

Abstract
Tumor suppressive miRNAs that target oncogenes are frequently downregulated in cancers, and this downregulation leads to oncogene pathway activation. Thus, tumor suppressive miRNAs and their target oncogenes have been proposed as useful targets in cancer treatment. miR-200 family downregulation has been reported in cancer progression and metastasis. The miR-200 family consists of two gene clusters, miR-200b/200a/429 and miR-200c/141, which are located on human chromosomes 1 and 12, respectively. Here, we identified that p53 response elements are located around both clusters of the miR-200 family and confirmed that miR-200s are transcriptional targets of the p53 family. In silico analyses of miRNA targets established the CRKL oncogene as a potential target for miR-200b/200c/429. Moreover, miR-200b/200c/429 inhibited CRKL mRNA and protein expression by directly targeting its 3′-UTR region. Importantly, endogenous CRKL expression was decreased in cancer cells through the introduction of p53 family and endogenous p53 activation. Moreover, the downregulation of CRKL by siRNA inhibited cancer cell growth. The Oncomine database demonstrates that CRKL is overexpressed in a subset of cancer types. Furthermore, CRKL is significantly overexpressed in primary breast cancer tissues harboring mutant TP53. Our results demonstrate that the p53 target miR-200b/200c/429 miRNAs are negative regulators of the CRKL oncogene.
Keywords: Cell growth, CRKL, miR-200 family, p53, p53 family
Several tumor suppressive miRNAs are downregulated in cancers, and this downregulation enhances the expression of target oncogenes. Thus, molecular targeted therapy may be useful in the treatment of cancers in which tumor suppressive miRNAs are downregulated. However, the molecular mechanisms that underlie the downregulation of tumor suppressive miRNAs in cancers are still unclear. The p53 tumor suppressor can act as a sequence-specific transcription factor that regulates target gene transcription and expression to modulate various cellular processes.1,2 Recent studies identified miRNAs induced by p53 that are important in cellular physiology, including cell growth, differentiation and death.3 For example, miRNAs, including miR-34, miR-143, miR-145, miR-192 and miR-215, mediate p53-induced cell growth inhibition.
The first identified p53-target miRNAs, the miR-34 family, target genes that are involved in cell cycle progression and apoptosis inhibition, such as CDK4, cyclin E2 and BCL2.4–6 miR-192 and miR-215 also promote cell cycle arrest in response to p53 activation by targeting a number of regulators of DNA synthesis and cell cycle checkpoints (CDC7, LMNB2, MAD2L1 and CUL5) and DHFR.7,8 p53 can repress c-Myc oncoprotein through the induction of miR-145 and miR-34.9,10 In addition, miR-143/145 and miR-192/194/215 decrease MDM2 expression, which increases acetylated p53 levels and p53 activity and, in turn, induces apoptosis in a p53-dependent manner.11,12 Thus, miRNAs are important components in p53-mediated gene repression. miR-200c, a p53-regulated miRNA, regulates epithelial to mesenchymal transition (EMT) by inhibiting ZEB1/2, which are transcriptional repressors of E-cadherin, a known epithelial cell marker.13,14
In the present study, we found that the p53 family downregulates the CRKL oncogene through miR-200b/200c/429 transactivation. miR-200b/200c/429 expression consistently downregulates CRKL via predicted binding sequences within the 3′-UTR of the CRKL gene. The CRKL gene encodes an adaptor protein containing Src homology 2 and 3 (SH2/SH3) domains.15 CRKL expression is increased in certain human solid tumors, including lung cancer, gastric cancer, breast cancer and bladder cancer as well as hematologic malignancies.16–19 Moreover, CRKL amplification was previously reported in non-small cell lung and gastric cancers, and CRKL protein overexpression contributes to oncogenic phenotypes in cancer cells.18,20 However, the mechanism underlying CRKL upregulation in solid tumors is largely unknown. Our data reveals that the p53 target miRNAs miR-200b/200c/429 are negative regulators of the actionable CRKL oncogene. Taken together, our results point toward a novel p53/miR-200/CRKL pathway in carcinogenesis and suggest that targeted therapy could be effective in this pathway, which includes an oncogene and tumor suppressive miRNAs.
Materials and Methods
Recombinant adenoviruses and plasmids
The construction, purification and infection of replication-deficient recombinant adenoviruses encoding human p53 family proteins fused to an amino-terminal FLAG epitope (Ad-p53, Ad-p73α, Ad-p73β, Ad-p63γ and Ad-p63α) or the bacterial lacZ gene (Ad-lacZ) were performed as previously described.21–23 Relative adenovirus infection efficiencies in each cell line were determined by subjecting cells that were infected with control Ad-lacZ to X-gal staining; 90–100% of the cells were infected at an MOI of 12.5–100. To construct CRKL-expressing plasmids lacking its 3′-UTR, the entire coding region of a human CRKL cDNA was inserted in-frame into the pF5K-CMV-neo or pFN28K with an N-terminal Halo epitope tag (Promega, Madison, WI, USA), and the resulting constructs were designated pF5K-CRKL and pFN28K-CRKL, respectively.
