

The catalytic mechanism and low pH optimum of C. bescii family 3 pectate lyase are explained.
Keywords: lyase, PL3, catalytic mechanism, Caldicellulosiruptor, thermostable
Abstract
The unique active site of the Caldicellulosiruptor bescii family 3 pectate lyase (PL3) enzyme has been thoroughly characterized using a series of point mutations, X-ray crystallography, pK a calculations and biochemical assays. The X-ray structures of seven PL3 active-site mutants, five of them in complex with intact trigalacturonic acid, were solved and characterized structurally, biochemically and computationally. The results confirmed that Lys108 is the catalytic base, but there is no clear candidate for the catalytic acid. However, the reaction mechanism can also be explained by an antiperiplanar trans-elimination reaction, in which Lys108 abstracts a proton from the C5 atom without the help of simultaneous proton donation by an acidic residue. An acidified water molecule completes the anti β-elimination reaction by protonating the O4 atom of the substrate. Both the C5 hydrogen and C4 hydroxyl groups of the substrate must be orientated in axial configurations, as for galacturonic acid, for this to be possible. The wild-type C. bescii PL3 displays a pH optimum that is lower than that of Bacillus subtilis PL1 according to activity measurements, indicating that C. bescii PL3 has acquired a lower pH optimum by utilizing lysine instead of arginine as the catalytic base, as well as by lowering the pK a of the catalytic base in a unique active-site environment.
1. Introduction
Nature has evolved complex systems to efficiently recycle plant biomass using combinations of enzymes that are specifically tailored to each of the various polymers that make up the plant cell wall. A cost-effective process to convert biomass to its component sugars is required to enable the commercial utilization of biomass as a feedstock for fuels and specialty chemicals. Recently, these processes have trended towards milder pretreatment chemistries, which leave the noncellulose polysaccharides, such as hemicellulose and pectin, largely intact. Thus, new digestion schemes require the development of enzyme cocktails with an increased diversity of activities. The need for new strategies is particularly clear for the consolidated bioprocessing approach (CBP), in which organisms are engineered to include all of the necessary enzymatic capabilities from unpretreated biomass degradation to the production of the desired end products.
Pectin is a structural polymer of higher plants (Carpita & Gibeaut, 1993 ▸), and compared with cellulose it has not received much attention in the biomass-conversion research community. This is largely owing to the fact that many pretreatments, such as high-temperature sulfuric acid, effectively hydrolyze the pectic backbones and side-chain linkages, making the survival of pectins in treated biomass unlikely. The recent shift towards milder pretreatments, as well as alternate chemistries and conditions, such as alkaline or peroxides, necessitates an increased focus on the enzymatic conversion of pectin to monomeric sugars. Pectate lyases are a class of pectinases that cleave linkages in the pectin backbone (Marín-Rodríguez et al., 2002 ▸). This activity makes them important candidates for addition to commercial enzyme cocktails and cloning into CBP microorganisms that digest unpretreated biomass where the cell-wall pectins have not been removed by chemical pretreatment. Deletion experiments with C. bescii have shown that the ability to grow on unpretreated biomass is reduced when a gene cluster containing pectin-degrading enzymes is deleted; however, the organism retains its wild-type ability to grow on soluble sugars if they are provided (Chung et al., 2014 ▸).
Pectins generally consist of α-1,4-linked polygalactosyluronic acid residues with regions of alternating galactosyluronic acid and rhamnosyl residues (Willats et al., 2001 ▸). Pectate lyases use an anti β-elimination reaction to degrade pectin. This reaction mechanism requires a basic residue (usually an arginine) to abstract the α-proton from the C5 atom of the substrate that has been acidified (by the Ca2+ ions at the active site) and concerted action by an acidic group that donates a proton to the carbonyl group, creating an enol intermediate (Gerlt et al., 1991 ▸; Gerlt & Gassman, 1992 ▸, 1993 ▸). Alternatively, the acidic group is not required for proton donation in an antiperiplanar trans-elimination reaction, where the C5 hydrogen and C4 hydroxyl groups must both be orientated in axial configurations, which is the case for galacturonic acid (Abbott et al., 2010 ▸). In the last step, the β-leaving group is eliminated through protonation of the O4 atom by another acidic group, while the enol intermediate is stabilized by the Ca atoms (Scavetta et al., 1999 ▸; Seyedarabi et al., 2010 ▸). In family 3 and 9 pectate lyases, lysine abstracts the α-proton at the beginning of the reaction (Alahuhta et al., 2013 ▸; Creze et al., 2008 ▸; Jenkins et al., 2004 ▸). Although lysine is an unlikely residue for this role, it is known to be able to perform similar functions. In enolases, a lysine abstracts the α-proton that has been acidified by two Mg2+ ions in a reaction mechanism similar to that of pectate lyases (Dinovo & Boyer, 1971 ▸; Pancholi, 2001 ▸). A simplified pectate lyase reaction mechanism is shown in Fig. 1 ▸.
Figure 1.

The classical reaction mechanism of pectate lyases.
The degradation of pectin is important for biomass saccharification, and thus pectinase incorporation into current commercial cocktails and CBP organisms is desirable. The optimum pH ranges for the major cellulase enzymes are typically lower than the optimum pH ranges reported for pectate lyases, complicating the synergy between pectate lyases and cellulases. The optimum activity for cellulose Cel6A occurs at pH 4–6 (Koivula et al., 1996 ▸) and that for cellulose Cel7A at pH 3.5–5.6 (Boer & Koivula, 2003 ▸). Conversely, pectate lyase enzymes are generally most active in alkaline conditions (Antranikian et al., 2005 ▸), with family 1 pectate lyases exhibiting maximum activity at pH values of 9 (Kluskens et al., 2003 ▸) or 10 (Soriano et al., 2000 ▸). The presence of a lysine (pK a 10.5) as the catalytic base in family 1 enzymes compared with an arginine (pK a 12.5) suggests that this mechanism can adapt to function in lower pH environments. Given that family 1 and family 3 pectate lyases share a parallel β-helix fold, the investigation of the differences in catalytic base residues, as well as other active-site characteristics, offers a better understanding of the full range of the effects of pH on activity and the physicochemical features that determine the optimal pH for a specific enzyme.
