

Abstract
Chicken is a major food source for humans, hence it is important to understand the mechanisms involved in nutrient absorption in chicken. In the gastrointestinal tract (GIT), the microbiota plays a central role in enhancing nutrient absorption and strengthening the immune system, thereby affecting both growth and health of chicken. There is little information on the diversity and functions of chicken GIT microbiota, its impact on the host, and the interactions between the microbiota and host. Here, we review the recent metagenomic strategies to analyze the chicken GIT microbiota composition and its functions related to improving metabolism and health. We summarize methodology of metagenomics in order to obtain bacterial taxonomy and functional inferences of the GIT microbiota and suggest a set of indicator genes for monitoring and manipulating the microbiota to promote host health in future.
Keywords: Chicken Gastrointestinal Tract, Microbiota, Metagenomics
INTRODUCTION
Domestic chickens are the most common poultry and are valuable sources of protein for humans. Recently, the dual roles of microbiota in the gastrointestinal tract (GIT) have attracted attention: they support digestion of food and also provide nutrients. In addition, the GIT microbiota may play crucial roles in the health and immune system of chickens (Brisbin et al., 2008; Stanley et al., 2014). However, the diverse and complex structure of GIT microbiota hindered the understanding of the roles of GIT microbiota in chicken’s growth and health. As relatively few cultivated specimens of chicken GIT microbes are available mainly due to their requirement for anaerobic conditions similar to those in the GIT, researchers have turned to new culture-independent approaches, metagenomics, to predict nutritional and ecological roles of GIT microbes. Metagenomics is the study of genetic materials from environmental or host-associated microbiota to identify the microbial diversity and its functions. Previously, conventional, low-throughput Sanger sequencing method has been limited in metagenomic studies of GIT microbiota due to high sequencing costs and insufficient sequence data. Recently, newly developed sequencing platforms such as next-generation sequencing (NGS) have allowed the substantial researches into the diversity and functions of microbiota from the guts of various livestock animals. High-throughput NGS generates large volumes sequence data containing genetic information (Metzker, 2010), and this allows hypothesis-driven researches on chicken GIT microbiota, thereby highlighting the roles of previously unknown and rare microbial GIT species (Sogin et al., 2006; Medinger et al., 2010). Furthermore, metagenomic data have raised new questions such as how microbiota stability and ecological shifts are influenced by nutrients and hosts (Andersson et al., 2008). Here, we review the current status on metagenomics-driven studies on chicken GIT’s microbiota and the involved methodology, and suggest possible approaches for better understanding of GIT microbiota to increase chicken productivity and overall health.
CHARACTERIZATION OF CHICKEN GIT MICROBIOTA
Bacterial community profiling based on 16S rRNA gene sequence and functional inference
When determining the composition of GIT microbiota using a gene-targeted-metagenomic approach, several marker genes such as those for ribosomal protein subunits, elongation factors, and RNA polymerase subunits represent distinct microbial populations. One of the most general choices for resolving the taxonomic composition of the microbial community is the hypervariable region in genes encoding 16S rRNA (Figure 1). Bacterial 16S rRNA gene possesses 9 hypervariable regions flanked by highly conserved regions, which are generally designated as polymerase chain reaction (PCR) primer sites. Sequence variations in hypervariable regions allow accurate bacterial taxonomic estimation by comparing against 16S rRNA gene sequences deposited in databases such as GreenGenes (DeSantis et al., 2006), the Ribosomal Database Project (Cole et al., 2014), and SILVA (Pruesse et al., 2007). Several bioinformatic pipelines of 16S rRNA gene NGS-based profiling (e.g., 16S amplicon sequencing: using 454 pyrosequencing, Illumina MiSeq, and Ion PGM Systems) have been proposed and designed for obtaining taxonomic information via processing raw sequences of 16S rRNA genes. These pipelines generally consist of the following steps: quality filtering of low-quality sequences and chimera checking (Edgar et al., 2011; Haas et al., 2011), removing pyrosequencing-oriented sequencing errors (Huse et al., 2007), and producing a cluster of nearly identical sequences referred to operational taxonomic units (OTUs) (Caporaso et al., 2010; Cole et al., 2014). OTUs are defined by two approaches i) unsupervised clustering on sequence similarity and ii) supervised clustering that directly assigns sequences to trained taxonomic clusters (Sul et al., 2011). For undefined microbiota, OTUs generated by unsupervised clustering are generally used to serve as individual phylotypes (Medinger et al., 2010). The representative sequences, which are either the most abundant sequences or sequence with the least distance summation within all sequences in the same OTUs, are then matched to those in the public database (e.g. the Ribosomal Database Project) to obtain taxonomic classification. This information has also been used to account for the degree of divergence between different communities or sample types. The latter approach is most useful for studying low-complexity microbiota and comparative analysis of the same type of samples under different experimental conditions.
Figure 1.
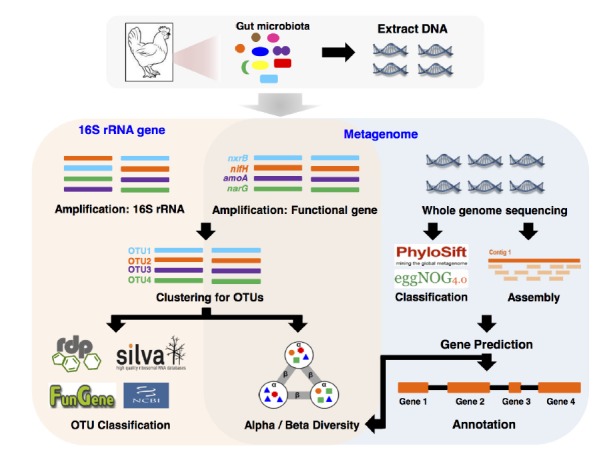
Metagenomic strategies for chicken GIT microbiota. GIT, gastrointestinal tract.