Luciferase assay
The CRKL 3′-UTR fragment containing the miR-200b/200c/429 seed sequence (5′-GTGCTATAAAATTAACAGTATTA-3′) and its mutant form (5′-GTGCTATAAAATTAAACTGCGGA-3′) were synthesized and cloned into the 3′ end of the pMIR-REPORT luciferase vector (Ambion, Austin, TX, USA) at the HindIII and SpeI sites, and the resulting constructs were named pMIR-CRKL-3′-UTR and pMIR-CRKL-3′-UTR-mut. The inserted fragments were confirmed by sequencing. Cells in 24-well plates were transfected with a pMIR-REPORT luciferase reporter vector (100 ng) together with miR-mimics (100 nM) or control miRNA as a negative control using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). A Renilla luciferase vector phRG-TK (2 ng) was co-transfected to normalize differences in transfection efficiency. Luciferase activity was measured using the Dual-Luciferase Reporter Assay (Promega).
Other methods are detailed in Data S1.
Results
The p53 family upregulates the expression of the miR-200 family
The miR-200 family consists of five members clustered in two genomic loci: chromosome 1p36.33 (miR-200b, miR-200a and miR-429) and chromosome 12p13.31 (miR-200c and miR-141). We searched for p53 motifs across the entire human genome using an in silico approach24 and determined that p53 motifs are located around both clusters of the miR-200 family (Fig. S1). We then analyzed interactions between the p53 family proteins and these candidate p53-binding sequences using ChIP in Saos2 osteosarcoma cells that were infected with adenoviruses expressing FLAG-tagged p53 family genes (Ad-p53, Ad-p73α, Ad-p73β, Ad-p63γ and Ad-p63α) and a control adenovirus. ChIP analysis revealed that the p53 family directly binds to the predicted p53-binding sequences on both chromosomes 1 and 12 (Fig. S2a). We designated these candidate p53 response elements 200b/200a/429-RE and 200c/141-RE, respectively (Fig. S1b). A reporter assay demonstrated that the p53 family significantly increased the luciferase activity of vectors containing both binding sites. In contrast, mismatches in these p53-binding sequences (200b/200a/429-RE-mut and 200c/141-RE-mut) significantly abolished transactivation by the p53 family (Fig. S2b). We also confirmed the transactivation of the miR-200 family clusters by the p53 family in human cancer cells by real-time RT-PCR (Fig.1). Taken together, these results indicate that the exogenous p53 family is a positive transcriptional regulator of the miR-200 family in several different cell types, which is consistent with previous findings.25–27
Figure 1.
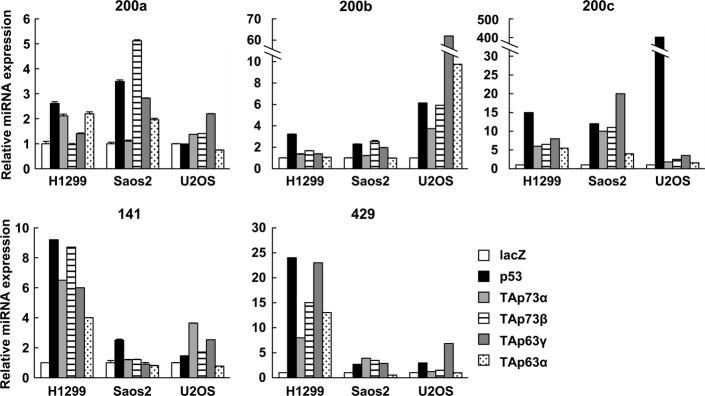
Upregulation of miR-200 family members by the p53 family in human cancer cell lines. Cells were infected with recombinant adenoviruses encoding human p53 family at an MOI of 25 (H1299), 12.5 (Saos2) or 50 (U2OS) and harvested 24 h after infection. The relative gene expression levels were quantified using the ΔΔCt method via real-time RT-PCR, and the results were normalized to the expression of the housekeeping gene RNU6B. The data are shown as the mean ± standard error of three independent experiments and were normalized to their respective controls as 1.
CRKL is downregulated by miR-200b/200c/429
We searched for putative target genes for the miR-200 family using three algorithms; miRanda,28 PicTar29 and TargetScan.30 The 3′-UTR of the CRKL gene contains a putative binding site for miR-200b/200c/429 (Fig.2a). We then examined whether CRKL was downregulated by the miR-200 family in cancer cells. We transfected miRNA mimic oligonucleotides into the H1299 lung cancer cell line. The introduction of miR-200b, miR-200c or miR-429 mimics downregulated CRKL protein levels (Fig.2b). Importantly, CRKL protein was not significantly decreased by miR-200a or miR-141 mimics, which have a one-base mismatch from the miR-200b/200c/429 seed sequence (Figs2b and S1a). Similar patterns were observed in the Saos2 and U2OS cell lines. In contrast, ZEB1, a known miR-200 family target, was downregulated by all members of the miR-200 family in both cells (Fig. S3). Consistent with the reduced protein level, real-time RT-PCR analysis demonstrated that the forced expression of miR-200b/200c/429 also remarkably reduced CRKL mRNA levels in H1299 cells (Fig.2c). A 3′-UTR luciferase reporter assay was also performed to demonstrate that the miRNA–mRNA interaction is direct. In this assay, the candidate miR-200b/200c/429 binding site in the CRKL 3′-UTR was cloned just downstream of the luciferase gene in the pMIR-REPORT vector (pMIR-CRKL-3′-UTR). The reporter plasmid and the miRNA mimics were co-transfected into H1299 cells, and luciferase reporter activity was monitored. Although the activity of the luciferase reporter with the CRKL 3′-UTR was downregulated upon co-transfection with miR-200b, miR-200c or miR-429 compared to transfection with control oligonucleotides, a reporter with a mutant miRNA binding site in the CRKL 3′-UTR abolished this regulation (CRKL-mut, Fig.2d). Taken together, these results indicate that miR-200b/200c/429 negatively regulates CRKL by directly targeting its 3′-UTR region.