Here, we report the thorough characterization of the PL3 catalytic mechanism from the hyperthermophilic and cellulolytic bacterium Caldicellulosiruptor bescii (Yang et al., 2010 ▸; Madigan & Marrs, 1997 ▸) using a series of point mutations, X-ray crystallography, pK a calculations and activity assays. We investigate the catalytic base in the context of the local chemical environment using in silico methods and compare the pK a of the lysine with the pK a of the arginine found in PL1 to identify features contributing to the optimal activity pH of the enzyme. With the combined conclusions from the structural, biochemical and computational results, we propose a revised reaction mechanism for family 3 pectate lyases.
2. Materials and methods
2.1. Cloning and mutagenesis
The pET-45b-PL3-cat encoding vector from our previous study (Alahuhta et al., 2013 ▸) was used as the basis for mutational analysis (Fig. 2 ▸). Mutations were generated using the QuikChange II XL kit (Agilent Technologies, Santa Clara, California, USA) according to the manufacturer’s instructions. Mutagenic primers were designed using Agilent’s mutagenic primer design software (http://www.genomics.agilent.com/primerDesignProgram.jsp). Double mutants were generated using the K108A construct as the platform for subsequent mutagenesis.
Figure 2.

Amino-acid sequence of the C. bescii PL3 catalytic module. Residues chosen for mutation are indicated in bold and vector-coded residues are underlined.
2.2. Expression and purification
The cells were grown to an OD600 of ∼0.6 in 1 l LB broth containing 100 µg ml−1 ampicillin at 37°C. At this point the temperature of the incubator was reduced to 19°C, and 1 h later expression was induced by the addition of 1 M IPTG to a final concentration of 1 mM. Cultures were incubated overnight and then harvested by centrifugation. Cell pellets were frozen overnight at ∼20°C, thawed at room temperature and lysed with BugBuster containing lysozyme and benzonase as per the manufacturer’s instructions (EMD Millipore, Billerica, Massachusetts, USA). Cell debris was removed by centrifugation at 15 000g for 15 min. The supernatant was diluted with one volume of buffer A (20 mM sodium phosphate, 500 mM NaCl, 20 mM imidazole pH 7.4) and loaded onto a 5 ml HisTrap FF column using an ÄKTAexplorer 100 FPLC system (GE Life Sciences, Piscataway, New Jersey, USA). After loading and washing the unbound proteins from the column, PL3 enzymes were eluted using a step gradient of 100% buffer B (20 mM sodium phosphate, 500 mM NaCl, 500 mM imidazole pH 7.4). The final purification was performed by size-exclusion chromatography using a HiLoad Superdex 75 (26/60) column (GE Healthcare, Piscataway, New Jersey, USA) in 20 mM acetic acid, 100 mM NaCl, 5 mM CaCl2 pH 5.0. Cell lysates and chromatography eluents were analysed by SDS–PAGE and Western blot using anti-His-tag mouse monoclonal antibody (Life Technologies) and goat anti-mouse alkaline phosphatase secondary antibody.
2.3. Crystallization
PL3 crystals were initially obtained by sitting-drop vapor diffusion using a 96-well plate with Crystal Screen HT from Hampton Research (Aliso Viejo, California, USA). 50 µl of well solution was added to the reservoir and drops were made with 0.2 µl well solution and 0.2 µl protein solution using a Phoenix crystallization robot (Art Robbins Instruments, Sunnyvale, California, USA). The crystals for all mutants were grown at 20°C using screens containing 0.1 M Tris pH 7.5–9.0 and 50–70%(v/v) (±)-2-methyl-2,4-pentanediol. The protein solutions contained 10–20 mg ml−1 protein, 20 mM acetic acid pH 5, 100 mM NaCl, 5 mM CaCl2 and 5 mM trigalacturonic acid (Sigma–Aldrich, St Louis, Missouri, USA).
2.4. Data collection and processing
The PL3-K108A and PL3-E84A crystals were flash-cooled in a nitrogen-gas stream at 100 K before home-source data collection using an in-house Bruker X8 MicroStar X-ray generator with Helios mirrors and a Bruker PLATINUM135 CCD detector. The crystals for the rest of the mutant PL3s were cooled in liquid nitrogen before being sent to SSRL for synchrotron data collection on beamline 11-1 (Soltis et al., 2008 ▸; McPhillips et al., 2002 ▸; Cohen et al., 2002 ▸). The home-source data sets were indexed and processed with the Bruker suite of programs v.2014.9 (Bruker AXS, Madison, Wisconsin, USA). The synchrotron data were indexed and processed with autoxds (Gonzalez & Tsai, 2010 ▸) and XDS (Kabsch, 2010a ▸,b ▸).
