by using cMonkey (24) that were enriched for KEGG pathways, the core proteome, and both gene sets, respectively (Fig. 3A). Furthermore, 46% of core genes were members of at least one core proteome-enriched bicluster (Fig. 3B). This percentage was statistically significantly higher than an equivalently defined percentage based on randomly chosen genes from the ME model (permutation test, P = 0.002). Finally, the 11 biclusters enriched for both KEGG pathways and the core proteome were enriched for eight KEGG pathways (Fig. 3C), of which six were consistently enriched across four biclustering runs (all except fatty acid biosynthesis; and glycine, serine, and threonine metabolism) (SI Appendix, SI Methods). In contrast, a similar analysis using either the Gil or Mushegian minimal gene sets led to only two KEGG pathways (ribosome and oxidative phosphorylation) enriched in biclusters that were also enriched in either minimal gene set. Thus, the core proteome is reflected better in functionally meaningful modules identified from high-throughput data, and may provide a more suitable basis for building genome-scale ME models in various environmental niches for a new prokaryotic organism. For example, functional homologs (25) of metabolic core genes in Bacillus subtilis were predicted to be significantly highly used across 171 simulated growth conditions (Wilcoxon rank-sum test, P = 5.2 × 10−7; median percentage of B. subtilis simulation conditions where core genes were predicted to be expressed was 84%) (SI Appendix, SI Methods).
Fig. 3.

Biclustering and enrichment analysis of omics data. (A) Venn diagram of biclusters significantly enriched (hypergeometric test, P < 0.01) in KEGG pathways or the core proteome. (B) Venn diagram of genes in four gene sets: all KEGG pathways, the core proteome, biclusters enriched for KEGG pathways, and biclusters enriched for the core proteome. (C) The 11 biclusters enriched in both KEGG pathways and the core proteome. Multiple KEGG groups may be enriched in a bicluster. The ratio is the number of genes in the bicluster that are also in the gene set (KEGG or core proteome), divided by the number of genes in the bicluster.
Toward Environment-Specific, Sufficient Proteomes.
In contrast to minimal gene sets, which are the smallest set of genes sufficient to allow growth under optimal conditions, the core proteome is the set of genes that are consistently used across a large number of conditions. This set of genes alone will not support growth, but rather, serves as the central core for optimally powering the cell. Thus, in addition to this core, there is a set of genes that are used in a condition-specific manner.
To understand groups of genes needed in a condition-specific manner, we clustered the 333 environmental perturbation simulations according to their predicted translation rates. This procedure resulted in 18 clusters of environments, and each of these was given loose categories based on the majority of nutrient perturbations (Dataset S7). Almost all of the environmental perturbations were to the carbon source in clusters 8–18, whereas most perturbations to the sulfur source showed up in clusters 5 and 7. Interestingly, clustering also separated the environments where nitrogenous bases were used as nitrogen, phosphorus, or carbon sources. For example, if pyrimidines were used as the sole nitrogen source, gene expression fell into cluster 2. However, if pyrimidines were used as the sole phosphorus or carbon source, gene expression fell into cluster 6 and cluster 16, respectively.
For each cluster of conditions, 525–684 genes were required for optimal growth across all environments within the cluster (Fig. 4). To investigate the sufficiency of each gene set, we checked them for inclusion of essential genes. To determine gene essentiality, we made use of two separate datasets. The first, containing 431 genes, was downloaded from EcoCyc (26), containing experimental observations largely from ref. 27 that used single-gene knockouts to study essentiality when grown on glucose/M9 minimal media. We also made use of a transposon mutagenesis of E. coli grown on glucose/M9 minimal media to generate a second list of 415 essential genes. Both datasets contain a significant overlap, but exhibit slight differences in essentiality calls due to strengths and limitations of each method (4). The removal of genes not represented in the ME model resulted in a list of 327 genes from EcoCyc and 300 genes from Tn-seq data (Dataset S8). Gene sets derived from each of the 18 nutrient perturbation clusters and the computational core were screened for the presence of essential genes. Each cluster was highly consistent with these essential genes, accounting for more than 80% for each gene essentiality dataset. The number of essential genes missing from each set is consistent with the 55 essential genes that the ME model falsely predicted as nonessential.
Fig. 4.
