

Abstract
In this article, we described a new database framework to perform integrative “gene-set, network, and pathway analysis” (GNPA). In this framework, we integrated heterogeneous data on pathways, annotated list, and gene-sets (PAGs) into a PAG electronic repository (PAGER). PAGs in the PAGER database are organized into P-type, A-type and G-type PAGs with a three-letter-code standard naming convention. The PAGER database currently compiles 44 313 genes from 5 species including human, 38 663 PAGs, 324 830 gene–gene relationships and two types of 3 174 323 PAG–PAG regulatory relationships—co-membership based and regulatory relationship based. To help users assess each PAG’s biological relevance, we developed a cohesion measure called Cohesion Coefficient (CoCo), which is capable of disambiguating between biologically significant PAGs and random PAGs with an area-under-curve performance of 0.98. PAGER database was set up to help users to search and retrieve PAGs from its online web interface. PAGER enable advanced users to build PAG–PAG regulatory networks that provide complementary biological insights not found in gene set analysis or individual gene network analysis. We provide a case study using cancer functional genomics data sets to demonstrate how integrative GNPA help improve network biology data coverage and therefore biological interpretability. The PAGER database can be accessible openly at http://discovery.informatics.iupui.edu/PAGER/.
Contact: jakechen@iupui.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 Introduction
To characterize complex patterns of genetic variations or RNA/protein expressions in surging Omics data sets, bioinformatics researchers have developed new data analysis approaches—gene-set analysis (Dinu et al., 2009; Nam and Kim, 2008) and network/pathway analysis (Wu et al., 2012). When compared with individual gene-based analysis approaches, “gene-set, network, and pathway analysis” (GNPA) have the potential advantage of increasing results reproducibility, model robustness, and data interpretability, while reducing biases in noisy Omics experimental data (Khatri et al., 2012). GNPA is essentially an integrative analysis strategy that takes advantage of a priori data structures acquired from many sources in: e.g. gene ontology (GO) (Kim et al., 2007), pathways (Luo et al., 2009), gene/protein mutation or expression signatures (Chuang et al., 2007; Zhang and Chen, 2013), chemical perturbations (Oprea et al., 2007), metabolomics signatures (Xia and Wishart, 2010), curated literature (Araki et al., 2012), computational predictions (Nam, 2010) or public databases for gene-sets, molecular signatures and pathway/network modules (Huang et al., 2012). Depending on the pathway details, these analyses can take as minimal information as possible, e.g. using gene list alone as in Gene-set Analysis (Dinu et al., 2009), incorporating gene-set databases with expression rank information as in Gene Set Enrichment Analysis (Subramania et al., 2005), or take as much pathway/network interaction detail as possible, e.g. using pathway reaction/regulation details as in EnrichNet (Glaab et al., 2012). For biomedical researchers, GNPA can significantly enhance their ability to annotate genes from Omics results (Ganter and Giroux, 2008), interpret heterogeneous genetic study results (Hale et al., 2012), identify disease subtypes and progression (Hung, 2013; Zhang and Chen, 2013), select or prioritize drug targets (Sivachenko and Yuryev, 2007) and understand biological mechanisms (Chen et al., 2006; Murohashi et al., 2010).
Heterogeneous bioinformatics tools have been developed to perform different aspects of GNPA Huang da et al., 2009). Computationally, GNPA can be categorized into over-representation analysis such as DAVID (Dennis et al., 2003), gene-set scoring and ranking such as GSEA (Subramania et al., 2005), multivariate machine learning Wu et al., 2010) and network topological analysis Martini et al., 2013). Common databases for GNPA include: manually curated functional or phenotypic databases such as Online Mendelian Inheritance in Man (OMIM) (Baxevanis, 2012), GAD (Becker et al., 2004)] and GO (Ashburner et al., 2000); curated signaling and regulatory pathway databases such as Reactome (Croft et al., 2011) and pathway interaction database (PID) (Schaefer et al., 2009); comprehensive curated gene signature database such as GeneSigDB (Culhane et al., 2012) and mSigDB (Liberzon et al., 2011); or comprehensive integrated knowledge repositories such as human pathway database (HPD) (Chowbina et al., 2009) and PAGED (Huang et al., 2012). Recently, multi-scale GNPA analysis tools such as Bioinformatics Enrichment Tools (Huang da, et al., 2009), MetaNet (Parikh et al., 2012) and EnrichNet (Glaab et al., 2012) have also been developed to explore network topological structures between pathways and gene-sets using a variety of computational strategies. However, even the most popular tools such as DAVID and GSEA lack major functionality or comprehensive data content found in other tools, forcing users to try out several tools and combining findings manually.
