

Summary
Circadian disruption is associated with obesity, implicating the central clock in body weight control. Our comprehensive screen of wild-type and three circadian mutant mouse models, with or without chronic jet-lag, shows that distinct genetic and physiologic interventions differentially disrupt overall energy homeostasis and Leptin signaling. We found that BMAL1/CLOCK generates circadian rhythm of C/EBPα-mediated leptin transcription in adipose. Per- and Cry-mutant mice show similar disruption of peripheral clock and deregulation of leptin in fat, but opposite body weight and composition phenotypes that correlate with their distinct patterns of POMC neuron deregulation in the arcuate nucleus. Chronic jet-lag is sufficient to disrupt the endogenous adipose clock and also induce central Leptin resistance in wild-type mice. Thus, coupling of the central and peripheral clocks controls Leptin endocrine feedback homeostasis. We propose that Leptin resistance, a hallmark of obesity in humans, plays a key role in circadian dysfunction-induced obesity and metabolic syndromes.
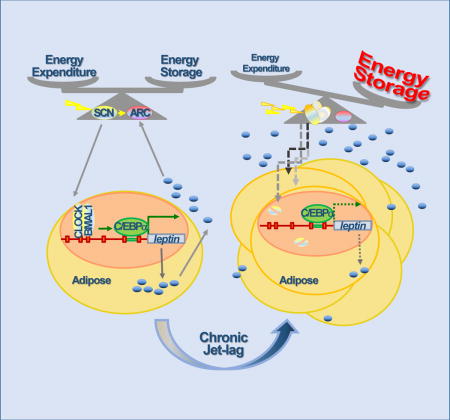
Graphical abstract
Introduction
Obesity and being overweight are associated with severe co-morbidities including type-2 diabetes mellitus, cardiovascular diseases and cancer (Calle et al., 2003; Grundy, 2004). Multiple contributors to the obesity epidemic have been proposed, and a combination of excessive food-intake and sedentary lifestyle is considered the main cause (Ng and Popkin, 2012). However, existing diet and physical exercise programs fail to maintain long-term weight loss (Sturm and Hattori, 2012), suggesting new insights into mechanisms controlling energy balance are needed.
Energy homeostasis in mammals is maintained by the interaction of peripheral signals with the central nervous system (CNS). One example of such interactions is the endocrine feedback loop regulated by Leptin, a potent appetite suppressant. This loop includes three key basic components: white adipose tissue (WAT) that produces Leptin, serum Leptin levels, and Leptin responsive neurons in various hypothalamic centers, especially the arcuate nucleus (ARC). The basal level of serum Leptin is proportional to body fat and rises following a meal or body weight gain. The subsequent activation of Leptin responsive neurons in the ARC suppresses food-intake and stimulates energy expenditure (Maffei et al., 1995; Spiegelman and Flier, 1996; Zhang et al., 1994).
The direct targets of Leptin in the ARC are NPY/AgRP and POMC neurons expressing the Leptin receptor LEPR-B. The NPY/AgRP neurons release orexigenic neuropeptide Y (NPY) and agouti-related peptide (AgRP). The POMC neurons secrete α-melanocyte-stimulating hormone (α-MSH), a cleaved product of pro-opiomelanocortin (POMC). α-MSH activates central melanocortin signaling to reduce food-intake and stimulate energy expenditure, while AgRP is an antagonist of the melanocortin receptors (Cone, 2005; Williams and Schwartz, 2005). An increase in plasma Leptin inhibits NPY/AgRP neurons but activates the Janus kinase 2-Signal Transducer and Activator of Transcription 3 (JAK2-STAT3) signaling pathway in POMC neurons, leading to STAT3-activated POMC transcription and the subsequent α-MSH synthesis and secretion. A decrease in plasma Leptin leads to activation of NPY/AgRP neurons but suppression of POMC neurons, resulting in increased energy storage by stimulating food-intake and suppressing energy expenditure (Elmquist and Scherer, 2012).
Mice lacking Leptin (ob/ob) or LEPR-B (db/db) develop severe obesity and metabolic syndromes (Coleman, 1978). Exogenous administration of Leptin decreases food-intake and body weight of ob/ob and diet-induced obese mice but not db/db mice that already have a high level of plasma Leptin (Campfield et al., 1995; Halaas et al., 1995). Since most obese humans display a similar reduced responsiveness to elevated levels of endogenous Leptin and exogenous Leptin administration as db/db mice, Leptin resistance is regarded as a key contributor to the obesity epidemic (Flier, 2004).
Serum Leptin levels display diurnal variation in both humans and rodents. Restriction of food access to the resting phase reverses serum Leptin circadian rhythm, while fasting abolishes Leptin diurnal rhythm and decreases plasma Leptin below the basal level (Ahima et al., 1996; Bodosi et al., 2004). Thus, external food cues have been thought to be the main factor controlling diurnal oscillation of Leptin signaling. However, recent studies have shown that circadian disruption can abolish diurnal oscillation of plasma Leptin in both humans and animal models without changes in external food cues (Kalsbeek et al., 2001; Scheer et al., 2009; Shea et al., 2005; Simon et al., 1998).
Circadian rhythm in mammals is generated by a central clock located in the hypothalamic suprachiasmatic nucleus (SCN) that constantly synchronizes to external solar light cues and controls subordinate clocks active in all peripheral tissues via circadian output pathways. At the molecular level, both central and peripheral clocks are operated by feedback loops of circadian genes driven by heterodimers of the bHLH-PAS transcription factors CLOCK/BMAL1 or NPAS2/BMAL1, which induce the expression of their own repressors Cryptochromes (Cry1 and 2) and Period (Per1–3) via E-box sequences in gene promoters. The rhythmic interaction of the core circadian genes over a 24 hour period defines the intrinsic circadian rhythmicity of the molecular clock (Mohawk et al., 2012).
The clock interacts reciprocally with energy homeostasis at multiple levels to couple physical activity, food-intake and energy expenditure with the external 24 hour solar day. In the CNS, the rhythmic expression of circadian genes combined with direct neuronal projections from the SCN is found in multiple hypothalamic centers including ARC (Huang et al., 2011). In peripheral tissues, the molecular clock not only regulates nutrient uptake, metabolism and energy storage by targeting genes controlling the key steps of these processes, but also responds to changes in food cues and nutrient sensors independent of the SCN clock (Buijs et al., 2013; Challet, 2013; Eckel-Mahan and Sassone-Corsi, 2013; Feng et al., 2011).
We report here that the circadian clock plays a critical role in long-term homeostasis of the Leptin neuroendocrine feedback loop. In adipose, the BMAL1/CLOCK heterodimer directly controls leptin expression by regulating the activity of CCAAT-enhancer-binding protein alpha (C/EBPα), the most potent transcriptional activator for leptin (Hollenberg et al., 1997; Hwang et al., 1996). Such regulation is sufficient to drive diurnal oscillation of serum Leptin. In the CNS, the SCN clock rhythmically potentiates the response of ARC neurons to circulating Leptin. The coordinated activity of the central and peripheral clocks generates a coupled circadian rhythm in food-intake, physical activity, plasma Leptin, LEPR-B-mediated STAT3-POMC signaling in the ARC and energy expenditure.
Ablation of Per or Cry genes in mice leads to opposite phenotypes of energy expenditure, body weight and body composition due to a differential disruption of Leptin-mediated central melanocortin signaling, but not leptin expression in WAT. Disrupting the SCN clock by chronic jet-lag shifts energy balance in both Per- and Cry-mutants but not in Bmal1-null mice that are insensitive to changes in environmental light cues (Bunger et al., 2000). Thus, energy homeostasis is a clock controlled dynamic physiological function in vivo. Importantly, we found that chronically jet-lagged Wt mice show a progressive gain in body fat and weight, a dramatic increase in serum Leptin, but dampened STAT3-POMC signaling in ARC neurons, and are also resistant to exogenous Leptin-induced STAT3-POMC activation in the ARC. These deficiencies of jet-lagged Wt mice are coupled with a decreased and arrhythmic energy expenditure profile independent of all previous identified obesity risk factors including gene mutations, diet choices and amounts of daily physical activity. Overall, genetic disruption of different core clock components or chronic jet lag induces similar dysregulation of leptin in adipose, but distinct body weight outcomes that correlate with ARC responses. We conclude that chronic jet-lag is sufficient to induce Leptin resistance in wild-type mice. Thus, chronic circadian disruption that is endemic to modern societies may contribute to the obesity pandemic.
