

Graphical abstract

Keywords: sGC (soluble guanylate cyclase), Nitric oxide, ODQ, Surface plasmon resonance
Abstract
Soluble guanylate cyclase (sGC) is a haem containing enzyme that regulates cardiovascular homeostasis and multiple mechanisms in the central and peripheral nervous system. Commonly used inhibitors of sGC activity act through oxidation of the haem moiety, however they also bind haemoglobin and this limits their bioavailability for in vivo studies. We have discovered a new class of small molecule inhibitors of sGC and have characterised a compound designated D12 (compound 10) which binds to the catalytic domain of the enzyme with a KD of 11 μM in a SPR assay.
1. Introduction
The heterodimeric enzyme soluble guanylate cyclase (sGC) is an endogenous receptor for nitric oxide (NO). NO binds to a haem prosthetic group resulting in a conformational change which activates the enzyme. Upon activation, sGC converts guanosine triphosphate (GTP) into cyclic guanosine monophosphate (cGMP). NO-induced activation of sGC is key to maintaining cardiovascular homeostasis and in the brain, NO—sGC acts as a neurotransmitter—receptor system.1–3
NO-induced signalling has been implicated in the modulation of synaptic transmission and to act in long-term potentiation, one of the major cellular mechanisms that underlie the processes of learning and memory.4 In rats, high cGMP levels promote neural stem cells differentiation to neurons whilst reduced cGMP levels promote differentiation to non-neuronal (mainly glial) cells, which consequently leads to impaired cognitive function.5
NO can mediate neurotoxicity and cause neuronal cell death. The rapid on–off-kinetics and desensitization profile of NO, combined with variations in the rate of cGMP breakdown, provide fundamental mechanisms for shaping cellular cGMP responses and is important in decoding NO signals under physiological and pathological conditions.6 The neurotransmitter is associated with pathogenic mechanisms involved in multiple neurodegenerative diseases, including Parkinson’s Disease (PD).7 The NO—sGC system is also involved in the etiology of migraine.8 Recent research has suggested the involvement of NO in PD is due to activation of sGC. Disruption of striatal NO-sGC-cGMP signalling cascades resulted in profound changes in behavioural, electrophysiological, and molecular responses to pharmacological manipulations of dopamine and glutamate transmission.9 Studies performed in animal models of PD with a sGC inhibitor, ODQ 1 (Fig. 1), have shown that the enzyme could be a new drug target for restoring basal ganglia dysfunction and attenuating motor symptoms associated with PD.9 ODQ 1 has been widely used to study the function of the NO-sGC-cGMP signal transduction pathway and it has been a valuable tool to distinguish signalling events mediated by sGC from those involving other nucleotide cyclases.10 The small molecule binds to the ferrous haem in the β-subunit of the enzyme, yielding ferric haem which cannot bind NO.11,12 Haem-binding compounds such as ODQ 1 and its 8-bromo-analogue NS2028, show activity against other haem containing proteins as such as haemoglobin, albeit at high concentrations.13,14 ODQ may also not be able to block cGMP signalling in all circumstances.15 Other known ways of inhibiting sGC activity include block of the catalytic site with ATP and GTP analogues16–20 though these have weak inhibitory potency. Inhibitors such as LY-83583 act indirectly by generating superoxide which reacts rapidly with NO.16 Previously we demonstrated that surface plasmon resonance (SPR) allied to biochemical screening was an effective way of discovering new sGC ligands. In this study we identified a new small molecule inhibitor of sGC, which does not act through oxidation of the haem.
Figure 1.

Chemical structures of sGC inhibitor ODQ 1, Lamotrigine 2, and analogues Sipatrigine 3 and 4.
2. Results
2.1. In silico similarity searching and screening
In the course of studies in our laboratory we observed activation of sGC with the anti-epileptic drug lamotrigine 2 and inhibition with lamotrigine analogues sipatrigine 3 and 4, all at low millimolar concentrations (data not shown) (Fig. 1). These compounds were used as the starting point for a screening study to find new inhibitors of sGC. The strategy was to conduct several rounds of similarity searching and screening to identify the best inhibitors. Some synthetic studies were conducted on the best hit to explore the structure–activity relationships.