In this work, we address the major challenge of constructing a comprehensive database infrastructure in performing integrative GNPA. The theoretical space of gene-sets for a given species with N genes can be extremely large, i.e. 2N gene-sets. Based on our practical experience in building comprehensive pathway and gene-set databases (Chowbina et al., 2009; Huang et al., 2012), different data sources collected for integrated GNPA are often partially overlapping yet complementary to one another. For example, GeneSigDB and mSigDB, two popular databases for Gene Set Analysis, have virtually no overlapping gene-sets, in which an overlap is defined as two gene-sets sharing >80% genes. KEGG (Ogata, et al., 1999) is the earliest effort in curating heterogeneous pathway data sources; however, its coverage in gene-set is missing. GO is widely used for gene set analysis; however, tools performing GO-based gene set analysis do not usually perform pathway/network analysis. HPD (Chowbina et al., 2009), PAGED (Huang et al., 2012) and GeneSetDB (Araki et al., 2012) are among the first attempts to integrate annotated gene lists from heterogeneous sources into a unified database. Although these databases provide significantly higher coverage of gene-sets and other annotated gene list, these databases lack pathway/network interaction or regulatory relationships details. In addition, increased gene-set coverage gives rise to gene-set data quality concerns that must be addressed. There is no report on how a new gene-set submitted from community users, e.g. from WikiPathways (Pico et al., 2008), should be evaluated for its quality before a database of gene-set should adopt it for data analysis. Moreover, there is a rising need to perform integrative network analysis both within gene-sets and between gene-sets; therefore, understanding how gene-sets relate to one another to build gene-set to gene-set relationships has become critical for ongoing integrative GNPA tools.
Our work has made the following contributions for future GNPA. First, we defined a new concept—PAG, which stands for Pathways (P-type), Annotated lists (A-type) and Gene-sets (G-type), to integrate heterogeneous gene-sets, networks, and pathways into a new comprehensive database called PAG Electronic Repository (PAGER). Each P-type PAG refers to a connected set of molecules (genes/proteins/metabolites), among which some detail of curated mechanism of actions, e.g. protein interactions, reactions, or gene regulations, are available (Biological Pathway, http://www.genome.gov/27530687). Each A-type PAG refers to a curated list of genes/proteins identified from a specific biological context, e.g. a shared GO category or a shared protein family. Each G-type PAG refers to a list of genes/proteins derived from any given high throughput Omics experiment, e.g. functional genomics, under a shared biological condition. PAGER collects and organizes 38 663 PAGs—the largest collection known to date—using a three-letter-code PAG classification system. Second, we developed a statistical measure called Cohesion Coefficient (CoCo) to help assess PAG data quality—the degree of biological relevance beyond random chance found among genes curated in each PAG. We demonstrated that the CoCo score can effectively separate biologically curated PAGs in PAGER from randomly generated PAGs with an sensitivity = 0.75 and specificity = 0.94, covering >94% of all PAGs compiled into the PAGER database, Third, we computed a novel type of relationship among PAGs called regulatory PAG–PAG relationships (r-type) in addition to computing the co-membership based PAG–PAG relationships described in earlier work (Chowbina et al., 2009). These r-type PAG–PAG relationships (n = 65 872 for human) significantly complement the single gene regulatory relationships that are known prior to this work (n = 22 127 for human). The PAGER database is accessible at http://discovery.informatics.iupui.edu/PAGER/ and the database content for PAGs may be downloaded for separate GNPA tools such as GSEA.
2 Methods
2.1 Source data collection and preprocessing of PAGs
In the PAGER database, we compiled PAGs from the following data sources (for download date and description details, refer to Supplementary Data S1): WikiPathway (Pico et al., 2008), from which we collected 202 public validated pathways; Reactome (Croft et al., 2011), from which we collected 651 peer-reviewed pathways via the HPD (Chowbina et al., 2009); BioCarta (Nishimura, 2001), from which we collected 253 pathways via the HPD; KEGG (Ogata et al., 1999), from which we collected 200 pathways via the HPD; PID (Schaefer et al., 2009), from which we collected 132 NCI-Nature curated pathways via the HPD; Protein Lounge (http://www.proteinlounge.com/Pathway), from which we collected 393 pathways; OMIM (Baxevanis, 2012), from which we collected 4409 manually-curated gene lists associating with phenotype terms; SPIKE (Elkon et al., 2008), from which we collected 28 signaling pathways from the Genetic Association Database; GAD (Becker et al., 2004), from which we collected 1679 unique phenotype/disease-related protein records; PharmGKB (Thorn et al., 2010), from which we collected 102 chemical-associated pathways; MSigDB (Liberzon et al., 2011), from which we collected 10 295 gene sets; GeneSigDB (Culhane et al., 2012), from which we collected 3515 gene signatures; NHGRI GWAS Catalog (Welter et al., 2014), from which we collected 1754 curated publications of 11 912 SNPs; and NGS Catalog (Xia et al., 2012), from which we collected 69 annotated gene list curated from next generation sequencing data analysis literature. During the data integration process, gene/protein identified obtained from different sources were all mapped to NCBI official gene symbols (Brown et al., 2015).
In the PAGER database, we also constructed a special type of PAGs, ‘singleton PAGs’ (sPAG), to refer to PAGs consisting of only one gene. sPAGs are essential for constructing PAG–PAG relationships (to be described later). The final PAGER database consists of 19 772 sPAGs, which represent all sPAGs from the underlying sources and an additional 15 161 human genes from the reviewed subset of the UniProt Knowledgebase (UniProt, 2013). In the PAGER web database, we mask out these sPAGs by default to avoid causing confusions to users who are primarily interested in performing analysis with regular PAGs (n = 18 607, among which 16 125 are for human).