2.5. Structure solution and refinement
Intensities were converted to structure factors and 5% of the reflections were flagged for R free calculations using F2MTZ, TRUNCATE, CAD and UNIQUE from the CCP4 package of programs (Winn et al., 2011 ▸). For the synchrotron structures, POINTLESS v.1.9.2 and AIMLESS v.0.2.17 were used to merge the data and provide statistics (Evans & Murshudov, 2013 ▸; Evans, 2006 ▸). MOLREP v.11.2.08 (Vagin & Teplyakov, 2010 ▸) was used for molecular replacement using the unliganded structure of the family 3 pectate lyase from C. bescii (PDB entry 3t9g; Alahuhta et al., 2011 ▸) as the search model. Refinement and manual correction was performed using REFMAC5 v.5.8.0073 (Murshudov et al., 2011 ▸) and Coot v.0.7.2 (Emsley et al., 2010 ▸). MolProbity (Chen et al., 2010 ▸) was used to analyze the Ramachandran plot, and root-mean-square deviations (r.m.s.d.s) of bond lengths and angles were calculated using the ideal values of stereochemical parameters of Engh & Huber (1991 ▸). The Wilson B factor was calculated using CTRUNCATE v.1.15.10 (Winn et al., 2011 ▸). Average B factors were calculated using ICM v.3.8-0 (Molsoft, La Jolla, California, USA). The data-collection and refinement statistics are shown in Table 1 ▸.
Table 1. X-ray data-collection and refinement statistics.
Values in parentheses are for the highest resolution bin.
| PL3-K108A | PL3-E84A | PL3-K108A/E39Q | PL3-K108A/Q111A | PL3-K108A/Q111N | PL3-K108A/D107N | PL3-K108A/R133A | |
|---|---|---|---|---|---|---|---|
| Space group | C2 | C2221 | C2 | C2 | C2 | C2 | C2 |
| Unit-cell parameters | |||||||
| a () | 137.9 | 135.3 | 138.3 | 138.8 | 137.7 | 138.3 | 137.7 |
| b () | 36.4 | 145.6 | 36.5 | 36.4 | 36.2 | 36.4 | 36.3 |
| c () | 99.6 | 160.1 | 100.1 | 100.5 | 99.5 | 100.1 | 99.8 |
| = () | 90.0 | 90.0 | 90.0 | 90.0 | 90.0 | 90.0 | 90.0 |
| () | 132.4 | 132.4 | 132.6 | 132.9 | 132.4 | 132.7 | 132.5 |
| Wavelength () | 1.54178 | 1.54178 | 0.98397 | 0.98397 | 0.98397 | 0.98398 | 0.98397 |
| Temperature (K) | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| Resolution () | 501.40 (1.501.40) | 501.50 (1.601.50) | 501.15 (1.171.15) | 501.25 (1.271.25) | 501.48 (1.511.48) | 501.25 (1.271.25) | 501.55 (1.581.55) |
| Unique reflections | 72222 (13269) | 66566 (11414) | 124577 (5327) | 100777 (4757) | 58991 (2736) | 96401 (4631) | 51894 (2536) |
| Observed reflections | 686831 (76960) | 427353 (36753) | 1282528 (10661) | 1167437 (29019) | 733863 (17716) | 657645 (32096) | 672396 (32812) |
| R int † | 0.0679 (0.4506) | 0.0596 (0.3388) | |||||
| R meas ‡ | 0.047 (0.656) | 0.054 (0.854) | 0.136 (2.665) | 0.066 (1.157) | 0.061 (0.667) | ||
| Average multiplicity | 9.5 (5.8) | 6.4 (3.2) | 10.3 (5.8) | 11.6 (6.1) | 12.4 (6.5) | 6.8 (6.9) | 13.0 (12.9) |
| I/(I) | 15.6 (2.3) | 17.3 (2.7) | 25.7 (3.0) | 25.6 (2.3) | 15.1 (0.8) | 15.5 (1.9) | 29.5 (5.2) |
| Completeness (%) | 99.8 (99.1) | 99.7 (98.4) | 95.5 (82.9) | 98.5 (95.0) | 97.0 (91.7) | 95.1 (93.1) | 97.7 (96.1) |
| R/R free | 0.122 (0.236)/0.184 (0.277) | 0.152 (0.264)/0.186 (0.336) | 0.116 (0.235)/0.148 (0.222) | 0.108 (0.261)/0.151 (0.275) | 0.162 (0.337)/0.199 (0.337) | 0.110 (0.250)/0.156 (0.299) | 0.160 (0.217)/0.200 (0.273) |
| No. of protein atoms | 3268 | 3250 | 3437 | 3369 | 3133 | 3336 | 3189 |
| No. of water molecules | 647 | 519 | 584 | 672 | 517 | 635 | 513 |
| No. of other atoms | 151 | 77 | 262 | 239 | 196 | 179 | 74 |
| R.m.s.d. from ideal§ | |||||||
| Bond lengths () | 0.019 | 0.027 | 0.022 | 0.020 | 0.022 | 0.020 | 0.021 |
| Bond angles () | 2.021 | 2.394 | 2.242 | 2.145 | 2.040 | 2.043 | 1.987 |
| Wilson B factor (2) | 8.3 | 9.0 | 8.4 | 8.4 | 16.4 | 8.3 | 11.6 |
| Average B factors (2) | |||||||
| Protein atoms | 14.8 | 14.2 | 11.3 | 13.3 | 15.1 | 15.3 | 15.6 |
| Water molecules | 29.2 | 29.9 | 25.3 | 26.8 | 27.6 | 27.2 | 28.4 |
| Ramachandran plot statistics¶ | |||||||
| Allowed (%) | 99.6 | 100 | 99.8 | 100.0 | 100.0 | 100.0 | 100.0 |
| Favored (%) | 93.2 | 94.6 | 93.8 | 93.3 | 94.4 | 93.9 | 93.6 |
| Outliers | 2 | 0 | 1 | 0 | 0 | 0 | 0 |
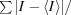
2.6. Structure analysis
Coot (Emsley et al., 2010 ▸), PyMOL (http://www.pymol.org) and ICM (Molsoft, La Jolla, California, USA) were used to compare and analyze structures. Figs. 3, 6 and 8 were created using PyMOL. Figs. 1 and 7 were created using ChemDoodle (http://web.chemdoodle.com/).
