

ABSTRACT
Pulmonary infections caused by Pseudomonas aeruginosa are a recalcitrant problem in cystic fibrosis (CF) patients. While the clinical implications and long-term evolutionary patterns of these infections are well studied, we know little about the short-term population dynamics that enable this pathogen to persist despite aggressive antimicrobial therapy. Here, we describe a short-term population genomic analysis of 233 P. aeruginosa isolates collected from 12 sputum specimens obtained over a 1-year period from a single patient. Whole-genome sequencing and antimicrobial susceptibility profiling identified the expansion of two clonal lineages. The first lineage originated from the coalescence of the entire sample less than 3 years before the end of the study and gave rise to a high-diversity ancestral population. The second expansion occurred 2 years later and gave rise to a derived population with a strong signal of positive selection. These events show characteristics consistent with recurrent selective sweeps. While we cannot identify the specific mutations responsible for the origins of the clonal lineages, we find that the majority of mutations occur in loci previously associated with virulence and resistance. Additionally, approximately one-third of all mutations occur in loci that are mutated multiple times, highlighting the importance of parallel pathoadaptation. One such locus is the gene encoding penicillin-binding protein 3, which received three independent mutations. Our functional analysis of these alleles shows that they provide differential fitness benefits dependent on the antibiotic under selection. These data reveal that bacterial populations can undergo extensive and dramatic changes that are not revealed by lower-resolution analyses.
IMPORTANCE
Pseudomonas aeruginosa is a bacterial opportunistic pathogen responsible for significant morbidity and mortality in cystic fibrosis (CF) patients. Once it has colonized the lung in CF, it is highly resilient and rarely eradicated. This study presents a deep sampling examination of the fine-scale evolutionary dynamics of P. aeruginosa in the lungs of a chronically infected CF patient. We show that diversity of P. aeruginosa is driven by recurrent clonal emergence and expansion within this patient and identify potential adaptive variants associated with these events. This high-resolution sequencing strategy thus reveals important intraspecies dynamics that explain a clinically important phenomenon not evident at a lower-resolution analysis of community structure.
INTRODUCTION
Cystic fibrosis (CF) is the most common fatal genetic disease among Caucasians (1, 2) and is characterized by chronic polymicrobial lung infections that are frequently dominated by opportunistic pathogens, such as Pseudomonas aeruginosa and species from the Burkholderia cepacia complex (3, 4). Polymicrobial communities in adult CF airways appear to be highly resilient and resistant to change, despite the administration of sequential antibiotic regimens targeting these dominant pathogens (5, 6). However, our perception that these communities are largely static is predominantly driven by culture-dependent studies of sputum and bronchoalveolar lavage fluid, microbiome studies using subregions of the 16S rRNA gene, and long-term longitudinal comparative genomics studies (7–9). While these approaches have tremendously informed our understanding of CF lung microbiology, they have not been performed at the temporal and taxonomic resolution required to characterize short-term intraspecies population dynamics. Since selection on microbial communities acts at the level of individual clones over the short temporal scale of antimicrobial treatments and lung disease exacerbations (10), a higher-resolution taxonomic and temporal approach is required to identify, understand, and ultimately manipulate the factors that control the composition and function of polymicrobial communities.
CF lung microbial communities persist in the face of powerful and complex pressures imposed by episodic exacerbations of lung inflammation and courses of antimicrobials, which typically include both long-term treatment with maintenance antibiotics as well as short-term, recurrent courses of other antimicrobials administered alone or in combination (11, 12). A number of studies have used comparative genomics to address how CF pathogens and P. aeruginosa in particular adapt to this dynamic lung environment. These have shown that airway-adapted isolates display very high levels of phenotypic diversity, strong mutational parallelism (recurrent mutations in the same genes among independent lineages), the emergence of hypermutable strains, mutations in global regulatory genes, and long-term persistence of a small number of clonal lineages (13–29). While these studies have furthered our understanding of the long-term evolutionary dynamics of P. aeruginosa in the CF lung, the large majority have been based on isolates collected several years apart—in some cases, 35 or more years apart—and none have focused on the immediate impact of antimicrobial therapies on the overall population of P. aeruginosa in the CF lung over a very short time span (19, 21, 24, 26, 27). This is a critical gap in our knowledge since clinical assessments and antimicrobial therapies are often modified as frequently as weekly (30).
A fundamental assumption of antimicrobial therapies is that they select against and thereby drive down the population size of target pathogens. A nearly unavoidable consequence of these therapies is the selection for resistant clones of the pathogen, which may ultimately dominate the population. The application of a selective pressure to microbial populations commonly leads to selective sweeps, which involve the spread of a clone carrying an adaptive mutation through the population and subsequent displacement of other preexisting and now less fit clones (31–34). Selective sweeps will periodically reoccur in a population as new beneficial mutations arise and new selected clones replace ancestral clones. This pattern of recurrent selective sweeps has historically been called periodic selection (35). While selective sweeps have been well documented in microbial populations (particularly with respect to viral populations, as reviewed in reference 36), the presence and significance of periodic selection in human health and infectious diseases are not known (37).
Clonal evolutionary dynamics such as selective sweeps have been largely overlooked in microbiome studies due to the lack of resolution and power. For example, 16S rRNA gene sequencing can only reliably resolve taxa to the genus level (38), while full metagenome sequencing suffers from the inability to phase genetic variation (assign variants into genotypes) (39). These limitations have restricted our ability to observe clonal evolutionary dynamics, identify their drivers, and assess their resulting impact on the overall microbial community as well as the host.
In the case of CF-associated lung infections, longitudinal population sampling (e.g., multiple clones assessed from each clinical specimen) performed over the short term will reveal the impact of intensive antimicrobial therapies on the dynamics of the target pathogen populations. Understanding intraspecies pathogen evolution over this time scale is essential to assess whether current treatment strategies effect desirable change of pathogen populations or if alternative treatment strategies, coupled with a better understanding of pathogen evolutionary potential, could lead to improved treatment outcomes.
In this study, we sequenced the genomes of 233 P. aeruginosa isolates from a single CF patient collected from 12 specimens over a 1-year period to assess very-short-term evolutionary dynamics. We identify the occurrence of a very recent common ancestor for the overall population, the emergence of multiple clonal lineages within a population that has remained relatively constant in size, and strong signals of positive selection—genomic features consistent with recurrent selective sweeps or periodic selection. We also find a very strong signal of parallel pathoadaptation at loci associated with virulence and antibiotic resistance and show how some of these variants confer fitness benefits that are dependent on the environment.
RESULTS
This study focuses on patient CF67, a 34-year-old female CF patient (of CF transmembrane conductance regulator [CFTR] genotype ΔF508/del2–3) who had been chronically infected with P. aeruginosa for at least 12 years prior to the initial study sample. The patient had advanced lung disease with a forced expiratory volume in 1 s (FEV1) of 21% of the predicted value throughout the course of the study. She provided 12 expectorated sputum specimens for evaluation over an ~1-year period (with the first specimen collected 350 days prior to the last specimen) (Fig. 1). The specimens were not collected uniformly over the course of the study, with 11 being collected within the last 126 days of the study.
FIG 1 .

Antibiotic treatment history and the relative abundance of the two clades over time. (A) Black bars indicate antibiotic administration, and hashed bars indicate intermittent exposure in that time block. The method of antibiotic administration is shown as intravenous (iv), inhaled (inh), or oral (po). Sputum samples were collected at the time points indicated by green lines, which extend to panels B and C. (B) Relative abundance at the genus level is shown as a percentage. (C) Relative abundance of clade A (blue shades) and clade B (red shades) genotypes over time. Genotypes are defined as whole-genome sequences differing by one or more SNPs segregating in at least two isolates. The same color and shading are used to identify specific genotypes across specimens.
During the study period, the patient was prescribed 28 distinct courses of antibiotics, which were administered by three different routes (oral, intravenous, and inhaled) in addition to continuous oral azithromycin (Fig. 1A; see Table S1 in the supplemental material). Despite this complex antimicrobial regimen, microbiome analysis via Illumina sequencing of the V5 to V7 hypervariable regions of the 16S rRNA gene (38) reveals that Pseudomonas remained the dominant genus throughout (Fig. 1B). Quantitative PCR of the 16S rRNA gene was used to quantify the bacterial load from each of the twelve specimens, revealing no appreciable change in the absolute level of bacteria recovered from each specimen over time (mean ± standard deviation [SD], 0.395 ± 0.185 ng/ml; regression slope, −0.0005x).
We collected multiple P. aeruginosa isolates from each of the 12 specimens (range, 18 to 20 per specimen) in order to obtain a finer-resolution analysis of the population-level dynamics. An effort was made to maintain overall morphotype relative abundance in our sampling as previously described (40).
Genomic diversity and phylogenetic analyses identify two genetically distinct clades.
A total of 233 P. aeruginosa isolates underwent whole-genome sequencing (7 of the total of 240 isolates were removed from the analysis due to quality control issues), yielding a median coverage depth of 54× (see Fig. S1 in the supplemental material). The genome of one isolate was assembled de novo for use as a reference for assembly of the remaining isolates. This assembly had an N50 of 329,133 bp and 46 contigs, with the smallest contig being 1,170 bp and a total genome size of 6,367,116 bp. We used a conservative pipeline to call single nucleotide polymorphisms (SNPs) that required the agreement of three algorithmically distinct reference assembly methods and then reevaluated each SNP position using a more relaxed inspection of the pileup data (see Fig. S2 in the supplemental material). The pipeline identified 107 segregating SNPs among the 233 isolates, of which 54 were phylogenetically informative (SNPs segregating in at least two isolates), while 39 were found in isolates collected from multiple time points (multispecimen SNPs). Additionally, we identified insertion/deletion (indel) variants by realigning the sequencing reads to initial candidates and then reviewing all putative indel positions in all isolates. This process revealed 58 indels, of which 28 were found in at least two isolates from different collection time points. Sequencing of the entire population of all P. aeruginosa isolates bulk extracted from one specimen did not reveal any mutations that were not present in the individual isolate sequencing, which indicated that our sampling of diversity is close to saturation (see Fig. S3 in the supplemental material).