Figure 2.
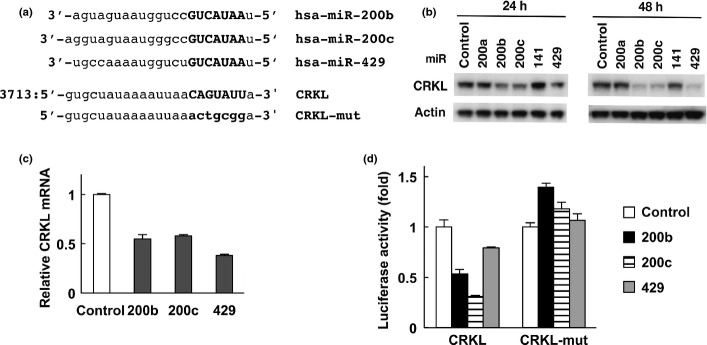
CRKL is downregulated by miR-200b/200c/429. (a) The putative miR-200b/200c/429 target site in the 3′-UTR of CRKL mRNA. Seed sequences are shown in bold type. (b) CRKL protein downregulation by miR-200b/200c/429 in H1299 cells. Cells were transfected with control or miRNA mimic oligonucleotides. CRKL and β-actin levels were analyzed by immunoblotting. (c) Downregulation of CRKL mRNA by miR-200b/200c/429 in H1299 cells. Cells were transfected with control or miRNA mimic oligonucleotides for 24 h. Relative gene expression levels were quantified using the ΔΔCt method, and the results were normalized to the expression of the housekeeping gene GAPDH. The data are shown as the mean ± standard errors of three independent experiments normalized to their respective controls as 1. (d) The CRKL 3′-UTR is responsive to miR-200b/200c/429 in H1299 cells. Cells were transfected with the pMIR-REPORT vector containing the CRKL 3′-UTR (pMIR-CRKL-3′-UTR) or a mutant (pMIR-CRKL-3′-UTR-mut) along with control plasmids expressing Renilla luciferase (phRG-TK). The cells were co-transfected with control or miRNA mimic oligonucleotides. After 24 h, luciferase activity was measured using the dual-luciferase reporter assay system. All of the experiments were performed in quadruplicate, and the mean and standard deviation are indicated by the bars .
The p53 family downregulates CRKL protein and mRNA expression levels
To determine whether CRKL is downregulated by the p53 family, we examined CRKL protein expression levels after overexpression of p53 family in H1299, Saos2, U2OS, Kiku and U87MG cells. CRKL was clearly downregulated by p53, TAp73β and TAp63γ in all cell lines tested (Fig.3a). CRKL mRNA was also decreased in the p53-transfected cells, TAp73-transfected cells and TAp63-transfected cells (Fig.3b). The reduction in CRKL protein levels was observed as early as 48 h after p53 or TAp63γ overexpression (Fig. S4). We then assessed the effect of the p53 family on the miR-200b/200c/429 regulatory element in the CRKL 3′-UTR. As demonstrated in Figure4, p53, TAp73 and TAp63 robustly decreased the activity of the CRKL 3′-UTR luciferase reporter. These observations indicate that p53 family members downregulate CRKL, at least partially through miR-200b/200c/429 transactivation, although we cannot rule out the possibility that p53 family downregulates CRKL through other mechanisms.
Figure 3.

Downregulation of CRKL by the p53 family. (a) Downregulation of CRKL protein by p53 family in human cancer cell lines. Cells were infected with replication-deficient recombinant adenoviruses encoding human p53 family proteins or the bacterial lacZ for 48 h. CRKL, p53, p73, p63, p21 and β-actin levels were analyzed by immunoblotting. (b) Downregulation of CRKL mRNA by the p53 family in human cancer cell lines. Cells were infected with adenoviruses, and relative gene expression levels were quantified as described above.
Figure 4.
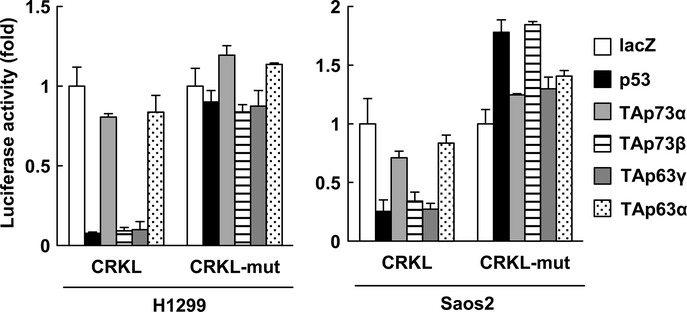
The CRKL 3′-UTR is regulated by the p53 family in H1299 and Saos2 cells. Cells were transiently transfected with the pMIR-REPORT vector containing CRKL 3′-UTR (pMIR-CRKL-3′-UTR) or a mutant (pMIR-CRKL-3′-UTR-mut) along with transfection control plasmids expressing Renilla luciferase. The cells were co-transfected with a control pcDNA3.1 vector or a vector that expressed the p53 family member 48 h prior to performing the luciferase assay. Luciferase activity was measured via the dual-luciferase reporter assay system. All of the experiments were performed in quadruplicate, and the mean and standard deviation are indicated by the bars.