2.7. Enzymatic assays
1 mg trigalacturonic acid was digested using a 5 µg ml−1 PL3 loading in the presence of 10 mM Ca2+ for each mutant in a 1 ml final volume. The buffer used was 20 mM Trizma base pH 8.5. All digestions were run in triplicate and pre-chilled on ice prior to addition of enzyme, and were then incubated in a floating water bath for 15 min at 35°C. Samples were then immediately placed into an ice bath and the pH was adjusted to pH 5 using 1 M sodium acetate to terminate the reaction. Samples were analyzed using HPLC (as explained below) and the data were normalized to represent 100% for wild-type PL3. The pH optimum was determined similarly to as described above; 20 mM Trizma base was used for the pH 7.5–12 range and bis-tris for the pH 5–7 range. The data were normalized to represent 100% for the highest value at pH 8.5.
2.8. HPLC/RID analysis of galacturonic acid samples
Analysis of samples was performed on an Agilent 1100 high-performance liquid-chromatography (HPLC) system equipped with a refractive-index detector (RID; Agilent Technologies, Palo Alto, California, USA). The Agilent ChemStation software v.B.04.02 was used to collect and quantitate the analytes of interest. 20 µl of 1.0 g l−1 samples were injected per run and fractionated using a Bio-Rad HPX-87H column with 0.01 N sulfuric acid as the mobile phase. The RID detector and column temperature was maintained at 55°C. The flow rate was held constant at 0.6 ml min−1, resulting in a run time of 26 min.
2.9. pK a calculations
The PROPKA 3.1 and H++ methods were used to evaluate the pK a values of the catalytic base residues, as well as other chargeable residues in the active site (Rostkowski et al., 2011 ▸; Søndergaard et al., 2011 ▸; Gordon et al., 2005 ▸). PROPKA 3.1 and H++ are both designed to account for ligand functional groups in the amino-acid pK a calculations. H++ calculations were performed using a salinity of 0.15, an internal dielectric constant of 10, an external dielectric constant of 80 and a pH of 8.5 to reflect the optimal activity pH for the PL3 presented here. The pK a values were evaluated for the apo and holo structures of C. bescii PL3 (PDB entries 3t9g and 4ew9, respectively; Alahuhta et al., 2011 ▸, 2013 ▸) as well as the apo and holo structures of B. subtilis PL1 (PDB entries 1bn8 and 2nzm, respectively; Pickersgill et al., 1994 ▸; Seyedarabi et al., 2010 ▸). The PL1 holo structure is inactive, with the catalytic arginine replaced by an alanine. The wild-type arginine was modeled into 2nzm using the Rosetta protein-design software (Kuhlman & Baker, 2000 ▸). Structural representations and distance measurements for active-site acidic residues were generated using PyMOL.
3. Results
3.1. C. bescii PL3 point mutant X-ray structures
Seven X-ray crystal structures of C. bescii PL3 point mutants have been solved, five of them in complex with intact trigalacturonic acid (Fig. 3 ▸; Table 1 ▸). The overall fold is a parallel β-helix, which is typical for pectate lyases (Marín-Rodríguez et al., 2002 ▸). All seven structures have two molecules in the asymmetric unit and have been deposited in the Protein Data Bank (PDB) with the following IDs: 4z03 (K108A), 4z05 (E84A), 4yz0 (K108A/E39Q), 4yzx (K108A/D107N), 4yzq (K108A/Q111N), 4yza (K108A/Q111A) and 4z06 (K108A/R133A).
Figure 3.

(a) The overall fold of the C. bescii PL3 K108A mutant catalytic module superimposed with the wild-type Lys108 (green C atoms) and the contents of the active site (cyan C atoms). The trigalacturonic acid is shown as sticks with yellow C atoms and red O atoms. Ca atoms are shown as red spheres. (b) The structure rotated 90°.
In our previous studies, we established that the most similar structure available to the C. bescii enzyme is Bacillus PL3, noting that the other structures were similar only at the level of the protein fold (Alahuhta et al., 2011 ▸). In fact, closer inspection of the PL3 catalytic module showed that its active site is shifted compared with PL1 structures and that the similar features of the active sites could only be correctly assigned by manual superimposition (Alahuhta et al., 2013 ▸).
Of the seven mutant structures studied, five have the substrate bound in the same way. The E84A mutant lacks two of the three calcium ions owing to the mutation and therefore does not bind the substrate. The R133A mutant is no longer properly able to stabilize the sugar at the −2 position. There is some scattered density for the trisaccharide in translated positions. As a result, we were only able to reliably model a single galacturonic acid at the −1 position. Other mutants have variations in the occupancies of the calcium ions caused by the mutations (Table 2 ▸).
Table 2. Structural and biochemical effects of point mutations and the proposed roles of these residues.