Results
Distinct disruptions of circadian dynamics differentially affect mouse body weight
A large number of studies have linked genetic or physiologic circadian disruption to diverse obesity and metabolic disorders in both rodents and humans (Bass and Takahashi, 2010; Evans and Davidson, 2013; Foster et al., 2013; Zhang et al., 2013). However, the circadian disruption experienced by human populations cannot be modeled by any individual circadian gene-mutant mouse model. To understand the role of the clock in energy homeostasis under physiological conditions, we first undertook a large scale, integrated study comparing energy homeostasis phenotypes of C57BL/6J inbred Wt mice with mice of the same strain lacking Bmal1 (Bmal1−/−), both Per1 and 2 (Per1−/−;Per2−/−), or both Cry1 and 2 (Cry1−/−;Cry2−/−) under both entrained and chronic jet-lag conditions.
When maintained in 24 hour light/dark cycles (24hr LD) and fed with a standard mouse chow, both male and female Per-mutants were significantly heavier but Cry-mutants displayed a dramatically reduced body weight relative to Wt controls until 35 to 40 weeks of age, before the detection of other abnormalities such as cancer (Fu et al., 2002; Lee et al., 2010), with changes in body weight more evident in males than in females (Fig. 1A and S1A). The average body weight of Bmal1−/− mice was similar to Wt controls until 12 weeks of age in male and 22 weeks in females and then steadily decreased (Fig. 1A and S1A). The age-dependent body weight phenotype of Bmal1−/− mice, also reported by others, was attributed to premature aging of Bmal1−/− mice (Bunger et al., 2005; Kondratov et al., 2006; Lee et al., 2010; Shi et al., 2013). Per- and Cry-mutants also showed changes in the ratios of gonadal fat to body weight proportional to the changes in their body weight at all ages studied. However, male Bmal1−/− mice showed a similar ratio of gonadal fat pad to body weight as Wt littermates until 20 weeks of age, despite an early and significant decrease in body weight. Bmal1−/− and Per1−/−;Per2−/− mice also had a significant higher total body fat composition than Wt controls in early adulthood (Fig. 1A–C and S1A–C). Surprisingly, although Cry-mutants of both sexes had a significantly reduced body weight and gonadal fat pad, they displayed similar total fat composition as 3 to 20 week old Wt controls (Fig. 1B–C and S1B–C). Thus, Cry-mutant mice are capable of compensating insufficient energy storage in adipose by increasing fat accumulation in other organs.
Figure 1. Circadian disruption abolishes body weight balance in mice.
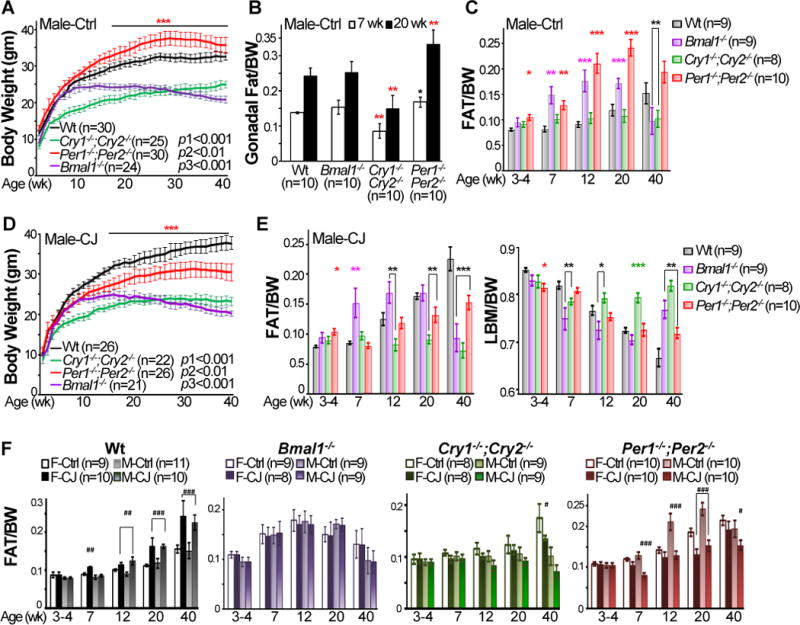
A. Body weights of male mice kept in 24hr LD cycles and fed with regular chow (Ctrl) from 4–40 weeks of age (p1: Wt vs. Cry-mutants from 3–40 weeks of age, p2: Wt vs. Per-mutants from 15–35 weeks of age, and p3: Wt vs. Bmal1−/− mice from 12–40 weeks of age). B. Ratios of gonadal fat mass vs. body weight (BW) of control male mice at 7 and 20 week of age (±SEM). C. Total fat composition of control male mice from 4–40 weeks of age (±SEM). D. Body weight of chronically jet-lagged (CJ) male mice from 4–40 weeks of age (p1: Wt vs. Cry-mutants from 3–40 weeks of age, p2: Wt vs. Per-mutants between 15–40 weeks of age, and p3: Wt vs. Bmal1−/− mice from 12 to 40 weeks of age). E. Fat (left panel) and lean body mass (LBM) (right panel) compositions of male jet-lagged mice from 4–40 weeks of age (±SEM). F. Fat composition in control and jet-lagged female (F) and male (M) mice from 4–40 weeks of age (±SEM). *Compare to control Wt mice at the same age. #Compare to control mice of the same genotype at the same age. */#p < 0.05, **/##p < 0.01 and ***/###p < 0.001. See also Figure S1 and S2.
We then treated Wt and mutant mice with chronic jet-lag from 4 to 40 weeks of age, based on an 8hr-phase advance in light onset at the beginning of each week (Monday) followed by an 8hr-phase delay in light onset in the middle of each week (Thursday) (Fig. S2A). Under jet-lag condition, Wt mice still maintained rhythmic wheel running activities coupled with environmental light cues, but Per- and Cry-mutants showed a significant increase in wheel running activity in the resting phase (Fig. S2B). Jet-lag significantly changed body weight of Wt and Per-mutant mice as well as body composition of all mouse models studied, except Bmal1−/− mice that are insensitive to chronic disruption of light cues. The changes in energy homeostasis were similar among males and females but more evident in males in Wt, Per- and Cry-mutant mice. Importantly, jet-lag significantly increased body weight and fat composition in Wt mice (Fig.1D–F, S1D–E and S2C).
Circadian dynamics of energy expenditure controls energy balance in vivo
We next studied energy balance of mice using a Comprehensive Lab Animal Monitoring System (CLAMS). Equal numbers of control and jet-lagged male Wt, Per- and Cry-mutant mice at 16–18 weeks of age were analyzed for 3 consecutive 24hr LD cycles at the same time. Loading mice on CLAMS caused a brief disruption of behavioral rhythm on the first day of analysis. However, control and jet-lagged Wt mice had similar total daily food-intake and physical activity as well as respiration exchanging rate (RER) that all displayed robust circadian rhythms coupled to the external light cues throughout 3 days of experiments, although these rhythms were slightly but significantly dampened in jet-lagged Wt mice (Fig. 2A–F and S3). Per- and Cry-mutants ate same amount food as Wt controls daily but display dampened or arrhythmic profiles of food-intake. The obesity-prone phenotypes of Per-mutants in steady 24hr LD cycles were coupled to their significantly lower levels of physical activity and RER, whereas the decrease in body weight and fat composition of these mice under jet-lag condition was associated with a dramatically increased physical activity and RER in the light phase. The body weight phenotypes of Cry-mutants under both entrained and jet-lagged conditions could only be explained by their abnormally high RER profiles but not low physical activity or normal amount of daily food-intake (Fig. 2A–F and S3).
Figure 2. Circadian dysfunction disrupts energy homeostasis.
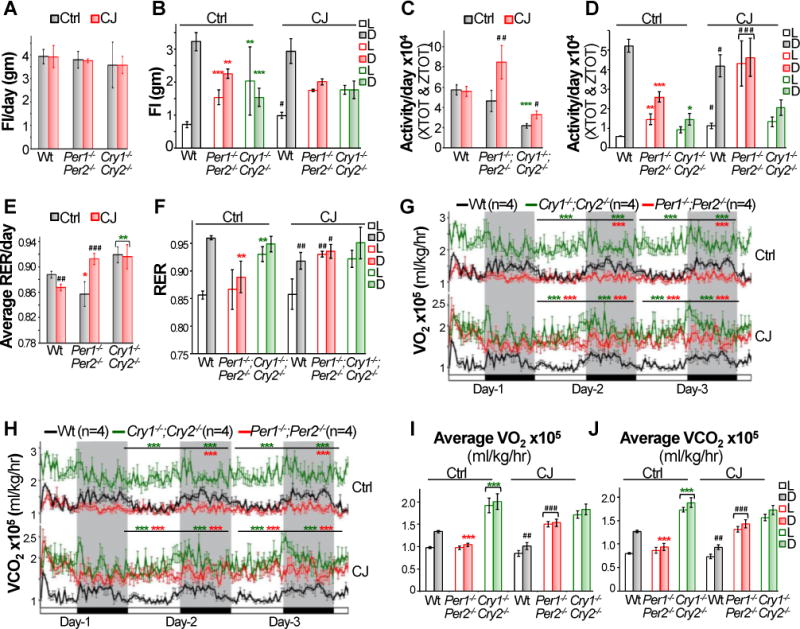
The average of total daily food-intake (FI) (A), or FI in light (L) or dark (D) phase (B) of control and jet-lagged Wt, Cry1−/−;Cry2−/− and Per1−/−;Per2−/− mice on day 2 and 3 of CLAMS study (±SEM). The average of total daily activity (C), or activity in light or dark phase of control and jet-lagged mice on day 2 and 3 of CLAMS study (±SEM). Average daily respiration exchanging rates (RER) (E) and RER in light or dark phase (F) in control and jet-lagged mice on day 2 and 3 of CLAMS study (±SEM). The circadian profiles of oxygen consumption (VO2) (G) and carbon dioxide production (VCO2) (H) in control and jet-lagged mice over 3 days of CLAMS analysis. Average rates of VO2 (I) and VCO2 (J) produced by control and jet-lagged mice in light and dark phases on day 2 and 3 of CLAMS study (±SEM). *Compare to control Wt mice under the same light/dark condition. #Compare to control mice of the same genotype. */#p < 0.05, **/##p < 0.01 and ***/###p < 0.001. See also Figure S3 and S4.