Virtual screening was performed by similarity searches using MACCS fingerprints at 85% and 75% Tanimoto of commercial libraries and the selected compounds were screened at 100 μM against purified bovine lung sGC using diethylamine NONOate (30 nM) as the NO donor. Enzyme activity was determined by measuring cGMP production using a standard cGMP [3H] radioimmunoassay.21,22 The initial structure searched (Fig. 2, substructure A) resulted in circa 500 structures, out of which 16 compounds were selected based upon diversity, molecular size, and availability. Compounds 5 and 6 showed inhibition of enzyme activity by 51% at 100 μM. Subsequent searches of substructures B and C resulted in the identification of [1,2,5]oxadiazolo[3,4-b]pyrazines 7 and 8, and N2,N3-diphenylquinoxaline-2,3-diamines 9 and 10 (designated D12) as inhibitors of sGC (Fig. 2).
Figure 2.

Virtual screening. Chemical structures of virtually searched substructures A–C and the most active compounds 5–10 of each series. The values in brackets correspond to the % inhibition of sGC activity at 100 μM compound.
2.2. Chemistry and structure activity studies
The hit compound 10 is formed of a quinoxaline scaffold with a nitro group in the 6-position of the heterocycle, and joined to two phenols via secondary amine linkers. A small set of analogues was synthesised to explore the binding role of the substituents, focusing on the nitro group and the phenols (Table 1).
Table 1.
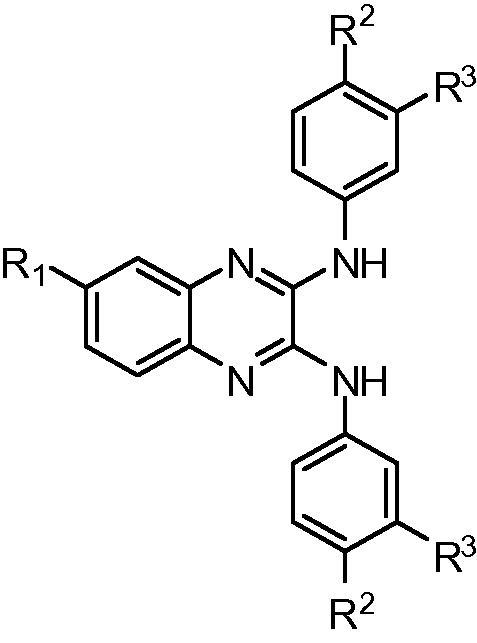
| Compd number | R1 | R2 | R3 | sGCcat (SPR) binding norm. resp. (RU) | sGC activity% inhibition at 100 μM |
|---|---|---|---|---|---|
| 10 | NO2 | OH | H | 14.4 ± 1.7 | 56.2 ± 22.4 |
| 14 | NO2 | H | OH | 11.6 ± 0.8 | 41.4 |
| 15 | NO2 | OCH3 | H | 1.0 ± 0.3 | — |
| 16 | NO2 | H | H | 2.6 ± 0.2 | — |
| 17 | NO2 | NHCOCH3 | H | 0.8 ± 0.3 | — |
| 18 | NO2 | H | NHCOCH3 | 3.0 ± 1.0 | — |
| 19 | NO2 | F | H | 1.9 ± 1.8 | — |
| 20 | H | OH | H | 12.4 ± 0.1 | — |
| 21 | CF3 | OH | H | 6.4 ± 0.5 | 44.4 |
| 22 | CF3 | H | OH | 3.0 ± 0.5 | 21.1 |
| 23 | CN | OH | H | 4.5 ± 2 | — |
| 24 | CN | H | OH | 10.5 ± 0.5 | — |
| 25 | Cl | OH | H | 9.7 ± 0.5 | — |
| 26 | Cl | H | OH | 9.0 ± 0.4 | — |
| 27 | CONHCH3 | OH | H | 1.1 ± 0.3 | — |
| 28 | CONHCH3 | H | OH | 9.6 ± 0.3 | — |
| 29 | NH2 | OH | H | 9.9 ± 0.2 | — |
Compound 10 and analogues 14–28 were synthesised via nucleophilic aromatic displacement using commercially available anilines and 2,3-dichloro-quinoxalines 12 when possible. In other cases 2,3-dichloroquinoxalines 12 were obtained via a known two-step synthesis, starting with the condensation of 1,2-diamines with oxalic acid.23 The resulting 2,3-dihydroxyquinoxalines 11 were chlorinated with thionyl chloride and a catalytic amount of DMF. The carboxamide substituted quinoxaline 13 was obtained after amidation of the carbonyl chloride intermediate 12a (Scheme 1).