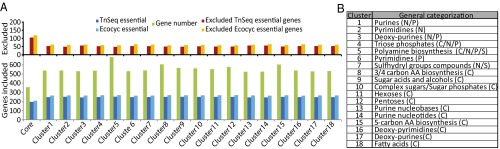
Consistency of core proteome and various cluster gene sets with gene essentiality. (A) Affinity propagation clustering of the 333 environmental perturbation simulations resulted in 18 clusters. Each environmental perturbation simulation consisted of changing either the carbon (C), nitrogen (N), phosphorus (P), or sulfur (S) nutrient source. Each cluster contained a different number of genes required for optimal growth across all conditions within the cluster (green bars). Two sets of gene essentiality calls were used, the first obtained via single-gene deletions (26) and the second via transposon sequencing. Only genes represented in the ME model were considered; essential genes included in each set of genes are shown in blue bars and genes excluded in red and gold bars. (B) Environmental perturbations in each cluster were loosely categorized, along with the major nutrient perturbation in each cluster.
Finally, we varied the number of simulated conditions a gene must be expressed in for it to be core (SI Appendix, Fig. S11). The resulting spectrum of minimal proteomes overlapped well with two minimal organisms [area under the resulting curves (AUC) = 0.80 for B. aphidicola, 0.76 for M. genitalium, and 0.78 for both] (SI Appendix, Fig. S7). For B. aphidicola, our gene overlap is similar to that found in ref. 11 by using genome reduction simulations, where a genome-scale model of E. coli metabolism is used, although, our gene overlap includes both metabolic and expression machinery genes. The best threshold, maximizing sensitivity plus specificity for both minimal organisms was 114 conditions (precision = 0.43, recall = 0.78) (SI Appendix, Fig. S7). The resulting minimal proteome of 517 genes may represent a good starting point for building ME models of organisms other than E. coli.
Discussion
In this work, we used validated genome-scale models (13) to identify and characterize a core proteome of E. coli metabolism, transcription, and translation by computing genome-wide metabolic, transcription, and translation rates across 333 different growth conditions. From these simulations, we identified a set of 356 genes that were always expressed. This systems biology-based core proteome definition was then compared with two minimal gene sets in the literature, which were defined based on comparative genomics (i.e., homology-based) (3, 4). The two homology-based minimal gene sets showed high similarity, having 60–77% gene overlap with the core proteome defined here. Conversely, the model-based core proteome was more distinctive because it showed only 40% overlap with the homology-based gene sets. Translation, amino acid metabolism and transport, and coenzyme metabolism accounted for more than half of the nonoverlapping model-based core genes. The majority of genes in the homology-based gene sets but not the model-based core proteome were genes outside the scope of the current ME model (e.g., replication) (13). However, more than 50% of the genes specific to the model-based core proteome were already nonmetabolic, highlighting the ability of the ME model-based approach to account for systemic gene interactions beyond metabolism. Therefore, as ME models continue to increase in biological scope (28, 29), systems-level understanding of the core proteome is expected to broaden as well.
We then characterized the defined core proteome in the context of transcriptomics or proteomics data across multiple growth conditions, strains, and genetic backgrounds. First, we found that the core proteome contains genes covering the majority of metabolic and expression-related functional categories (19/23 COGs) and was composed of a significantly highly expressed set of genes. Transcriptomics of eight adaptive laboratory evolution (ALE) endpoint strains (16) showed that the core proteome was not only significantly enriched for commonly up-regulated genes but also significantly depleted for commonly down-regulated genes. The core proteome also showed 2–5 times higher correlation of protein abundances than the noncore proteome between MG1655 and four strains of BW25113. These results indicate that the E. coli core proteome comprises a highly expressed and tightly regulated set of genes. Furthermore, genes that were differentially expressed, when grown on acetate and fructose (relative to glucose), were significantly depleted in the model-based core proteome but not in the homology-based minimal gene sets. Finally, the core proteome was more often enriched together with KEGG pathways in biclusters identified across 444 microarray experiments. These results suggest that constraint-based ME models offer additional classificatory power for comparing relative transcript abundances across different growth conditions that purely homology-based methods may lack.