Calcium occupancies are reported for both molecules in the asymmetric unit.
| Mutation | Substrate bound? | Calcium 1 occupancy (%) | Calcium 2 occupancy (%) | Calcium 3 occupancy (%) | Remaining activity (%) | Proposed role |
|---|---|---|---|---|---|---|
| K108A | Yes | 87/100 | 100/100 | 100/100 | 0 | Catalytic base |
| E84A | No | 0/0 | 0/0 | 100/86 | 0 | Calcium 1/2 coordination |
| E39Q | Yes | 75/83 | 63/69 | 100/100 | 6 | Calcium 2 coordination |
| Q111A | Yes | 89/65 | 100/99 | 100/100 | 1 | Substrate binding, transition-state stabilization for the second part of the reaction |
| Q111N | Yes | 76/86 | 96/100 | 98/100 | 8 | Substrate binding, transition-state stabilization for the second part of the reaction |
| D107N | Yes | 50/56 | 100/100 | 100/100 | 0 | Calcium 1 coordination |
| R133A | Weakly | 56/69 | 71/91 | 80/92 | 9 | Substrate binding |
3.2. Enzymatic assays
The effect of mutations on the enzymatic activity was measured to better understand their importance and specific role in substrate binding and the reaction mechanism. We also determined the wild-type C. bescii PL3 activity as a function of pH to find the optimum and the extent of pH values that can be used. The pH profile is shown in Fig. 4 ▸ and the enzymatic activity relative to the wild type is shown in Fig. 5 ▸.
Figure 4.
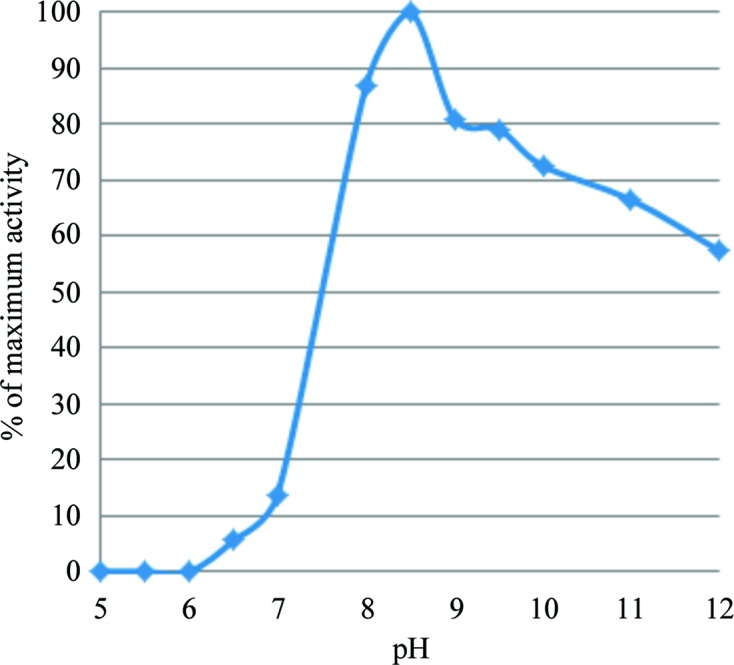
C. bescii PL3 activity as a function of pH.
Figure 5.

C. bescii PL3 wild-type and mutant relative activities with trigalacturonic acid as a substrate.
The wild-type C. bescii PL3 has optimal activity at pH 8.5, but retains over 10% of activity at pH 7. The E84A, D107N and K108A mutations result in a complete loss of activity. The E39Q, Q111A, Q111N and R133A mutations have residual activity.
3.3. pK a calculations
PROPKA and H++ were used to calculate the pK a values of the catalytic base residues of the C. bescii PL3 and B. subtilis PL1 structures with and without ligands. The pK a values of the apo structures calculated by the H++ program are suspect at values greater than 12; however, the values for the holo structures agree with those from PROPKA. The results are shown in Table 3 ▸.
Table 3. The pK a values for the catalytic base residues of C. bescii PL3 and B.subtilis PL1 indicate that the local chemical environment, as well as the amino-acid identity, determine the optimal pH ranges for each enzyme.
| PDB code | Apo/holo | Organism | PROPKA | H++ |
|---|---|---|---|---|
| C. bescii PL3, Lys108 | ||||
| 3t9g | Apo | C. bescii | 9.7 | >12 |
| 4ew9 | Holo† | C. bescii | 8.7 | 7.2 |
| B. subtilis PL1, Arg279 | ||||
| 1bn8 | Apo | B. subtilis | 12.0 | >12 |
| 2nzm | Holo | B. subtilis | 9.1 | 9.1 |
4ew9 represents PL3 with reacted ligand or product.
4. Discussion
4.1. The structural and enzymatic effects of point mutations
Structural comparison between the different mutants did not show structural changes beyond the immediate vicinity of the mutated residues; however, these changes were significant for the activity observed. For example, the inactivating K108A mutation caused Arg133 to adopt two conformations to fill the void left by the missing lysine side chain, the glutamine side chain of the E39Q point mutant had two slightly rotated conformations compared with the wild type, and the Ca atoms and the substrate lost occupancy. Characterization of the mutants revealed two kinds of changes affecting the function of the enzyme: (i) a loss of binding to trigalacturonic acid and (ii) a partial or total loss of active-site calcium ions. These changes result in a variable loss of enzymatic activity. Using these structural and activity results, we were able to propose a role for each mutated residue (Table 2 ▸).
From the data shown in Table 2 ▸, Lys108 is clearly the catalytic base. Its mutation to an alanine causes a complete loss of activity, but no other changes were found. The catalytic acid proposed earlier, Glu84, is needed for activity; however, structural results (Table 2 ▸, Fig. 6 ▸) indicate that it is crucial for calcium binding. Closer inspection of its immediate environment (Fig. 6 ▸) shows that it is behind the calcium ions, preventing it from donating a proton. Moreover, Glu39 and Asp107, which are the only other possible candidates for this role, are both a little too distant to be proton donors (∼3 Å) and have an unfavorable angle to be effective (Fig. 6 ▸). Both Glu39 and Asp107 are needed for proper calcium binding.
Figure 6.