Under entrained conditions, Wt mice displayed a robust circadian rhythm of oxygen consumption (VO2) and carbon dioxide production (VCO2), which was coupled with their daily food-intake and physical activity. Per-mutants showed a flat and low energy expenditure profile over a 24hr period due to lack the peak of VO2 and VCO2 in the active phase. Cry-mutants displayed dramatically elevated VO2 and VCO2 levels especially in the resting phase (Fig. 2G–J). Chronic jet-lag significantly changed the rate of energy expenditure in Wt mice and Per-mutants but not Cry-mutants. The 24hr VO2 and VCO2 profiles in jet-lagged Wt mice resembled those found in control Per-mutants, which, in jet-lagged Per-mutants, became similar to that of control Cry-mutants (Fig. 2G–J and S4).
Overall, the distinct body weight phenotypes of Wt, Per- and Cry-mutant mice under entrained and jet-lagged conditions can be explained by their unique energy expenditure profiles and the shifts of these profiles in response to the disruption of environmental light cues but not the amount of daily food-intake or physical activity (Fig. 1–2 and S1–4). Thus, distinct genetic and physiologic disruptions of circadian homeostasis can have quite different impacts on energy homeostasis.
The endogenous circadian clock drives the circadian rhythm of plasma Leptin
The loss of energy homeostasis in jet-lagged Wt mice suggests that circadian homeostasis of neuroendocrine function is essential for long-term energy balance. To explore this, we focused on the best characterized neuroendocrine pathway for body weight control: the Leptin-mediated central melanocortin signaling. In accord with prior studies (Ahima et al., 1996; Bodosi et al., 2004), we found that plasma Leptin displays a robust circadian rhythm coupled with the diurnal profiles of food-intake, physical activity and energy expenditure in control Wt mice. Ablation of both Per or Cry genes abolished diurnal oscillation of plasma Leptin although Per-mutants still maintained a dampened circadian rhythm of food-intake and physical activity. The basal serum level of Leptin in Per- and Cry-mutants correlated with the ratios of their fat mass versus body weights but not energy expenditure profiles (Fig. 1B, 2, 3A and S3–5). The circadian rhythm of plasma Leptin was maintained in Wt mice in constant darkness (24hr DD cycles) (Fig. 3B). Fasting reduced plasma Leptin below the basal level in the active phase in Wt but not Per-mutant mice (Fig. 3C). Strikingly, acute jet-lag prevented the rise of plasma Leptin in the active phase in Wt mice, despite the fact that these mice maintained a robust circadian rhythm in food-intake and physical activity. Chronic jet-lag resulted in a high and arrhythmic serum level of Leptin over a 24hr period in Wt mice, which correlated with a significant increase in fat gain but not their diurnal profiles of food-intake, physical activity and energy expenditure (Fig. 1D–F, 2, 3D, S2C and S3–5). Jet-lag decreased basal serum Leptin levels in both Per- and Cry-mutants, which correlated with a decrease in their fat mass but not changes in energy expenditure profiles (Fig. 1F, 2 and S3–5). Together, these findings strongly suggest that the circadian clock controls the diurnal oscillation of plasma Leptin independent of external food cues.
Figure 3. Circadian control of leptin expression in adipose and serum.
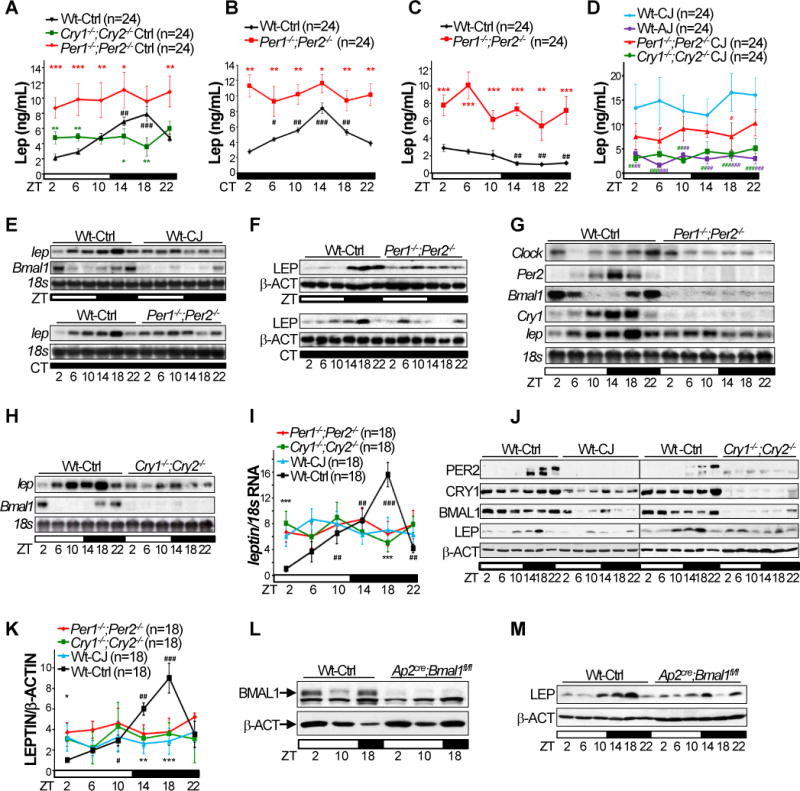
Serum levels of Leptin in Wt, Cry1−/−;Cry2−/− and Per1−/−;Per2−/− mice in 24hr LD cycles (A), in Wt and Per1−/−;Per2−/− mice in 24hr DD cycles (B) and during 22hr fasting (C), and in acutely jet-lagged (AJ) Wt mice and chronically jet-lagged Wt, Cry1−/−;Cry2−/− and Per1−/−;Per2−/− mice (D) (±SEM). E. The leptin mRNA (E) and protein (F) expression follow a robust circadian rhythm in both LD (top panel) and DD (bottom panel) cycles in WAT of control Wt mice, but not in WAT from Per-mutants or jet-lagged Wt mice. G–H. Northern blots show disruption of adipose clock and circadian expression of leptin mRNA in Per- (G) and Cry-mutant (H) mice. I. A summary of 3 independent Northern blotting on leptin mRNA expression in mouse WAT in 24hr LD cycles (±SEM). J. Western blotting on PER2, BMAL1, CRY1, Leptin (LEP) and β-Actin (β-ACT) in WAT of control and jet-lagged Wt (left panel) and Cry-mutants (right panel). K. A summary of 3 independent Western blotting on Leptin expression in mouse WAT in 24hr LD cycles (±SEM). Western blotting show lack of BMAL1 expression in nuclear extracts (L) and dampened and arrhythmic Leptin expression (M) in WAT of Ap2Cre;Bmal1fl/fl mice. ZT: zeitgeber time with light on at ZT0 and off at ZT12. CT: circadian time with CT0 as the beginning of a subjective day and CT12 as the beginning of a subjective night. *: Compare to control Wt samples at the same time, or jet-lagged Wt samples in D. #: Compare to Wt at ZT2; */#p < 0.05, **/##p < 0.01 and ***/###p < 0.001. See also Figure S5.
The peripheral clock directly regulates leptin transcription in adipose
We then studied whether the clock controls leptin expression in adipose to drive serum Leptin levels. We found that leptin mRNA and protein expression followed a robust circadian rhythm in both 24hr LD and DD cycles in WAT of Wt mice (Fig. 3E–F), which peaked at the mid of active phase and coupled with the diurnal profiles of food-intake, activity, energy expenditure and plasma Leptin (Fig. 2 and 3A). Jet-lagged Wt mice, Per- and Cry-mutants, and adipose-specific Bmal1 knock-outs (Ap2cre;Bmal1fl/fl) showed a similar disruption of the endogenous adipose clock and circadian expression of leptin mRNA and/or protein (Fig. 3E–M), despite their unique phenotypes of food-intake, body weight, fat composition, and energy expenditure and activity profiles (Fig.1 and 2) (Paschos, 2012). Thus, the endogenous clock rather than external food cues drives circadian leptin expression in adipose.