Scheme 1.

General synthetic route to compounds 10 and 14–29. (a) 4 N HCl, reflux, 2 h; (b) SOCl2, DMF, reflux, 2 h; (c) aniline, NMP, 150 °C, 5 min, μW; (d) CH3NH2, EtOH, DCM, rt, 24 h.
Reduction of the nitro group in compound 10 was successfully achieved using tin(II) chloride in the presence of sodium borohydride to give compound 29 (Scheme 2).
Scheme 2.

Reduction of nitro group to a primary amine. (a) NaBH4, SnCl2·2H2O, EtOH, reflux, 2 h.
We have previously described a surface plasmon resonance-based assay for the detection of binding of small molecules to full-length sGC and a smaller construct of the catalytic domain (sGCcat).24 This assay was used to measure the binding of the compounds to sGCcat.24 In general, compounds with hydroxyls on the anilino phenyl rings (10, 14, 20, 21, 23–26, 28, 29) showed higher binding to the enzyme, suggesting they might be involved in hydrogen bonding. The position of the hydroxyl group can be changed whilst retaining binding strength and activity. However, other groups such as acetamide 17, 18, methoxy 15, or fluoro 19, rendered the compound biochemically inactive and reduced its binding strength.
The electron-withdrawing nitro group commonly presents as a challenge in drug design. Nitro-aromatics are commonly associated with toxicity, but identifying suitable replacements has proven difficult.25 A series of 6-substituted-2,3-dichloroquinoxalines was synthesised. Further modifications included the reduction of the nitro group to a primary amine, and the conversion of an acyl chloride into an amide (Schemes 1 and 2). Changes in the 6-position of the quinoxaline have resulted in compounds which show high or medium binding to the receptor, but poor or null inhibition of its activity, with the exception of the trifluoromethyl compounds 21 and 22. Removal of the nitro group (compound 20) showed only a small decrease in binding, but showed no significant inhibition of the enzyme. This would suggest that binding of the compounds is not greatly influenced by the group in the 6-position of the quinoxaline, but this is however required for activity. Insertion of amine 29, chloro 25, 26, and nitrile 23, 24 groups also resulted in compounds which bind to the receptor but do not inhibit the enzyme activity.
2.3. Molecular modelling
A possible binding mode was investigated by molecular modelling, using the available crystal structure of sGCcat (pdb code 3uvj). Compound 10 was docked into the allosteric site of sGC and possible binding modes were analysed based on their Goldscore fitness. Most binding modes are similarly docked at the interface of the two subunits. The docking poses show four key interactions with the receptor: one oxygen of the nitro group may act as H-bond acceptor for two backbone NHs, being the only interaction with residues of the β-subunit; the two NH groups attached to the quinoxaline may act as H-bond donors for the side chain of a glutamic acid; one or two OH groups can act as a H-bond donors; and a π–cation interaction is suggested between a charged lysine and one of the aromatic rings. In addition, the lysine and arginine residues present in the site could show π–cation interactions with other aromatic rings of the ligand (Fig. 3).
Figure 3.