A total of 324 830 gene–gene relationship data are also imported into the PAGER database from various data sources. In total, 205 185 molecular association data are derived from the STRING version 9.1 (Szklarczyk et al., 2011) database after removing those with confidence score of 800 or less. A total of 93 713 human protein–protein interaction (PPI) data are derived from the HAPPI database (quality ≥ 3-star ratings) (Chen et al., 2009). A total of 25 932 gene regulation data are derived from the TRANSFAC (Wingender et al., 1996) (with quality defined as having five-binding sites or more), TRED (Jiang et al., 2007) and SPIKE (Elkon et al., 2008) databases. The PPI data are used to build PAG biological relevance measures. The gene regulation data are used to construct regulatory PAG–PAG relationships later.
We follow the PAG ID standard naming convention as shown in Figure 1. Every PAG begins with a three-letter-code followed by six digits. The first letter position is used to signify the PAG data type category. The second letter position is used to signify the PAG derivation category. The third letter position is used to signify PAG relationship details. The human-readable naming convention may make it easy for end users to understand how the PAGs are constructed.
Fig. 1.

PAG ID standard naming convention and three types of PAGs
2.2 Defining CoCo measures to evaluate the biological relevance of each PAG
To evaluate the biological relevance of PAGs, we developed three PAG cohesion measures, i.e. Cohesion by Interaction enrichment (CoI), Cohesion by Triangle enrichment (CoT) and CoCo. Conventional tests such as chi-square, hypergeometric test or Fisher’s exact test have all been used to assess gene-set enrichment results. However, none of these statistical tests may generate true statistical P-values in practice (Huang da et al., 2009). Therefore, for simplicity of calculation, we calculate all three cohesion measures based on signed probability mass function of hypergeometric distribution. Mathematical notations are defined as the following: Π is the set of genes in the reference PPI database; i and j are PAG indexes; Ii or Ij is the set of PPIs in PAGi and PAGj, respectively; IΠ is the set of all PPIs in the referenced database; Ti or Tj is the set of PPI triangles in PAGi and PAGj, respectively given IΠ; TΠ is the set of all PPI triangles in Π. For an overview of how each cohesion measure is calculated, refer to the illustrated example shown in Figure 2.
Fig. 2.
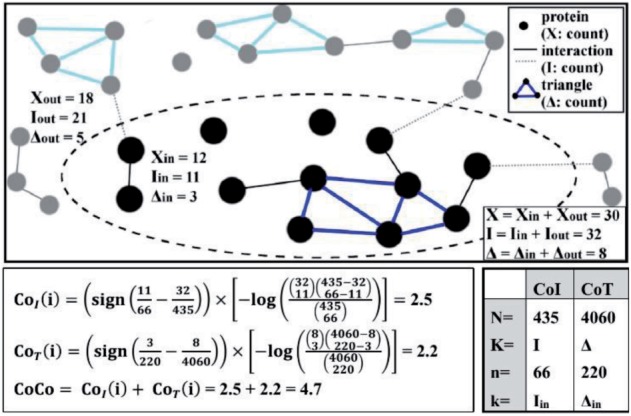
Example of calculation of each Cohesion measure
2.2.1 CoI definition
We developed CoI to measure the statistical significance of observing a given number of PPIs among all genes within a PAG. We define CoI from a hypergeometric probability mass function as:
in which is the number of theoretical PPIs in Π, K = |IΠ| is the number of actual PPIs in the referenced database, is the theoretical number of PPIs inside PAG i, and k = |Ii| is the number of actual PPIs inside PAG i. The sign function compares the expected PPI count ratio K/N and PAG i’s PPI count ratio k/n. It returns 1 for over-representation if the PPI count ratio inside PAG i is more than the expected ratio and returns −1 for under-representation if the interaction ratio inside PAG i is less than the expected ratio. For PAGs containing no PPIs, we will not calculate a CoI. A high positive CoI score implies that genes inside the PAG are strongly linked while a high negative CoI score implies that genes inside the PAG are impossibly linked. Randomly generated PAGs should have a mean CoI close to 0.
2.2.2 CoT definition
We developed CoT to measure the statistical significance of observing a given number of triangles (PPIs forming a connected loop with three exact nodes/edges) among all genes within a PAG. Similar to CoI, CoT is also calculated using the same hypergeometric distribution probability mass function (hence we will not repeat here) but with different parameters for counting PPI triangles instead: is the number of theoretical triangles in Π, K = |TΠ| is the number of actual triangles in the referenced database, is the theoretical number of triangles inside PAG_i, and k = |Ti| is the number of actual triangles inside PAG_i. When compared with CoI, it has similar characteristics for highly positive CoT or highly negative CoT cases. For PAGs containing no PPIs, we will not calculate a CoT. A high positive CoT score implies that genes inside the PAG are strongly linked while a high negative CoT score implies that genes inside the PAG are impossibly linked. Randomly generated PAGs should have a mean CoT close to 0.
2.2.3 CoCo definition
We define CoCo = CoI + CoT for PAGs that contain at least one PPI triangle, and CoCo = CoI for PAGs that contain no PPI triangle but at least one PPI. When compared with CoI and CoT, the combined CoCo score has similar characteristics for highly positive CoCo or highly negative CoCo cases. For PAGs without valid calculated CoI, there will not be a CoCo score. In practice, since we used a high-coverage PPI database (the HAPPI database), the portion of PAGs without any PPIs within is relatively low.