Although the core proteome is meant to comprise a necessary set of genes for growth, it is not intended nor expected to be sufficient. As such, it accounted for 64–65% of essential genes identified from single-gene deletion studies (30) and Tn-seq experiments, respectively. To characterize the core proteome in the context of a proteome predicted to be sufficient for growth, we defined 18 C/N/P/S nutrient clusters representing different environmental niches by clustering the expression profiles from 333 simulations. The ME model predicted the addition of 169–328 additional genes to the core proteome to sustain optimal growth in these 18 clusters. These sufficient proteomes were up to 81% consistent for both essentiality datasets. Furthermore, by systematically adding ∼160 genes based on ME simulations, we could closely approximate the genomes of the minimal organisms, B. aphidicola APS and M. genitalium (AUC = 0.78). B. aphidicola is a well-studied endosymbiont whereas M. genitalium has the smallest genome of an organism that can be grown in pure culture—collectively, they are distinct representatives of minimal organisms. Therefore, we expect the model-based core proteome definition and its characterization presented here to accelerate the development of future models of metabolism and expression for a broader range of organisms, growing in various environmental niches.
Finally, we found that transcriptional regulation of the core proteome is significantly more complex than noncore genes, in both E. coli and K. pneumoniae, as they had more TSSs per gene, and had significantly longer 5′UTRs. These observations suggest that understanding the regulation of the core proteome may be a crucial first step toward reconstructing integrated genome-scale models of regulation (O), metabolism (M), and expression (E), or OME models (31).
This work can thus be seen as a first step toward a model-driven, systems-level characterization of the core metabolic and gene expression functions necessary for life, and a way to begin to elucidate the mechanisms that control them. The core and sufficient proteomes defined serve as a platform for generating ME models for new bacterial species and a template with which to develop new algorithms for various ME model computations.
Methods
Transcriptomics and Proteomics Data Analysis.
HTSeq. (32) and DESeq2 (33) were used to obtain normalized counts and to identify differentially expressed genes. The P values were combined by using Stouffer’s method (34) and adjusted for multiple testing by using the Benjamini–Hochberg correction (35). RNA-Seq data were converted into Z scores after log2 transformation and included growth on 3 carbon sources (sbrg.ucsd.edu/Downloads/SupplementalData), and 8 ALE endpoints on glucose minimal medium (16). Proteomics comparisons were performed pairwise between MG1655 and four strains of BW25113 (WT, ΔpKa, ΔpKaΔarcA, ΔarcA). See SI Appendix, SI Methods for details.
Statistical and Multivariate Analysis.
Affinity propagation (36) was used to cluster simulated translation flux profiles (37). Enrichment analysis was performed by using hypergeometric P values, with the Benjamini–Hochberg correction for multiple testing (29). Statistical tests of difference between Pearson correlation coefficients were performed by using Fisher’s Z-transform method (38) and Zou’s confidence interval method (39). Statistical test of difference between Spearman rank correlations was performed by using a permutation test with 10,000 permutations.
Analysis of B. aphidicola and M. genitalium.
We varied the number of conditions a gene was required to be expressed in ME simulations to be added to the core (i.e., cutoffs ranging from 1 to 333 conditions). For each cutoff, we computed the fraction of true-positive predictions (expressed gene set present in B. aphidicola) and false-positive predictions (expressed genes not present in B. aphidicola). We then computed the AUC. See SI Appendix, SI Methods for details.
ME Simulation Across 333 Growth Conditions and Core Proteome Definition.
Simulations were carried out by using iOL1554-ME, the genome-scale model of E. coli K-12 MG1655 metabolism and expression (13), for a base medium of M9 minimal medium + glucose. In each simulation, the main carbon, nitrogen, phosphate or sulfate source in the medium was changed, with the other three nutrient sources held constant. In total, 333 simulations were performed, corresponding to 180 different carbon sources, 49 phosphorus sources, 93 nitrogen sources, and 11 sulfur sources. We analyzed the trend in core proteome size as a function of the number of required conditions for expression (gene’s translation flux > 0) (SI Appendix, Fig. S11). To avoid an ambiguous intermediate threshold, we used the strictest definition of the core proteome: gene expressed in all 333 conditions.
Tn-seq Experiments and Data Analysis.
Genes were considered essential if the adjusted P value was <0.05 and the log2 ratio of (normalized) measured-over-expected number of reads per gene was below the optimal cutoff as predicted by ESSENTIALS (40). See SI Appendix, SI Methods for details.