In addition to providing taxonomic information, the OTUs provide information on population diversity, indicating richness and evenness of individual species in a sample as alpha diversity (Colwell, 2009). Both richness and evenness of the diversity of the microbiota are often important indicators of the health of livestock. For instance, the number of microbes in the gut decreases during antibiotic treatment or nutrient imbalance (Lozupone et al., 2012). Several indices, including Chao, abundance-based coverage estimators, Shannon, and Simpson, calculate the abundance or distribution of OTUs within a particular population. For example, low richness suggests low number of species in the community and low evenness indicates that the sample consists of a few dominating taxa. These alpha diversity indices could be obtained using a bioinformatics pipeline such as MOTHUR (Hughes et al., 2011; Schloss et al., 2009) and Qiime (Caporaso et al., 2010). MOTHUR output consists of numeric data presented in a table or as rarefaction curves, which can then be compared with the values for the microbial community samples. Alpha diversity is generally considered to provide a poor reflection of microbial functional inference as an ecosystem. Although alpha diversity is not consistent with different samples, functionality can be maintained and replaced by the closely related and/or functionally similar species (Purschke et al., 2013). However, recent reports showed that alpha diversity is strongly correlated with diversity in a specific gene functions (Johnson et al., 2014). In addition, a few studies suggested that gut microbiota with high diversity could be more stable or healthier than those with low diversity (Clemente et al., 2012; Lozupone et al., 2012). These studies indicate that alpha diversity can contribute functional inference about the underlying microbiota mechanism.
Beta diversity is used when comparing multiple communities to determine the number of OTUs or taxa shared among them. Beta diversity integrates information about the degree of functional dissimilarity between multiple populations. Beta diversity calculated as a similarity index (e.g., Jaccard, Bray-Curtis) that shows shared populations between the microbial profiles of multiple communities, whereas alpha diversity shows a statistical summary of diversity in a single population (Martiny et al., 2011). Two approaches to measure beta diversity are widely used: unweighted UniFrac (qualitative) and weighted UniFrac (quantitative) (Lozupone and Knight 2005; 2007). UniFrac is a method to calculate a distance between microbial communities using phylogenetic diversity. Many studies using UniFrac show that beta diversity is strongly influenced by environmental factors such as substrates, temperature, and pH.
Inflation of microbial diversity results could occur during a sequencing process due to sample preparation and preservation, DNA extraction, and choice of PCR primers (Lauber et al., 2010; Cruaud et al., 2014; Hang et al., 2014). The diversity results can be statistically varied even though the samples were stored at 4°C or lower. DNA extraction methods seem to affect the diversity results in different sample types. The choice of 16S rRNA gene’s hypervariable regions to be amplified by selected PCR primers introduces significant variation in the microbial diversities in the samples, because different regions show considerable differences in taxonomic coverage. These artifacts clearly indicate that the diversity results with different sample preparations should be carefully interpreted.
In diversity analysis, it is important to account for the contribution of sequencing errors on dramatically different outputs (Huse et al., 2007; Kunin et al., 2010). These errors can be caused by regions of 16S rRNA, read length and sequencing technology (Gilles et al., 2011). Recently, Illumina MiSeq and HiSeq are becoming widely popular sequencing platform for high-throughput sequencing since sequencing errors produced in these systems are easier to manage computationally than those from 454 pyrosequencing (Claesson et al., 2010; Luo et al., 2012). Another type of error that occurs during gene amplification is generation of chimeric sequences from two unique parental 16S rRNA gene sequences. Various computation programs have been developed to remove chimeric sequences by using reference sequences or self-query sequences (Edgar et al., 2011; Haas et al., 2011). Quality sequence filtering process is essential before analysis of microbial diversity.
Functional gene-targeted metagenomics
In contrast to 16S rRNA gene profiling, analysis of genes encoding proteins that function to regulate metabolic pathways can help to explore the diversity of particular ecological functions in the GIT sample (Figure 1). Further, protein-coding genes can be used as phylogenetic markers (most likely with 16S rRNA gene), although the high evolutionary rate of functional genes can mislead to linking with functional and taxonomic results (Case et al., 2007). Functional Gene Pipeline and Repository (FunGene; http://fungene.cme.msu.edu/) provides databases of biogeochemical, biodegradation, and antibiotic functional genes, as well as a pipeline that allows analysis of functional diversity based on OTUs (Fish et al., 2013). Using FunGene, the sequences are analyzed through two approaches: reference-based approaches or unsupervised approaches. The reference-based approaches use a tool, named FrameBot, to correct a sequencing error generated frameshift by matching the sequences against curated reference sequences (Wang et al., 2013). The unsupervised approaches cluster the aligned sequences generated by HMMER3, which is based on implementation of hidden Markov model for searching similar protein sequences and for making protein sequence alignment (Finn et al., 2011). All clustered sequences can be submitted into calculation of alpha and beta diversity, and classification of representative sequences extracted from each cluster using FunGene database. Approaches to further analysis of functional genes would be similar to analysis of 16S rRNA genes, but abundance of functional genes can be highly influenced by the function of interest in the ecosystem (Pereyra et al., 2010). Therefore, the accurate and fast quantification of functional genes in the ecosystem is a critical step to understand the potential of microbial function in the ecosystem. Multiplex PCR combined with q-PCR (Multiplex q-PCR) is an ideal tool to be used in this context, since not only quantitative results about the target functional gene, but also the distribution of multiple genes in the ecosystem is crucial for understanding microbial function in different ecosystems (Elnifro et al., 2000; Smith and Osborn, 2009). The multiplex q-PCR can amplify more than one targeted gene by addition of more than one pair of primers in the same reaction. This technique shows strong potential to save time and labor for multiple samples and genes targeted in different areas (Stedtfeld et al., 2008). For example, 18 antibiotic resistance types (total 141 primer sets) were successfully tested to evaluate the abundance and diversity of antibiotic resistance genes in swine gut microbiota using multiplex q-PCR (Looft et al., 2012). Recently, 3rd generation PCR technology, named digital PCR (dPCR), has developed for directly quantifying and clonally amplifying nucleic acids. Although dPCR is carried out a single reaction within a sample, the sample is distributed into a hundred thousands of partitions and PCR reactions are carried out in parallel. This feature provides for more accurately quantifying results than conventional q-PCR. Microarrays have been developed to use gene expression studies and monitor environmental processes. GeoChip is one of well-known comprehensive microarray chips in microbial ecology, especially useful to study ecosystem processes and functions (He et al., 2007). Current version of GeoChip contains more than 50,000 gene probes involved in biogeochemical cycles, metal reduction, resistance and organic compound degradation. It is a promising tool for high-throughput and cost-effective analysis in quantifying functional genes.