2.2.4 Performance evaluations
To compare and evaluate the performance of three cohesion measures, we plot Receiver Operator Characteristic (ROC) curves for each of the measures, CoI, CoT, and CoCo. To create the positive set, we use true PAGs (size>1 for CoI and size>2 for CoT or CoCo) from the PAGER database. To create the negative set, we substitute all genes in each PAGi with genes randomly picked from the PAGER database. After all the cohesion measures are calculated, we calculate true positive rate (true positives over all positives cases) and true negative rate (true negatives over all negatives cases) for each possible cohesion measure threshold that exists in the data before plotting them on the ROC curve.
2.3 Inferring PAG–PAG relationships
We computationally derive two types of PAG–PAG relationships: co-membership based PAG–PAG relationships (m-type) and regulatory PAG–PAG relationships (r-type). These two types of PAG–PAG relationships may be calculated by Fisher’s exact test (Al-Shahrour et al., 2004; Li et al., 2008; Parikh et al., 2012) using a 2 × 2 contingency table. In this study, however, we used hypergeometric distribution probability mass function instead to score each PAG–PAG relationship. Mathematical notations are defined as the following: Ω is the number of genes covered in PAGER, R_ini is the set of gene regulations coming into genes in PAGi, R_outi is the set of gene regulations coming out from genes in PAGi, Ri->j is the set of gene regulations from genes in PAGi to genes in PAGj, Ri<->j is the set of gene regulations from genes in PAGi to genes in PAGj if and only if the genes belong to PAGi ∩ PAGj.
2.3.1 Inferring m-type PAG–PAG relationships
We define the m-type relationship as the significance of observing the number of shared genes among PAGi and PAGj given the gene regulation data related to the two PAGs, or membership strength score in short. The hypergeometric distribution parameters for m-type relationships are defined as: N = Ω, K = |PAGi|, n = |PAGj| and k = |PAGi ∩ PAGj|.
2.3.2 Inferring r-type PAG–PAG relationships
We define the r-type relationship from PAGi to PAGj as the significance of observing the number of gene regulations from genes in PAGi to genes in PAGj, excluding the genes in PAGi ∩ PAGj, or regulatory strength score in short. The hypergeometric distribution parameters for m-type relationships are defined as: N = |R_ini| + |R_outi| + |R_inj| + |R_outj| − |Ri->j| is the total number of gene regulations in PAGi and PAGj, K = |R_outi|, n = |R_inj| and k = |Ri->j| − |Ri<->j| is the number of gene regulation from genes in PAGi to genes in PAGj, excluding the genes in PAGi ∩ PAGj.
2.4 Developing the web application and user interface
We developed a web interface for the PAGER database, which is located at http://discovery.informatics.iupui.edu/PAGER/, for users to retrieve PAGs and PAG–PAG relationships with gene or keyword based queries. We used PHP5 and Codeigniter version 2.1.3 (EllisLab, 2014) as the web presentation framework and Oracle 11g as the database backend. Real-time calculation of hypergeometric probability mass function was implemented with PDL (Meagher et al., 2013), a PHP library for statistics. Cytoscape.js (http://js.cytoscape.org), an open-source graph library, and jQuery were used to visualize gene and PAG networks. D3.js (http://d3js.org/) was used to perform matrix visualizations. We also implemented advanced features such as batch gene search, matrix or network visualization, gene or PAG transaction management to hold temporary user-selected contents, and data bulk download.
2.5 Case study: application of PAGER in myeloid-derived suppressor cells expression data analysis
Myeloid-derived suppressor cell (MDSC) microarray data analysis between tumor and normal control conditions were performed at Purdue University Center for Cancer Research. The study aims to establish a hypothesis on what factors were essential in promoting MDSC during cancer progression to transition from tumor-suppressor cells to tumor-helper cells. We collected tumor MDSC at the peritoneal cavity (T0) and compared control MDSC at the spleen (N0) of inflamed mice. Using the pulsed electroacoustic method, we extracted two cell sub-populations: CD11b+ Gr-1low (PC Glow) and CD11b+ Gr-1low (Sp Glow). Standard differential expression analysis of individual genes were performed using SAM (Tusher et al., 2001). After the data analysis using pmf < 0.05 as the filter, we set the minimum log fold-change of 2.5 to select 1105 differentially expressed genes (N0), which includes 576 over-expressed genes (N+) and 529 under-expressed genes (N−).
To generate an MDSC tumor versus control (T0 versus N0) gene regulatory network, we queried the N0 gene-set against the available gene regulation data in the PAGER database to obtain the seeded gene regulatory network. In addition, we queried N+ and N− gene-sets against the PAGER database to obtain N+ associated PAGs and N− associated PAGs. Using the r-type PAG–PAG regulatory relationships in PAGER, we also acquired the MDSC-specific network.