We next examined whether the activation of endogenous p53 can inhibit CRKL expression. Indeed, adriamycin-mediated p53 activation in the p53 wild-type osteosarcoma cell lines Kiku and U2OS decreased CRKL protein and mRNA expression levels (Fig.5a,b), which was associated with an induction of miR-200b/200c/429 levels (Fig.5c). This effect was abolished when p53-null Saos2 and p53-mutated HOS cells were treated with adriamycin. We then examined CRKL expression levels in human lung cancer cell lines upon adriamycin or Nutlin-3 treatment. Although CRKL mRNA was slightly decreased in p53-null H1299 cells after adriamycin treatment, adriamycin or Nutlin-3 treatment resulted in an apparent decrease in CRKL expression in p53-wild type A549 cells (Fig. S5a,b). Therefore, wild type p53 is necessary for the downregulation of CRKL expression after DNA damage.
Figure 5.
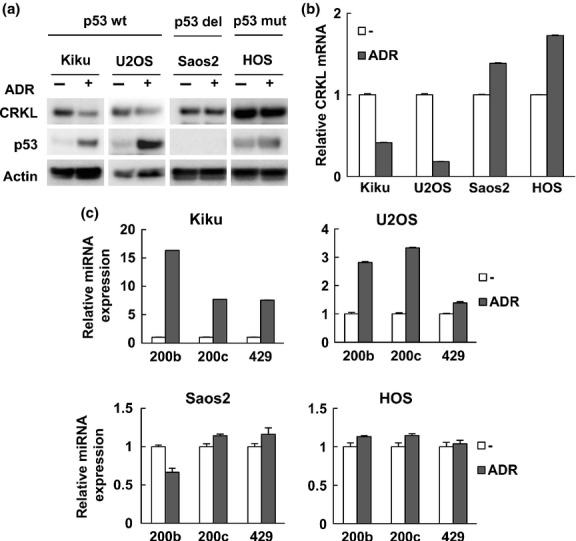
Endogenous p53 downregulates CRKL mRNA and protein levels. (a) Cells were treated with adriamycin (ADR, 0.5 μg/mL) for 24 h. CRKL and p53 protein levels were determined by immunoblot analysis. β-actin was included as a loading control. Kiku and U2OS have wild type p53, Saos2 cells are deleted for p53, and HOS cells have mutant p53. (b) CRKL mRNA levels were assessed by real-time RT-PCR. (c) The indicated cells were treated with ADR for 16 h. Relative miRNA expression levels were quantified by real-time RT-PCR.
CRKL regulates cancer cell growth and invasion
To examine the effects of CRKL on cancer cell growth, we transfected CRKL expression vectors into A549 lung cancer cells that express low levels of endogenous CRKL (Fig.6a). Using colony formation and MTT assays, we demonstrated that the ectopic expression of CRKL significantly promoted cancer cell growth (Fig.6b,c). Conversely, we knocked down the CRKL gene with siRNA vectors targeting CRKL in H1299, Saos2, U2OS, Kiku and U87MG cells (Fig. S6a). CRKL silencing suppressed the proliferation of cancer cells as assessed by MTT assays (Fig. S6b).
Figure 6.
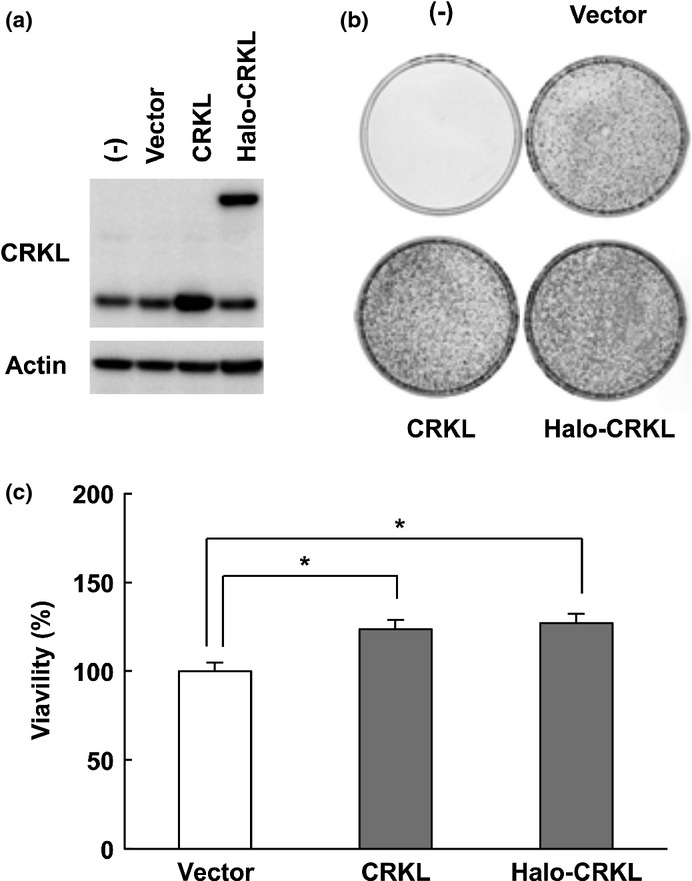
CRKL overexpression promotes cancer cell growth. (a) A549 cells were transfected with pF5K-CRKL (CRKL), Halo-tagged CRKL expression vector pFN28K-CRKL (Halo-CRKL) or empty vector (vector). Cell lysates were prepared 24 h post-transfection, and CRKL protein levels were determined by immunoblot analysis. β-actin was included as a loading control. (b) Colony formation assay of A549 cells transfected with CRKL expression vectors. After 14 days of geneticin selection, the cells were fixed and Giemsa-stained. (c) Cells were transfected as described above. After 48 h, the cell survival was subsequently determined by MTT assay. Data are the mean and standard deviation from three independent experiments. *P < 0.05 relative to empty vector (vector).