Although amplicon-based profiling is the most common approach to study environmental microbial diversity, it showed several limitations including amplicon size, primer (or probe) sensitivity, and errors during amplification and contamination. However, metagenome analysis without the PCR amplification step can be a powerful alternative to amplicon-based profiling for investigating all metabolic processes of microorganisms found in environmental samples. The process of metagenomic analysis compared with amplicon-based profiling is summarized in Figure 1. The simplest way to analyze the metagenome is by using short reads directly after the quality sequence filtering step (Thomas et al., 2012). These short reads can be used as taxonomic markers and matched against a marker gene database (e.g. Phylosift, EggNOG) and functional markers quantifying abundance of genetic pathways (Darling et al., 2014; Powell et al., 2014). These analyses provide taxonomic (e.g. MEGAN) and functional (e.g. KEGG pathway) contents of single data sets to rapidly search for interesting research targets and test hypothesis generated from experimental design. Alternatively, assembly algorithms can be used to reconstruct short reads into a sequence contig, which is a set of overlapping sequences representing the contiguous DNA fragment (Mende et al., 2012; Thomas et al., 2012). Although obtaining a complete individual genome from metagenomic sequences is still challenging unless high sequencing coverage, it is sufficient to characterize the major functions of the microbial communities as well as to identify their taxon by assigning to public genome reference database (Howe et al., 2014; Nielsen et al., 2014).
THE DIFFERENT ROLES OF CHICKEN GIT MICROBIOTA IN NUTRIENT METABOLISM IN HOST
The chicken GIT is like a machine for metabolizing nutrients. In most recent studies, researchers have not focused on culture-dependent studies but on culture-independent phylogenetic profiling against chicken GIT. Taking advantages of NGS, metagenomic approaches revealed the bacterial composition of the chicken GIT and also functional genes highly related to host digestion and nutrient absorption mechanisms. This technique brings us understanding of the taxonomic variety of the co-metabolizing micro-community within the host and the gene level of target nutrients metabolism. Therefore, we reviewed the recent reports about the candidates of target bacteria or genes to understand how they can help in host metabolism.
Polysaccharide metabolism
The caecum is a key organ for fermentation of various forms of polysaccharides and is highly associated with host health and performance (Stanley et al., 2014). Cecal microbiota play a central role in metabolizing polysaccharides because chickens do not possess a complete metabolic cycle for producing readily absorbable forms of polysaccharides. Recently, Sergeant et al. (2014) reported numerous polysaccharide degrading enzymes in the cecal metagenome, and Qu et al. (2008) showed that the genes for carbohydrate metabolism occupied about 20% genes in the GIT microbiota metagenome (Qu et al., 2008; Sergeant et al., 2014) including key enzymes like: carbohydrate esterase, polysaccharide lyase, and glycoside hydrolase which are lacking in chickens (Beckmann et al., 2006; Yeoman et al., 2012). During the digestion of polysaccharides, GIT microbiota produced various short chain volatile fatty acids (SCFA), mostly acetate, while propionate, butylate, valerate, isobutylate, and isovalerate are other forms also found (Dunkley et al., 2007; Yeoman et al., 2012). The ratio of SCFA production was lower during the uptake and digestion of soluble polysaccharides than during the uptake and digestion of insoluble polysaccharides (Lei et al., 2012). In addition, carbohydrate metabolism-related proteins were most commonly found in the metaproteomic analysis of chicken GIT microbiota (Tang et al., 2014). Recently, Tang et al. (2014) showed the positive correlation between metagenomic and metaproteomic analyses for chicken microbiome composition and protein expression.
Nitrogen metabolism
Recently, Qu et al. (2008) reported that the environmental gene tags of the subsystems-based annotations (SEED) database showed that the GIT microbiota has genes involved in metabolism of protein and amino acids (9%), and nitrogen (1%) as nutrient sources (Qu et al., 2008). In case of host leaked or incompletely metabolized dietary protein, the GIT microbiota digested further amino acids as host nutrient because chickens unlike some other animals lack the enzymes required for complete nitrogen metabolism (Latshaw and Zhao, 2011). As a result, effective nitrogen metabolism by GIT microbiota has produced healthier and more productive chickens (Stanley et al., 2014). When chicken GIT microbiota metabolized nitrogen sources, half of the nitrogen sources were converted into ammonia, which was excreted subsequently and this loss adversely affected the health of the chicken (McCubbin et al., 2002; Xin et al., 2011). Undissolved uric acid, which itself is not toxic, affects induction or degradation of bacterial proteins in chicken gut (Tang et al., 2014).