The data were analyzed by both network-based (neighbor-net) and traditional (maximum likelihood) phylogenetic methods (Fig. 2). For the latter, the position of the root is very similar, regardless of whether it is identified by incorporating P. aeruginosa LESB58 into the analysis, selecting it based on the position of the strains isolated in specimen 1, or through midpoint rooting. Visual inspection of both trees revealed an unusual topology. Approximately one-third of the isolates share more structured relationships with relatively long branches, and these are distinctly separated from another set of isolates that have relatively short branches originating from a very recent common ancestor. Based on this observation, we designate two clades: clades A and B. While the clades are discrete and distinct in the network-based analyses, the rooted phylogenetic analysis shows that clade B is nested inside clade A, making clade A technically paraphyletic since it is not monophyletic without the clade B strains. Despite this, we will refer to the two groups as clades for simplicity. Importantly, all analyses comparing the two clades are performed on discrete sets of strains, where all strains are designated as belonging to either clade A or clade B as determined by the network-based phylogenetic analysis.
FIG 2.

Maximum likelihood and network-based (neighbor-net) phylogenetic analyses. The phylogenetic structure of 233 P. aeruginosa isolates was characterized based on genome-wide single nucleotide polymorphisms (SNPs) using the maximum likelihood (A) and neighbor-net (B) algorithms. The structure of the resulting tree revealed two populations, clades A (blue) and B (red). The 152 isolates in clade B produce a star phylogeny consistent with a recent expansion of a clonal population, while the 81 isolates in clade A show longer branches with more phylogenetic structure. (A) The scale bar shows genetic distance using the maximum composite distance and all segregating SNPs. The strain names shown on the tree are a composite of the specimen number from which the clone was isolated, an arbitrary clone letter, and the clade designation. (B) Individual strain names at the tips of each branch have been replaced with pie charts indicating the distribution of dates during which the strains were sampled (indicated by the circular legend). The scale bar indicates genetic distances.
Clade A has a lower overall relative abundance than clade B (81 isolates versus 152 isolates for clades A and B, respectively [Fig. 1 and Table 1]) but is significantly more genetically diverse, with an average of 4.80 ± 3.97 (mean ± SD) pairwise differences among clade A isolates versus 2.84 ± 1.60 differences for clade B isolates (P <<< 0.0001, t test) (Fig. 3). Neither clade shows a significant recombination signal, with a four-gamete recombination test identifying a minimum of only one recombination event in the entire sample (41). This is supported by the nearly complete lack of reticulation in the neighbor-net phylogenetic analysis. The relatively uniform distance of clade B strains from their common ancestor seen in the phylogenetic analyses is reminiscent of a “star phylogeny” pattern of divergence, which is often associated with recent purges of genetic variation, such as those seen during population expansions or selective sweeps (42). There are six segregating mutations (four SNPs and two indels) that have variants fixed between the two clades. This is significant since fixed differences reflect population subdivision.
TABLE 1 .
Genetic diversity
| Variable | No. (%) |
||
|---|---|---|---|
| Clade A | Clade B | Total | |
| Isolates | 81 (34.8) | 152 (65.2) | 233 |
| SNPs | 72 (67.3) | 35 (32.7) | 107 |
| Indels | 31 (53.4) | 28 (48.3) | 58a |
| Phylogenetically informative SNPs | 36 (64.3) | 20 (35.7) | 56 |
| Multi-time SNPs | 21 (53.8) | 18 (46.2) | 39 (57.1) |
| Multi-time indels | 13 (46.4) | 16 (57.1) | 28a |
| Genetic distance, b avg ± SD | 0.052 ± 0.04 | 0.029 ± 0.03 | 0.047 ± 0.04 |
| No. of SNP differences, avg ± SD | 4.80 ± 3.97 | 2.84 ± 1.60 | 6.49 ± 4.41 |
| No. of indel differences, avg ± SD | 4.69 ± 3.11 | 3.05 ± 1.91 | 5.03 ± 3.09 |
One indel segregates in clade A and clade B.
Maximum composite likelihood distance.
FIG 3 .

Frequency spectrum of SNPs in clades A and B. (A) The frequencies of the number of SNP differences between isolates within clade A and B reveal different distributions. While in clade A, most pairwise SNP differences occurred evenly, in clade A, pairwise SNP differences lower than 5 are overrepresented. (B) Distribution of pairwise SNP differences within clades A and B shows different profiles (P <<< 0.0001, t test).
Time to common ancestry.
We used the Bayesian approach implemented in BEAST (43, 44) to infer the time to most recent common ancestry (tMRCA) for the total population and each clade individually. The total population of 233 isolates coalesces to an MRCA of 993.24 ± 9.09 days (2.72 years) prior to the last specimen, while clade A isolates coalesce to a tMRCA of 956.75 ± 8.93 days (2.70 years) prior to the last specimen, and clade B isolates coalesce to an tMRCA of 329.42 ± 1.80 days (0.90 years) prior to the last specimen. We compared our isolates to other fully sequenced P. aeruginosa isolates (see Fig. S4 in the supplemental material) and found the Liverpool epidemic strain LESB58 to be the closest reference strain, with a tMRCA of 24.06 years prior to the last specimen (45, 46). The probability that our sample of 233 isolates captures the true tMRCA of the population can be calculated by (n − 1)/(n + 1) = 0.991, where n is the sample size (47).
Our analysis supports a population model in which clade A is a longer-lived ancestral population that has accumulated more genetic variation than clade B. Clade B, on the other hand, appears to be a more recently emerged population that arose between collection of the first and second specimens. The relatively recent coalescence of the total sample and clade B in particular suggests that we may have captured two periods of clonal emergence, expansion, and replacement. This is supported by the fact that clade A is more abundant in the earlier specimens, while clade B dominates in the vast majority of the later specimens (Fig. 1).
Selection analysis.
The emergence and spread of a new clone are called a selective sweep when driven by positive selection; therefore, we sought to identify evidence of selective pressure driving the emergence of clade B. Analysis of the 107 segregating sites in the total population gave a ratio of nonsynonymous to synonymous mutations (dN/dS ratio) of 1.01, which is consistent with the null model of neutral evolution but which does not rule out more complex selective patterns (Fig. 4 and Table 2). Importantly, this value is also consistent with a nonequilibrium population, in which the short time frame of the study allows us to capture mutations as they arise but before their fate has been determined by natural selection or genetic drift. Consequently, it is probable that some of the observed SNPs are deleterious to some degree and will be culled from the population in the longer term. To minimize this confounding effect, we elected to focus on only those 39 SNPs that segregated over at least two sampling time points (multispecimen SNPs). It would be expected that removing transient deleterious mutations from the analysis should decrease the relative number of nonsynonymous mutations, thus decreasing dN/dS ratio. Despite this, we find a dN/dS ratio of 1.43, consistent with weak positive selection acting on mutations maintained over multiple time points. When we split the sample based on the inferred clade structure, we find that the clade A, multispecimen segregating SNPs gave a dN/dS ratio of 1.04, compared to a dN/dS ratio of 2.42 for clade B. Thus, clade B is under greater positive selective pressure than clade A. When we further break down the analysis into the 12 individual specimens, we find a very consistent pattern, with clade B maintaining a higher dN/dS ratio relative to clade A throughout, with the exception of specimen 9 (Fig. 4). It is also intriguing the dN/dS ratio observed in clade A starts at 1.3 and then drops to 1.0 or below after specimen 1, once again with the exception of specimen 9. While there are many potential factors that can influence dN/dS ratios, and their calculation is highly dependent on sampling variance, the overall pattern observed in these data supports stronger and more sustained positive selection in clade B than clade A. Notably, the reversal of the dN/dS ratio trend noted in specimen 9 coincided with the cessation of aztreonam during that sampling period.
FIG 4 .

dN/dS ratio calculation over time. Shown is the nonsynonymous over synonymous substitution rate (dN/dS) for multispecimen SNPs calculated for each sample time and partitioned based on clade membership. Clade B arose between sampling specimens 1 and 2; therefore, there are no data for clade B.
TABLE 2 .
Selection analysis results
| Variable | No. (%) |
dN/dS ratio | ||
|---|---|---|---|---|
| Nonsynonymous | Synonymous | Intergenic | ||
| All SNPs | 73 (68.2) | 25 (23.4) | 9 (8.4) | 1.01 |
| Multitime SNPs | ||||
| All | 29 (74.4) | 7 (17.9) | 3 (7.7) | 1.43 |
| Clade A | 15 (71.4) | 5 (23.8) | 1 (4.8) | 1.04 |
| Clade B | 14 (77.8) | 2 (11.1) | 2 (11.1) | 2.42 |
The dN/dS results supporting positive selection are further bolstered by the significantly negative result for both a Tajima’s D test (48) and Fu and Li’s test (49). Tajima’s D = to −2.08 (P = 0.002), while Fu and Li’s D* = −5.58 (P = 0.001) and D = to −5.60 (P < 0.001, using the P. aeruginosa LESB58 strain as an outgroup). No significant differences were observed between the two clades in these analyses. These results indicate that there has been a recent population expansion after a selective sweep or population bottleneck. Given that there was no change in the absolute size of the Pseudomonas population in this patient, the best explanation for this expansion is the replacement of one clone by another.
Parallel pathoadaptation.
A striking pattern in the data is the number of loci that were independently mutated multiple times. The occurrence of multiple independent mutations within the same locus can be a very strong indication of adaptive parallel evolution and strong selection on the locus (50–52). Nineteen loci were mutated multiple times (Table 3). Thirty-three of the 107 SNPs (30.8%) occur in these loci, with 15 (38.5%) being multispecimen SNPs, while 20 of the 58 indels (34.5%) are found in loci with multiple independent mutations, 11 of which are multispecimen indels (39.3%).
TABLE 3 .