C. bescii PL3 K108A mutant active site (cyan C atoms) with intact substrate superimposed with the wild-type Lys108 (green C atoms). The trigalacturonic acid is shown as sticks with yellow C atoms and red O atoms. Ca atoms are shown as red spheres.
Mutation of Gln111 to alanine or asparagine or of Arg133 to alanine all result in a greater than 90% loss of activity. Arg133 is clearly involved in substrate binding and unsurprisingly results in an almost complete loss of trigalacturonic acid binding without other significant conformational changes in the vicinity of the active site. For the R133A mutation some residual density could be observed for trigalacturonic acid binding at the active site in positions where it has moved either one step forwards or backwards. Interestingly, loss of substrate leads to partial loss of calcium ions 1 and 2 at the active site, indicating that these catalytically necessary ions are properly bound only in the presence of ligands. The role of Gln111 also seems to be one of substrate stabilization, but mutating this residue to alanine does not lead to a loss of substrate binding. Thus, it must have a more specific role, such as stabilizing the O4 atom during the last step of the reaction.
Overall, the mutations cause three different activity-decreasing structural changes: (i) loss of the catalytic base removes all activity when Lys108 is mutated to alanine, (ii) a decrease of activity owing to weakened substrate or transition-state stabilization (Q111N/A and R133A), (iii) a partial to complete loss of activity caused by disrupted calcium coordination (E84A, E39Q and D107N).
4.2. Revision of the catalytic mechanism of PL3
After the role of the nearby residues had been investigated at the structural level and with enzyme-activity measurements, we concluded that we still did not have a candidate for the catalytic base. Although this task could be performed by a water molecule, an antiperiplanar trans-elimination reaction would better explain the first part of the β-elimination reaction without the need for a proton donor (Abbott et al., 2010 ▸). In antiperiplanar trans-elimination reactions, the C5 hydrogen and C4 hydroxyl group both must be orientated in axial configurations, which is true for galacturonic acid. In Fig. 7 ▸, we propose a revised reaction mechanism for C. bescii PL3 that agrees with the mechanism proposed by Abbott et al. (2010 ▸) for family 22 pectate lyases.
Figure 7.

The revised reaction mechanism of C. bescii PL3.
4.3. pK a calculations and calcium binding
Many PL1 enzymes display pH optima at pH 9–10 (Tardy et al., 1997 ▸), utilizing a catalytic arginine with a pK a of 12.5 for the side chain in solution. The C. bescii PL3 investigated here exhibits a lower optimum pH of 8.5, utilizing a lysine as the catalytic base with a lower relative pK a of 10.5 in solution. The pK a for the catalytic lysine of C. bescii PL3 and the catalytic arginine of B. subtilis PL1 have been evaluated to identify how local chemical effects perturb the pK a values of these residues (Table 3 ▸). The catalytic cycle for C. bescii PL3 is considered by evaluating the apo (PDB entry 3t9g) and reacted (PDB entry 4ew9) enzymatic states. For comparison, the catalytic cycle for B. subtilis PL1 is evaluated with the apo (PDB entry 1bn8) and holo (PDB entry 2nzm) states.
The computed pK a values for the catalytic base residues in all considered structures appear to be estimated on the high side. The H++ program in particular calculates pK a values for the catalytic bases from both the PL3 and PL1 apo structures as greater than 12, precluding a comparison of the two pectate lyase family members using H++. However, PROPKA values suggest the microenvironments of both apo PL1 and PL3 lower the pK a of the catalytic base residues from their solution values, allowing them to operate as general bases at the relative pH optimum. Furthermore, both the PROPKA and the H++ results indicate that the pK a of Lys108 in the holo PL3 structure (PDB entry 4ew9) decreases upon binding Ca2+, indicating that the role of the primary Ca2+ is the priming of the catalytic lysine. The pK a of Arg279 in the holo PL1 structure (PDB entry 2nzm) also decreases upon binding Ca2+, but is relatively higher using both PROPKA and H++, in agreement with the higher optimal activity pH. Thus, while the absolute pK a values appear to be high using both PROPKA and H++, the same trend in pK a values upon binding calcium is seen using both methods.
Pectate lyase isozymes with different pH optima provide an opportunity to understand how enzymes performing the same catalytic mechanism can evolve to operate under different conditions. Comparing enzyme kinetics includes considering both the turnover number (k cat) and binding of the substrate (K m). For pectate lyase enzymes, the affinity for Ca2+ is weak. Evaluating Ca2+ binding to a pectate lyase suggests that the optimum pH for Ca2+/ligand binding has evolved to agree with the optimum pH for enzyme activity (Herron et al., 2003 ▸). Importantly, a striking difference is seen between the PL1 and PL3 structures when considering the entire calcium-binding site. The acidic residues comprising the calcium-binding site of B. subtilis PL1 are further apart. The acidic residues of the C. bescii PL3 calcium-binding site are, by contrast, much closer (Fig. 8 ▸). The close proximity of like-charged acidic residues raises the pK a as the need for counterions is increased. Given the high Ca2+ concentrations in plant cell walls, the closely spaced acidic residues seen in PL3 serve to enhance Ca2+ binding. Thus, the four carboxyl groups of the PL3 calcium-binding site spaced closely together increases the nucleophilicity of the entire site compared with that observed in the binding site of PL1.
Figure 8.

The arrangement of the active-site acidic residues, based on distance between residues, indicates that the shape of the calcium-binding site plays a role in the affinity for calcium ions and ligand. The acidic residues of the calcium-binding site of (a) B. subtilis PL1 and (b) C. bescii PL3 are shown in stick form, with calcium ions shown as grey spheres and the backbone shown in cartoon representation. Distances between the Cα atoms for each residue are shown, indicating the shape and distance of the acidic residues comprising the calcium-binding sites.