Because ectopic expression of the miR-200 family induced EMT and inhibited invasion of cancer cells,25,26 we addressed whether CRKL can block miR-200c-mediated inhibition of cancer cell invasion. Transfection of miR-200c in H1299 cells resulted in reduction of CRKL and ZEB1 as expected (Fig. S7a). Matrigel invasion assay revealed that miR-200c also inhibited cell invasion. This invasion-inhibiting effect of miR-200c was largely abolished by cotransfection with Halo-tagged CRKL transcript lacking its 3′-UTR (Fig. S7b). These results suggest that miR-200c regulates cancer cell invasion partially through targeting CRKL.
Association between CRKL mRNA levels and TP53 status in human cancer tissues
Using a collection of human cancer microarray data (Oncomine Database), we determined that CRKL is significantly overexpressed in cancer tissues in multiple microarray datasets (Fig. S8a). We also examined the relationship between CRKL expression and TP53 mutational status using the Oncomine Database. A large published dataset that includes 251 clinically annotated cases of breast cancer with RNA microarray and TP53 mutation data was interrogated from the Gene Expression Omnibus (GEO) database (dataset GSE3494). CRKL mRNA expression in these breast cancers was significantly correlated with mutant TP53 status (Fig. S8b, P = 0.0025). Survivin and hepatoma-derived growth factor (HDGF), known targets that are negatively regulated by p53,31 are also significantly overexpressed in breast cancer tissues with mutant TP53 compared with wild type p53 breast cancer tissue (Fig. S8b and data not shown). Taken together, these data suggest that CRKL expression is increased at least partially in a mutant TP53-dependent manner in human cancers. The relationship between the expression of CRKL and prognosis in cancer patients was examined using the PrognoScan database, which is a large collection of publicly available cancer microarray datasets with clinical annotation.32 As shown in Figure S9, the upregulation of CRKL correlated with poor prognosis in certain cancers.
Discussion
In addition to gene transactivation, p53 downregulates multiple genes that are involved in cell cycle progression, cell growth and anti-apoptosis. Several mechanisms can explain p53-mediated gene repression.33 We recently determined that p53 repressed HDGF transcription by altering HDAC-dependent chromatin remodeling.31 In addition, a growing list of p53 target miRNAs and their target mRNAs have been identified as newly downstream players of the p53 pathway. In the current study, we identified p53 response elements located around two miR-200 family clusters, both of which interacted with p53 family proteins (Fig. S2a,b). All five members of the miR-200 family (miR-200a, miR-200b, miR-200c, miR-141 and miR-429) were increased by p53, TAp73 and TAp63 in different cell types, albeit at varying degrees. Therefore, the p53 family might regulate two miR-200 clusters in a cell-specific manner, but our results suggest that the miR-200 family is a common target of p53 family members.
The five miRNAs in the miR-200 family contain very similar seed sequences (Fig. S1a). The conservation of their seed sequences suggests that miR-200b/200c/429 share identical mRNA targets, whereas miR-141/200a targets may have slightly different sequences. Several software programs for target prediction indicate that a sequence complementary to the miR-200b/200c/429 seed sequence is present in the 3′-UTR of CRKL mRNA. miR-200b/200c/429 expression suppressed CRKL levels in several cancer cells and negatively regulated the CRKL 3′-UTR in reporter assays. We also determined that the overexpression of CRKL stimulated cancer cell growth. Conversely, the inactivation of CRKL inhibited cell growth, indicating that CRKL is a novel target for miR-200b/200c/429 regulation. We additionally discovered that CRKL could block miR-200c-mediated inhibition of cancer cell invasion.
To date, several studies have demonstrated that the members of the miR-200 family are important mediators of tumor suppression. They have been implicated in the regulation of various cancer-related processes, such as proliferation, apoptosis, EMT, migration and invasion. miR-200 family members regulate these processes through the downregulation of their multiple targets. In a previous study, we identified the forkhead box transcription factor FOXF1 as a direct p53 target; FOXF1 regulates E-cadherin transcriptional activity by interacting with its consensus binding site located upstream of the CDH1 gene.34 These data suggest that p53 may contribute to EMT suppression through multiple pathways.