The leptin promoter contains multiple consensus E-boxes that are potential targets of BMAL1/CLOCK heterodimers. As expected, a luciferase reporter driven by the mouse Per1 promoter (Per1-Luc) showed dose-dependent transactivation by the circadian heterodimers in co-transfection assays, which was suppressed by CRY1 (Fig. 4A). In contrast, a reporter driven by a 2.7 kb mouse leptin promoter containing 18 consensus E-boxes (lep2762-Luc) only showed a marginal response to a low level of heterodimers, which was suppressed by increasing levels of heterodimers. CRY1 had little effect on the inhibitory effect of higher doses of heterodimers on the lep2762-Luc reporter (Fig. 4A). The dual role of heterodimers on lep-Luc reporter activity was affected by the length of leptin promoter: the lep2762-Luc reporter was most sensitive to the stimulatory effect of the heterodimer at a low concentration, while the lep450-Luc reporter containing 0.45 kb of the minimal leptin promoter and 3 consensus E-boxes was most efficiently inhibited by a high expression level of the heterodimer (Fig. S6A–B).
Figure 4. The heterodimer controls leptin promoter activity in vitro.
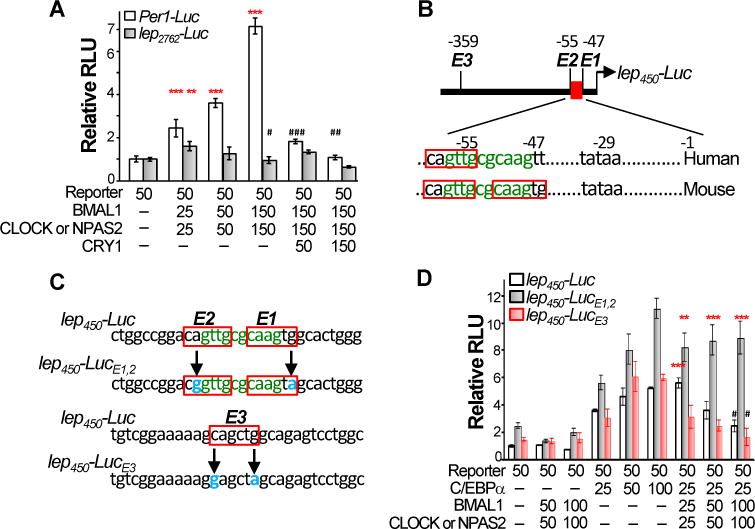
A. Circadian heterodimers marginally stimulate a leptin promoter-driven luciferase (lep2762-Luc) reporter at a low concentration but inhibit the reporter activity at higher concentrations independent of CRY1. Numbers indicate the amounts of reporter and expression vector DNA (ng) used in each transfection (±SEM, *: stimulation and #: suppression). B. The C/EBPα binding sites (green) overlaps with consensus E-boxes (red boxes) in both human and mouse leptin promoters. C. The strategy for generating mutant leptin promoter-driven luciferase reporters with mutated bases shown in blue. D. The heterodimer potentiates C/EBPα-mediated lep450-Luc reporter activation at a low concentration but inhibits C/EBPα activity at higher concentrations. Mutation of E1 and 2 abolishes the inhibitory effect of heterodimer. Mutation of E3 abolishes the stimulatory effect of heterodimer (±SEM, *: stimulation and #: suppression of by the heterodimers). */#p < 0.05, **/##p < 0.01 and ***p < 0.001. See also Figure S6A–D.
Further analysis revealed an overlap of the two proximal E-boxes with a C/EBPα binding site immediately upstream of the TATA box in the mouse leptin promoter. One of the E-boxes is conserved in mouse and human (Fig. 4B). C/EBPα is known as the most potent transcriptional activator of leptin (Fig. S6C) (Hollenberg et al., 1997; Hwang et al., 1996). In addition, although BMAL1/CLOCK and BMAL1/NPAS2 had the exactly same effect on the lep2762-Luc reporter activity in co-transfection assays, NPAS2 is not expressed adipose, and a direct interaction of the heterodimer with C/EBPα is not detected (Fig. S6D and data not shown). Thus, we hypothesized that the heterodimer is not a leptin specific transcriptional regulator. However, it may stimulate C/EBPα-mediated leptin transcription when binding to remote E-boxes, possibly by affecting chromatin structures (Menet et al., 2014), but suppress C/EBPα function independent of CRY when competing with C/EBPα to bind at the proximal E boxes. To test this, we inactivated both proximal E boxes (E1 and 2; lep450-LucE1,2), or the upstream E3 site (lep450-LucE3) without affecting the C/EBPα site in the leptin promoter (Fig. 4C). The C/EBPα-mediated activation of the wild-type lep450-Luc reporter was stimulated by a low level of the heterodimers, but this was not observed with lep450-LucE3. Higher doses of the heterodimer strongly suppressed C/EBPα transactivation of the lep450-Luc and lep450-LucE3 reporters, as predicted, but this inhibitory effect was completely absent with the lep450-LucE1,2 reporter (Fig. 4D).
The levels of c/ebpα mRNA and protein did not display clear circadian oscillation (Fig. 5A–B). However, the binding of BMAL1/CLOCK heterodimer to its target genes varies robustly over a circadian cycle and peaks in the mid sleeping phase (Koike et al., 2012), suggesting a rhythmic competition of the heterodimer with C/EBPα for binding the proximal leptin promoter in vivo. We tested this hypothesis by chromatin immunoprecipitation (ChIP) after validating BMAL1- and C/EBPα-specific antibodies (Fig. 5C and S6E–F), using primers flanking the E1, E2 and C/EBPα-binding site, or the upstream E7 and E8, in the leptin promoter, and nuclear extracts from WAT of control and jet-lagged Wt mice and Ap2cre;Bmal1fl/fl mice at ZT2, 10 and 18 (Fig. 3L and 5C–D). We confirmed that BMAL1 directly binds to both regions of the leptin promoter following a similar circadian rhythm as its binding to the Per1 promoter, which peaked at ZT10 and was lowest at ZT18, with a higher affinity to E-box 1–2 than E-box 7–8 at ZT10 and a low affinity to E-box 1–2 than E-box 7–8 at ZT2 on leptin promoter. Chronic jet-lag abolished the circadian rhythm of BMAL1 binding to leptin and Per1 promoters (Fig. 5E).
Figure 5. Leptin is a clock-controlled gene in vivo.
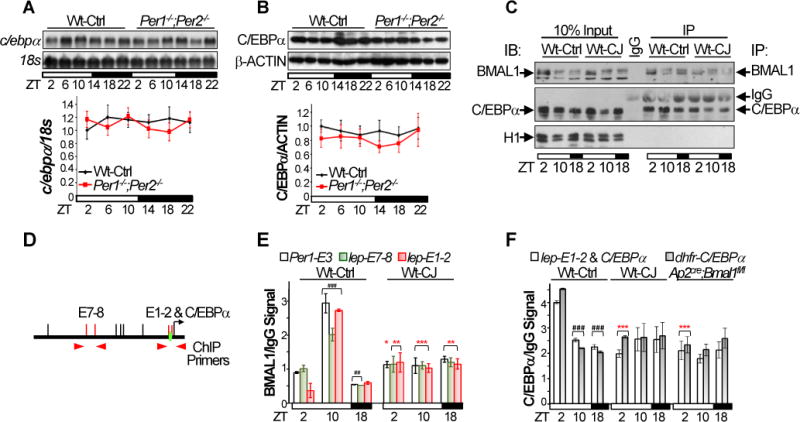
The levels of c/ebpα mRNA and protein in WAT of control Wt and Per1−/−;Per2−/− mice. Top panels: representative Northern (A) and Western (B) blots (±SEM). Bottom panels: summaries of 3 independent Northern (A) and Western (B) blotting (±SEM). C. Representative Western blots show the success of immunoprecipitation of BMAL1 and C/EBPα from WAT nuclear extracts of control and jet-lagged Wt mice at ZT2, 10 and 18 in ChIP assays. D. The positions of qPCR primers on the leptin promoter used in ChIP. E. BMAL1/IgG ChIP signals on Per1 and leptin promoters in WAT of control and jet-lagged Wt mice blotting (±SEM). F. C/EBPα/IgG ChIP signals on leptin and dhfr promoters in WAT of control and jet-lagged Wt mice and Ap2Cre;Bmal1fl/fl mice (±SEM). Primers interacting with an untranslated region in mouse genome that does not contain an E-box or a C/EBPα binding site were also used as negative controls in qPCR, which did not detect any BMAL1 or C/EBPα ChIP signals (supplemental information). *:Compare to control Wt at the same time. #: Compare to control Wt at ZT2; */#p < 0.05, **/##p < 0.01 and ***/###p < 0.001. See also Figure S6E–F.