Multiply mutated loci
| Encoded protein | Locus | No. of SNPs/indels |
Probability a |
|---|---|---|---|
| Murein tripeptide ligase (Mpl) | PA4020 | 1/5 | 1.20 × 10−19 |
| Anti-sigma factor (MucA)/sigma factor (AlgU)b | PA0763/PA0762 | 2/3 | 1.17 × 10−14 |
| Alginate-C5-mannuronan epimerase (AlgG) | PA3545 | 5/0 | 7.31 × 10−16 |
| Conserved hypothetical protein | PA4701 | 2/2 | 3.56 × 10−11 |
| Transcriptional regulator (AmpR)/β-lactamase precursor (AmpC) | PA4109/PA4110 | 4/0 | 3.56 × 10−11 |
| Penicillin-binding protein 3 (PBP3) | PA4418 | 3/0 | 2.70 × 10−08 |
| Multidrug efflux membrane fusion protein (MexA)/multidrug efflux transporter (MexB) | PA0425/PA0426 | 0/3 | 1.08 × 10−07 |
| Transcriptional regulator (MexT) | PA2492 | 2/0 | 1.64 × 10−04 |
| Outer membrane lipoprotein precursor (OprI)c | PA2853 | 2/0 | 1.64 × 10−04 |
| Hypothetical protein | PA3093 | 2/0 | 1.64 × 10−04 |
| Hypothetical proteind | NCGM23404 | 2/0 | 1.64 × 10−04 |
| Aminopeptidase P (PepP)/ubiquinone biosynthesis protein (UbiH)c | PA5224/PA5223 | 0/2 | 3.29 × 10−04 |
| ld-Carboxypeptidase (LdcA) | PA5198 | 1/1 | 1.64 × 10−04 |
| Conserved hypothetical protein | PA5133 | 1/1 | 1.64 × 10−04 |
| Conserved hypothetical protein/hypothetical protein | PA4962/PA4961 | 0/2 | 3.29 × 10−04 |
| Transcriptional regulator (LasR) | PA1430 | 1/1 | 1.64 × 10−04 |
| Multidrug resistance operon repressor (MexR) | PA0424 | 1/1 | 1.64 × 10−04 |
| Probable transcriptional regulator | PA0535 | 1/1 | 1.64 × 10−04 |
| d-Amino acid dehydrogenase, small subunit (DadA) | PA5304 | 0/2 | 1.64 × 10−04 |
Calculated based on the probability of resampling with replacement any locus n times, given a genome size of N. P = (1/N)^(n − 1). We used (n − 1) since we are calculating the probability for any locus, rather than a specific locus.
Mutations that occurred in two adjacent loci and which may be part of a common regulon according to the Pseudomonas Genome Database (97). The probability calculation was adjusted by dividing the total number of loci in the genome in half.
One mutation occurred in the intergenic region flanking this locus.
This locus is not found in PAO1. The homolog with highest similarity is in P. aeruginosa NCGM2.S1 (100).
We compared the mutational class spectrum (relative frequency of nonsynonymous, synonymous, and intergenic changes) in our entire data set to that seen in loci with multiple mutations to determine if the parallel evolution is due to selection or simply a higher local mutation rate. We would expect an elevated mutation rate to maintain the same ratio of nonsynonymous to synonymous changes, while adaptive parallel evolution should predominantly be driven by nonsynonymous changes. We observed a mutational class spectrum of 68.2% nonsynonymous, 23.4% synonymous, and 8.4%, intergenic mutations in our full data set and a spectrum of 91.7%, 0.0%, and 8.3%, respectively, for the loci with multiple mutations. This highly significant skew toward more nonsynonymous mutations (chi-square test, P = 1.5 × 10−7) strongly supports selection-driven parallel evolution.
Many of the loci with multiple mutations are known to play important roles in P. aeruginosa virulence in the setting of CF. Among them is the gene that encodes a murein peptide ligase (Mpl), which has been shown to increase expression of AmpC and resistance to the β-lactam antibiotics piperacillin and ceftazidime (53). This locus carries five independent indels and one SNP. Given a genome size of 6,082 genes and making some simplifying assumptions concerning uniformity of gene size, the probability of any gene being mutated six times is 1.2 × 10−19. Similarly, genes involved in alginate biosynthesis, such as the MucA and AlgU cluster and the locus that encodes AlgG, were each mutated five times (P = 1.17 × 10−14 and P = 7.3 × 10−16, respectively). Many other loci carrying multiple independent mutations also influence antimicrobial resistance, such as the operon encoding multidrug resistance-related loci MexA and MexB, which was mutated four times (P = 1.08 × 10−07), the transcriptional regulator AmpR and its neighboring β-lactamase precursor, AmpC, which were mutated four times (P = 3.6 × 10−11), and the gene encoding penicillin-binding protein 3 (PBP3), which was mutated three times (P = 2.7 × 10−8). Six other loci were mutated twice (P = 1.6 × 10−4 for any one locus to receive two independent mutations), including the gene encoding the transcriptional regulator MexT.
Identification of potential adaptive variants.
To detect possible adaptive variants, we determined the ontology of multispecimen segregating variants. Many loci with segregating variation in our sample are implicated in antibiotic resistance and adaptation to the host (see Table S2 in the supplemental material). The six fixed differences distinguishing the two clades include SNPs in genes encoding the two-component regulator NarX, a phosphomannose isomerase, an arginine/ornithine succinyltransferase, and the fimbrial subunit CupC1. All but the last one are nonsynonymous substitutions. One of the fixed indels leads to a frameshift mutation in the locus that encodes AlgF, which controls the addition of acetyl groups to alginate. The second fixed indel is located in the intergenic region upstream from the gene encoding DNA polymerase I (polA). Beyond these fixed mutations, there are SNPs in the gene encoding a probable nonribosomal peptide synthetase and alginate-C5-mannuronan epimerase AlgG that are also fixed between the two clades, with the exception of a single clade A specimen 1 isolate that carries the clade B variant at both sites. Additionally, there are SNPs in genes encoding a β-lactamase precursor and the transcriptional regulator MexT that are found only in clade B isolates and that first appear in specimen 2, which is the first specimen containing clade B isolates. These SNPs are found in 29.0% and 17.8% of clade B strains, respectively. Finally, an indel located in the gene that encodes a murein tripeptide ligase (Mpl) segregates exclusively in clade B, where it is found in 76.3% of the isolates. All of the isolates lacking this mutation in clade B also have a second indel in the same locus 1,240 bases upstream.
Given the relatively large number of variants that distinguish clades A and B, it is impossible to conclusively identify the beneficial mutation that initially gave rise to clade B. Nevertheless, it may be possible to identify SNPs that helped maintain one or both populations. As discussed above, three independent nonsynonymous mutations arose during the course of this study in the pbpB locus, encoding PBP3. Two derived alleles are found exclusively in clade B isolates (one at very low frequency), and one derived allele is found exclusively in clade A isolates (Fig. 5; see Fig. S5 and S6 in the supplemental material). The ancestral PBP3 allele is found in both clades.
FIG 5 .

Distribution of pbpB alleles in clade A (A) and clade (B) over the 12 sampling time points. Clades A and B share an ancestral major allele. A minor allele is observed specifically in clade A, and two others segregate only in clade B.
We noted that aztreonam and piperacillin-tazobactam were first administered intravenously during the time that roughly coincides with the emergence of clade B (Fig. 1). Since aztreonam has high affinity for PBP3 (54), we examined the in vitro fitness of the clade A-derived pbpB allele, the most common clade B-derived pbpB allele, and the ancestral pbpB allele using standard MIC assays against four antipseudomonal β-lactam antibiotics (Fig. 6) (55). The ancestral allele is present in both clades but carried in different genetic backgrounds; nevertheless, the resistance it confers does not differ significantly among strains, regardless of their clade of origin (P = 0.15 to 0.44). The derived PBP3 alleles from both clades A and B increased strain resistance relative to the ancestral allele in the presence of both aztreonam and cefsulodin. In contrast, when tested on ceftazidime and piperacillin, while the clade A allele increased resistance, the clade B allele was no more resistant than the ancestral allele. These results indicate that the in vitro fitness contribution of each derived PBP3 allele (as measured by antibiotic resistance) is dependent on the specific antibiotic environment in which it is found.
FIG 6 .
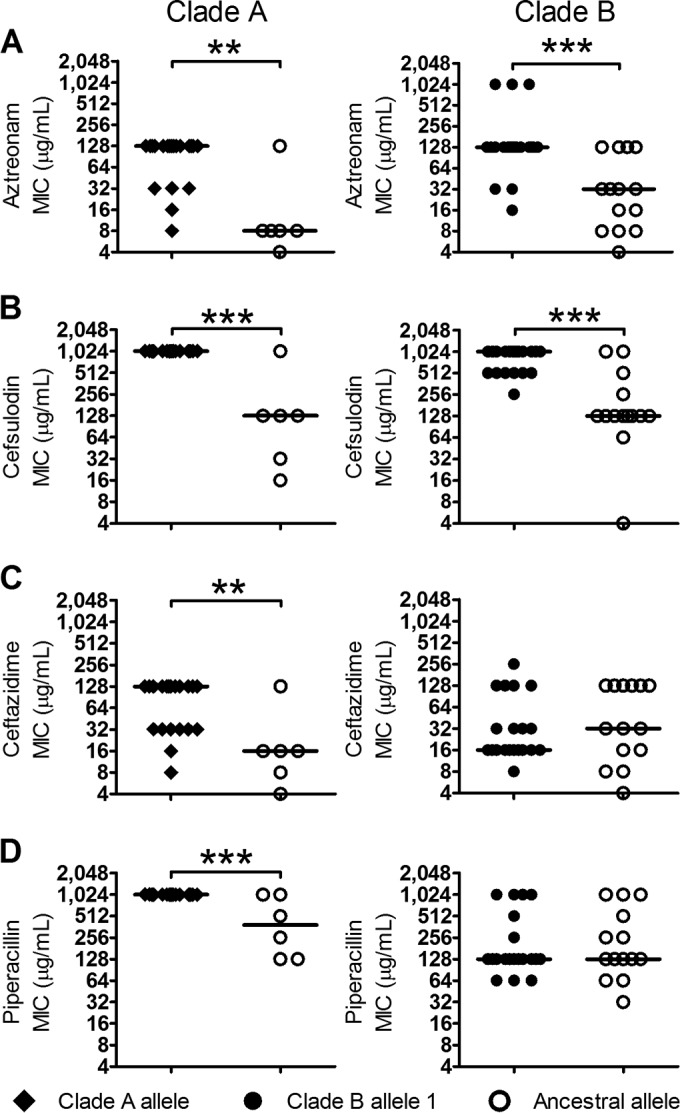
Relationship between pbpB alleles and MICs to selected antipseudomonal antibiotics. Pseudomonas aeruginosa isolates were grouped by pbpB genotype based on the presence of major and minor alleles, as well as clade membership in clade A (ancestral) and clade B (sweep) populations. Antibiotic MICs were assayed for aztreonam (A), cefsulodin (B), ceftazidime (C), and piperacillin (D). The presence of the minor alleles was associated with significantly increased resistance to aztreonam and cefsulodin in both clades but to ceftazidime and piperacillin only for the clade A minor allele. Statistical significance was determined by Mann-Whitney U test and is indicated with asterisks. (**, P ≤ 0.01; ***, P ≤ 0.001).