CRKL is a member of the CRK family (CRKI, CRKII, CRKIII and CRKL) of adapter proteins that bind to upstream proteins via their SH2 domain and to downstream proteins via their N-terminal SH3 domain. CRK family proteins thus transmit cellular signals to target subcellular pathways.35,36 CRKL plays a role in several signal transduction pathways in response to growth factors and cytokines and is best known as a substrate of the BCR-ABL kinase in CML.35,37 Recent studies have demonstrated that CRKL plays an important role in cell proliferation and correlates with the invasion and metastasis of a wide variety of solid cancers such as lung, stomach, liver, bladder and breast tumors.16,18–20,38 Increased CRKL protein levels have been associated with advanced-stage human cancers with high-grade or enhanced proliferation.19,39,40 CRKL amplification was previously observed in non-small cell lung and gastric cancers, strongly suggesting that CRKL is an oncogene in multiple cancers. Consistent with the data from previous papers that CRKL expression correlates with poor prognosis,17,38,41 Prognoscan, a publicly available database, indicates that high expression of CRKL may serve as a poor prognostic factor for liposarcoma and lung adenocarcinoma patients (Fig. S9). We also analyzed CRKL levels in Oncomine and found frequent overexpression in multiple solid tumors (Fig. S8a). However, the mechanism that underlies CRKL upregulation in cancer is largely unknown. Our data reveal that the CRKL gene is downregulated by p53 through miR-200s. We also determined that breast cancer patients with mutant p53 have significantly higher CRKL levels than those with wild type p53 (Fig. S8b). This finding supports an inverse correlation between CRKL expression and wild type p53. Taken together, these data suggest that the CRKL oncogene is a target for downregulation in the p53 pathway.
miR-126 downregulation has been reported in various cancer types, including breast, pancreatic and gastric cancer.42–45 Wang et al.45 also observed that miR-126 inhibits CRKL by targeting the 3′-UTR region of CRKL mRNA. Our miRNA microarray analysis indicated that miR-126 was not induced by p53, TAp73β and TAp63γ (data not shown); however, p53 family may downregulate CRKL via transactivation of other unidentified p53-target miRNAs. CRKL has been demonstrated to activate the RAS signaling pathways and its expression is associated with resistance to kinase inhibitors in NSCLC patients.40 We determined genetic alterations in genes involved in the receptor tyrosine kinase (RTK)/RAS-BRAF signal transduction pathway in cell lines used in this study using a next-generation sequencer (Table S1). We showed the miR-200s induction and CRKL suppression by p53 in cancer cells independent of the activation of this pathway. We also found that knockdown of CRKL significantly inhibited the growth of cell lines without RAS family mutations. Our study provides evidence that miR-200b/200c/429 are directly involved in the regulation of a key anticancer target CRKL.
A peptide inhibitor that selectively targets the CRKL SH3 domain can block primary CML blast cell proliferation by interacting with a target kinase BCR-ABL.46 Moreover, the dual Src/BCR-ABL inhibitor dasatinib can suppress the viability of gastric cancer cells with the CRKL gene amplification by inhibiting CRKL phosphorylation.18 Dasatinib affected the stability of protein interactions downstream of BCR-ABL, including CRKL, suggesting that the treatment of live cells affects both direct target proteins and downstream effectors.47 These studies suggest that CRKL can serve as an “actionable” therapeutic target in p53-mutated cancers. The studies outlined herein clearly identify the CRKL oncogene as a key downstream effector of the p53 pathway in cancer (Fig. S10).
Acknowledgments
This research was supported in part by MEXT KAKENHI Grant Number 221S0001 and JSPS KAKENHI Grant Number 25430115.
Disclosure Statement
The authors have no conflict of interest to declare.
Supporting Information
Fig. S1. miR-200 family members.
Fig. S2. miR-200 family members are direct targets of the p53 family.
Fig. S3. CRKL protein is downregulated by miR-200b/200c/429 in Saos2 and U2OS cells.
Fig. S4. Downregulation of CRKL by p53 and TAp63γ.
Fig. S5. Endogenous p53 downregulates CRKL protein and mRNA levels in human lung cancer cells.
Fig. S6. Downregulation of CRKL by siRNA inhibits cancer cell growth.
Fig. S7. CRKL prevents miR-200c-mediated inhibition of cell invasion.
Fig. S8. CRKL expression profiles in human tumors using published human oncology microarray data in Oncomine.
Fig. S9. The correlation between CRKL expression and prognosis among cancer patients.
Fig. S10. A model for CRKL downregulation by p53 via miR-200s in normal and cancer cells.
Table S1. Summary of mutated genes detected in cell lines used in this study.
Data S1. Supplementary materials and methods.