Supplementary Material
Acknowledgments
This work was supported by the National Institute of General Medical Sciences of the National Institutes of Health Grants U01 GM102098 and R01 GM057089, Novo Nordisk Foundation Grant 1R01 GM098105, and a Marie Curie International Outgoing Fellowship within the European Commission’s 7th Framework Programme for Research and Technological Development (Grant 332020, project AMBiCon).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. J.T.L. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1501384112/-/DCSupplemental.
References
- 1.Fleischmann RD, et al. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science. 1995;269(5223):496–512. doi: 10.1126/science.7542800. [DOI] [PubMed] [Google Scholar]
- 2.Pruitt KD, Tatusova T, Maglott DR. NCBI reference sequences (RefSeq): A curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007;35(Database issue):D61–D65. doi: 10.1093/nar/gkl842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mushegian AR, Koonin EV. A minimal gene set for cellular life derived by comparison of complete bacterial genomes. Proc Natl Acad Sci USA. 1996;93(19):10268–10273. doi: 10.1073/pnas.93.19.10268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gil R, Silva FJ, Peretó J, Moya A. Determination of the core of a minimal bacterial gene set. Microbiol Mol Biol Rev. 2004;68(3):518–537. doi: 10.1128/MMBR.68.3.518-537.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Klein CC, et al. Exploration of the core metabolism of symbiotic bacteria. BMC Genomics. 2012;13:438. doi: 10.1186/1471-2164-13-438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grosjean H, et al. Predicting the minimal translation apparatus: Lessons from the reductive evolution of mollicutes. PLoS Genet. 2014;10(5):e1004363. doi: 10.1371/journal.pgen.1004363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Monk JM, et al. Genome-scale metabolic reconstructions of multiple Escherichia coli strains highlight strain-specific adaptations to nutritional environments. Proc Natl Acad Sci USA. 2013;110(50):20338–20343. doi: 10.1073/pnas.1307797110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Danchin A. Scaling up synthetic biology: Do not forget the chassis. FEBS Lett. 2012;586(15):2129–2137. doi: 10.1016/j.febslet.2011.12.024. [DOI] [PubMed] [Google Scholar]
- 9.Acevedo-Rocha CG, Fang G, Schmidt M, Ussery DW, Danchin A. From essential to persistent genes: A functional approach to constructing synthetic life. Trends Genet. 2013;29(5):273–279. doi: 10.1016/j.tig.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feist AM, et al. A genome-scale metabolic reconstruction for Escherichia coli K-12 MG1655 that accounts for 1260 ORFs and thermodynamic information. Mol Syst Biol. 2007;3:121. doi: 10.1038/msb4100155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pál C, et al. Chance and necessity in the evolution of minimal metabolic networks. Nature. 2006;440(7084):667–670. doi: 10.1038/nature04568. [DOI] [PubMed] [Google Scholar]
- 12.Thiele I, et al. Multiscale modeling of metabolism and macromolecular synthesis in E. coli and its application to the evolution of codon usage. PLoS One. 2012;7(9):e45635. doi: 10.1371/journal.pone.0045635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.O’Brien EJ, Lerman JA, Chang RL, Hyduke DR, Palsson BØ. Genome-scale models of metabolism and gene expression extend and refine growth phenotype prediction. Mol Syst Biol. 2013;9(1):693. doi: 10.1038/msb.2013.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tatusov RL, et al. The COG database: An updated version includes eukaryotes. BMC Bioinformatics. 2003;4:41. doi: 10.1186/1471-2105-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37(1):1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.LaCroix RA, et al. Use of adaptive laboratory evolution to discover key mutations enabling rapid growth of Escherichia coli K-12 MG1655 on glucose minimal medium. Appl Environ Microbiol. 2015;81(1):17–30. doi: 10.1128/AEM.02246-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taniguchi Y, et al. Quantifying E. coli proteome and transcriptome with single-molecule sensitivity in single cells. Science. 2010;329(5991):533–538. doi: 10.1126/science.1188308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peebo K, et al. Coordinated activation of PTA-ACS and TCA cycles strongly reduces overflow metabolism of acetate in Escherichia coli. Appl Microbiol Biotechnol. 2014;98(11):5131–5143. doi: 10.1007/s00253-014-5613-y. [DOI] [PubMed] [Google Scholar]