Fatty acids and lipids metabolism
Commercial chickens feed on grains which usually contain non-starch polysaccharides (NSPs) and the metabolism of NSPs produces SCFA. Despite the fact that Firmicutes, Bacteroides, and Proteobacteria were usually isolated or detected in the chicken ceca, Clostridiales contributed the most to SCFA metabolism (Oakley et al., 2014). SCFA can inhibit the growth of acid-sensitive pathogens by inducing low-pH environment and can improve both mineral absorption in host and growth of epithelial cells (Oakley et al., 2014; Sergeant et al., 2014). Generally, fatty acid and lipid metabolism related genes were detected to be only about 1% to 2% in the whole gut metagenome (Qu et al., 2008). Conjugated linoleic acid (CLA) is a by-product during metabolism of polyunsaturated linoleic acid and presence of CLA in the diet was reported to affect the death rate of embryo and quality of yolk (Aydin et al., 2001; Yeoman et al., 2012).
Host health
The presence of bacterial pathogens such as Salmonella, Clostridium, Campylobacter, Staphylococcus, and E. coli within chicken microbiota is a critical issue for host well-being (Oakley et al., 2014). Bacteria that are non-pathogenic to chickens and are found in chicken microbiota are sometimes pathogenic to humans (Newell et al., 2011). For example, recent reports suggest that most of Campylobacter infection cases in humans resulted from handling and eating raw or undercooked poultry meat (Kaakoush et al., 2014). Despite the presence of a pathogen (for humans) at a detectable level in the microbiota of healthy chickens, there were no significant changes in microbial community structure and diversity (Skraban et al., 2013). Similarly, even infection with a pathogen will not cause major changes in the remaining chicken resident microbiota (Videnska et al., 2013). Oakley et al. (2014) suggested that competitive exclusion is one of the most effective treatments for blocking pathogens, usually Salmonella, leading to enrichment in intestinal regulation (Oakley et al., 2014). The other approach is false signal binders, which mimic pathogen docking sites to gut epithelia that can prevent gut pathogens from recognizing and landing on these sites (Oakley et al., 2014).
The antibiotic effect on the GIT microbiome depends upon the dose of antibiotics and the age of host (Zhou et al., 2007; Stanley et al., 2014). In recent studies, no significant changes were detected in different flocks grown with or without antibiotics (Pedroso et al., 2013). Although, after discontinuing input of antibiotics, GIT microbiota was found to struggle to recover and to resist compositional change (Dethlefsen et al., 2008; Stanley et al., 2014). The mannan-oligosaccharide (MOS) treatment raised the Firmicutes population, whereas, there was no effect on the Bacteroidetes population. An increased ratio of Firmicutes/Bacteroidetes is known to be related to obesity in mouse and human, owing to improved energy harvesting capacity for Firmicutes species (Turnbaugh et al., 2006; Stanley et al., 2014). MOS has been shown to reduce the abundance of E. coli (Baurhoo et al., 2007) and Salmonella in cecal contents of experimentally infected chickens (Fernandez et al., 2002). This reduction may be due to the ability of mannose in MOS to bind to mannose-binding lectins of Gram-negative bacteria expressing type 1 fimbriae, which consequently, reduces bacterial attachment to the intestinal epithelial cells (Ganner and Schatzmayr, 2012). The recent study suggested that MOS promotes a greater increase of the villi heights and goblet cell numbers of birds under mild environmental stress than when under antibiotic treatment. Finally, MOS can increase mucins, a main component of mucus, production so that it can better maintain host intestinal health. The intestinal mucus thickness is a key for defending against invasion of enteric pathogens into epithelial cells and is reduced by antibiotics (Stanley et al., 2014). Unlike MOS, Fructooligosaccharide directly improves host performance, and while it does not inhibit pathogens activity, it promotes beneficial bacteria activity (Xu et al., 2003; Stanley et al., 2014). Several studies have reported that feed with probiotics containing Lactobacillus cultures can enhance chicken to gain weight more and create efficient feed absorption (Oakley et al., 2014). For example, two members of the Lactobacilli, Bifidobacterium thermophilum, and Enterococcus faecium, increased the intestinal muscle thickness and the jejuna villus perimeter and height (Chichlowski et al., 2007). Twelve Lactobacillus strains complex reduced serum cholesterol level in broilers (Jin et al., 1998). As shown in human and pig cases, Firmicutes also helps chickens to increase nutrient absorption, whereas Bacteriodetes decrease nutrient absorption (Jumpertz et al., 2011).
FUTURE TRENDS
Increase knowledge of the chicken GIT microbiome via culture-independent metagenomic analysis has helped to understand the dynamics of microbial communities and their role in metabolism and health state of chicken. Further GIT microbiota research will focus on finding the microbe’s role in mediating chicken growth under various environmental conditions, such as nutrients, stress and welfare to determine better quantity and quality of productivity. From metagenomic data of chicken GIT microbiota, identifying and tracking the key enzymatic genes (suggested in Table 1) related to metabolism and health states valuations may find an approach to improve the growth and productivity of chickens by combating deficiencies by administering in diet or doses, and introducing useful or less harmful bacteria possessing nutrient digesting genes. In addition, development of metagenomic approaches and bioinformatic tools will be crucial for progress in an analysis of chicken gut microbiota. By focusing on functional genes of chicken GIT microbiota, strategies for improving nutrient metabolism can be designed. For example, identifying the more effective polysaccharides degrading probiotic bacteria which then can be used to facilitate chicken metabolism. Strain-specific 16S rRNA gene sequencing with improved bioinformatic tools (Eren et al., 2014) can trace how each strain will be changed under various growth and environmental conditions and then monitor the abundance changes of target strains by food treatment. In addition, metabolism specific real-time PCR, performed with primer sets to detect each nutrient gene allows us to monitor the state of GIT microbiota. Metagenomic analyses in combination with other approaches will allow the identification of the microorganisms and real players among GIT microbiota that help in host metabolism and in maintaining its health.