5. Conclusions
In this manuscript, we have described the X-ray structures of seven PL3 active-site mutants, five of which are complexed with intact trigalacturonic acid. Analyses of the properties of these mutations using pK a calculations and enzyme-activity assays were also conducted. Our results confirm that Lys108 is the catalytic base which abstracts a proton from the C5 atom in an antiperiplanar trans-elimination reaction and an acidified water completes the anti β-elimination reaction by protonating the O4 atom of the substrate. By combining activity measurements and structural results, we were able to determine the biological role of the other mutated active-site residues. The acidic residues Glu84, Glu39 and Asp107 are involved in calcium coordination, with Glu84 being necessary for the binding of two of the three active-site Ca atoms. According to the structural results, Gln111 and Arg133 play a role in substrate binding. Arg133 is especially important in correct substrate positioning, whereas Gln111 has a smaller effect on substrate binding but seems to stabilize the O4 atom during the last step of the reaction.
We have determined that the pH optimum of C. bescii PL3 is lower than the pH optimum of B. subtilis PL1 (8.5 versus 9–10). This difference is most likely owing to different catalytic bases with different intrinsic pK a values (lysine, pK a 10.5, versus arginine, pK a 12.5). We verified that this hypothesis is true for the special environment of the active site by computing pK a values for the catalytic base residue with and without substrate. In absence of ligand, both PL1 and PL3 lower the pK a of the catalytic base. In the presence of ligand and Ca atoms, PL3 lowers the pK a of Lys108 by almost two pH units to pH 8.7, illustrating the importance of the calcium ions for priming the active site. Clearly, calcium stabilization at the active site is critically linked to activity. Furthermore, our structural results show that loss of substrate leads to partial loss of active-site calcium ions 1 and 2, indicating that these catalytically critical ions are properly bound only when ligands are present. Structural comparison between C. bescii PL3 and B. subtilis PL1 shows that the acidic residues coordinating the active-site calcium ions are closer to each other, making the cluster more compact. This configuration increases the nucleophilicity of the entire site.
Taken together, our structural, computational and biochemical results permit us to conclude that C. bescii PL3 has evolved a lower catalytic optimum using lysine, instead of arginine, as the catalytic base, as well as by further lowering its pK a by changing the environment of the active site. We note that this adaptation is critical for C. bescii, which grows optimally near neutral pH in hot springs.
Supplementary Material
PDB reference: C. bescii family 3 pectate lyase, K108A/Q111A double mutant, complex with trigalacturonic acid, 4yza
PDB reference: K108A/Q111N double mutant, complex with trigalacturonic acid, 4yzq
PDB reference: K108A/D107N double mutant, complex with trigalacturonic acid, 4yzx
PDB reference: K108A/E39Q double mutant, complex with trigalacturonic acid, 4yz0
PDB reference: K108A mutant, complex with trigalacturonic acid, 4z03
PDB reference: E84A mutant, 4z05
PDB reference: K108A/R133A double mutant, complex with α-d-galactopyranuronic acid, 4z06
Acknowledgments
This work was funded by the US DOE Office of Science, Biological and Environmental Research Program, Bioenergy Research Center (BioEnergy Science Center, BESC) managed by Oak Ridge National Laboratory. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory is supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS or NIH.
References
- Abbott, D. W., Gilbert, H. J. & Boraston, A. B. (2010). J. Biol. Chem. 285, 39029–39038. [DOI] [PMC free article] [PubMed]
- Alahuhta, M., Brunecky, R., Chandrayan, P., Kataeva, I., Adams, M. W. W., Himmel, M. E. & Lunin, V. V. (2013). Acta Cryst. D69, 534–539. [DOI] [PubMed]
- Alahuhta, M., Chandrayan, P., Kataeva, I., Adams, M. W. W., Himmel, M. E. & Lunin, V. V. (2011). Acta Cryst. F67, 1498–1500. [DOI] [PMC free article] [PubMed]
- Antranikian, G., Vorgias, C. E. & Bertoldo, C. (2005). Adv. Biochem. Eng. Biotechnol. 96, 219–262. [DOI] [PubMed]
- Boer, H. & Koivula, A. (2003). Eur. J. Biochem. 270, 841–848. [DOI] [PubMed]
- Carpita, N. C. & Gibeaut, D. M. (1993). Plant J. 3, 1–30. [DOI] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Chung, D., Pattathil, S., Biswal, A. K., Hahn, M. G., Mohnen, D. & Westpheling, J. (2014). Biotechnol. Biofuels, 7, 147. [DOI] [PMC free article] [PubMed]
- Cohen, A. E., Ellis, P. J., Miller, M. D., Deacon, A. M. & Phizackerley, R. P. (2002). J. Appl. Cryst. 35, 720–726. [DOI] [PMC free article] [PubMed]
- Creze, C., Castang, S., Derivery, E., Haser, R., Hugouvieux-Cotte-Pattat, N., Shevchik, V. E. & Gouet, P. (2008). J. Biol. Chem. 283, 18260–18268. [DOI] [PubMed]
- Diederichs, K. & Karplus, P. A. (1997). Nature Struct. Mol. Biol. 4, 269–275. [DOI] [PubMed]
- Dinovo, E. C. & Boyer, P. D. (1971). J. Biol. Chem. 246, 4586–4593.
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Engh, R. A. & Huber, R. (1991). Acta Cryst. A47, 392–400.
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- Evans, P. R. & Murshudov, G. N. (2013). Acta Cryst. D69, 1204–1214. [DOI] [PMC free article] [PubMed]
- Gerlt, J. A. & Gassman, P. G. (1992). J. Am. Chem. Soc. 114, 5928–5934.