DISCUSSION
We have characterized the fine-scale evolutionary dynamics of P. aeruginosa during human CF lung infection and have observed a temporal shift in intraspecific population structure consistent with recurrent emergence and expansion of new clonal lineages. Since there is no change in the absolute size of the bacterial population over time, and Pseudomonas makes up the bulk of each of these populations (as shown by quantitative PCR [qPCR] analysis and microbiome sequencing, respectively), it is likely that the expansion of any new clonal lineage comes at the cost of preexisting clonal linages. Further, since there is strong evidence for positive selection in the population, we believe the best explanation for these data are recurrent selective sweeps, also known as periodic selection.
Although this patient has been infected with P. aeruginosa for at least 12 years, the entire population of P. aeruginosa coalesced to a common ancestor less than 3 years before the last specimen sampled for this study, indicating that this sweep went to fixation. It is possible that our sputum-based sampling did not capture the entirety of the P. aeruginosa population living within this patient and that there are additional distinct subpopulations of P. aeruginosa located in other anatomical compartments. However, if this were the case, then the migration rate between these subpopulations would have to be so low that we did not detect a single migrant over the year of sampling. Any rare migration from a divergent subpopulation would have been seen in the genealogical structure and time to most recent common ancestry of the overall population. On balance, we believe that such a low rate of migration between anatomical compartments is fairly unlikely and that we have therefore likely sampled the overall diversity of P. aeruginosa in this patient.
A second clonal lineage arose ~2 years after the first, giving rise to clade B. We cannot conclusively identify the “origin mutation” that gave rise to this lineage since there are a number of mutations that are fixed between the two clades. Consequently, no single variant is uniquely associated with the origin of clade B. This is perhaps not surprising since clade B originated in the nearly 6-month period between the collection of the first and second specimens. Any mutations that occurred in the emergent clade B common ancestor would be held in linkage disequilibrium with the causative mutation due to the very low rate of recombination.
Incomplete selective sweeps, such as those we observe in clade B, have been documented in other systems and generated substantial experimental and theoretical interest (56–69). A number of hypotheses can be proposed to explain why clade B has not swept to fixation. For example, we may have simply caught the sweep at an intermediate stage on its way to fixation, or the short-term persistence of the two clades could be due to stochastic processes. It is also possible that the ancestor of clade B colonized a distinct anatomical space and has since stayed isolated from its ancestral lineage. However, perhaps the most interesting explanation is clonal interference (70) driven by either competing beneficial mutations, negative epistasis between beneficial and deleterious mutations, or changing selection pressures driven by environmental heterogeneity. An example of the latter can be seen in our PBP3 data. While the clade A PBP3-derived allele confers a fitness advantage relative to the ancestral allele under all antibiotics tested, the clade B-derived allele is more fit than the ancestral allele only under half of the tested antibiotics. This is an example of latent potential for selection (71), in which the selective advantage of an allele is dependent on the environment. Even if the clade B allele is beneficial under certain conditions (such as during aztreonam treatment), it may be disadvantageous in other environments (such as during ceftazidime treatment), thereby, promoting the maintenance of the ancestral population and potentially explaining an underlying mechanism for clonal interference.
The analysis of natural selection within population data is complicated by short-term segregation of genetic polymorphisms, which can confound the more appropriate measure of fixed differences between populations (72). We have attempted to compensate for this issue by comparing dN/dS signals that emerge in the full data set versus the data set containing only the multispecimen SNPs. While the full data set provides a dN/dS value that is consistent with the expectation of a purely random mutational process (both synonymous and nonsynonymous mutations occurring at a rate consistent with the number of potential sites), the SNPs that are recovered from multiple specimens (multiple time points) over the course of the study show a signal consistent with strong positive selection in clade B, as would be expected in the case of a selective sweep. Again, it is not possible to use this analysis to identify the causative variant since the sweep population has had very little time for recombination to disentangle the selected substitution from the genomic background.
While dN/dS values can be difficult to interpret, a particularly notable indicator of selection is the number of loci that have accumulated multiple mutations over the short time since the most recent common ancestor arose. Our population of 233 strains has seven Mpl alleles, six AlgG and MucA/AlgU alleles, five AmpR/AmpC alleles, four PBP3 and MexA/MexB alleles, and three MexT and MucA alleles segregating (Table 3). The probability of even two mutations occurring at the same locus (giving rise to three alleles) is 1.64 × 10−4, or 0.027 when Bonferroni corrected for 165 independent mutations (107 SNPs and 58 indels). This pattern of multiple independent mutations is a powerful signal of parallel pathoadaptation (73) and mirrors the findings of Marvig and colleagues (28), who found extensive evidence for parallel evolution of P. aeruginosa in the CF lung. A number of multiply mutated loci overlapped between studies, including those that encode Mpl, MucA, AlgU, MexA/MexB, and LasR. A recent study by Williams et al. (29) also identified the mpl locus as carrying homoplasious (parallel) mutations in multiple strains, possibly due to homologous recombination. These patterns of parallel pathoadaptation are particularly striking given that nearly all of the affected loci are involved in either antimicrobial resistance or biofilm production.
The use of deep population sequencing in a single CF patient over a short period reveals community dynamics that are not evident by 16S rRNA gene microbiome sequencing or long-term longitudinal studies. Analyses of CF microbiota using culture-independent techniques (which are typically limited to genus-level taxonomic identification) have demonstrated that Pseudomonas commonly dominates microbial communities, is highly resilient, is associated with poor lung function and disease aggressiveness, and is a determinant of community dynamics (5, 6, 74–77). However, these analyses have been limited in their ability to predict temporal variation in patient status, such as the origins of exacerbations, and have not been able to explain the apparent stability of Pseudomonas through treatment. Our analysis shows that, although apparently stable at the genus level, Pseudomonas (P. aeruginosa specifically) can be a highly dynamic pathogen, rapidly adapting to environmental stress, which thus enables it to persist and resist the intended effects of antibiotic therapy.
The rapid evolution of P. aeruginosa revealed by deep sequencing in this study may help explain why significant changes in the antimicrobial treatment regimen did not result in any significant change in bacterial community composition. The apparent microbiologic stability observed belies a highly dynamic P. aeruginosa population that carries extensive genetic variation in a range of virulence- and resistance-associated loci, which are under strong positive selection. These population dynamics, which are undetectable using lower-resolution methods (e.g., microbiome- or culture-based methods), allow the pathogen to rapidly respond to therapeutic changes and could explain why some changes in treatment do not result in clinical improvement.
The PBP3 data reveal that changes in the selective environment (e.g., different antibiotics) can influence the fitness of strains in unpredictable ways. This observation may even be extended to the microbiome level, where changes in the relative abundance of Pseudomonas do not correlate with the anticipated response to antibiotics administered based on their antipseudomonal properties (such as the drop in Pseudomonas relative abundance following metronidazole administration at time point 9). Ultimately, we are left with a number of critical questions about the relationships between disease state, treatment approaches, and pathogen evolution. To what extent do irregular and variable antimicrobial treatments drive pathogen population dynamics? Are highly dynamic pathogen populations associated with specific clinical prognoses? Are there population structures or pathogen lineages that would favor better clinical outcomes? Is it possible to select for these lineages, and how could this be accomplished?
This study is limited to a single bacterial species in one infected individual over a defined time period, and thus its results may not generalize to other CF patients, disease stages, or bacterial species. Selective stress in human infection is highly heterogeneous and includes a broad array of differences in host response, microbial milieu, and treatment administration that would be expected to produce various temporal and spatial selective pressures, resulting in multiple patterns of intraspecies evolution. Future efforts to characterize the prevalence and dynamics of different patterns of selection across pathogens, individuals, and disease states are needed to understand their significance in health, disease, and treatment response.
MATERIALS AND METHODS
Specimen collection and microbiological analysis.
All protocols for the collection and use of patient sputum were approved by the Research Ethics Boards at the University Health Network (Toronto) and St. Michael’s Hospital (Toronto). Sputum samples were voluntarily produced by expectoration over a 350-day period by a single female study patient, CF67. Sputa were processed and P. aeruginosa cultured as previously described (40).
Following 72 h of incubation, all cultures were visually inspected and sampled for overall diversity in colony morphology. Twenty colonies resembling P. aeruginosa that were oxidase positive and exhibited growth on cetrimide and MacConkey agars (Becton, Dickinson, Hunt Valley, MD) were identified and cryopreserved for further analysis as previously described (38).
To study the bulk P. aeruginosa population, patient sputum was also spread plated directly onto P. aeruginosa-selective cetrimide agar and incubated at 37°C for 72 h. The resulting lawn of P. aeruginosa colonies was scraped from each replicate plate using a sterile inoculating loop and pooled in a single tube of sterile H2O for population sequencing.
DNA isolation.
Genomic DNA was extracted for whole-genome sequencing from 233 individual P. aeruginosa isolates and the pooled populations using the DNeasy blood and tissue kit (Qiagen, Ontario, Canada) as per the manufacturer’s protocol for Gram-negative bacteria.
Antimicrobial susceptibility testing of pbpB variants.