3 Results
3.1 An overview of summary statistics of the new PAGER database
From Table 1, we can obtain several data characteristics for the new PAGER database. First, there are 38 379 unique PAGs, which consists of 19 772 sPAGs and 18 607 mPAGs (regular PAGs) with a size variation ranging between 1 and 4939 (refer to Supplementary Fig. S1 for additional details of PAG size distribution categorized into different data sources). When compared with MSigDB—the largest gene-set database prior to this publication, PAGER represents a 181% increase of gene-set data coverage. In addition, we noticed an unusual spike in the size distribution for data derived only from MSigDB (with PAG sizes in the 200–212 range, Supplementary Fig. S1). This suggests potential PAG data curation size bias, i.e. taking only the top 200 genes from an expression signature, in the popular MSigDB. Second, there is a high percentage of PAGs determined to be biologically relevant as determined from the cohesion measure, CoCo score. In total, 14 701 (79%) of the 18 607 mPAGs are computed with a valid CoCo score and 13 856 (94%) of these mPAGs also have a CoCo score > 1. This finding suggests fairly high biological relevance in the majority of PAGs that we integrated into the PAGER database. Third, we also noticed that PAGs are more likely to overlap with each other than to regulate on one another. In the data statistics table, there 3 101 499 m-type PAG–PAG relationships—a number significantly higher than that of the total 72 824 r-type PAG–PAG relationships identified.
Table 1.
Basic statistics of the PAGER database
| In PAGER DB | In PAGER (Human) | |
|---|---|---|
| Genes in PAGs | 44 313 | 40 476 |
| Gene–gene relationships | 324 830 | 306 066 |
| Molecular association | 205 185 | 190 226 |
| PPI | 93 713 | 93 713 |
| Gene regulation | 25 932 | 25 932 |
| PAGs | 38 379 | 35 897 |
| Singleton (n = 1) | 19 772 | 19 772 |
| Regular (n > 1) | 18 607 | 16 125 |
| with CoCo scores (n > 1) | 14 701 | 12 496 |
| with CoCo score ≥1 | 13 856 | 11 784 |
| PAG–PAG pairs | ||
| m-type (P-value < 1e-5) | 3 101 499 | 2 230 614 |
| r-type (pmf < 0.05) | 72 824 | 65 983 |
| sPAG to mPAG | 7250 | 7022 |
| mPAG to mPAG | 39 253 | 32 966 |
| sPAG to sPAG | 23 842 | 23 842 |
| mPAG to sPAG | 2479 | 2153 |
3.2 Cohesion scores to classify PAGs based on biological relevance
We evaluated PAG classification performance (biological relevant YES/NO classes) using three different PAG cohesion measures, i.e. CoI, CoT and CoCo. First, using the ROC curve (Fig. 3), we observed that all these measures can classify true PAGs from randomly generated PAGs effectively. The area-under-curves (AUCs) of classification performance for balanced positive class (integrated PAGs from the PAGER database) and negative class (randomly generated PAGs) are 0.96, 0.95 and 0.98 respectively for each of the cohesion measures CoI, CoT and CoCo. Second, we compared the positive class with the negative class using these cohesion measures’ score distributions. Measurement score distributions between samples from the two classes are statistically significant at P-values of 8.5e-138 (for CoI), 3.0e-10 (for CoT) and 2.4e-62 (for CoCo), respectively, when two-sample t-test analysis is used. Third, we observed variable effects between PAG size and cohesion measurements’ classification performance. For example, the AUC performance using CoT is slightly better than that for CoI among small PAGs (n < 100), based on Table 2. These observation justifies the use of the combined score CoCo whenever CoT may be calculated (size>2 and minimal PPI triangle = 1).
Fig. 3.
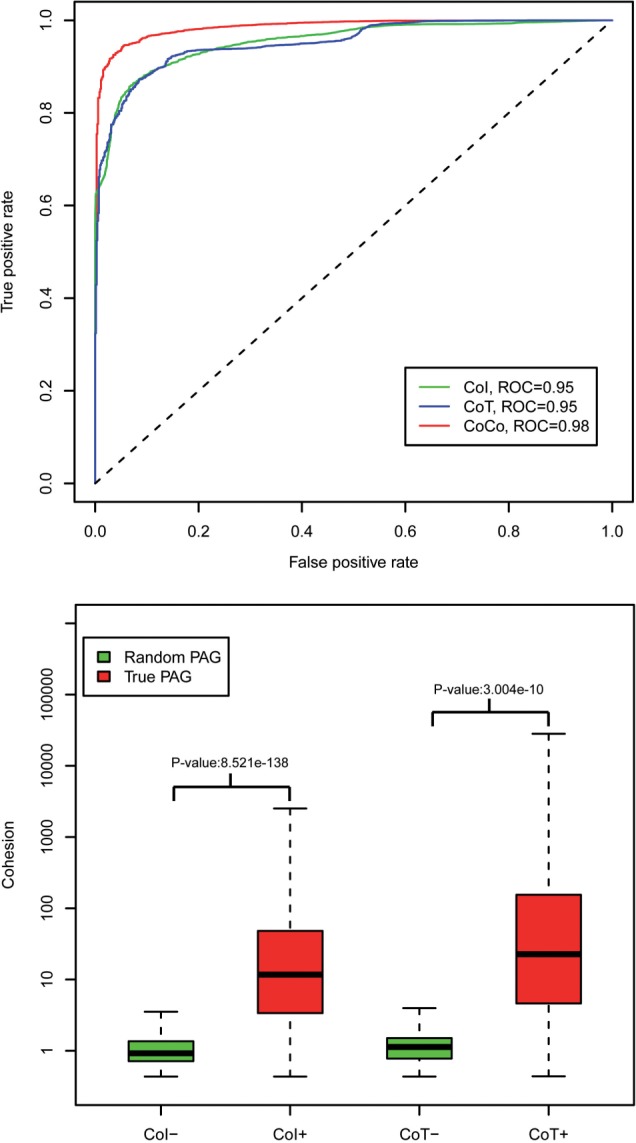
Cohesions (CoI, CoT and CoCo) performance: (a) ROC curves, (b) comparison boxplot. CoI+, CoI in the true PAGs; CoI−, CoI in the random PAGs; CoT+, CoT in the true PAGs; CoT−, CoT in the random PAGs
Table 2.