The binding of C/EBPα to the leptin and control dhfr promoters peaked at ZT2 and was low at ZT10 and ZT18 in WAT of Wt control mice. The rhythmic binding of C/EBPα to these two promoters was completely abolished in WAT of jet-lagged Wt and Ap2cre;Bmal1fl/fl mice (Fig. 5F). Overall, BMAL1 and C/EBPα showed the expected reciprocal occupancy on the proximal leptin promoter during the sleeping phase (ZT2 and ZT10), but reached the nadir for both in the active phase (ZT18). Together with the functional promoter analysis, our ChIP results suggest that BMAL1/CLOCK stimulates C/EBPα-mediated leptin transcription at the early sleeping phase but suppresses C/EBPα transactivation function at the late sleeping phase, following the same rhythm observed with Per and Cry genes (Fig. 4 and 5) (Koike et al., 2012). This results in a similar circadian expression profile for leptin, Per and Cry mRNAs, with peaks in the active phase (Fig. 3G).
The SCN clock rhythmically potentiates Leptin-mediated central melanocortin signaling
The results presented in figures 3–5 demonstrate a direct control of leptin expression by the peripheral clock, but cannot account for the distinct body weight phenotypes of the different circadian mutants as they all lacked the adipose clock in WAT (Fig. 3G–M). We therefore studied the circadian control of Leptin-mediated central melanocortin signaling. We found that STAT3 phosphorylation (pSTAT3) in POMC neurons, which is a direct consequence of LEPR-B-mediated JAK2 signaling and is required for LEPR-B-controlled POMC activation (Bates et al., 2003), peaked at ZT2. This follows the peak of serum Leptin at ZT18 but precedes the peak of POMC expression at ZT10 in the ARC of control Wt mice. Both pSTAT3 and POMC expression were arrhythmic and dampened in the ARC of obesity-prone Per-mutant and jet-lagged Wt mice that display high serum levels of Leptin. In contrast, the lean Cry-mutants showed a high constitutive level of pSTAT3-POMC signaling in ARC despite their low plasma Leptin throughout the 24hr period (Fig. 3A, 3D, 6A–B S5 and S7A). Thus, the differential disruption of Leptin-mediated central melanocortin signaling drives the body weight and composition phenotypes of all mouse models studied.
Figure 6. Disruption of circadian homeostasis induces Leptin resistance in Wt mice.
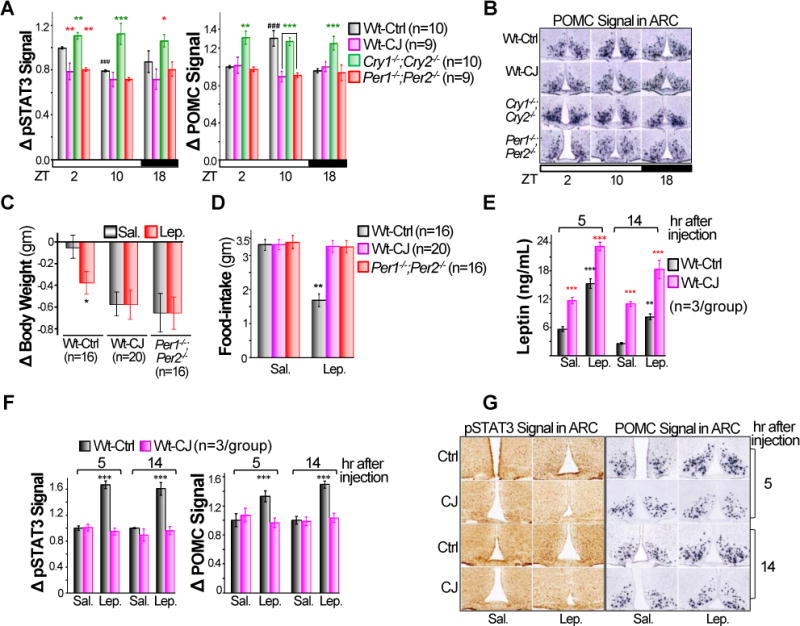
A. A summary of pSTAT3 signals detected by immunohistochemistry (IHC) (left) and POMC signal detected by in situ (right) in ARC of control Wt, Cry1−/−;Cry2−/− and Per1−/−;Per2−/− mice and jet-lagged Wt mice, with signals detected in the ARC of control Wt mice at ZT2 as arbitrary unit 1 (±SEM). B. Representative POMC in situ images in ARC of mice described in A. Changes in body weight (C) and food-intake (D) of control Wt and Per1−/−;Per2−/− mice and jet-lagged Wt mice over a 24hr period in response to a single intraperitoneal injection of recombinant mouse Leptin (±SEM). E. The serum levels of Leptin at 5 and 14hr after a single injection of exogenous Leptin in control and jet-lagged Wt mice (±SEM). F. A summary of pSTAT3 (left) and POMC (right) signals in the ARC of control and jet-lagged Wt mice at 5 and 14hr after a single administration of exogenous Leptin, with signals detected in the ARC of saline injected control Wt mice at 5hr after the injection as arbitrary unit 1 (±SEM). G. Representative pSTAT3 IHC (left) and POMC in situ (right) images in the ARC of Wt control and jet-lagged mice after a single administration of exogenous Leptin. *: Compare to control Wt samples at the same time in A and B, or control Wt mice injected with Saline in C–F. #: Compare to Wt samples at ZT2 in A and B; *p < 0.05, **p < 0.01 and ***/###p < 0.001. See also Figure S7.
To further study the role of the circadian clock in controlling the central melanocortin signaling, we intraperitoneally injected recombinant mouse Leptin into Wt control and jet-lagged mice as well as Per-mutants. A single exogenous Leptin administration significantly reduced body weight and food-intake of control Wt mice over a 24hr period, but had no effect on food-intake of Per1−/−;Per2−/− or jet-lagged Wt mice (Fig. 6C–D). Per1−/−;Per2−/− and jet-lagged Wt mice showed the same body weight reduction in 24hrs in response to Leptin or saline injection (Fig. 6C), suggesting that body weight decrease in these mice was caused by their reduced ability to cope with the stress of injection but not the action of exogenous Leptin.
To explore the mechanism of these differential responses, we studied STAT3-POMC signaling in the ARC of Wt control and jet-lagged mice at 5 or 14hr after Leptin injection. Although jet-lagged Wt mice displayed a significantly higher serum Leptin than control Wt mice after exogenous Leptin administration, they failed to show the activation of STAT3-POMC pathway at all times studied (Fig. 6E–G and S7B). We conclude that chronic circadian disruption induces Leptin resistance in the CNS of Wt mice.
Discussion
Virtually all key metabolic pathways in peripheral tissues display a robust circadian rhythm in vivo. Whole body circadian gene knock-out mouse models show deregulation of these pathways and also distinct energy homeostasis phenotypes (Bass and Takahashi, 2010). However, tissue-specific ablation of Bmal1 in metabolic tissues such as adipose, liver, skeletal muscle or pancreas, which locally inactivates the peripheral clock, does not abolish energy homeostasis at the organismal level, despite the deregulation of key metabolic pathways in tissues affected, such as loss of liver glucose homeostasis, hypo- or hyperglycemia, impaired glucose tolerance and insulin secretion, and elevated oxidative stress, etc. (Lamia et al., 2008; Lee et al., 2013; Marcheva et al., 2010; Paschos et al., 2012; Shimba et al., 2011).
Our extensive characterization of the impact of circadian disruption via genetic and jet-lag strategies in matched cohorts of mice reveals several important discoveries. First, Wt, Bmal1−/−,Cry1−/−;Cry2−/− and Per1−/−;Per2−/− mice display distinct phenotypes of energy homeostasis under both entrained and jet-lagged conditions, which are similar between male and females in each model. Second, energy homeostasis changes in response to disruption of environmental light cues in all mouse models, except for Bmal1−/− mice that lack a functional central clock. Third, jet-lag induced changes in energy homeostasis are independent of food-intake or physical activity but coupled with the changes in energy expenditure. Finally, jet-lag-induced change in energy expenditure is sufficient to account for increased body weight and fat gain in Wt mice.
Further studies lead us to define the role of dysfunction of Leptin neuroendocrine loop in circadian disruption induced obesity risk. We found that the expression of leptin is directly regulated by the molecular clock in adipose, independent of food cues. However, the peripheral deregulation of leptin in Per- and Cry-mutants cannot account for their opposite body weight phenotypes (Fig. 1, 3–5, S1–2 and S6). We also found that the energy homeostasis phenotypes of all mouse models studied do correlate with dysregulation of POMC neurons in the hypothalamus (Fig. 1–2, 6, S1–4 and S7A). Perhaps most importantly, we showed that chronic jet-lag is sufficient to induce Leptin resistance in Wt mice. Their decreased energy expenditure in active phase and complete lack of a feeding response to exogenous Leptin is associated with loss of STAT3 activation in ARC (Fig.6 and S7B). Together, these results demonstrate that normal control of body weight requires the coordinated functions of the central and peripheral clocks and is dynamic in response to changes in both external and internal cues.