MICs for the β-lactam antibiotics aztreonam (Alfa-Aesar, Ward Hill, MA), ceftazidime (Sigma-Aldrich, Ontario, Canada), cefsulodin (A.G. Scientific, Inc., San Diego, CA), and piperacillin (Sigma-Aldrich, Ontario, Canada) were determined for 60 pbpB variants (20 per genotype) by agar dilution, as described by the Clinical and Laboratory Standards Institute guidelines (78). Significant differences in fitness defined by variations in MIC associated with clade A or clade B isolates carrying the pbpB ancestral major allele, ancestral minor allele, or sweep minor allele were assessed in GraphPad Prism 5.0 (GraphPad Software, Inc., San Diego, CA) using a Mann-Whitney U test with a 95% confidence interval.
Microbiome analysis and 16S quantitation.
Microbiome analysis was performed by analysis of the V5 to V7 hypervariable regions of the 16S rRNA gene using the SI-Seq protocol (38). Sequencing was performed at the University of Toronto, Centre for the Analysis of Genome Evolution and Function (CAGEF). An in-house Java script was used to prepare the sequence for downstream analysis using MACQIIME (79). Chimeric reads were removed using reference-based chimera detection with USEARCH (80). Operational taxonomic units (OTU) were picked against an SI-Seq structured reference set using a similarity of 0.87 (corresponding to the genus level by the SI-Seq method), with premature termination turned off. Taxonomy was assigned using an RDP structured data set using UCLUST with --max_accepts set to 0 and similarity set to 0.97 (empirically determined to produce the highest consistency identification at the genus level using P. aeruginosa controls) (80, 81). Unassigned OTU were identified with BLASTN (82). OTU representing less than 0.005% of overall sequence were removed. Quantitative PCR of the 16S rRNA gene was performed as previously described (83).
Sequencing and quality control.
Whole-genome sequencing of sampled P. aeruginosa isolates was performed on the GAIIx, HiSeq, and MiSeq Illumina platforms. The distribution of the number of bases sequenced per isolate ranged from 167 to 1,426 million bases, and the median was 402 million bases (see Fig. S1A in the supplemental material). This sequencing depth yielded a median read depth per position of 54× (range 25× to 183× [see Fig. S1B]). Sequencing reads were quality trimmed employing Quake and its native k-mer counter (84), and adapters were removed in the CLC Genomics Workbench v. 6.5.1 (Aarhus, Denmark).
De novo and reference mapping assembly.
Isolate CF67-71, which was sequenced with 250-bp-long paired-end reads, was assembled de novo using the CLC Genomics Workbench (Aarhus, Denmark), using its default assembly settings. Contigs with a scaffolding depth of lower than 10× and/or with a size smaller than 1 kb were removed from this analysis. Contigs that passed this filter were annotated at the RAST server using the native gene caller and Classic RAST as the annotation scheme (85). CF67-71 contigs were used as the reference for mapping assembly of each remaining isolate. We performed six different reference-mapping assemblies using BWA v. 0.7.4 (86), LAST v. 284 (87), and novoalign v. 2/8/03 (Novocraft Technologies), using sequencing reads with and without quality trimming (Fig. 2).
SNP and indel calling.
The preliminary list of variants was extracted from the reference-mapping step using SAMtools and BCFtools v. 0.1.19 (88). We then employed a two-step SNP calling process similar to that of Lieberman et al. (89). In the first step, we generated a set of 108 polymorphic positions by identifying the overlapping set of variants called by all six reference-mapping approaches using a custom Java pipeline (available at https://github.com/DSGlab/SNPCallingPipeline). This step used the following criteria: (i) variant Phred quality score of ≥30 and (ii) variants must be found at least 150 bp away from the edge of the reference contig and/or an indel. In the second step, we revised each polymorphic position from the previous step in all of the isolates and included calls that had a Phred score of at least 25. Finally, we tested the sequencing support for either the SNP or the reference base. This multihypothesis correction required that at least 80% of the reads supported the SNP or the reference. If the data did not support either base, then the position was called as an ambiguous base (“N”).
We used Dindel v. 1.01 (90) to realign the sequencing reads (mapped and unmapped) and test whether candidate indels produced by BWA and SAMtools were supported by those reads. Putative indel positions were selected as having an indel call in at least one nonreference isolate with the following criteria: (i) a variant Phred quality score of ≥35, (ii) at least 2 forward and 2 reverse reads, and (iii) sequencing coverage of ≥10. Each of these putative indel positions was then reviewed in each of the 233 isolates. An indel was called if the variant had a Phred quality score of ≥25 and the allele frequency was ≥80%. Indel calls where the allele frequency was ≤20% were labeled as ambiguous. All other indel calls were discarded.
Population and single-genome-sequencing evaluation.
To confirm that our sampling design adequately sampled the underlying P. aeruginosa population, we compared the major allele frequency for each segregating site from individual isolates with that obtained from bulk population sequencing of P. aeruginosa isolates collected from 1 of the 12 specimens (see Fig. S3 in the supplemental material).
Sequencing reads from each isolate from specimen 9 were rarified to 1/20th of the total number of reads in the population sequencing experiment such that the overall number of reads would be the same. Reads from both experiments were mapped to the contigs of isolate CF67-71 as described above. Major and minor allele frequencies were calculated using the quality thresholds described by Lieberman et al. (89).
Phylogenetic, population genetic, and coalescent analyses.
An alignment of the 107 SNPs was employed to reconstruct the phylogeny of the 233 isolates by both maximum likelihood and neighbor-joining methods using maximum composite likelihood distances and gamma correction (α = 1) in MEGA6 (91). The two trees gave nearly identical results. The phylogeny was also investigated with a network-based phylogenetic analysis. SplitsTree4 (92, 93) with the Jukes Cantor distance matrix was used to create a neighbor-net phylogenetic tree.
The time to the most recent common ancestor (tMRCA) among our 233 isolates and between our isolates and the P. aeruginosa strain LESB58 was calculated by Bayesian Markov chain Monte Carlo (MCMC) analysis using BEAUTi and BEAST v. 1.75 (43, 44). The parameters deduced from prior information were days between isolation dates and nucleotide substitution model and frequencies. The nucleotide substitution model that best fit our data according to FindModel (http://www.hiv.lanl.gov/content/sequence/findmodel/findmodel.html), which is a web application of ModelTest (94), was the general time-reversible (GTR) model (Log likelihood [LnL] = −927.007482, and Akaike information criterion [AIC] = 1,870.014964). The nucleotide substitution frequencies AC = 0.0661, AG = 0.3306, AT = 0.0275, CG = 0.1858, GT = 0.0657, and CT = 0.3241 were estimated employing the Maximum Likelihood Estimate of Substitution Matrix tool in MEGA6 (91). Preliminary MCMC analyses consisting of 10 million generations were employed to infer the appropriate molecular clock and demographic models (95). We inferred the log-normal relaxed uncorrelated molecular clock for the constant size coalescent analysis. Given the estimated priors, the final analysis was run in duplicate through 1 billion MCMC generations sampled every 1,000 MCMC generations, and the burn-in period was set at 100 million MCMC generations. The inferred molecular clock was consistent with the number of mutations observed in our isolates through time (see Fig. S7 in the supplemental material).
Population genetic tests were performed with DnaSP v. 5.10.01 (96). P values were determined via coalescent simulations as implemented in DnaSP.
Interstrain whole-genome alignment and phylogeny.
The full genomic sequences of the P. aeruginosa strains LESB58, PAO1, DK2, PA7, NCGM2, UCBPP, B136, and M18 were obtained from the Pseudomonas Genome Database (97). These sequences and the contig sequences of the de novo-assembled isolate CF67-71 were aligned using Mugsy v. 1.2.3 (98). The output alignment file in MAF format was converted to the more broadly used FASTA format in the Galaxy platform (99). Finally, the neighbor-joining phylogeny (see Fig. S4 in the supplemental material) was inferred using the CLC Genomics Workbench v. 6.5.1 (Aarhus, Denmark) with a bootstrap value of 500.
SUPPLEMENTAL MATERIAL
Whole-genome sequencing of 233 isolates of P. aeruginosa with Illumina. (A) Distribution of number of bases sequenced per isolate. The distribution ranges from 167 to 1,426 million sequenced bases, and the median is 402 million sequenced bases. (B) Distribution of median read depth per position. Download
Flowchart of the SNP calling pipeline. All sequencing reads were quality trimmed and assembled de novo. The isolate with the highest N50 was used as the reference to which the sequencing reads of all isolates were mapped using three algorithmically distinct methods. SNPs called by all of these methods were selected to avoid biases from sequencing quality or coverage. Download
Individual isolate sequencing reflects whole-population sequencing adequately. The frequency of the major allele of 21 SNPs among the 20 isolates collected in the 9th time point is plotted against the frequency of the alleles as determined by population sequencing of the specimen. No alleles were recovered in population sequencing that were not found in the individual isolate sequencing. Download
CF67 isolates in the context of other P. aeruginosa complete genomes. Shown is the unrooted neighbor-joining (NJ) phylogeny of CF67 isolate 71 and 8 P. aeruginosa complete genomes based on genome-wide alignment. All nodes have 100% bootstrap support. Download
Maximum likelihood tree annotated with PBP3 alleles. The tree is constructed as described in the legend to Fig. 2. The identity of the PBP3 allele carried by each strain is indicated by the color of the box to the left of the isolate name. Download
Location of nonsynonymous mutations in the pbpB gene in the 3D crystal structure of PBP3. (A) Multiple sequence alignment of the PBP3 protein sequences of the ancestral allele, minor allele of clade A, and the most common minor allele of clade B. The mutated amino acid is shaded in red and green for clades A and B, respectively. (B) The crystal structure of PBP3 complexed with aztreonam (PDB ID 3PBS [42, 43]) was customized on the PyMOL Molecular Graphics System v. 1.5.0.4 (Schrödinger, LLC). The aztreonam structure is in white, and the mutated positions are colored in red for the clade A minor allele and green for the clade B minor allele. Download
Temporal signal compared to the inferred substitution rate of the 233 isolates. (A) Regression analysis of the root-to-tip distance as a function of time of isolation using the Path-O-Gen program (http://tree.bio.ed.ac.uk/software/pathogen/) as described in reference 101. Each circle represents the average root-to-tip distance of the isolates from the respective sampling time point. The resulting trend shows that the inferred molecular clock was consistent with the changes seen in our isolates through time (R2 = 0.79, P = 0.0001). (B) Rate of substitution as inferred by the coalescent analysis (2.92 ± 0.70 SNPs per year). The line is set to intersect the x axis at the tMRCA of all isolates of this study. Download
Patient details for the study period.