AUC performance comparison between CoI and CoT for small and large PAGs
| AUC (size <100) | AUC (size >300) | |
|---|---|---|
| CoI | 0.953 | 0.987 |
| CoT | 0.962 | 0.979 |
To decide the threshold of high informative PAG, we choose the CoCo score as threshold to minimize the sum of false positive and false negative. We observe the threshold of 1 has the minimum sum of false positive and false negative counts equal to 2490 (Supplementary Table S2) with the false negative rate equal to 0.057 and the false positive rate equal to 0.252. We also show the PAG coverage decreases when the threshold increases, which means we need to balance the precision and PAGs recall (Supplementary Fig S2 and Supplementary Table S3).
3.3 PAG-PAG regulatory relationship network characterization
The PAG regulatory network in PAGER has the following characteristics. First, the network only connects a small portion of all PAGs. The network only covers 3304 regular PAGs, or 20.49% of the human regular PAGs, and contains 6783 directed PAG–PAG regulation; therefore, on average each PAG has the regulatory degree of 2.05. Second, the regulatory network is well-connected. This network has 39 connected components; however, the largest connected component covers 3236 PAGs, or 97.94% of the network size. Third, the PAG regulatory network node degree strictly follows the power law, achieves R2 = 0.884 for in-degree analysis, and R2 = 0.855 for out-degree analysis. Due to the sparsity of high-quality PAG-PAG regulatory relationships identified in PAGER, we therefore suggest using both gene regulatory relationship data from conventional methods and new PAG-PAG regulatory relationship data from PAGER for GNPA.
3.4 Web interface to access PAGER data
The PAGER web interface provides four basic features: basic search, advanced search, matrix and network view, and data download. The basic search allows the users to enter terms, such as a disease name or a gene symbol. The advanced search allows the users to enter a list of gene symbols. The users can decide specific options to filter out poor quality PAGs. When the users enter a list of terms, PAGER returns genes and PAGs associating with this list. When the users search a list of genes, PAGER returns the related PAGs, showing PAGs’ gene membership, cohesions, p-value and FDR. The users can construct, expand both m-PAG and r-PAG networks and compare PAGs using a similarity matrix view. The users can sort the matrix view by name, frequency. The site also allows a user to download the data used to construct the networks and images of the matrix. Details on what features the web application provides and how to use them are provided in Supplementary File S1.
3.5 PAGER analysis of MDSC gene expression data
MDSCs have been identified in most cancer patients and tumor mice models based on their ability to suppress T-cell activation (Ostrand-Rosenberg and Sinha, 2009). MDSC are induced by tumor-secreted factors, many of which are known pro-inflammation markers. In this study, we performed a functional genomics study using microarrays, by comparing MDSC at tumor site versus at control spleens, to identify detailed molecular mechanisms that trigger the MDSC’s immunity inhibition functions. To compare the effectiveness of performing integrative GNPA with the new PAGER database, we set up a control experiment to examine the gene regulatory network that can be constructed following microarray data analysis. Among the 1105 differentially expressed genes, there are only 256 genes (23% coverage) with direct gene regulation relationships between them in the MDSC gene regulatory network (Table 3 and Supplementary Fig S3). On the other hand, there are 972 genes (88% coverage) enriched in 91 connected mPAGs through newly defined r-type PAG–PAG relationship in the MDSC PAG regulatory network. Although one can try to characterize the MDSC-derived tumor versus control (TvN) individual gene biomarkers from the gene regulatory network formed within the 256 differentially expressed genes, the poor network data coverage due to limited availability of gene regulation data can lead to significant bias in the knowledge discovery process (refer to Table 1 for summary statistics). From the new PAGER database, we are able to identify 91 enriched mPAGs (13 up-regulated PAGs and 78 PAGs, all of which have CoCo score > 1), the majority of which come from diverse data sources. Although conventional GNPA relies on gene-set enrichment analysis, we demonstrate how to perform multi-scale integrative GNPA, using regulatory PAG–PAG network analysis (Fig. 4). In the figure, we show each PAG as a node of different sizes and shapes, with size proportional to the CoCo score (in log scales) for each PAG. The MDSC inflammatory regulatory PAG network shows a significantly suppressed PAG—FEX001153 (size n = 4568)—which includes many genes responsible for immunity functions. Upstream of the immunity-suppressed process are many activated cell growth and differentiation signal molecules, such as BCL2L1, a potent inhibitor of cell death; FGFR1, Tyrosine-protein kinase that acts as cell-surface receptor that can regulate embryonic development, cell proliferation, differentiation and migration; ID1, which regulates a variety of cellular processes, including cellular growth, senescence, differentiation, apoptosis, angiogenesis and neoplastic transformation; and AHR that are involved in cell-cycle regulation. Downstream of the immunity-suppressed process are many processes responsible for cell differentiation (e.g. FEX00813) and cancer progression (e.g. FEX002152 and EPHA2), and chemokine signaling (e.g. CCR5). GEX001173, the closest downstream PAG of FEX001153, is a nephrolithiasis-related gene set acquired from GAD, and is associated with the seeded overexpressed sub gene list. This anti-immunity and pro-cell proliferation signaling can be clearly explored in the PAG/gene multi-scale regulatory network consisting of both genes and PAGs implicated in the MDSC functional genomics data in tumor.