Table 1.
Target genes for detecting GIT microbiota’s role metagenome in chicken metabolism
| Name | Metabolism | Function (protein) |
|---|---|---|
| Glucoside hydrolase | Carbohydrate | Hydrolysis of carbohydrate |
| Polysaccharide lyase | ||
| Carbohydrate esterase | ||
| Galactose transport protein | ||
| Glucosamine-6-phosphate deaminase | ||
| Methylmalonyl decarboxylase | Fatty acids | Propionate production |
| Butyryl-CoA:acetate-CoA transferase | Butyrate production | |
| 3-hydroxybutyryl-CoA dehydrolase | Butyrate fermentation | |
| Acetate kinase | Acetate fermentation | |
| Phosphate acetyltransferase | Acetate fermentation | |
| Methylmalonyl-CoA epimerase | Propionate production | |
| Dipeptidyl aminopeptidase | Nitrogen | Protein hydrolysis |
GIT, gastrointestinal tract.
ACKNOWLEDGMENTS
This research was supported by the Chung-Ang University Research Grants in 2013 and by a grant from the Strategic Initiative for Microbiomes in Agriculture and Food, Ministry of Agriculture, Food and Rural Affairs, Republic of Korea (as part of the [multi-ministerial] Genome Technology to Business Translation Program).
REFERENCES
- Andersson AF, Lindberg M, Jakobsson H, Bäckhed F, Nyrén P, Engstrand L. Comparative analysis of human gut microbiota by barcoded pyrosequencing. PLoS One. 2008;3(7):e2836. doi: 10.1371/journal.pone.0002836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aydin R, Pariza MW, Cook ME. Olive oil prevents the adverse effects of dietary conjugated linoleic acid on chick hatchability and egg quality. J Nutr. 2001;131:800–806. doi: 10.1093/jn/131.3.800. [DOI] [PubMed] [Google Scholar]
- Baurhoo B, Phillip L, Ruiz-Feria CA. Effects of purified lignin and mannan oligosaccharides on intestinal integrity and microbial populations in the ceca and litter of broiler chickens. Poult Sci. 2007;86:1070–1078. doi: 10.1093/ps/86.6.1070. [DOI] [PubMed] [Google Scholar]
- Beckmann L, Simon O, Vahjen W. Isolation and identification of mixed linked beta-glucan degrading bacteria in the intestine of broiler chickens and partial characterization of respective 1,3-1,4-beta-glucanase activities. J Basic Microbiol. 2006;46:175–185. doi: 10.1002/jobm.200510107. [DOI] [PubMed] [Google Scholar]
- Brisbin JT, Gong J, Sharif S. Interactions between commensal bacteria and the gut-associated immune system of the chicken. Anim Health Res Rev. 2008;9:101–110. doi: 10.1017/S146625230800145X. [DOI] [PubMed] [Google Scholar]
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case RJ, Boucher Y, Dahllöf I, Holmström C, Doolittle WF, Kjelleberg S. Use of 16S rRNA and rpoB genes as molecular markers for microbial ecology studies. Appl Environ Microbiol. 2007;73:278–288. doi: 10.1128/AEM.01177-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chichlowski M, Croom WJ, Edens FW, McBride BW, Qiu R, Chiang CC, Daniel LR, Havenstein GB, Koci MD. Microarchitecture and spatial relationship between bacteria and ileal, cecal, and colonic epithelium in chicks fed a direct-fed microbial, PrimaLac, and salinomycin. Poult Sci. 2007;86:1121–1132. doi: 10.1093/ps/86.6.1121. [DOI] [PubMed] [Google Scholar]
- Claesson MJ, Wang Q, O’Sullivan O, Greene-Diniz R, Cole JR, Ross RP, O’Toole PW. Comparison of two next-generation sequencing technologies for resolving highly complex microbiota composition using tandem variable 16S rRNA gene regions. Nucl Acids Res. 2010;38:e200. doi: 10.1093/nar/gkq873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemente JC, Ursell LK, Parfrey LW, Knight R. The impact of the gut microbiota on human health: An integrative view. Cell. 2012;148:1258–1270. doi: 10.1016/j.cell.2012.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole JR, Wang Q, Fish JA, Chai B, McGarrell DM, Sun Y, Brown CT, Porras-Alfaro A, Kuske CR, Tiedje JM. Ribosomal Database Project: data and tools for high throughput rRNA analysis. Nucl Acids Res. 2014;42:D633–642. doi: 10.1093/nar/gkt1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colwell RK. Biodiversity: concepts, patterns, and measurement. In: Levin SA, editor. The Princeton Guide to Ecology. Princeton University Press; Princeton, NJ, USA: 2009. pp. 257–263. [Google Scholar]
- Cruaud R, Vigneron A, Lucchetti-Miganeh C, Ciron PE, Godfroy A, Cambon-Bonavita MA. Influence of DNA extraction method, 16S rRNA targeted hypervariable regions, and sample origin on microbial diversity detected by 454 pyrosequencing in marine chemosynthetic ecosystems. Appl Environ Microbiol. 2014;80:4626–4639. doi: 10.1128/AEM.00592-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling AE, Jospin G, Lowe E, Matsen FA, 4th, Bik HM, Eisen JA. PhyloSift: phylogenetic analysis of genomes and metagenomes. PeerJ. 2014;2:e243. doi: 10.7717/peerj.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol. 2006;72:5069–5072. doi: 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dethlefsen L, Huse S, Sogin ML, Relman DA. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol. 2008;6(11):e280. doi: 10.1371/journal.pbio.0060280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunkley KD, Dunkley CS, Njongmeta NL, Callaway TR, Hume ME, Kubena LF, Nisbet DJ, Ricke SC. Comparison of in vitro fermentation and molecular microbial profiles of high-fiber feed substrates incubated with chicken cecal inocula. Poult Sci. 2007;86:801–810. doi: 10.1093/ps/86.5.801. [DOI] [PubMed] [Google Scholar]
- Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27:2194–2200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elnifro EM, Ashshi AM, Cooper RJ, Klapper PE. Multiplex PCR: optimization and application in diagnostic virology. Clin Microbiol Rev. 2000;13:559–570. doi: 10.1128/cmr.13.4.559-570.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eren AM, Maignien L, Sul WJ, Murphy LG, Grim SL, Morrison HG, Sogin ML. Oligotyping: Differentiating between closely related microbial taxa using 16S rRNA gene data. Methods Ecol Evol. 2014;4:1111–1119. doi: 10.1111/2041-210X.12114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez F, Hinton M, Van Gils B. Dietary mannan-oligosaccharides and their effect on chicken caecal microflora in relation to Salmonella Enteritidis colonization. Avian Pathol. 2002;31:49–58. doi: 10.1080/03079450120106000. [DOI] [PubMed] [Google Scholar]
- Fish JA, Chai B, Wang Q, Sun Y, Brown CT, Tiedje JM, Cole JR. FunGene: the functional gene pipeline and repository. Front Microbiol. 2013;4:291. doi: 10.3389/fmicb.2013.00291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RD, Clements J, Eddy SR. HMMER web server: interactive sequence similarity searching. Nucl Acids Res. 2011;39:W29–37. doi: 10.1093/nar/gkr367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganner A, Schatzmayr G. Capability of yeast derivatives to adhere enteropathogenic bacteria and to modulate cells of the innate immune system. Appl Microbiol Biotechnol. 2012;95:289–297. doi: 10.1007/s00253-012-4140-y. [DOI] [PubMed] [Google Scholar]
- Gilles A, Meglécz E, Pech N, Ferreira S, Malausa T, Martin JF. Accuracy and quality assessment of 454 GS-FLX Titanium pyrosequencing. BMC Genomics. 2011;12:245. doi: 10.1186/1471-2164-12-245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas BJ, Gevers D, Earl AM, Feldgarden M, Ward DV, Giannoukos G, Ciulla D, Tabbaa D, Highlander SK, Sodergren E, Methé B, DeSantis TZ, Petrosino JF, Knight R, Birren BW Human Microbiome Consortium. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 2011;21:494–504. doi: 10.1101/gr.112730.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hang J, Desai V, Zavajevski N, Yang Y, Lin X, Satya RV, Martinez LJ, Blaylock JM, Jarman RG, Thomas SJ, Kuschner RA. 16S rRNA gene pyrosequencing of reference and clinical samples and investigation of the temperature stability of microbiome profiles. Microbiome. 2014;2:31. doi: 10.1186/2049-2618-2-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Z, Gentry TJ, Schadt CW, Wu L, Liebich J, Chong SC, Huang Z, Wu W, Gu B, Jardine P, Criddle C, Zhou J. GeoChip: A comprehensive microarray for investigating biogeochemical, ecological and environmental processes. ISME J. 2007;1:67–77. doi: 10.1038/ismej.2007.2. [DOI] [PubMed] [Google Scholar]
- Howe AC, Jansson JK, Malfatti SA, Tringe SG, Tiedje JM, Brown CT. Tackling soil diversity with the assembly of large, complex metagenomes. Proc Natl Acad Sci USA. 2014;111:4904–4909. doi: 10.1073/pnas.1402564111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes JB, Hellmann JJ, Ricketts TH, Bohannan BJ. Counting the uncountable: statistical approaches to estimating microbial diversity. Appl Environ Microbiol. 2011;67:4399–4406. doi: 10.1128/AEM.67.10.4399-4406.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huse SM, Huber JA, Morrison HG, Sogin ML, Welch DM. Accuracy and quality of massively parallel DNA pyrosequencing. Genome Biol. 2007;8:R143. doi: 10.1186/gb-2007-8-7-r143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin LZ, Ho YW, Abdullah N, Jalaludin S. Growth performance, intestinal microbial populations, and serum cholesterol of broilers fed diets containing Lactobacillus cultures. Poult Sci. 1998;77:1259–1265. doi: 10.1093/ps/77.9.1259. [DOI] [PubMed] [Google Scholar]
- Johnson DR, Lee TK, Park J, Fenner K, Helbling DE. The functional and taxonomic richness of wastewater treatment plant microbial communities are associated with each other and with ambient nitrogen and carbon availability. Environ Microbiol. 2014 doi: 10.1111/1462-2920.12429. [DOI] [PubMed] [Google Scholar]
- Jumpertz R, Le DS, Turnbaugh PJ, Trinidad C, Bogardus C, Gordon JI, Krakoff J. Energy-balance studies reveal associations between gut microbes, caloric load, and nutrient absorption in humans. Am J Clin Nutr. 2011;94:58–65. doi: 10.3945/ajcn.110.010132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaakoush NO, Sodhi N, Chenu JW, Cox JM, Riordan SM, Mitchell HM. The interplay between Campylobacter and Helicobacter species and other gastrointestinal microbiota of commercial broiler chickens. Gut Pathog. 2014;6:18. doi: 10.1186/1757-4749-6-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunin V, Engelbrektson A, Ochman H, Hugenholtz P. Wrinkles in the rare biosphere: Pyrosequencing errors can lead to artificial inflation of diversity estimates. Environ Microbiol. 2010;12:118–123. doi: 10.1111/j.1462-2920.2009.02051.x. [DOI] [PubMed] [Google Scholar]