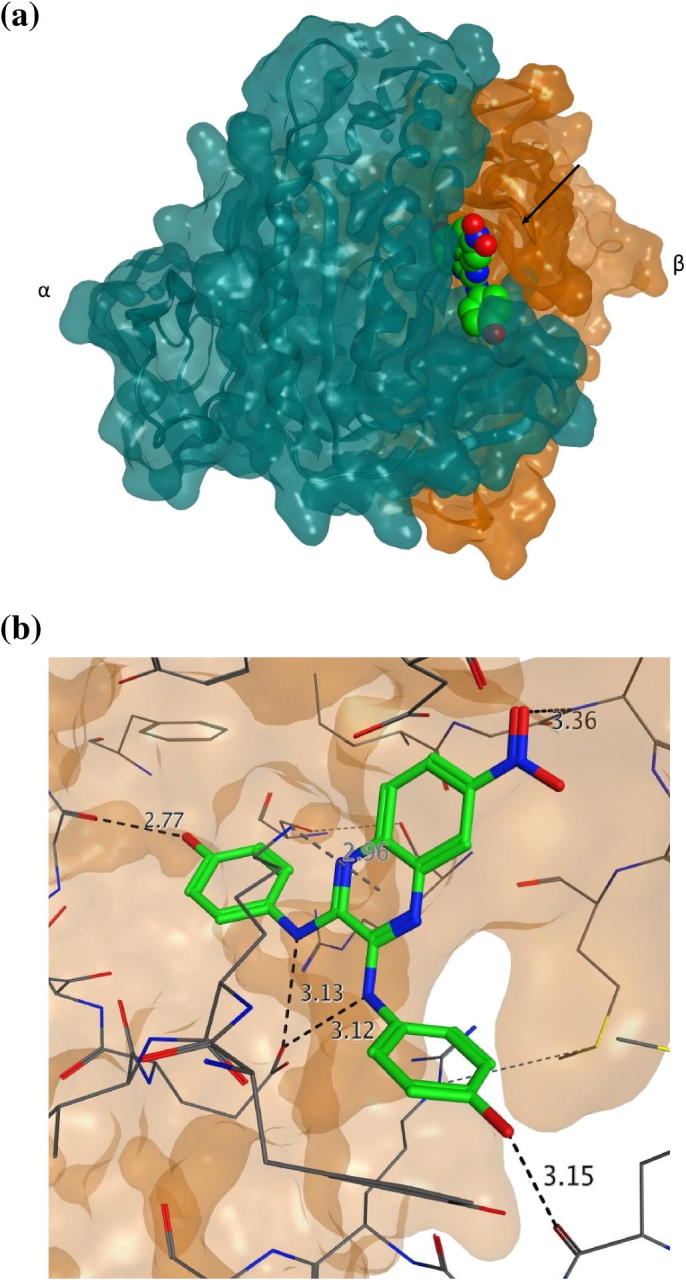
Docking of compound 10 into the allosteric binding site of sGC. (a) Cartoon representation of compound 10 docked at the interface of the two subunits of the catalytic domain of sGC (PDB code 3uvj), in which the α-subunit is coloured green and the β-subunit coloured orange; (b) representation of the ligand interaction between the compound and the residues of sGC.
2.4. Biological and biophysical characterisation of compound 10
The activity of compound 10 on sGC was tested in the presence of increasing concentrations of GTP (Fig. 4a) and NO (Fig. 4b). Concentration–response curves did not show appreciable variation amongst different conditions, suggesting activity of the compound is not through competition with NO or GTP. Activation of sGC can also be induced by the haem-mimetic drug cinaciguat in the absence of NO.26 Cinaciguat binds to the haem-deficient protein, simulating the NO-activated state.
Figure 4.

Biochemical characterisation of compound 10 activity. Compound 10 inhibited NO-stimulated cGMP production by isolated sGC with varying concentrations of GTP (50, 200, 1000 μM) and clamped NO concentrations at (a) 0.3 nM and (b) 10 nM.