- 19.Mar JC, et al. Variance of gene expression identifies altered network constraints in neurological disease. PLoS Genet. 2011;7(8):e1002207. doi: 10.1371/journal.pgen.1002207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carrera J, et al. An integrative, multi-scale, genome-wide model reveals the phenotypic landscape of Escherichia coli. Mol Syst Biol. 2014;10:735. doi: 10.15252/msb.20145108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim D, et al. Comparative analysis of regulatory elements between Escherichia coli and Klebsiella pneumoniae by genome-wide transcription start site profiling. PLoS Genet. 2012;8(8):e1002867. doi: 10.1371/journal.pgen.1002867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lewis NE, Cho BK, Knight EM, Palsson BO. Gene expression profiling and the use of genome-scale in silico models of Escherichia coli for analysis: providing context for content. J Bacteriol. 2009;191:3437–3444. doi: 10.1128/JB.00034-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lewis NE, et al. Omic data from evolved E. coli are consistent with computed optimal growth from genome-scale models. Mol Syst Biol. 2010;6:390. doi: 10.1038/msb.2010.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reiss DJ, Baliga NS, Bonneau R. Integrated biclustering of heterogeneous genome-wide datasets for the inference of global regulatory networks. BMC Bioinformatics. 2006;7:280. doi: 10.1186/1471-2105-7-280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aziz RK, et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genomics. 2008;9:75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Keseler IM, et al. EcoCyc: Fusing model organism databases with systems biology. Nucleic Acids Res. 2013;41(Database issue):D605–D612. doi: 10.1093/nar/gks1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baba T, et al. Construction of Escherichia coli K‐12 in‐frame, single‐gene knockout mutants: The Keio collection. Mol Syst Biol. 2006;2(1):2006.0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu JK, et al. Reconstruction and modeling protein translocation and compartmentalization in Escherichia coli at the genome-scale. BMC Syst Biol. 2014;8(1):110. doi: 10.1186/s12918-014-0110-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.O'Brien EJ, Palsson BO. Computing the functional proteome: recent progress and future prospects for genome-scale models. Curr Opin Biotech. 2015;34:125–134. doi: 10.1016/j.copbio.2014.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yamamoto N, et al. Update on the Keio collection of Escherichia coli single-gene deletion mutants. Mol Syst Biol. 2009;5:335. doi: 10.1038/msb.2009.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Feist AM, Herrgård MJ, Thiele I, Reed JL, Palsson BØ. Reconstruction of biochemical networks in microorganisms. Nat Rev Microbiol. 2009;7(2):129–143. doi: 10.1038/nrmicro1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Anders S, Pyl PT, Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31(2):166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Whitlock MC. Combining probability from independent tests: The weighted Z-method is superior to Fisher’s approach. J Evol Biol. 2005;18(5):1368–1373. doi: 10.1111/j.1420-9101.2005.00917.x. [DOI] [PubMed] [Google Scholar]
- 35.Benjamini Y, Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol. 1995;57:289–300. [Google Scholar]
- 36.Frey BJ, Dueck D. Clustering by passing messages between data points. Science. 2007;315(5814):972–976. doi: 10.1126/science.1136800. [DOI] [PubMed] [Google Scholar]
- 37.Bodenhofer U, Kothmeier A, Hochreiter S. APCluster: An R package for affinity propagation clustering. Bioinformatics. 2011;27(17):2463–2464. doi: 10.1093/bioinformatics/btr406. [DOI] [PubMed] [Google Scholar]
- 38.Diedenhofen B, Musch J. cocor: A comprehensive solution for the statistical comparison of correlations. PLoS One. 2015;10(3):e0121945. doi: 10.1371/journal.pone.0121945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zou GY. Toward using confidence intervals to compare correlations. Psychol Methods. 2007;12(4):399–413. doi: 10.1037/1082-989X.12.4.399. [DOI] [PubMed] [Google Scholar]
- 40.Zomer A, Burghout P, Bootsma HJ, Hermans PWM, van Hijum SAFT. ESSENTIALS: Software for rapid analysis of high throughput transposon insertion sequencing data. PLoS One. 2012;7(8):e43012. doi: 10.1371/journal.pone.0043012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.