The circadian clock controls key components of the Leptin feedback loop to maintain the homeostasis of energy balance. In WAT, the rhythmic binding of the BMAL1/CLOCK heterodimer to leptin promoter potentiates C/EBPα-mediated leptin transcription in early sleeping phase but suppresses C/EBPα function at late sleeping phase. This is followed by the peak of leptin mRNA and protein accumulation in the mid of active phase (Fig. 3–5). Jet-lagged Wt mice, Per- and Cry-mutants, and adipose-specific Bmal1 knockouts, all show a similar disruption of the fat clock and inhibition of BMAL1 expression, which is correlated with the suppression of leptin expression in adipose regardless of their distinct food-intake profiles (Fig. 2–5). The rhythmic expression of leptin driven by the fat clock in WAT is sufficient to drive the circadian rhythm of serum Leptin. Only Wt control mice display a coupled circadian rhythm of plasma Leptin and food-intake. Per- and Cry-mutants and chronic jet-lagged Wt mice show a similar arrhythmic profile of serum Leptin with a basal level proportional to their fat mass but not food-intake activity (Fig. 1–3).
In Wt mice, the peaks of plasma Leptin, the activation of STAT3 and POMC in ARC, and the rate of energy expenditure are at ZT18, ZT2, ZT10 and ZT18, respectively. This sequential activation of the key components of Leptin feedback loop is completely abolished in jet-lagged Wt mice and Per- and Cry-mutants. However, Per-mutants show dampened STAT3-POMC signaling in the ARC despite a high level of serum Leptin, while Cry-mutants show constitutive activation of STAT3-POMC signaling in ARC independent of circulating Leptin. The differential disruption of STAT3-POMC signaling in ARC correlates well with the distinct body weight, fat mass and energy expenditure phenotypes of Per- and Cry-mutants (Fig. 1–3, 6, S1–4 and S7A). Together with the finding that adipose-specific ablation of Bmal1 does not significantly change energy balance in mice when kept in 24hr LD cycles and fed with regular chow (Paschos et al., 2012), our studies suggest that although the adipose clock drives diurnal oscillation of serum Leptin, the circadian potentiation of Leptin responsive neurons in the CNS to circulating Leptin plays a dominant role in the homeostasis of Leptin endocrine feedback loop.
The basis for the strikingly differential effects of loss of Per or Cry on the CNS remains to be established. These two genes may play different roles in controlling LEPR-B-STAT3-POMC\ signaling in ARC as both PER and CRY have been shown to directly interact with various transcriptional factors in peripheral tissues (Zhao et al., 2014). Alternatively, differential uncoupling of individual SCN oscillators in Per- and Cry-mutants may differentially deregulate the LEPR-B-STAT3-POMC pathway in ARC by affecting SCN efferent connections in the hypothalamus (Mohawk et al., 2012; Moore, 2013). Importantly, chronically jet-lagged Wt mice, like Per-mutants, show arrhythmic and dampened LEPR-B-STAT3-POMC signaling in ARC in the presence of high levels of plasma Leptin, and are resistant to exogenous Leptin suppression of food-intake (Fig. 3D, 6, S5 and S7). Thus, chronic disruption of circadian homeostasis induces Leptin resistance independent of other known risk factors including gene mutations, diet choices, excess food-intake and sedentary lifestyles.
Based on our findings, we conclude that the circadian homeostasis of Leptin-mediated neuroendocrine feedback loop is a key mechanism for the clock to control long-term energy balance at the organismal level. In the adipose, BMAL1/CLOCK directly controls circadian leptin transcription that drives the rhythm of serum Leptin. In the CNS, the SCN clock potentiates the response of LEPR-B expressing ARC neurons to circulating Leptin to maintain the balance of food-intake and energy expenditure. Under normal physiological conditions, the activities of central and peripheral clock are coupled with environmental cues. Acute circadian disruption abolishes peripheral clock-controlled leptin expression in adipose to suppress plasma Leptin levels, leading to a shift in energy balance and weight gain. Increased fat mass elevates circulating Leptin. However, chronic circadian dysfunction desensitizes the LEPR-B expressing ARC neurons to increased Leptin signaling, resulting in Leptin resistance, a hallmark of obesity in humans (Fig. 7).
Figure 7. A model of circadian control of Leptin neuroendocrine feed-back loop.
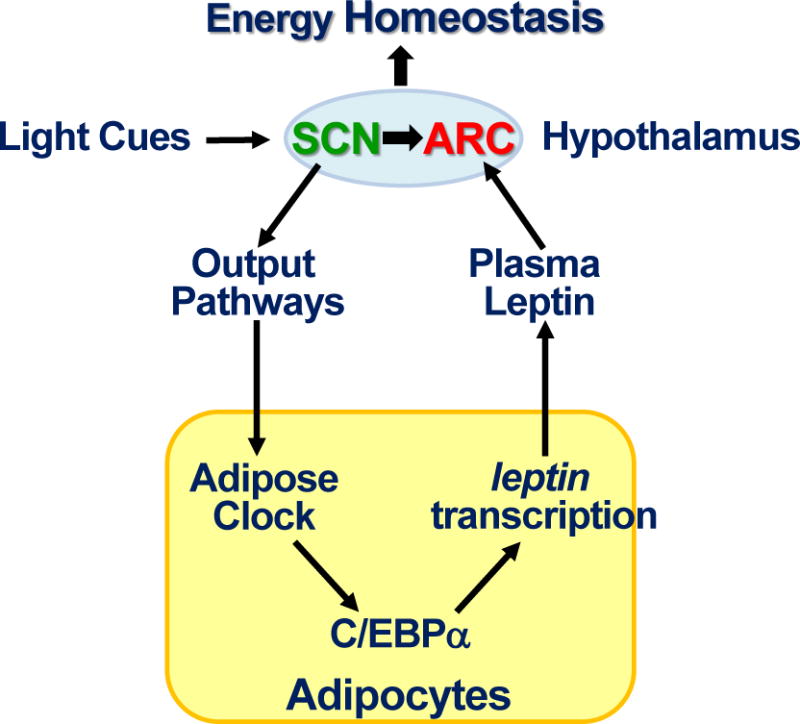
The circadian homeostasis of Leptin endocrine feedback loop controls long-term energy balance at the organismal level as explained in the discussion.
Experimental Procedures
Animal Maintenance
All animal experiments were approved by the Institutional Animal Care and Use Committee at Baylor College of Medicine. Detailed experimental procedures for mouse maintenance, tissue and serum sample isolation, body weight and composition as well as energy homeostasis analyses are described in Supplemental Experimental Procedures.
ELISA
Serum was collected as described in Supplemental Experimental Procedures. Plasma Leptin levels were determined by a Mouse Leptin ELISA kit (BioVendor).
RNA Analysis
Northern blotting followed standard procedure using p32-labled cDNA probes for Bmal1, Clock, Per1, Per2, Cry1, Naps2 and 18s rRNA as described previously (Fu et al., 2005). The leptin cDNA probe was from a mouse leptin coding sequence (Genebank number: NM_008493.3, nucleotides +60 to +560). Signals detected were quantified using a Molecular Dynamics Storm 860 PhosphorImager/Fluorima.
Plasmid Construction
A 2762bp mouse leptin promoter was cloned by screening a mouse lambda genomic library (Strategene) using a p32-labled probe containing the 3′ end of leptin promoter (−10 to −365) obtained by PCR amplification of C57BL/6J mouse genomic DNA using a forward primer 5′ ggaaaaagcagctggcag 3′ and a reverse primer 5′ gctccatgcctgcccctc 3′, and inserted between the Hind III and Kpn I sites in the pGL-3 vector. The resulting reporter was named lep2762-Luc. Further deletion and religation of the lep2762-Luc generated lep1545- and lep450-Luc reporters containing a 1545 and 450bp leptin promoter upstream of the luciferase cDNA in the pGL-3 vector, respectively.
Site-direct Mutagenesis
Mutant leptin report plasmids were generated using the Lep450-Luc plasmid as a template, a site-direct mutagenesis kit (Invitrogen) and primers 5′ ctggccggacggttgcgcaagtagcactggggc 3′ (forward) and 5′ gccccagtgctacttgcgcaaccgtccggccag 3′ (reverse) for the lep450-LucE1,2 vector, 5′ cggaaaaagctgctggcagagtcctggc 3′ (forward) and 5′ gccaggactctgccagcagctttttccg 3′ (reverse) for the lep450-LucE3 vector, respectively.