References
- Nakamura Y. Isolation of p53-target genes and their functional analysis. Cancer Sci. 2004;95:7–11. doi: 10.1111/j.1349-7006.2004.tb03163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–10. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- Feng Z, Zhang C, Wu R, et al. Tumor suppressor p53 meets microRNAs. J Mol Cell Biol. 2011;3:44–50. doi: 10.1093/jmcb/mjq040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bommer GT, Gerin I, Feng Y, et al. p53-mediated activation of miRNA34 candidate tumor-suppressor genes. Curr Biol. 2007;17:1298–307. doi: 10.1016/j.cub.2007.06.068. [DOI] [PubMed] [Google Scholar]
- He L, He X, Lim LP, et al. A microRNA component of the p53 tumour suppressor network. Nature. 2007;447:1130–4. doi: 10.1038/nature05939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tazawa H, Tsuchiya N, Izumiya M, et al. Tumor-suppressive miR-34a induces senescence-like growth arrest through modulation of the E2F pathway in human colon cancer cells. Proc Natl Acad Sci USA. 2007;104:15472–7. doi: 10.1073/pnas.0707351104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georges SA, Biery MC, Kim SY, et al. Coordinated regulation of cell cycle transcripts by p53-Inducible microRNAs, miR-192 and miR-215. Cancer Res. 2008;68:10105–12. doi: 10.1158/0008-5472.CAN-08-1846. [DOI] [PubMed] [Google Scholar]
- Song B, Wang Y, Kudo K, et al. miR-192 Regulates dihydrofolate reductase and cellular proliferation through the p53-microRNA circuit. Clin Cancer Res. 2008;14:8080–6. doi: 10.1158/1078-0432.CCR-08-1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christoffersen NR, Shalgi R, Frankel LB, et al. p53-independent upregulation of miR-34a during oncogene-induced senescence represses MYC. Cell Death Differ. 2010;17:236–45. doi: 10.1038/cdd.2009.109. [DOI] [PubMed] [Google Scholar]
- Sachdeva M, Zhu S, Wu F, et al. p53 represses c-Myc through induction of the tumor suppressor miR-145. Proc Natl Acad Sci USA. 2009;106:3207–12. doi: 10.1073/pnas.0808042106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichiorri F, Suh SS, Rocci A, et al. Downregulation of p53-inducible microRNAs 192, 194, and 215 impairs the p53/MDM2 autoregulatory loop in multiple myeloma development. Cancer Cell. 2010;18:367–81. doi: 10.1016/j.ccr.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Zhang J, Sun Q, Zhang Z, et al. Loss of microRNA-143/145 disturbs cellular growth and apoptosis of human epithelial cancers by impairing the MDM2-p53 feedback loop. Oncogene. 2013;32:61–9. doi: 10.1038/onc.2012.28. [DOI] [PubMed] [Google Scholar]
- Shimono Y, Zabala M, Cho RW, et al. Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell. 2009;138:592–603. doi: 10.1016/j.cell.2009.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellner U, Schubert J, Burk UC, et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol. 2009;11:1487–95. doi: 10.1038/ncb1998. [DOI] [PubMed] [Google Scholar]
- Feller SM. Crk family adaptors-signalling complex formation and biological roles. Oncogene. 2001;20:6348–71. doi: 10.1038/sj.onc.1204779. [DOI] [PubMed] [Google Scholar]
- Han B, Luan L, Xu Z, et al. Clinical significance and biological roles of CRKL in human bladder carcinoma. Tumour Biol. 2013;35:4101–6. doi: 10.1007/s13277-013-1536-y. [DOI] [PubMed] [Google Scholar]
- Lin F, Chengyao X, Qingchang L, et al. CRKL promotes lung cancer cell invasion through ERK-MMP9 pathway. Mol Carcinog. 2014;S1:E35–44. doi: 10.1002/mc.22148. [DOI] [PubMed] [Google Scholar]
- Natsume H, Shinmura K, Tao H, et al. The CRKL gene encoding an adaptor protein is amplified, overexpressed, and a possible therapeutic target in gastric cancer. J Transl Med. 2012;10:97. doi: 10.1186/1479-5876-10-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao T, Miao Z, Wang Z, et al. Overexpression of CRKL correlates with malignant cell proliferation in breast cancer. Tumour Biol. 2013;34:2891–7. doi: 10.1007/s13277-013-0851-7. [DOI] [PubMed] [Google Scholar]
- Kim YH, Kwei KA, Girard L, et al. Genomic and functional analysis identifies CRKL as an oncogene amplified in lung cancer. Oncogene. 2010;29:1421–30. doi: 10.1038/onc.2009.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fomenkov A, Zangen R, Huang YP, et al. RACK1 and stratifin target DeltaNp63alpha for a proteasome degradation in head and neck squamous cell carcinoma cells upon DNA damage. Cell Cycle. 2004;3:1285–95. doi: 10.4161/cc.3.10.1155. [DOI] [PubMed] [Google Scholar]
- Stiewe T. The p53 family in differentiation and tumorigenesis. Nat Rev Cancer. 2007;7:165–8. doi: 10.1038/nrc2072. [DOI] [PubMed] [Google Scholar]
- Wu N, Rollin J, Masse I, et al. p63 regulates human keratinocyte proliferation via MYC-regulated gene network and differentiation commitment through cell adhesion-related gene network. J Biol Chem. 2012;287:5627–38. doi: 10.1074/jbc.M111.328120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idogawa M, Ohashi T, Sasaki Y, et al. Identification and analysis of large intergenic non-coding RNAs regulated by p53 family members through a genome-wide analysis of p53-binding sites. Hum Mol Genet. 2014;23:2847–57. doi: 10.1093/hmg/ddt673. [DOI] [PubMed] [Google Scholar]