Compound 10 was capable of inhibiting cGMP production after enzyme activation with cinaciguat, further demonstrating that its mechanism of action is not NO-dependent. Activity against membrane-bound GCs was measured using purified rat lung membranes in the presence of 10 μM ODQ 1 to inhibit NO-activated sGC. cGMP generation was measured after stimulation with atrial natriuretic peptide and compound 10 showed inhibition of pGC-generated cGMP, indicating an action through the catalytic domain of the enzyme (Table 2).
Table 2.
sGC activity of compound 10
| GC activator | Compound 10 IC50 (μM) ± SE |
Hill slope | |
|---|---|---|---|
| Cinaciguat | 1 μM | 37 ± 40 | 4 |
| 20 nM | 25 ± 4 | 4 | |
| ANP | 1 μM | 25 ± 13 | 1 |
| 10 nM | 21 ± 3 | 2 | |
| Basal | 15 ± 2 | 2 | |
The ability of compound 10 to inhibit sGC in tissues was explored using NMDA-stimulated cGMP production in cerebellar slices, which depends on NO formation. Rat cerebellar slices were pre-incubated with compound 10 then treated with NMDA. Compound 10 inhibited the ensuing cGMP generation almost completely at 100 μM and to a lesser extent at 1 μM (Fig. 5).
Figure 5.

Activity of compound 10 in rat cerebellum. Compound 10 inhibits cGMP production induce by NMDA in rat cerebellar slices at 100 μM. Results are means ± SEM (n = 3); *p <0.02, **p <0.001 versus NMDA control (2-sample t-test).
The binding affinity of compound 10 to sGCcat was explored in detail using SPR, and suggests a 1:1 interaction between the compound and the dimer, with a calculated KD of 11.4 ± 1.8 μM (Fig. 6). Further characterisation with full-length enzyme gave a KD of 19.4 ± 2.1 μM.
Figure 6.

Binding of compound 10 to sGCcat. Compound 10 binds to the catalytic domain of sGC (KD = 11.4 ± 1.8). (a) SPR sensorgram and (b) dose–response of compound 10 binding to sGCcat.
3. Discussion
The sGC heterodimer is composed of a α-subunit and a shorter β-subunit which presents in the N-terminal the prosthetic haem group bound to histidine-105.27 Each subunit is composed of a haem-nitric oxide binding domain, a PAS-like domain, a coiled-coil bundle, and the C-terminal catalytic domain where turnover of GTP into cGMP occurs.28 The catalytic domain of sGC shows two sites at the interface of the α- and β-subunits that can accommodate small molecules: the GTP binding site, and an allosteric regulatory binding site.29–31 The two sites are different, and small molecules can bind to one or to both sites, thus showing a 1:1 or 1:2 binding ratio. We have previously shown by surface plasmon resonance (SPR) that ATP binds to both sites and may inhibit sGC activity via competition with GTP, highlighting the potential for allosteric regulation of the enzyme.24 GC domains are also present in the membrane-bound natriuretic peptide receptors type-A and type-B. There receptors, also referred to as particulate GCs (pGCs), are activated by binding of natriuretic peptides to the extracellular domain of the receptors, leading to an increase in GTP turnover at the intracellular GC domain.32–34 In this study we observed by SPR that compound 10 bound to the catalytic domain of sGC in a 1:1 ratio. Inhibition of particulate GC was also observed and supported the hypothesis of interaction at the catalytic domain. Binding of compound 10 to an allosteric site that is also capable of binding ATP could suggest these compounds might also act on other ATP- and cyclic-nucleotide—binding domains. We tested compound 10 against adenylyl cyclase, an enzyme that converts ATP into cAMP, and it showed no activity on basal or forskolin-stimulated adenylyl cyclase (data not shown). However, activity against kinases or phosphodiesterases was not investigated.