SNPs implicated in antibiotic resistance and host adaptation.
ACKNOWLEDGMENTS
We thank Leena Rizvi and Katherine Griffin at St. Michael’s Hospital (Toronto, Ontario, Canada) for collecting clinical specimens and metadata and Fern Parisian (Hospital for Sick Children, Toronto, Ontario, Canada) for guidance with antimicrobial susceptibility testing.
This research was funded by scientific grants from the National Sanitarium Association and the Canadian Institutes of Health Research (CIHR)/Cystic Fibrosis Canada Emerging Team grant (CMF108027). J.D.C. is a recipient of an Ontario Trillium Scholarship. S.T.C. is a recipient of an Ontario Graduate Scholarship. B.C. is a recipient of a CIHR Fellowship.
Footnotes
Citation Diaz Caballero J, Clark ST, Coburn B, Zhang Y, Wang PW, Donaldson SL, Tullis DE, Yau YCW, Waters VJ, Hwang DM, Guttman DS. 2015. Selective sweeps and parallel pathoadaptation drive Pseudomonas aeruginosa evolution in the cystic fibrosis lung. mBio 6(5):e00981-15. doi:10.1128/mBio.00981-15.
Contributor Information
Bill Hanage, Harvard Medical School.
Gerald B. Pier, Harvard Medical School.
REFERENCES
- 1.Gibson RL, Burns JL, Ramsey BW. 2003. Pathophysiology and management of pulmonary infections in cystic fibrosis. Am J Respir Crit Care Med 168:918–951. doi: 10.1164/rccm.200304-505SO. [DOI] [PubMed] [Google Scholar]
- 2.Lipuma JJ. 2010. The changing microbial epidemiology in cystic fibrosis. Clin Microbiol Rev 23:299–323. doi: 10.1128/CMR.00068-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Al-Aloul M, Crawley J, Winstanley C, Hart CA, Ledson MJ, Walshaw MJ. 2004. Increased morbidity associated with chronic infection by an epidemic Pseudomonas aeruginosa strain in CF patients. Thorax 59:334–336. doi: 10.1136/thx.2003.014258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ciofu O, Hansen CR, Høiby N. 2013. Respiratory bacterial infections in cystic fibrosis. Curr Opin Pulm Med 19:251–258. doi: 10.1097/MCP.0b013e32835f1afc. [DOI] [PubMed] [Google Scholar]
- 5.Tunney MM, Klem ER, Fodor AA, Gilpin DF, Moriarty TF, McGrath SJ, Muhlebach MS, Boucher RC, Cardwell C, Doering G, Elborn JS, Wolfgang MC. 2011. Use of culture and molecular analysis to determine the effect of antibiotic treatment on microbial community diversity and abundance during exacerbation in patients with cystic fibrosis. Thorax 66:579–584. doi: 10.1136/thx.2010.137281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fodor AA, Klem ER, Gilpin DF, Elborn JS, Boucher RC, Tunney MM, Wolfgang MC. 2012. The adult cystic fibrosis airway microbiota is stable over time and infection type, and highly resilient to antibiotic treatment of exacerbations. PLoS One 7:e45001. doi: 10.1371/journal.pone.0045001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Palmer KL, Mashburn LM, Singh PK, Whiteley M. 2005. Cystic fibrosis sputum supports growth and cues key aspects of Pseudomonas aeruginosa physiology. J Bacteriol 187:5267–5277. doi: 10.1128/JB.187.15.5267-5277.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harris JK, De Groote MA, Sagel SD, Zemanick ET, Kapsner R, Penvari C, Kaess H, Deterding RR, Accurso FJ, Pace NR. 2007. Molecular identification of bacteria in bronchoalveolar lavage fluid from children with cystic fibrosis. Proc Natl Acad Sci U S A 104:20529–20533. doi: 10.1073/pnas.0709804104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stokell JR, Gharaibeh RZ, Hamp TJ, Zapata MJ, Fodor AA, Steck TR. 2015. Analysis of changes in diversity and abundance of the microbial community in a cystic fibrosis patient over a multiyear period. J Clin Microbiol 53:237–247. doi: 10.1128/JCM.02555-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lieberman TD, Flett KB, Yelin I, Martin TR, McAdam AJ, Priebe GP, Kishony R. 2014. Genetic variation of a bacterial pathogen within individuals with cystic fibrosis provides a record of selective pressures. Nat Genet 46:82–87. doi: 10.1038/ng.2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Conway SP, Brownlee KG, Denton M, Peckham DG. 2003. Antibiotic treatment of multidrug-resistant organisms in cystic fibrosis. Am J Respir Med 2:321–332. doi: 10.1007/BF03256660. [DOI] [PubMed] [Google Scholar]
- 12.Waters V, Ratjen F. 2006. Multidrug-resistant organisms in cystic fibrosis: management and infection-control issues. Expert Rev Anti Infect Ther 4:807–819. doi: 10.1586/14787210.4.5.807. [DOI] [PubMed] [Google Scholar]
- 13.Smith EE, Buckley DG, Wu Z, Saenphimmachak C, Hoffman LR, D’Argenio DA, Miller SI, Ramsey BW, Speert DP, Moskowitz SM, Burns JL, Kaul R, Olson MV. 2006. Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. Proc Natl Acad Sci U S A 103:8487–8492. doi: 10.1073/pnas.0602138103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jelsbak L, Johansen HK, Frost AL, Thøgersen R, Thomsen LE, Ciofu O, Yang L, Haagensen JA, Høiby N, Molin S. 2007. Molecular epidemiology and dynamics of Pseudomonas aeruginosa populations in lungs of cystic fibrosis patients. Infect Immun 75:2214–2224. doi: 10.1128/IAI.01282-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoboth C, Hoffmann R, Eichner A, Henke C, Schmoldt S, Imhof A, Heesemann J, Hogardt M. 2009. Dynamics of adaptive microevolution of hypermutable Pseudomonas aeruginosa during chronic pulmonary infection in patients with cystic fibrosis. J Infect Dis 200:118–130. doi: 10.1086/599360. [DOI] [PubMed] [Google Scholar]
- 16.Huse HK, Kwon T, Zlosnik JE, Speert DP, Marcotte EM, Whiteley M. 2010. Parallel evolution in Pseudomonas aeruginosa over 39,000 generations in vivo. mBio 1(4):e00199-10. doi: 10.1128/mBio.00199-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rau MH, Hansen SK, Johansen HK, Thomsen LE, Workman CT, Nielsen KF, Jelsbak L, Høiby N, Yang L, Molin S. 2010. Early adaptive developments of Pseudomonas aeruginosa after the transition from life in the environment to persistent colonization in the airways of human cystic fibrosis hosts. Environ Microbiol 12:1643–1658. doi: 10.1111/j.1462-2920.2010.02211.x. [DOI] [PubMed] [Google Scholar]
- 18.Cramer N, Klockgether J, Wrasman K, Schmidt M, Davenport CF, Tümmler B. 2011. Microevolution of the major common Pseudomonas aeruginosa clones C and PA14 in cystic fibrosis lungs. Environ Microbiol 13:1690–1704. doi: 10.1111/j.1462-2920.2011.02483.x. [DOI] [PubMed] [Google Scholar]
- 19.Yang L, Jelsbak L, Marvig RL, Damkiær S, Workman CT, Rau MH, Hansen SK, Folkesson A, Johansen HK, Ciofu O, Høiby N, Sommer MO, Molin S. 2011. Evolutionary dynamics of bacteria in a human host environment. Proc Natl Acad Sci U S A 108:7481–7486. doi: 10.1073/pnas.1018249108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chung JC, Becq J, Fraser L, Schulz-Trieglaff O, Bond NJ, Foweraker J, Bruce KD, Smith GP, Welch M. 2012. Genomic variation among contemporary Pseudomonas aeruginosa isolates from chronically infected cystic fibrosis patients. J Bacteriol 194:4857–4866. doi: 10.1128/JB.01050-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rau MH, Marvig RL, Ehrlich GD, Molin S, Jelsbak L. 2012. Deletion and acquisition of genomic content during early stage adaptation of Pseudomonas aeruginosa to a human host environment. Environ Microbiol 14:2200–2211. doi: 10.1111/j.1462-2920.2012.02795.x. [DOI] [PubMed] [Google Scholar]
- 22.Dettman JR, Rodrigue N, Aaron SD, Kassen R. 2013. Evolutionary genomics of epidemic and nonepidemic strains of Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 110:21065–21070. doi: 10.1073/pnas.1307862110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hogardt M, Heesemann J. 2013. Microevolution of Pseudomonas aeruginosa to a chronic pathogen of the cystic fibrosis lung. Curr Top Microbiol Immunol 358:91–118. doi: 10.1007/82_2011_199. [DOI] [PubMed] [Google Scholar]
- 24.Marvig RL, Johansen HK, Molin S, Jelsbak L. 2013. Genome analysis of a transmissible lineage of Pseudomonas aeruginosa reveals pathoadaptive mutations and distinct evolutionary paths of hypermutators. PLoS Genet 9:e1003741. doi: 10.1371/journal.pgen.1003741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Workentine ML, Sibley CD, Glezerson B, Purighalla S, Norgaard-Gron JC, Parkins MD, Rabin HR, Surette MG. 2013. Phenotypic heterogeneity of Pseudomonas aeruginosa populations in a cystic fibrosis patient. PLoS One 8:e60225. doi: 10.1371/journal.pone.0060225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feliziani S, Marvig RL, Luján AM, Moyano AJ, Di Rienzo JA, Krogh Johansen H, Molin S, Smania AM. 2014. Coexistence and within-host evolution of diversified lineages of hypermutable Pseudomonas aeruginosa in long-term cystic fibrosis infections. PLoS Genet 10:e1004651. doi: 10.1371/journal.pgen.1004651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Markussen T, Marvig RL, Gómez-Lozano M, Aanæs K, Burleigh AE, Høiby N, Johansen HK, Molin S, Jelsbak L. 2014. Environmental heterogeneity drives within-host diversification and evolution of Pseudomonas aeruginosa. mBio 5(5):e01592-14. doi: 10.1128/mBio.01592-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marvig RL, Sommer LM, Molin S, Johansen HK. 2015. Convergent evolution and adaptation of Pseudomonas aeruginosa within patients with cystic fibrosis. Nat Genet 47:57–64. doi: 10.1038/ng.3148. [DOI] [PubMed] [Google Scholar]