- Gerlt, J. A. & Gassman, P. G. (1993). J. Am. Chem. Soc. 115, 11552–11568.
- Gerlt, J. A., Kozarich, J. W., Kenyon, G. L. & Gassman, P. G. (1991). J. Am. Chem. Soc. 113, 9667–9669.
- Gonzalez, A. & Tsai, Y. (2010). A Quick XDS Tutorial for SSRL. http://smb.slac.stanford.edu/facilities/software/xds/#autoxds_script.
- Gordon, J. C., Myers, J. B., Folta, T., Shoja, V., Heath, L. S. & Onufriev, A. (2005). Nucleic Acids Res. 33, W368–W371. [DOI] [PMC free article] [PubMed]
- Herron, S. R., Scavetta, R. D., Garrett, M., Legner, M. & Jurnak, F. (2003). J. Biol. Chem. 278, 12271–12277. [DOI] [PubMed]
- Jenkins, J., Shevchik, V. E., Hugouvieux-Cotte-Pattat, N. & Pickersgill, R. W. (2004). J. Biol. Chem. 279, 9139–9145. [DOI] [PubMed]
- Kabsch, W. (2010a). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Kabsch, W. (2010b). Acta Cryst. D66, 133–144. [DOI] [PMC free article] [PubMed]
- Kluskens, L. D., van Alebeek, G. J., Voragen, A. G., de Vos, W. M. & van der Oost, J. (2003). Biochem. J. 370, 651–659. [DOI] [PMC free article] [PubMed]
- Koivula, A., Reinikainen, T., Ruohonen, L., Valkeajärvi, A., Claeyssens, M., Teleman, O., Kleywegt, G. J., Szardenings, M., Rouvinen, J., Jones, T. A. & Teeri, T. T. (1996). Protein Eng. Des. Sel. 9, 691–699. [DOI] [PubMed]
- Kuhlman, B. & Baker, D. (2000). Proc. Natl Acad. Sci. USA, 97, 10383–10388. [DOI] [PMC free article] [PubMed]
- Madigan, M. T. & Marrs, B. L. (1997). Sci. Am. 276, 82–87. [DOI] [PubMed]
- Marín-Rodríguez, M. C., Orchard, J. & Seymour, G. B. (2002). J. Exp. Bot. 53, 2115–2119. [DOI] [PubMed]
- McPhillips, T. M., McPhillips, S. E., Chiu, H.-J., Cohen, A. E., Deacon, A. M., Ellis, P. J., Garman, E., Gonzalez, A., Sauter, N. K., Phizackerley, R. P., Soltis, S. M. & Kuhn, P. (2002). J. Synchrotron Rad. 9, 401–406. [DOI] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Pancholi, V. (2001). Cell. Mol. Life Sci. 58, 902–920. [DOI] [PMC free article] [PubMed]
- Pickersgill, R., Jenkins, J., Harris, G., Nasser, W. & Robert-Baudouy, J. (1994). Nature Struct. Biol. 1, 717–723. [DOI] [PubMed]
- Rostkowski, M., Olsson, M. H. M., Sondergaard, C. R. & Jensen, J. H. (2011). BMC Struct. Biol. 11, 6. [DOI] [PMC free article] [PubMed]
- Scavetta, R. D., Herron, S. R., Hotchkiss, A. T., Kita, N., Keen, N. T., Benen, J. A., Kester, H. C., Visser, J. & Jurnak, F. (1999). Plant Cell, 11, 1081–1092. [DOI] [PMC free article] [PubMed]
- Seyedarabi, A., To, T. T., Ali, S., Hussain, S., Fries, M., Madsen, R., Clausen, M. H., Teixteira, S., Brocklehurst, K. & Pickersgill, R. W. (2010). Biochemistry, 49, 539–546. [DOI] [PubMed]
- Soltis, S. M. et al. (2008). Acta Cryst. D64, 1210–1221. [DOI] [PMC free article] [PubMed]
- Søndergaard, C. R., Olsson, M. H. M., Rostkowski, M. & Jensen, J. H. (2011). J. Chem. Theory Comput. 7, 2284–2295. [DOI] [PubMed]
- Soriano, M., Blanco, A., Díaz, P. & Pastor, F. I. (2000). Microbiology, 146, 89–95. [DOI] [PubMed]
- Tardy, F., Nasser, W., Robert-Baudouy, J. & Hugouvieux-Cotte-Pattat, N. (1997). J. Bacteriol. 179, 2503–2511. [DOI] [PMC free article] [PubMed]
- Vagin, A. & Teplyakov, A. (2010). Acta Cryst. D66, 22–25. [DOI] [PubMed]
- Willats, W. G., McCartney, L., Mackie, W. & Knox, J. P. (2001). Plant Mol. Biol. 47, 9–27. [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Yang, S.-J., Kataeva, I., Wiegel, J., Yin, Y. B., Dam, P., Xu, Y., Westpheling, J. & Adams, M. W. W. (2010). Int. J. Syst. Evol. Microbiol. 60, 2011–2015. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: C. bescii family 3 pectate lyase, K108A/Q111A double mutant, complex with trigalacturonic acid, 4yza
PDB reference: K108A/Q111N double mutant, complex with trigalacturonic acid, 4yzq
PDB reference: K108A/D107N double mutant, complex with trigalacturonic acid, 4yzx
PDB reference: K108A/E39Q double mutant, complex with trigalacturonic acid, 4yz0
PDB reference: K108A mutant, complex with trigalacturonic acid, 4z03
PDB reference: E84A mutant, 4z05
PDB reference: K108A/R133A double mutant, complex with α-d-galactopyranuronic acid, 4z06