Table 3.
The statistic for gene regulatory network and PAG regulatory network for MDSCs gene expression data
| Gene regulatory network | PAG regulatory network | |
|---|---|---|
| Gene (n = 1105) | 256 | 972 |
| Up-regulated (n = 576) | 110 | 489 |
| Down-regulated (n = 529) | 146 | 483 |
| PAGs (n = 1196) | 0 | 133 |
| m-type | 0 | 91 |
| Up-regulated | 0 | 13 |
| Down-regulated | 0 | 78 |
| Regulations | 501 | 136 |
| Gene regulations (gene-gene) | 501 | 0 |
| PAG regulations (mPAG-mPAG) | 0 | 94 |
| PAG regulations (sPAG-mPAG) | 0 | 42 |
Fig. 4.

The PAG regulatory network constructed for myeloid-derived suppressor cells gene expression data
4 Conclusion
In this work, we presented a new conceptual framework that unifies pathways, annotated gene lists, and gene-sets into a standardized representation called PAGs. We constructed a comprehensive data repository called PAGER for PAG data from heterogeneous sources including ontology, pathway, omics, and public databases. The new PAGER database collects 80% more PAG data than that of the popular MSigDB database. We showed that a significant benefit for using PAGER data is the benchmarking of each PAG with cohesion measures—scores that can help evaluate the biological relevance of all genes/proteins in a given PAG in high confidence. To address the practical challenges of performing GNPA, we define new PAG–PAG relationships as m-type for those with shared members between PAGs and t-type for those with strong supporting gene regulatory relationships pointing from one PAG to the next. We demonstrated through an integrative cancer genomics study how integrative multi-scale GNPA could help gain significant biological insights of the Omics data.
We expect future researchers in the field to focus on addressing several key questions related to PAG data management and integrative GNPA. First, there should be sufficient focus on curating PAGs with rich meta-data, e.g. information on individual genetic background, life-style, and electronic medical records. Second, there should be comprehensive linking of genomics and functional genomics data from public databases into PAG data structure. Therefore, researchers may further investigate the biological relevance of enriched PAGs and regulatory PAG relationships derived from GNPA. Last, we expect to develop novel literature mining and crowd sourcing within the biomedical research community to continue rapid extension of data for the PAGER database.
Supplementary Material
Acknowledgements
The authors thank Indiana University Information Technology Support team at IUPUI for their generous database and web server support.
Funding
The authors appreciate the partial grant support from the National Institute of Health to Dr Tim Ratliff (R21CA173918 and DK084454) and Dr Jake Chen to complete this work.
Conflict of Interest: none declared.
References
- Al-Shahrour F., et al. (2004) FatiGO: a web tool for finding significant associations of Gene Ontology terms with groups of genes. Bioinformatics, 20, 578–580. [DOI] [PubMed] [Google Scholar]
- Araki H., et al. (2012) GeneSetDB: a comprehensive meta-database, statistical and visualisation framework for gene set analysis. FEBS Open Biol. 2, 76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M., et al. (2000) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. , 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxevanis A.D. (2012) Searching Online Mendelian Inheritance in Man (OMIM) for information on genetic loci involved in human disease. Current Protocols in Human Genetics/Editorial Board, Jonathan L Haines [et al] 2012, Chapter 9:Unit 9 13 11-10. [DOI] [PubMed] [Google Scholar]
- Becker K.G., et al. (2004) The genetic association database. Nat. Genet. 36, 431–432. [DOI] [PubMed] [Google Scholar]
- Brown G.R., et al. (2015) Gene: a gene-centered information resource at NCBI. Nucleic Acids Res. , 43 (Database issue), D36–D42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.Y., et al. (2006) A systems biology case study of ovarian cancer drug resistance. Computational Systems Bioinformatics/Life Sciences Society Computational Systems Bioinformatics Conference 2006:389–398. [PubMed] [Google Scholar]
- Chen J.Y., et al. (2009) HAPPI: an online database of comprehensive human annotated and predicted protein interactions. BMC Genomics , 10 (Suppl 1), S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowbina S.R., et al. (2009) HPD: an online integrated human pathway database enabling systems biology studies. BMC Bioinformatics, 10 (Suppl 11), S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang H.Y., et al. (2007) Network-based classification of breast cancer metastasis. Mol. Syst. Biol. 3, 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croft D., et al. (2011) Reactome: a database of reactions, pathways and biological processes. Nucleic Acids Res. , 39 (Database issue), D691–D697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culhane A.C., et al. (2012) GeneSigDB: a manually curated database and resource for analysis of gene expression signatures. Nucleic Acids Res., 40 (Database issue): D1060–D1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis G., et al. (2003) DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 4, P3. [PubMed] [Google Scholar]
- Dinu I., et al. (2009) Gene-set analysis and reduction. Brief. Bioinform. , 10, 24–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkon R., et al. (2008) SPIKE—a database, visualization and analysis tool of cellular signaling pathways. BMC Bioinformatics, 9, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EllisLab (2014) CodeIgniter. In: British Columbia Institute of Technology. [Google Scholar]
- Ganter B., Giroux C.N. (2008) Emerging applications of network and pathway analysis in drug discovery and development. Curr. Opin. Drug Discov. Develop. 11, 86–94. [PubMed] [Google Scholar]