- 29.Williams D, Evans B, Haldenby S, Walshaw MJ, Brockhurst MA, Winstanley C, Paterson S. 2015. Divergent, coexisting Pseudomonas aeruginosa lineages in chronic cystic fibrosis lung infections. Am J Respir Crit Care Med 191:775–785. doi: 10.1164/rccm.201409-1646OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aaron SD, Ferris W, Henry DA, Speert DP, Macdonald NE. 2000. Multiple combination bactericidal antibiotic testing for patients with cystic fibrosis infected with Burkholderia cepacia. Am J Respir Crit Care Med 161:1206–1212. doi: 10.1164/ajrccm.161.4.9907147. [DOI] [PubMed] [Google Scholar]
- 31.Smith M, Haigh J. 1974. The hitch-hiking effect of a favourable gene. Genet Res 23:23–35. doi: 10.1017/S0016672300014634. [DOI] [PubMed] [Google Scholar]
- 32.Elena SF, Cooper VS, Lenski RE. 1996. Punctuated evolution caused by selection of rare beneficial mutations. Science 272:1802–1804. doi: 10.1126/science.272.5269.1802. [DOI] [PubMed] [Google Scholar]
- 33.Notley-McRobb L, Ferenci T. 2000. Experimental analysis of molecular events during mutational periodic selections in bacterial evolution. Genetics 156:1493–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Santiago E, Caballero A. 2005. Variation after a selective sweep in a subdivided population. Genetics 169:475–483. doi: 10.1534/genetics.104.032813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Atwood KC, Schneider LK, Ryan FJ. 1951. Periodic selection in Escherichia coli. Proc Natl Acad Sci U S A 37:146–155. doi: 10.1073/pnas.37.3.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Volz EM, Koelle K, Bedford T. 2013. Viral phylodynamics. PLoS Comput Biol 9:e1002947. doi: 10.1371/journal.pcbi.1002947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Folkesson A, Jelsbak L, Yang L, Johansen HK, Ciofu O, Høiby N, Molin S. 2012. Adaptation of Pseudomonas aeruginosa to the cystic fibrosis airway: an evolutionary perspective. Nat Rev Microbiol 10:841–851. doi: 10.1038/nrmicro2907. [DOI] [PubMed] [Google Scholar]
- 38.Maughan H, Wang PW, Diaz Caballero J, Fung P, Gong Y, Donaldson SL, Yuan L, Keshavjee S, Zhang Y, Yau YC, Waters VJ, Tullis DE, Hwang DM, Guttman DS. 2012. Analysis of the cystic fibrosis lung microbiota via serial Illumina sequencing of bacterial 16S rRNA hypervariable regions. PLoS One 7:e45791. doi: 10.1371/journal.pone.0045791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mande SS, Mohammed MH, Ghosh TS. 2012. Classification of metagenomic sequences: methods and challenges. Brief Bioinform 13:669–681. doi: 10.1093/bib/bbs054. [DOI] [PubMed] [Google Scholar]
- 40.Clark ST, Diaz Caballero J, Cheang M, Coburn B, Wang PW, Donaldson SL, Zhang Y, Liu M, Keshavjee S, Yau YC, Waters VJ, Tullis DE, Guttman DS, Hwang DM. 2015. Phenotypic diversity within a Pseudomonas aeruginosa population infecting an adult with cystic fibrosis. Sci Rep 5:10932. doi: 10.1038/srep10932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hudson RR, Kaplan NL. 1985. Statistical properties of the number of recombination events in the history of a sample of DNA sequences. Genetics 111:147–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McVean G. 2007. The structure of linkage disequilibrium around a selective sweep. Genetics 175:1395–1406. doi: 10.1534/genetics.106.062828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Drummond AJ, Rambaut A. 2007. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Drummond AJ, Suchard MA, Xie D, Rambaut A. 2012. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol 29:1969–1973. doi: 10.1093/molbev/mss075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Salunkhe P, Smart CH, Morgan JA, Panagea S, Walshaw MJ, Hart CA, Geffers R, Tümmler B, Winstanley C. 2005. A cystic fibrosis epidemic strain of Pseudomonas aeruginosa displays enhanced virulence and antimicrobial resistance. J Bacteriol 187:4908–4920. doi: 10.1128/JB.187.14.4908-4920.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jeukens J, Boyle B, Kukavica-Ibrulj I, Ouellet MM, Aaron SD, Charette SJ, Fothergill JL, Tucker NP, Winstanley C, Levesque RC. 2014. Comparative genomics of isolates of a Pseudomonas aeruginosa epidemic strain associated with chronic lung infections of cystic fibrosis patients. PLoS One 9:e87611. doi: 10.1371/journal.pone.0087611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saunders IW, Tavaré S, Watterson GA, Tavare S. 1984. On the genealogy of nested subsamples from a haploid population. Adv Appl Probab 16:471–491. doi: 10.2307/1427285. [DOI] [Google Scholar]
- 48.Tajima F. 1989. Statistical methods for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fu YX, Li WH. 1993. Statistical tests of neutrality of mutations. Genetics 133:693–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wood TE, Burke JM, Rieseberg LH. 2005. Parallel genotypic adaptation: when evolution repeats itself. Genetica 123:157–170. doi: 10.1007/s10709-003-2738-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Arendt J, Reznick D. 2008. Convergence and parallelism reconsidered: what have we learned about the genetics of adaptation? Trends Ecol Evol 23:26–32. doi: 10.1016/j.tree.2007.09.011. [DOI] [PubMed] [Google Scholar]
- 52.Bailey SF, Rodrigue N, Kassen R. 2015. The effect of selection environment on the probability of parallel evolution. Mol Biol Evol 32:1436–1448. doi: 10.1093/molbev/msv033. [DOI] [PubMed] [Google Scholar]
- 53.Tsutsumi Y, Tomita H, Tanimoto K. 2013. Identification of novel genes responsible for overexpression of ampC in Pseudomonas aeruginosa PAO. Antimicrob Agents Chemother 57:5987–5993. doi: 10.1128/AAC.01291-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Davies TA, Shang W, Bush K, Flamm RK. 2008. Affinity of doripenem and comparators to penicillin-binding proteins in Escherichia coli and Pseudomonas aeruginosa. Antimicrob Agents Chemother 52:1510–1512. doi: 10.1128/AAC.01529-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gotoh N, Nunomura K, Nishino T. 1990. Resistance of Pseudomonas aeruginosa to cefsulodin: modification of penicillin-binding protein 3 and mapping of its chromosomal gene. J Antimicrob Chemother 25:513–523. doi: 10.1093/jac/25.4.513. [DOI] [PubMed] [Google Scholar]
- 56.Hermisson J, Pennings PS. 2005. Soft sweeps: molecular population genetics of adaptation from standing genetic variation. Genetics 169:2335–2352. doi: 10.1534/genetics.104.036947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sabeti PC, Schaffner SF, Fry B, Lohmueller J, Varilly P, Shamovsky O, Palma A, Mikkelsen TS, Altshuler D, Lander ES. 2006. Positive natural selection in the human lineage. Science 312:1614–1620. doi: 10.1126/science.1124309. [DOI] [PubMed] [Google Scholar]
- 58.Voight BF, Kudaravalli S, Wen X, Pritchard JK. 2006. A map of recent positive selection in the human genome. PLoS Biol 4:e72. doi: 10.1371/journal.pbio.0040072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Campos PR, Neto PS, de Oliveira VM, Gordo I. 2008. Environmental heterogeneity enhances clonal interference. Evolution 62:1390–1399. doi: 10.1111/j.1558-5646.2008.00380.x. [DOI] [PubMed] [Google Scholar]
- 60.Pickrell JK, Coop G, Novembre J, Kudaravalli S, Li JZ, Absher D, Srinivasan BS, Barsh GS, Myers RM, Feldman MW, Pritchard JK. 2009. Signals of recent positive selection in a worldwide sample of human populations. Genome Res 19:826–837. doi: 10.1101/gr.087577.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Campos PR, Wahl LM. 2010. The adaptation rate of asexuals: deleterious mutations, clonal interference and population bottlenecks. Evolution 64:1973–1983. doi: 10.1111/j.1558-5646.2010.00981.x. [DOI] [PubMed] [Google Scholar]
- 62.Chou HH, Chiu HC, Delaney NF, Segrè D, Marx CJ. 2011. Diminishing returns epistasis among beneficial mutations decelerates adaptation. Science 332:1190–1192. doi: 10.1126/science.1203799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Khan AI, Dinh DM, Schneider D, Lenski RE, Cooper TF. 2011. Negative epistasis between beneficial mutations in an evolving bacterial population. Science 332:1193–1196. doi: 10.1126/science.1203801. [DOI] [PubMed] [Google Scholar]
- 64.Burke MK. 2012. How does adaptation sweep through the genome? Insights from long-term selection experiments. Proc Biol Sci 279:5029–5038. doi: 10.1098/rspb.2012.0799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Orozco-terWengel P, Kapun M, Nolte V, Kofler R, Flatt T, Schlötterer C. 2012. Adaptation of Drosophila to a novel laboratory environment reveals temporally heterogeneous trajectories of selected alleles. Mol Ecol 21:4931–4941. doi: 10.1111/j.1365-294X.2012.05673.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Strelkowa N, Lässig M. 2012. Clonal interference in the evolution of influenza. Genetics 192:671–682. doi: 10.1534/genetics.112.143396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Barrick JE, Lenski RE. 2013. Genome dynamics during experimental evolution. Nat Rev Genet 14:827–839. doi: 10.1038/nrg3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lee MC, Marx CJ. 2013. Synchronous waves of failed soft sweeps in the laboratory: remarkably rampant clonal interference of alleles at a single locus. Genetics 193:943–952. doi: 10.1534/genetics.112.148502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Messer PW, Petrov DA. 2013. Population genomics of rapid adaptation by soft selective sweeps. Trends Ecol Evol 28:659–669. doi: 10.1016/j.tree.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Miralles R, Gerrish PJ, Moya A, Elena SF. 1999. Clonal interference and the evolution of RNA viruses. Science 285:1745–1747. doi: 10.1126/science.285.5434.1745. [DOI] [PubMed] [Google Scholar]