Cell Culture and Reporter Assays
COS-7 cells were maintained in DMEM supplemented with 10% FBS and grown at 37°C with 5% CO2. Co-transfections were performed using Lipofectamine 2000 (Invitrogen) following the standard protocol, and the same amounts of reporter and β-gal expression vectors, variable amounts of expression vectors, and an empty pcDNA3 vector to normalize the total amount DNA. Cell extracts were prepared 48hr after transfection and used for luciferase assay with β-gal co-transfection serving as an internal control (Fu et al., 2005).
Chromatin immunoprecipitation (ChIP)
Nuclear extracts prepared from WAT of mice at ZT2, 10 and 18, and antibodies recognizing BMAL1 and C/EBPα were used for immunoprecipitation. qPCRs were performed to detect BMAL1 and C/EBPα ChIP signals on the leptin, Per1 and dhfr promoters. Detailed procedure is described in Supplemental Experimental Procedures.
Protein Expression Studies
Western blotting using total WAT protein or nuclear extracts were performed following the standard procedure using antibodies as described in Supplemental information.
In situ hybridization (ISH)
Coronal brain sections through arcuate nucleus were processed for highly sensitive non radioactive colorimetric ISH using an antisense mouse pro-opiomelanocortin cDNA as a probe (Genebank number: NM_008895.3, nucleotides +29 to +944) (McGill et al., 2006). A sense NPY probe was used as a negative control (http://mouse.brain-map.org/gene/show/73806). Image quantification was performed using the Celldetekt and Image J software (Carson et al., 2005).
Immunohistochemistry (IHC)
IHC was performed as described (Fukuda et al., 2011). Coronal brain sections placed on slides were incubated with an anti-pSTAT3 antibody (Cell Signaling) overnight, washed in PBS, and incubated with a biotinylated anti-rabbit antibody (Vector Laboratories). Slides were then treated with ABC solution (Vector Laboratories) followed by treatment with ImmPACT DAB (Vector Laboratories). The images obtained were analyzed using the Celldetekt and Image J software (Carson et al., 2005).
Intraperitoneal injection of Leptin
Single-housed control or jet-lagged male mice were used for intraperitoneal injection of saline or recombinant mouse Leptin. Detailed procedure is described in Supplemental Experimental Procedures.
Statistical Analysis
Statistical significance was assessed by Student’s two-tailed t test. Values were considered statistically significant at p < 0.05.
Supplementary Material
Acknowledgments
We thank Drs. A Sancar and C Bradfield for kindly providing Cry1−/−;Cry2+/− and Bmal1+/− mice, ML Fiorotto for assisting CLAMS analyses, CM Ljungberg for in situ hybridization, and M Fukuda for judicious suggestions. This work is supported by grants from USDA/ARS (6250-51000-055) and NIH/NCI (R01 CA137019-01A) to L. Fu, a NIH/NIDDK training grant to NM Kettner (5T32 DK007696-20), and partially by shared NIH equipment grants to CM Liungberg (1 S10 OD016167-01 and DK56338), and a NIH/NICHD grant (U54HD083092) to the Baylor IDDRC Neuropathology Core for immunohistochemistry studies.
Footnotes
Author Contributions
L.F. conceived research. L.F. and D.D.M. designed experiments. N.M.K., L.F., S.A.M., and J.H. performed experiments. C.L. contributed reagents and assisted with ChIP troubleshooting. L.F., N.M.K., and D.D.M. analyzed data. L.F., D.D.M., and N.M.K. wrote the paper.
Literature Cited
- Ahima RS, Prabakaran D, Mantzoros C, Qu D, Lowell B, Maratos-Flier E, Flier JS. Role of leptin in the neuroendocrine response to fasting. Nature. 1996;382:250–252. doi: 10.1038/382250a0. [DOI] [PubMed] [Google Scholar]
- Bass J, Takahashi JS. Circadian integration of metabolism and energetics. Science. 2010;330:1349–1354. doi: 10.1126/science.1195027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates SH, Stearns WH, Dundon TA, Schubert M, Tso AW, Wang Y, Banks AS, Lavery HJ, Haq AK, Maratos-Flier E, Neel BG, Schwartz MW, Myers MG., Jr STAT3 signalling is required for leptin regulation of energy balance but not reproduction. Nature. 2003;421:856–859. doi: 10.1038/nature01388. [DOI] [PubMed] [Google Scholar]
- Bodosi B, Gardi J, Hajdu I, Szentirmai E, Obal F, Jr, Krueger JM. Rhythms of ghrelin, leptin, and sleep in rats: effects of the normal diurnal cycle, restricted feeding, and sleep deprivation. Am J Physiol Regul Integr Comp Physiol. 2004;287:R1071–1079. doi: 10.1152/ajpregu.00294.2004. [DOI] [PubMed] [Google Scholar]
- Buijs R, Salgado R, Sabath E, Escobar C. Peripheral circadian oscillators: time and food. Progress in molecular biology and translational science. 2013;119:83–103. doi: 10.1016/B978-0-12-396971-2.00004-X. [DOI] [PubMed] [Google Scholar]
- Bunger MK, Walisser JA, Sullivan R, Manley PA, Moran SM, Kalscheur VL, Colman RJ, Bradfield CA. Progressive arthropathy in mice with a targeted disruption of the Mop3/Bmal-1 locus. Genesis. 2005;41:122–132. doi: 10.1002/gene.20102. [DOI] [PubMed] [Google Scholar]
- Bunger MK, Wilsbacher LD, Moran SM, Clendenin C, Radcliffe LA, Hogenesch JB, Simon MC, Takahashi JS, Bradfield CA. Mop3 is an essential component of the master circadian pacemaker in mammals. Cell. 2000;103:1009–1017. doi: 10.1016/s0092-8674(00)00205-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. The New England journal of medicine. 2003;348:1625–1638. doi: 10.1056/NEJMoa021423. [DOI] [PubMed] [Google Scholar]
- Campfield LA, Smith FJ, Guisez Y, Devos R, Burn P. Recombinant mouse OB protein: evidence for a peripheral signal linking adiposity and central neural networks. Science. 1995;269:546–549. doi: 10.1126/science.7624778. [DOI] [PubMed] [Google Scholar]
- Carson JP, Eichele G, Chiu W. A method for automated detection of gene expression required for the establishment of a digital transcriptome-wide gene expression atlas. J Microsc. 2005;217:275–281. doi: 10.1111/j.1365-2818.2005.01450.x. [DOI] [PubMed] [Google Scholar]
- Challet E. Circadian clocks, food intake, and metabolism. Progress in molecular biology and translational science. 2013;119:105–135. doi: 10.1016/B978-0-12-396971-2.00005-1. [DOI] [PubMed] [Google Scholar]
- Coleman DL. Obese and diabetes: two mutant genes causing diabetes-obesity syndromes in mice. Diabetologia. 1978;14:141–148. doi: 10.1007/BF00429772. [DOI] [PubMed] [Google Scholar]
- Cone RD. Anatomy and regulation of the central melanocortin system. Nat Neurosci. 2005;8:571–578. doi: 10.1038/nn1455. [DOI] [PubMed] [Google Scholar]
- Eckel-Mahan K, Sassone-Corsi P. Epigenetic regulation of the molecular clockwork. Progress in molecular biology and translational science. 2013;119:29–50. doi: 10.1016/B978-0-12-396971-2.00002-6. [DOI] [PubMed] [Google Scholar]
- Elmquist JK, Scherer PE. The cover. Neuroendocrine and endocrine pathways of obesity. JAMA: the journal of the American Medical Association. 2012;308:1070–1071. doi: 10.1001/jama.2012.3209. [DOI] [PubMed] [Google Scholar]
- Evans JA, Davidson AJ. Health consequences of circadian disruption in humans and animal models. Progress in molecular biology and translational science. 2013;119:283–323. doi: 10.1016/B978-0-12-396971-2.00010-5. [DOI] [PubMed] [Google Scholar]
- Feng D, Liu T, Sun Z, Bugge A, Mullican SE, Alenghat T, Liu XS, Lazar MA. A circadian rhythm orchestrated by histone deacetylase 3 controls hepatic lipid metabolism. Science. 2011;331:1315–1319. doi: 10.1126/science.1198125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flier JS. Obesity wars: molecular progress confronts an expanding epidemic. Cell. 2004;116:337–350. doi: 10.1016/s0092-8674(03)01081-x. [DOI] [PubMed] [Google Scholar]
- Foster RG, Peirson SN, Wulff K, Winnebeck E, Vetter C, Roenneberg T. Sleep and circadian rhythm disruption in social jetlag and mental illness. Progress in molecular biology and translational science. 2013;119:325–346. doi: 10.1016/B978-0-12-396971-2.00011-7. [DOI] [PubMed] [Google Scholar]
- Fu L, Patel MS, Bradley A, Wagner EF, Karsenty G. The molecular clock mediates leptin-regulated bone formation. Cell. 2005;122:803–815. doi: 10.1016/j.cell.2005.06.028. [DOI] [PubMed] [Google Scholar]