- Latshaw JD, Zhao L. Dietary protein effects on hen performance and nitrogen excretion. Poult Sci. 2011;90:99–106. doi: 10.3382/ps.2010-01035. [DOI] [PubMed] [Google Scholar]
- Lei F, Yin Y, Wang Y, Deng B, Yu HD, Li L, Xiang C, Wang S, Zhu B, Wang X. Higher-level production of volatile fatty acids in vitro by chicken gut microbiotas than by human gut microbiotas as determined by functional analyses. Appl Environ Microbiol. 2012;78:5763–5772. doi: 10.1128/AEM.00327-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauber CL, Zhou N, Gordon JI, Knight R, Fierer N. Effect of storage conditions on the assessment of bacterial community structure in soil and human-associated samples. FEMS Microbiol Lett. 2010;307(1):80–86. doi: 10.1111/j.1574-6968.2010.01965.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei F, Yin Y, Wang Y, Deng B, Yu HD, Li L, Xiang C, Wang S, Zhu B, Wang X. Higher-level production of volatile fatty acids in vitro by chicken gut microbiotas than by human gut microbiotas as determined by functional analyses. Appl Environ Microbiol. 2012;78:5763–5772. doi: 10.1128/AEM.00327-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Looft T, Johnson TA, Allen HK, Bayles DO, Alt DP, Stedtfeld RD, Sul WJ, Stedtfeld TM, Chai B, Cole JR, Hashsham SA, Tiedje JM, Stanton TB. In-feed antibiotic effects on the swine intestinal microbiome. Proc Natl Acad Sci USA. 2012;109:1691–1696. doi: 10.1073/pnas.1120238109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone CA, Knight R. UniFrac: A new phylogenetic method for comparing microbial communities. Appl Environ Microbiol. 2005;71:8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone CA, Knight R. Global patterns in bacterial diversity. Proc Natl Acad Sci USA. 2007;104:11436–11440. doi: 10.1073/pnas.0611525104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. Diversity, stability and resilience of the human gut microbiota. Nature. 2012;489:220–230. doi: 10.1038/nature11550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo C, Tsementzi D, Kyrpides N, Read T, Konstantinidis KT. Direct comparisons of Illumina vs. Roche 454 sequencing technologies on the same microbial community DNA sample. PLoS One. 2012;7(2):e30087. doi: 10.1371/journal.pone.0030087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martiny JBH, Eisen JA, Penn K, Allison SD, Horner-Devine MC. Drivers of bacterial beta-diversity depend on spatial scale. Proc Natl Acad Sci USA. 2011;108:7850–7854. doi: 10.1073/pnas.1016308108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCubbin DR, Apelberg BJ, Roe S, Divita F. Livestock ammonia management and particulate-related health benefits. Environ Sci Technol. 2002;36:1141–1146. doi: 10.1021/es010705g. [DOI] [PubMed] [Google Scholar]
- Medinger R, Nolte V, Pandey RV, Jost S, Ottenwälder B, Schlötterer C, Boenigk J. Diversity in a hidden world: potential and limitation of next-generation sequencing for surveys of molecular diversity of eukaryotic microorganisms. Mol Ecol. 2010;1:32–40. doi: 10.1111/j.1365-294X.2009.04478.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mende DR, Waller AS, Sunagawa S, Järvelin AI, Chan MM, Arumugam M, Raes J, Bork P. Assessment of metagenomic assembly using simulated next generation sequencing data. PLoS One. 2012;7(2):e31386. doi: 10.1371/journal.pone.0031386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzker ML. Sequencing technologies - the next generation. Nat Rev Genet. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- Morris SC. Life’s Solution: Inevitable Humans in a Lonely Universe. Cambridge University Press; Cambridge, UK: 2003. [Google Scholar]
- Newell DG, Elvers KT, Dopfer D, Hansson I, Jones P, James S, Gittins J, Stern NJ, Davies R, Connerton I, Pearson D, Salvat G, Allen VM. Biosecurity-based interventions and strategies to reduce Campylobacter spp. on poultry farms. Appl Environ Microbiol. 2011;77:8605–8614. doi: 10.1128/AEM.01090-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen HB, Almeida M, Juncker AS, Rasmussen S, Li J, Sunagawa S, Plichta DR, Gautier L, Pedersen AG, Le Chatelier E, et al. Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes. Nat Biotechnol. 2014;32:822–828. doi: 10.1038/nbt.2939. [DOI] [PubMed] [Google Scholar]
- Oakley BB, Lillehoj HS, Kogut MH, Kim WK, Maurer JJ, Pedroso A, Lee MD, Collett SR, Johnson TJ, Cox NA. The chicken gastrointestinal microbiome. FEMS Microbiol Lett. 2014;360:100–112. doi: 10.1111/1574-6968.12608. [DOI] [PubMed] [Google Scholar]
- Pedroso AA, Hurley-Bacon AL, Zedek AS, Kwan TW, Jordan APO, Avellaneda G, Hofacre CL, Oakley BB, Collett SR, Maurer JJ, Lee MD. Can probiotics improve the environmental microbiome and resistome of commercial poultry production? Int J Environ Res Public Health. 2013;10:4534–4559. doi: 10.3390/ijerph10104534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereyra LP, Hiibel SR, Prieto Riquelme MV, Reardon KF, Pruden A. Detection and quantification of functional genes of cellulose-degrading, fermentative, and sulfate-reducing bacteria and methanogenic archaea. Appl Environ Microbiol. 2010;76:2192–2202. doi: 10.1128/AEM.01285-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell S, Forslund K, Szklarczyk D, Trachana K, Roth A, Huerta-Cepas J, Gabaldón T, Rattei T, Creevey C, Kuhn M, Jensen LJ, von Mering C, Bork P. eggNOG v4.0: nested orthology inference across 3686 organisms. Nucl Acids Res. 2014;42:D231–239. doi: 10.1093/nar/gkt1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, Glöckner FO. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007;35:7188–7196. doi: 10.1093/nar/gkm864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purschke O, Schmid BC, Sykes MT, Poschlod P, Michalski SG, Durka W, Kuhn I, Winter M, Prentice HC. Contrasting changes in taxonomic, phylogenetic and functional diversity during a long-term succession: Insights into assembly processes. J Ecol. 2013;101:857–866. [Google Scholar]