Activity of compound 10 in rat brain tissue highlights the potential to use this new class of allosteric sGC inhibitors to study the role of the NO—sGC—cGMP signalling pathway in the brain. Reducing amounts of cGMP in cells would have implications in downstream signalling proteins, such as cGMP-dependent protein kinases and phosphodiesterases.35
4. Conclusion
We have identified a new type of small molecule inhibitors of sGC, which are thought to be the first class to act through allosteric regulation of the enzyme catalytic domain, rather than oxidation of the haem or through the purine p-site.36 It is possible that the inhibitors presented bind to the catalytic domain of sGC inducing a conformational change, or ‘locking’ the enzyme in a basal conformation, that is not favourable to activation by NO or GTP binding. The inhibitor also inhibits particulate GC but not the related adenylate cyclase. Compound 10 (D12), with an IC50 and KD in the micromolar range, may be used as a new tool to inhibit the NO—sGC—cGMP signalling pathway, and further study its implications and regulatory functions in the brain.
5. Experimentals
Full-length human recombinant guanylyl cyclase (soluble) and bovine lung guanylyl cyclase (soluble) were obtained from Enzo lifesciences (catalogue numbers ALX-201-177 and ALX-202-039-C005). Biacore consumables were purchased from GE Healthcare (UK), including buffer stock solutions. All other reagents used were obtained from Sigma. Starting materials were either commercially available or synthesized according to methods reported in the literature. 1H and 13C NMR spectra were recorded on a Bruker AMX-300 or a Bruker AMX-500 spectrometer. Chemical shifts are reported as ppm relative to TMS internal standard. Mass spectra were recorded on a Fisons VG70-SE spectrometer (EI, FAB) or an Agilent 6100 Series LC-mass spectrometer using C-18 or C-4 columns. Microwave reactions were carried out using a CEM Discover microwave.
5.1. Surface plasmon resonance
Surface plasmon resonance (SPR) experiments were performed with a Biacore T200 instrument at 25 °C. Data processing and analysis were performed using BIAevaluation software and Scrubber2. All sensorgrams were double referenced by subtracting the response in a reference flowcell and a blank sample. Materials and methods used were the same as previously published.24
5.2. Soluble guanylate cyclase assay
In general, bovine lung soluble guanylyl cyclase (5 ng/ml) was incubated with 50 mM Tris, 0.3 mM MgCl, 100 μM EGTA, 0.045% BSA, and 1000 units/mL SOD at pH 7.4 and 1 mM MgGTP at 37 °C. Compounds were added at 100 μM prior to adding the NO donor DEA/NO (30 nM) or clamped NO concentrations. cGMP generation was allowed to proceed for two minutes, after which aliquots were withdrawn and inactivated by boiling in 50 mM Tris, 4 mM EDTA buffer. cGMP formation was measured by a standard radioimmunoassay.
5.3. Particulate guanylate cyclase assay
Purified rat lung membranes (20 μg protein/200 μL) were incubated in Tris buffer with 10 mM phosphocreatine, 10 units/mL creatine phosphokinase, and 10 μM ODQ to inhibit NO-activated GC. The reaction took place with 0.3 mM MgCl, 0.5 mM MgATP, and 1 mM MgGTP, at 37 °C. The compound was added and 1 μM ANP used to activate pGC. cGMP formation was allowed to proceed for 10.5–12.5 min and was subsequently measured by radioimmunoassay.
5.4. cGMP measurement in rat cerebellar slices
Brain slices methods were conducted as described in detail.37,38 Briefly, 0.4 mm thick sagittal cerebellar slices from 10-day-old animals were cut using a McIlwain tissue chopper. Experiments were carried out in the presence of the broad spectrum phosphodiesterase inhibitor IBMX (1 mM; 10 min preincubation) and, when appropriate, antagonists were added 10 min prior to IBMX. Following treatment, brain slices were inactivated in boiling Tris–HCl buffer (50 mM, pH 7.5) containing EDTA (4 mM) and homogenised by sonication after which aliquots were removed for measurement of protein (bicinchoninic acid method) and cGMP (radioimmunoassay).
5.5. Virtual screening
Similarity searching using MACCS fingerprints at 85% and 75% Tanimoto similarity was performed in 2007 using the following databases: Abinitio, Acros, Actimol, Akos/Akl, Akox/Akx, Akos/Owh, Asinex, Chembridge, Chemstar, LifeChemicals, MoscowMedChemLabs, Pharmeks, Sigma, SPECS, Vitas/Dah, Vitas/Stk. The available structures were selected by hand based upon diversity, molecular size, and apparent availability.