- Chang CJ, Chao CH, Xia W, et al. p53 regulates epithelial–mesenchymal transition and stem cell properties through modulating miRNAs. Nat Cell Biol. 2011;13:317–23. doi: 10.1038/ncb2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T, Veronese A, Pichiorri F, et al. p53 regulates epithelial–mesenchymal transition through microRNAs targeting ZEB1 and ZEB2. J Exp Med. 2011;208:875–83. doi: 10.1084/jem.20110235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knouf EC, Garg K, Arroyo JD, et al. An integrative genomic approach identifies p73 and p63 as activators of miR-200 microRNA family transcription. Nucleic Acids Res. 2012;40:499–510. doi: 10.1093/nar/gkr731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John B, Enright AJ, Aravin A, et al. Human MicroRNA targets. PLoS Biol. 2004;2:e363. doi: 10.1371/journal.pbio.0020363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krek A, Grun D, Poy MN, et al. Combinatorial microRNA target predictions. Nat Genet. 2005;37:495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- Sasaki Y, Negishi H, Idogawa M, et al. p53 negatively regulates the hepatoma growth factor HDGF. Cancer Res. 2011;71:7038–47. doi: 10.1158/0008-5472.CAN-11-1053. [DOI] [PubMed] [Google Scholar]
- Mizuno H, Kitada K, Nakai K, et al. PrognoScan: a new database for meta-analysis of the prognostic value of genes. BMC Med Genomics. 2009;2:18. doi: 10.1186/1755-8794-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho J, Benchimol S. Transcriptional repression mediated by the p53 tumour suppressor. Cell Death Differ. 2003;10:404–8. doi: 10.1038/sj.cdd.4401191. [DOI] [PubMed] [Google Scholar]
- Tamura M, Sasaki Y, Koyama R, et al. Forkhead transcription factor FOXF1 is a novel target gene of the p53 family and regulates cancer cell migration and invasiveness. Oncogene. 2014;33:4837–46. doi: 10.1038/onc.2013.427. [DOI] [PubMed] [Google Scholar]
- Birge RB, Kalodimos C, Inagaki F, et al. Crk and CrkL adaptor proteins: networks for physiological and pathological signaling. Cell Commun Signal. 2009;7:13. doi: 10.1186/1478-811X-7-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prosser S, Sorokina E, Pratt P, et al. CrkIII: a novel and biologically distinct member of the Crk family of adaptor proteins. Oncogene. 2003;22:4799–806. doi: 10.1038/sj.onc.1206714. [DOI] [PubMed] [Google Scholar]
- Mayer BJ, Hamaguchi M, Hanafusa H. A novel viral oncogene with structural similarity to phospholipase C. Nature. 1988;332:272–5. doi: 10.1038/332272a0. [DOI] [PubMed] [Google Scholar]
- Liu CH, Chen TC, Chau GY, et al. Analysis of protein-protein interactions in cross-talk pathways reveals CRKL protein as a novel prognostic marker in hepatocellular carcinoma. Mol Cell Proteomics. 2013;12:1335–49. doi: 10.1074/mcp.O112.020404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Dong QZ, Fu L, et al. Overexpression of CRKL correlates with poor prognosis and cell proliferation in non-small cell lung cancer. Mol Carcinog. 2013;52:890–9. doi: 10.1002/mc.21935. [DOI] [PubMed] [Google Scholar]
- Cheung HW, Du J, Boehm JS, et al. Amplification of CRKL induces transformation and epidermal growth factor receptor inhibitor resistance in human non-small cell lung cancers. Cancer Discov. 2011;1:608–25. doi: 10.1158/2159-8290.CD-11-0046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng S, Guo J, Yang Q, et al. Crk-like adapter protein regulates CCL19/CCR7-mediated epithelial-to-mesenchymal transition via ERK signaling pathway in epithelial ovarian carcinomas. Med Oncol. 2015;32:47. doi: 10.1007/s12032-015-0494-1. [DOI] [PubMed] [Google Scholar]
- Feng R, Chen X, Yu Y, et al. miR-126 functions as a tumour suppressor in human gastric cancer. Cancer Lett. 2010;298:50–63. doi: 10.1016/j.canlet.2010.06.004. [DOI] [PubMed] [Google Scholar]
- Hamada S, Satoh K, Fujibuchi W, et al. MiR-126 acts as a tumor suppressor in pancreatic cancer cells via the regulation of ADAM9. Mol Cancer Res. 2012;10:3–10. doi: 10.1158/1541-7786.MCR-11-0272. [DOI] [PubMed] [Google Scholar]
- Tavazoie SF, Alarcon C, Oskarsson T, et al. Endogenous human microRNAs that suppress breast cancer metastasis. Nature. 2008;451:147–52. doi: 10.1038/nature06487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Chen X, Li P, et al. CRKL promotes cell proliferation in gastric cancer and is negatively regulated by miR-126. Chem Biol Interact. 2013;206:230–8. doi: 10.1016/j.cbi.2013.09.003. [DOI] [PubMed] [Google Scholar]
- Kardinal C, Konkol B, Schulz A, et al. Cell-penetrating SH3 domain blocker peptides inhibit proliferation of primary blast cells from CML patients. FASEB J. 2000;14:1529–38. doi: 10.1096/fj.14.11.1529. [DOI] [PubMed] [Google Scholar]
- Savitski MM, Reinhard FB, Franken H, et al. Proteomics. Tracking cancer drugs in living cells by thermal profiling of the proteome. Science. 2014;346:1255784. doi: 10.1126/science.1255784. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. miR-200 family members.
Fig. S2. miR-200 family members are direct targets of the p53 family.
Fig. S3. CRKL protein is downregulated by miR-200b/200c/429 in Saos2 and U2OS cells.
Fig. S4. Downregulation of CRKL by p53 and TAp63γ.
Fig. S5. Endogenous p53 downregulates CRKL protein and mRNA levels in human lung cancer cells.
Fig. S6. Downregulation of CRKL by siRNA inhibits cancer cell growth.
Fig. S7. CRKL prevents miR-200c-mediated inhibition of cell invasion.
Fig. S8. CRKL expression profiles in human tumors using published human oncology microarray data in Oncomine.
Fig. S9. The correlation between CRKL expression and prognosis among cancer patients.
Fig. S10. A model for CRKL downregulation by p53 via miR-200s in normal and cancer cells.
Table S1. Summary of mutated genes detected in cell lines used in this study.
Data S1. Supplementary materials and methods.