- Glaab E., et al. (2012) EnrichNet: network-based gene set enrichment analysis. Bioinformatics , 28, i451–i457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale P.J., et al. (2012) Genome-wide meta-analysis of genetic susceptible genes for Type 2 Diabetes. BMC Syst. Biol. , 6 (Suppl 3), S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W., et al. (2009) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. , 37, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H., et al. (2012) PAGED: a pathway and gene-set enrichment database to enable molecular phenotype discoveries. BMC Bioinformatics, 13 (Suppl 15), S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung J.H. (2013) Gene Set/Pathway enrichment analysis. Methods Mol. Biol., 939, 201–213. [DOI] [PubMed] [Google Scholar]
- Jiang C., et al. (2007) TRED: a transcriptional regulatory element database, new entries and other development. Nucleic Acids Res. , 35 (Database issue), D137–D140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatri P., et al. (2012) Ten years of pathway analysis: current approaches and outstanding challenges. PLoS Comput. Biol. , 8, e1002375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SB., et al. (2007) GAzer: gene set analyzer. Bioinformatics, 23, 1697–1699. [DOI] [PubMed] [Google Scholar]
- Li Y., et al. (2008) A global pathway crosstalk network. Bioinformatics , 24, 1442–1447. [DOI] [PubMed] [Google Scholar]
- Liberzon A., et al. (2011) Molecular signatures database (MSigDB) 3.0. Bioinformatics, 27, 1739–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W., et al. (2009) GAGE: generally applicable gene set enrichment for pathway analysis. BMC Bioinformatics , 10, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martini P., et al. (2013) Along signal paths: an empirical gene set approach exploiting pathway topology. Nucleic Acids Res., 41, e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meagher P., et al. (2013) Probability Distributions Library (PDL). In. [Google Scholar]
- Murohashi M., et al. (2010) Gene set enrichment analysis provides insight into novel signalling pathways in breast cancer stem cells. Br. J. Cancer , 102, 206–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam D. (2010) De-correlating expression in gene-set analysis. Bioinformatics , 26, i511–i516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam D., Kim S.Y. (2008) Gene-set approach for expression pattern analysis. Brief. Bioinform. , 9, 189–197. [DOI] [PubMed] [Google Scholar]
- Nishimura D. (2001) BioCarta. Biotech Softw. Internet Rep., 2, 117–120. [Google Scholar]
- Ogata H., et al. (1999) KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. , 27, 29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oprea T.I., et al. (2007) Systems chemical biology. Nat. Chem. Biol. , 3, 447–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrand-Rosenberg S., Sinha P. (2009) Myeloid-derived suppressor cells: linking inflammation and cancer. J. Immunol., 182, 4499–4506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh J.R., et al. (2012) Multi-edge gene set networks reveal novel insights into global relationships between biological themes. PloS One , 7, e45211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pico A.R., et al. (2008) WikiPathways: pathway editing for the people. PLoS Biol., 6, e184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer C.F., et al. (2009) PID: the pathway interaction database. Nucleic Acids Res. , 37 (Database issue), D674–D679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivachenko A.Y., Yuryev A. (2007) Pathway analysis software as a tool for drug target selection, prioritization and validation of drug mechanism. Exp. Opin. Ther. Targets , 11, 411–421. [DOI] [PubMed] [Google Scholar]
- Subramania A., et al. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA , 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D., et al. (2011) The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. , 39 (Database issue):D561–D568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorn C.F., et al. (2010) Pharmacogenomics and bioinformatics: PharmGKB. Pharmacogenomics , 11, 501–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tusher V.G., et al. (2001) Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. USA , 98, 5116–5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt C. (2013) Update on activities at the Universal Protein Resource (UniProt) in 2013. Nucleic Acids Res. , 41 (Database issue), D43–D47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welter D., et al. (2014) The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. , 42 (Database issue), D1001–D1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingender E., et al. (1996) TRANSFAC: a database on transcription factors and their DNA binding sites. Nucleic Acids Res., 24, 238–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M.C., et al. (2010) Powerful SNP-set analysis for case-control genome-wide association studies. Am. J. Hum. Genet. , 86, 929–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X., et al. (2012) Pathway and network analysis in proteomics. J. Theor. Biol. , 362, 44–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J., Wishart D.S. (2010) MetPA: a web-based metabolomics tool for pathway analysis and visualization. Bioinformatics, 26, 2342–2344. [DOI] [PubMed] [Google Scholar]
- Xia J., et al. (2012) NGS catalog: a database of next generation sequencing studies in humans. Hum. Mut., 33, E2341–E2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F., Chen J.Y. (2013) Breast cancer subtyping from plasma proteins. BMC Med. Genom. , 6 (Suppl 1), S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.