5.6. Molecular modelling
Docking was performed using the software Gold 5.1. The pdb file (3uvj) was imported and analysed, and missing residue side chains were added to the structure. The binding site was selected by importing a list of residues correspondent to the allosteric binding site. The fitness function selected was GOLDSCORE and the genetic algorithm search was performed at medium speed. Results were visualised in CCG MOE software.
5.7. Synthesis
5.7.1. General synthesis of 2,3-dichloroquinoxalines 12
A solution of oxalic acid in 4 N HCl (5 mL) was added to a stirring solution of the diamine (1:1) in 4 N HCl (15 mL) and the reaction mixture was refluxed for 2 h. The solution was cooled to room temperature and the precipitate filtered by suction, washed with water and freeze-dried. The resulting 2,3-dihydroxyquinoxaline 11 was dissolved in thionyl chloride (5 mL). A catalytic amount of N,N-dimethylformamide (0.01 equiv) was added to the solution which was stirred under reflux for 2 h. The reaction mixture was cooled to room temperature and concentrated under vacuum. Water (10 mL) was added to the solid on an ice bath and the slurry was stirred for 30 min at room temperature, after which the solid was filtered and washed with water. The precipitate was taken up in a minimum amount of DCM and filtered through a silica column and eluted with DCM.
5.7.2. 2,3-Dichloro-N-methylquinoxaline-6-carboxamide 13
Methylamine solution (33 wt % in absolute ethanol, 256 μL) was added to DCM (3 mL) on ice and the resulting mixture was added to a solution of 2,3-dichloroquinoxaline-6-carbonyl chloride (261 mg, 1 mmol) in DCM (3 mL) whilst stirring at room temperature. The mixture was stirred at room temperature overnight after which it was diluted with DCM (10 mL) and washed with saturated sodium bicarbonate (3 × 10 mL) and brine (3 × 10 mL). The organics were dried over MgSO4, filtered, and the solved evaporated. The crude compound was purified by flash column chromatography using a gradient of 30–60% ethyl acetate in cyclohexane to yield the named compound as a white powder (89 mg, 35%). 1H NMR (500 MHz, d6-DMSO) δ: 8.84 (s, 1H, NH), 8.48 (s, 1H, ArH), 8.29 (d, J = 8.6 Hz, 1H, ArH), 8.14 (d, J = 8.6 Hz, 1H, ArH), 2.85 (d, J = 3.7 Hz, 3H, CH3). 13C NMR (126 MHz, d6-DMSO) δ: 165.0 (ArC), 146.0 (ArC), 145.6 (ArC), 141.1 (ArC), 139.6 (ArC), 136.8 (ArC), 129.9 (ArCH), 128.1 (ArCH), 126.6 (ArCH), 26.5 (CH3). MS-EI (m/z): [M] calculated for C10H7Cl2N3O, 256.089; found 255.90.
5.7.3. General synthesis of compounds 10 and 14–29
The quinoxaline and aniline (1:4) were transferred into a 10 mL microwave vial containing a stirrer bar and NMP (1 mL). The mixture was stirred at 160 °C for 5 min under microwave irradiation. The residue was taken up in water (5 mL) and extracted with ethyl acetate (3 × 5 mL). The organics were washed with water (3 × 5 mL) and brine (3 × 5 mL), dried over Mg2SO4, filtered and concentrated in vacuo. The final compound was purified by flash column chromatography.