- Fu L, Pelicano H, Liu J, Huang P, Lee C. The circadian gene Period2 plays an important role in tumor suppression and DNA damage response in vivo. Cell. 2002;111:41–50. doi: 10.1016/s0092-8674(02)00961-3. [DOI] [PubMed] [Google Scholar]
- Fukuda M, Williams KW, Gautron L, Elmquist JK. Induction of leptin resistance by activation of cAMP-Epac signaling. Cell metabolism. 2011;13:331–339. doi: 10.1016/j.cmet.2011.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy SM. Obesity, metabolic syndrome, and cardiovascular disease. The Journal of clinical endocrinology and metabolism. 2004;89:2595–2600. doi: 10.1210/jc.2004-0372. [DOI] [PubMed] [Google Scholar]
- Halaas JL, Gajiwala KS, Maffei M, Cohen SL, Chait BT, Rabinowitz D, Lallone RL, Burley SK, Friedman JM. Weight-reducing effects of the plasma protein encoded by the obese gene. Science. 1995;269:543–546. doi: 10.1126/science.7624777. [DOI] [PubMed] [Google Scholar]
- Hollenberg AN, Susulic VS, Madura JP, Zhang B, Moller DE, Tontonoz P, Sarraf P, Spiegelman BM, Lowell BB. Functional antagonism between CCAAT/Enhancer binding protein-alpha and peroxisome proliferator-activated receptor-gamma on the leptin promoter. The Journal of biological chemistry. 1997;272:5283–5290. doi: 10.1074/jbc.272.8.5283. [DOI] [PubMed] [Google Scholar]
- Huang W, Ramsey KM, Marcheva B, Bass J. Circadian rhythms, sleep, and metabolism. The Journal of clinical investigation. 2011;121:2133–2141. doi: 10.1172/JCI46043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang CS, Mandrup S, MacDougald OA, Geiman DE, Lane MD. Transcriptional activation of the mouse obese (ob) gene by CCAAT/enhancer binding protein alpha. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:873–877. doi: 10.1073/pnas.93.2.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalsbeek A, Fliers E, Romijn JA, La Fleur SE, Wortel J, Bakker O, Endert E, Buijs RM. The suprachiasmatic nucleus generates the diurnal changes in plasma leptin levels. Endocrinology. 2001;142:2677–2685. doi: 10.1210/endo.142.6.8197. [DOI] [PubMed] [Google Scholar]
- Koike N, Yoo SH, Huang HC, Kumar V, Lee C, Kim TK, Takahashi JS. Transcriptional architecture and chromatin landscape of the core circadian clock in mammals. Science. 2012;338:349–354. doi: 10.1126/science.1226339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondratov RV, Kondratova AA, Gorbacheva VY, Vykhovanets OV, Antoch MP. Early aging and age-related pathologies in mice deficient in BMAL1, the core componentof the circadian clock. Genes & development. 2006;20:1868–1873. doi: 10.1101/gad.1432206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamia KA, Storch KF, Weitz CJ. Physiological significance of a peripheral tissue circadian clock. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:15172–15177. doi: 10.1073/pnas.0806717105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Moulik M, Fang Z, Saha P, Zou F, Xu Y, Nelson DL, Ma K, Moore DD, Yechoor VK. Bmal1 and beta-cell clock are required for adaptation to circadian disruption, and their loss of function leads to oxidative stress-induced beta-cell failure in mice. Molecular and cellular biology. 2013;33:2327–2338. doi: 10.1128/MCB.01421-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Donehower LA, Herron AJ, Moore DD, Fu L. Disrupting circadian homeostasis of sympathetic signaling promotes tumor development in mice. PloS one. 2010;5:e10995. doi: 10.1371/journal.pone.0010995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maffei M, Halaas J, Ravussin E, Pratley RE, Lee GH, Zhang Y, Fei H, Kim S, Lallone R, Ranganathan S, et al. Leptin levels in human and rodent: measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nature medicine. 1995;1:1155–1161. doi: 10.1038/nm1195-1155. [DOI] [PubMed] [Google Scholar]
- Marcheva B, Ramsey KM, Buhr ED, Kobayashi Y, Su H, Ko CH, Ivanova G, Omura C, Mo S, Vitaterna MH, Lopez JP, Philipson LH, Bradfield CA, Crosby SD, JeBailey L, Wang X, Takahashi JS, Bass J. Disruption of the clock components CLOCK and BMAL1 leads to hypoinsulinaemia and diabetes. Nature. 2010;466:627–631. doi: 10.1038/nature09253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGill BE, Bundle SF, Yaylaoglu MB, Carson JP, Thaller C, Zoghbi HY. Enhanced anxiety and stress-induced corticosterone release are associated with increased Crh expression in a mouse model of Rett syndrome. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:18267–18272. doi: 10.1073/pnas.0608702103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menet JS, Pescatore S, Rosbash M. CLOCK:BMAL1 is a pioneer-like transcription factor. Genes & development. 2014;28:8–13. doi: 10.1101/gad.228536.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohawk JA, Green CB, Takahashi JS. Central and peripheral circadian clocks in mammals. Annu Rev Neurosci. 2012;35:445–462. doi: 10.1146/annurev-neuro-060909-153128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore RY. The suprachiasmatic nucleus and the circadian timing system. Progress in molecular biology and translational science. 2013;119:1–28. doi: 10.1016/B978-0-12-396971-2.00001-4. [DOI] [PubMed] [Google Scholar]
- Ng SW, Popkin BM. Time use and physical activity: a shift away from movement across the globe. Obes Rev. 2012;13:659–680. doi: 10.1111/j.1467-789X.2011.00982.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paschos GK, Ibrahim S, Song WL, Kunieda T, Grant G, Reyes TM, Bradfield CA, Vaughan CH, Eiden M, Masoodi M, Griffin JL, Wang F, Lawson JA, Fitzgerald GA. Obesity in mice with adipocyte-specific deletion of clock component Arntl. Nature medicine. 2012;18:1768–1777. doi: 10.1038/nm.2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheer FA, Hilton MF, Mantzoros CS, Shea SA. Adverse metabolic and cardiovascular consequences of circadian misalignment. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:4453–4458. doi: 10.1073/pnas.0808180106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shea SA, Hilton MF, Orlova C, Ayers RT, Mantzoros CS. Independent circadian and sleep/wake regulation of adipokines and glucose in humans. The Journal of clinical endocrinology and metabolism. 2005;90:2537–2544. doi: 10.1210/jc.2004-2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi SQ, Ansari TS, McGuinness OP, Wasserman DH, Johnson CH. Circadian disruption leads to insulin resistance and obesity. Current biology: CB. 2013;23:372–381. doi: 10.1016/j.cub.2013.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimba S, Ogawa T, Hitosugi S, Ichihashi Y, Nakadaira Y, Kobayashi M, Tezuka M, Kosuge Y, Ishige K, Ito Y, Komiyama K, Okamatsu-Ogura Y, Kimura K, Saito M. Deficient of a clock gene, brain and muscle Arnt-like protein-1 (BMAL1), induces dyslipidemia and ectopic fat formation. PloS one. 2011;6:e25231. doi: 10.1371/journal.pone.0025231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon C, Gronfier C, Schlienger JL, Brandenberger G. Circadian and ultradian variations of leptin in normal man under continuous enteral nutrition: relationship to sleep and body temperature. The Journal of clinical endocrinology and metabolism. 1998;83:1893–1899. doi: 10.1210/jcem.83.6.4864. [DOI] [PubMed] [Google Scholar]
- Spiegelman BM, Flier JS. Adipogenesis and obesity: rounding out the big picture. Cell. 1996;87:377–389. doi: 10.1016/s0092-8674(00)81359-8. [DOI] [PubMed] [Google Scholar]
- Sturm R, Hattori A. Morbid obesity rates continue to rise rapidly in the United States. International journal of obesity. 2012 doi: 10.1038/ijo.2012.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams DL, Schwartz MW. The melanocortin system as a central integrator of direct and indirect controls of food intake. Am J Physiol Regul Integr Comp Physiol. 2005;289:R2–3. doi: 10.1152/ajpregu.00226.2005. [DOI] [PubMed] [Google Scholar]
- Zhang L, Ptacek LJ, Fu YH. Diversity of human clock genotypes and consequences. Progress in molecular biology and translational science. 2013;119:51–81. doi: 10.1016/B978-0-12-396971-2.00003-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- Zhao X, Cho H, Yu RT, Atkins AR, Downes M, Evans RM. Nuclear receptors rock around the clock. EMBO reports. 2014;15:518–528. doi: 10.1002/embr.201338271. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.