- Qu A, Brulc JM, Wilson MK, Law BF, Theoret JR, Joens LA, Konkel ME, Angly F, Dinsdale EA, Edwards RA, Nelson KE, White BA. Comparative metagenomics reveals host specific metavirulomes and horizontal gene transfer elements in the chicken cecum microbiome. PLoS One. 2008;3(8):e2945. doi: 10.1371/journal.pone.0002945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergeant MJ, Constantinidou C, Cogan TA, Bedford MR, Penn CW, Pallen MJ. Extensive microbial and functional diversity within the chicken cecal microbiome. PLoS ONE. 2014;9(3):e91941. doi: 10.1371/journal.pone.0091941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skraban J, Dzeroski S, Zenko B, Tusar L, Rupnik M. Changes of poultry faecal microbiota associated with Clostridium difficile colonisation. Vet Microbiol. 2013;165:416–424. doi: 10.1016/j.vetmic.2013.04.014. [DOI] [PubMed] [Google Scholar]
- Smith CJ, Osborn AM. Advantages and limitations of quantitative PCR (Q-PCR)-based approaches in microbial ecology. FEMS Microbiol Ecol. 2009;67:6–20. doi: 10.1111/j.1574-6941.2008.00629.x. [DOI] [PubMed] [Google Scholar]
- Sogin ML, Morrison HG, Huber JA, Mark Welch D, Huse SM, Neal PR, Arrieta JM, Herndl GJ. Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proc Natl Acad Sci USA. 2006;103:12115–12120. doi: 10.1073/pnas.0605127103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley D, Hughes RJ, Moore RJ. Microbiota of the chicken gastrointestinal tract: Influence on health, productivity and disease. Appl Microbiol Biotechnol. 2014;98:4301–4310. doi: 10.1007/s00253-014-5646-2. [DOI] [PubMed] [Google Scholar]
- Stedtfeld RD, Baushke SW, Tourlousse DM, Miller SM, Stedtfeld TM, Gulari E, Tiedje JM, Hashsham SA. Development and experimental validation of a predictive threshold cycle equation for quantification of virulence and marker genes by high-throughput nanoliter-volume PCR on the OpenArray platform. Appl Environ Microbiol. 2008;74:3831–3838. doi: 10.1128/AEM.02743-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sul WJ, Cole JR, Jesus ED, Wang Q, Farris RJ, Fish JA, Tiedje JM. Bacterial community comparisons by taxonomy-supervised analysis independent of sequence alignment and clustering. Proc Natl Acad Sci USA. 2011;108:14637–14642. doi: 10.1073/pnas.1111435108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Underwood A, Gielbert A, Woodward MJ, Petrovska L. Metaproteomics analysis reveals the adaptation process for the chicken gut microbiota. Appl Environ Microbiol. 2014;80:478–485. doi: 10.1128/AEM.02472-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas T, Gilbert J, Meyer F. Metagenomics - A guide from sampling to data analysis. Microb Inform Exp. 2012;2:3. doi: 10.1186/2042-5783-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, Egholm M, Henrissat B, Heath AC, Knight R, Gordon JI. A core gut microbiome in obese and lean twins. Nature. 2008;457:480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Videnska P, Sisak F, Havlickova H, Faldynova M, Rychlik I. Influence of Salmonella enterica serovar Enteritidis infection on the composition of chicken cecal microbiota. BMC Vet Res. 2013;9:140. doi: 10.1186/1746-6148-9-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Quensen JF, Fish JA, Lee TK, Sun Y, Tiedje JM, Cole JR. Ecological patterns of nifH genes in four terrestrial climatic zones explored with targeted metagenomics using FrameBot, a new informatics tool. mBio. 2013;4:e00592–13. doi: 10.1128/mBio.00592-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin H, Gates RS, Green AR, Mitloehner FM, Moore PA, Jr, Wathes CM. Environmental impacts and sustainability of egg production systems. Poult Sci. 2011;90:263–277. doi: 10.3382/ps.2010-00877. [DOI] [PubMed] [Google Scholar]
- Xu ZR, Hu CH, Xia MS, Zhan XA, Wang MQ. Effects of dietary fructooligosaccharide on digestive enzyme activities, intestinal microflora and morphology of male broilers. Poult Sci. 2003;82:1030–1036. doi: 10.1093/ps/82.6.1030. [DOI] [PubMed] [Google Scholar]
- Yeoman CJ, Chia N, Jeraldo P, Sipos M, Goldenfeld ND, White BA. The microbiome of the chicken gastrointestinal tract. Anim Health Res Rev. 2012;13:89–99. doi: 10.1017/S1466252312000138. [DOI] [PubMed] [Google Scholar]
- Zhou H, Gong J, Brisbin JT, Yu H, Sanei B, Sabour P, Sharif S. Appropriate chicken sample size for identifying the composition of broiler intestinal microbiota affected by dietary antibiotics, using the polymerase chain reaction-denaturing gradient gel electrophoresis technique. Poult Sci. 2007;86:2541–2549. doi: 10.3382/ps.2007-00267. [DOI] [PubMed] [Google Scholar]