- 71.Hartl DL, Dykhuizen DE. 1981. Potential for selection among nearly neutral allozymes of 6-phosphogluconate dehydrogenase in Escherichia coli. Proc Natl Acad Sci U S A 78:6344–6348. doi: 10.1073/pnas.78.10.6344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kryazhimskiy S, Plotkin JB. 2008. The population genetics of dN/dS. PLoS Genet 4:e1000304. doi: 10.1371/journal.pgen.1000304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sokurenko EV, Hasty DL, Dykhuizen DE. 1999. Pathoadaptive mutations: gene loss and variation in bacterial pathogens. Trends Microbiol 7:191–195. doi: 10.1016/S0966-842X(99)01493-6. [DOI] [PubMed] [Google Scholar]
- 74.Stressmann FA, Rogers GB, van der Gast CJ, Marsh P, Vermeer LS, Carroll MP, Hoffman L, Daniels TW, Patel N, Forbes B, Bruce KD. 2012. Long-term cultivation-independent microbial diversity analysis demonstrates that bacterial communities infecting the adult cystic fibrosis lung show stability and resilience. Thorax 67:867–873. doi: 10.1136/thoraxjnl-2011-200932. [DOI] [PubMed] [Google Scholar]
- 75.Zhao J, Schloss PD, Kalikin LM, Carmody LA, Foster BK, Petrosino JF, Cavalcoli JD, VanDevanter DR, Murray S, Li JZ, Young VB, LiPuma JJ. 2012. Decade-long bacterial community dynamics in cystic fibrosis airways. Proc Natl Acad Sci U S A 109:5809–5814. doi: 10.1073/pnas.1120577109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Carmody LA, Zhao J, Schloss PD, Petrosino JF, Murray S, Young VB, Li JZ, LiPuma JJ. 2013. Changes in cystic fibrosis airway microbiota at pulmonary exacerbation. Ann Am Thorac Soc 10:179–187. doi: 10.1513/AnnalsATS.201211-107OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Coburn B, Wang PW, Diaz Caballero J, Clark ST, Brahma V, Donaldson S, Zhang Y, Surendra A, Gong Y, Elizabeth Tullis D, Yau YC, Waters VJ, Hwang DM, Guttman DS. 2015. Lung microbiota across age and disease stage in cystic fibrosis. Sci Rep 5:10241. doi: 10.1038/srep10241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Clinical and Laboratory Standards Institute 2012. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; approved standard, 9th ed. CLSI document M07-A09 Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 79.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 81.Cole JR, Chai B, Farris RJ, Wang Q, Kulam-Syed-Mohidee AS, McGarrell DM, Bandela AM, Cardenas E, Garrity GM, Tiedje JM. 2007. The Ribosomal Database Project (RDP-II): introducing myRDP space and quality controlled public data. Nucleic Acids Res 35:D169–D172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nadkarni MA, Martin FE, Jacques NA, Hunter N. 2002. Determination of bacterial load by real-time PCR using a broad-range (universal) probe and primers set. Microbiology 148:257–266. [DOI] [PubMed] [Google Scholar]
- 84.Kelley DR, Schatz MC, Salzberg SL. 2010. Quake: quality-aware detection and correction of sequencing errors. Genome Biol 11:R116. doi: 10.1186/gb-2010-11-11-r116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M, Meyer F, Olsen GJ, Olson R, Osterman AL, Overbeek RA, McNeil LK, Paarmann D, Paczian T, Parrello B, Pusch GD, Reich C, Stevens R, Vassieva O, Vonstein V, Wilke A, Zagnitko O. 2008. The RAST server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shrestha AM, Frith MC. 2013. An approximate Bayesian approach for mapping paired-end DNA reads to a reference genome. Bioinformatics 29:965–972. doi: 10.1093/bioinformatics/btt073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup . 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lieberman TD, Michel JB, Aingaran M, Potter-Bynoe G, Roux D, Davis MR Jr, Skurnik D, Leiby N, LiPuma JJ, Goldberg JB, McAdam AJ, Priebe GP, Kishony R. 2011. Parallel bacterial evolution within multiple patients identifies candidate pathogenicity genes. Nat Genet 43:1275–1280. doi: 10.1038/ng.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Albers CA, Lunter G, MacArthur DG, McVean G, Ouwehand WH, Durbin R. 2011. Dindel: accurate indel calls from short-read data. Genome Res 21:961–973. doi: 10.1101/gr.112326.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Huson DH. 1998. SplitsTree: analyzing and visualizing evolutionary data. Bioinformatics 14:68–73. doi: 10.1093/bioinformatics/14.1.68. [DOI] [PubMed] [Google Scholar]
- 93.Huson DH, Bryant D. 2006. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol 23:254–267. doi: 10.1093/molbev/msj030. [DOI] [PubMed] [Google Scholar]
- 94.Posada D, Crandall KA. 1998. ModelTest: testing the model of DNA substitution. Bioinformatics 14:817–818. doi: 10.1093/bioinformatics/14.9.817. [DOI] [PubMed] [Google Scholar]
- 95.Drummond AJ, Ho SY, Phillips MJ, Rambaut A. 2006. Relaxed phylogenetics and dating with confidence. PLoS Biol 4:e88. doi: 10.1371/journal.pbio.0040088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Librado P, Rozas J. 2009. DnaSP V5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- 97.Winsor GL, Van Rossum T, Lo R, Khaira B, Whiteside MD, Hancock RE, Brinkman FS. 2009. Pseudomonas Genome Database: facilitating user-friendly, comprehensive comparisons of microbial genomes. Nucleic Acids Res 37:D483–D488. doi: 10.1093/nar/gkn861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Angiuoli SV, Salzberg SL. 2011. Mugsy: fast multiple alignment of closely related whole genomes. Bioinformatics 27:334–342. doi: 10.1093/bioinformatics/btq665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Blankenberg D, Von Kuster G, Coraor N, Ananda G, Lazarus R, Mangan M, Nekrutenko A, Taylor J. 2010. Galaxy: a web-based genome analysis tool for experimentalists. Curr Protoc Mol Biol 89:19.10:19.10.1–1.10.21. doi: 10.1002/0471142727.mb1910s89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Miyoshi-Akiyama T, Kuwahara T, Tada T, Kitao T, Kirikae T. 2011. Complete genome sequence of highly multidrug-resistant Pseudomonas aeruginosa NCGM2.S1, a representative strain of a cluster endemic to Japan. J Bacteriol 193:7010. doi: 10.1128/JB.06312-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Firth C, Kitchen A, Shapiro B, Suchard MA, Holmes EC, Rambaut A. 2010. Using time-structured data to estimate evolutionary rates of double-stranded DNA viruses. Mol Biol Evol 27:2038–2051. doi: 10.1093/molbev/msq088. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Whole-genome sequencing of 233 isolates of P. aeruginosa with Illumina. (A) Distribution of number of bases sequenced per isolate. The distribution ranges from 167 to 1,426 million sequenced bases, and the median is 402 million sequenced bases. (B) Distribution of median read depth per position. Download
Flowchart of the SNP calling pipeline. All sequencing reads were quality trimmed and assembled de novo. The isolate with the highest N50 was used as the reference to which the sequencing reads of all isolates were mapped using three algorithmically distinct methods. SNPs called by all of these methods were selected to avoid biases from sequencing quality or coverage. Download
Individual isolate sequencing reflects whole-population sequencing adequately. The frequency of the major allele of 21 SNPs among the 20 isolates collected in the 9th time point is plotted against the frequency of the alleles as determined by population sequencing of the specimen. No alleles were recovered in population sequencing that were not found in the individual isolate sequencing. Download
CF67 isolates in the context of other P. aeruginosa complete genomes. Shown is the unrooted neighbor-joining (NJ) phylogeny of CF67 isolate 71 and 8 P. aeruginosa complete genomes based on genome-wide alignment. All nodes have 100% bootstrap support. Download
Maximum likelihood tree annotated with PBP3 alleles. The tree is constructed as described in the legend to Fig. 2. The identity of the PBP3 allele carried by each strain is indicated by the color of the box to the left of the isolate name. Download
Location of nonsynonymous mutations in the pbpB gene in the 3D crystal structure of PBP3. (A) Multiple sequence alignment of the PBP3 protein sequences of the ancestral allele, minor allele of clade A, and the most common minor allele of clade B. The mutated amino acid is shaded in red and green for clades A and B, respectively. (B) The crystal structure of PBP3 complexed with aztreonam (PDB ID 3PBS [42, 43]) was customized on the PyMOL Molecular Graphics System v. 1.5.0.4 (Schrödinger, LLC). The aztreonam structure is in white, and the mutated positions are colored in red for the clade A minor allele and green for the clade B minor allele. Download
Temporal signal compared to the inferred substitution rate of the 233 isolates. (A) Regression analysis of the root-to-tip distance as a function of time of isolation using the Path-O-Gen program (http://tree.bio.ed.ac.uk/software/pathogen/) as described in reference 101. Each circle represents the average root-to-tip distance of the isolates from the respective sampling time point. The resulting trend shows that the inferred molecular clock was consistent with the changes seen in our isolates through time (R2 = 0.79, P = 0.0001). (B) Rate of substitution as inferred by the coalescent analysis (2.92 ± 0.70 SNPs per year). The line is set to intersect the x axis at the tMRCA of all isolates of this study. Download
Patient details for the study period.
SNPs implicated in antibiotic resistance and host adaptation.