5.7.3.1. 4,4′-((6-Nitroquinoxaline-2,3-diyl)bis(azanediyl))diphenol 10
1H NMR (500 MHz, d6-DMSO) δ: 9.37 (s, 1H, OH), 9.31 (s, 1H, OH), 9.26 (s, 1H, NH), 9.07 (s, 1H, NH), 8.18 (d, J = 2.5 Hz, 1H, ArH), 8.01 (dd, J = 8.9, 2.5 Hz, 1H, ArH), 7.64 (dd, J = 10.9, 8.9 Hz, 4H, 4 × ArH), 7.51 (d, J = 8.9 Hz, 1H, ArH), 6.83 (d, J = 8.9 Hz, 4H, 4 × ArH). 13C NMR (126 MHz, d6-DMSO) δ 154.2 (ArC), 153.8 (ArC), 143.2 (ArC), 142.6 (ArC), 141.5 (ArC), 140.6 (ArC), 135.3 (ArC), 130.7 (ArC), 130.3 (ArC), 125.4 (ArCH), 123.7 (ArCH), 123.2 (ArCH), 120.1 (ArCH), 118.6 (ArCH), 115.5 (ArCH), 115.2 (ArCH). HRMS-ES (m/z): [M+H]+ calculated for C20H16N5O4, 390.1202; found, 390.1205.
5.7.3.2. 3,3′-((6-Nitroquinoxaline-2,3-diyl)bis(azanediyl))diphenol 14
1H NMR (500 MHz, d6-DMSO) δ: 9.50 (s, 1H, OH), 9.48 (s, 1H, OH), 9.41 (s, 1H, NH), 9.24 (s, 1H, NH), 8.32 (d, J = 2.6 Hz, 1H, ArH), 8.11 (dd, J = 9.0, 2.6 Hz, 1H, ArH), 7.66 (d, J = 2.0 Hz, 1H, ArH), 7.64 (d, J = 9.0 Hz, 1H, ArH), 7.51 (t, J = 7.3 Hz, 1H, ArH), 7.27 (dd, J = 18.4, 8.0 Hz, 2H, 2 × ArH), 7.22–7.16 (m, 2H, 2 × ArH), 6.54 (ddd, J = 17.0, 8.0, 1.5 Hz, 2H, 2 × ArH). 13C NMR (126 MHz, d6-DMSO) δ: 155.89, 155.55, 143.29, 143.23, 142.52, 141.43, 135.29, 132.27, 131.87, 125.53, 123.46, 122.95, 120.18, 118.80, 113.95, 55.25, 55.22. HRMS-ES (m/z): [M+H]+ calculated for C22H16N5O4, 390.1202; found, 390.1189.
5.7.3.3. N2,N3-Bis(4-methoxyphenyl)-6-nitroquinoxaline-2,3-diamine 15
1H NMR (500 MHz, d6-DMSO) δ: 9.39 (s, 1H, NH), 9.20 (s, 1H, NH), 8.21 (d, J = 2.6 Hz, 1H, ArH), 8.04 (dd, J = 8.9, 2.6 Hz, 1H, ArH), 7.82–7.74 (m, 4H, ArH), 7.54 (d, J = 8.9 Hz, 1H, ArH), 7.05–6.99 (m, 4H, ArH), 3.78 (s, 6H, 2 × OCH3). 13C NMR (126 MHz, d6-DMSO) δ 155.9 (ArC), 155.6 (ArC), 143.3 (ArC), 143.2 (ArC), 142.5 (ArC), 141.4 (ArC), 135.3 (ArC), 132.3 (ArC), 131.9 (ArC), 125.5 (ArCH), 123.5 (ArCH), 122.9 (ArCH), 120.2 (ArCH), 118.8 (ArCH), 113.9 (ArCH), 55.3 (OCH3), 55.2 (OCH3). HRMS-ES (m/z): [M+H]+ calculated for C22H20N5O4, 418.1515; found, 418.1531.
5.7.3.4. 6-Nitro-N2,N3-diphenylquinoxaline-2,3-diamine 16
1H NMR (500 MHz, d6-DMSO) δ: 9.51 (s, 1H, NH), 9.34 (s, 1H, NH), 8.27 (d, J = 2.2 Hz, 1H, ArH), 8.07 (dd, J = 8.8, 2.2 Hz, 1H, ArH), 7.95–7.85 (m, 4H, 4 × ArH), 7.62 (d, J = 8.8 Hz, 1H, ArH), 7.47–7.39 (m, 4H, 4 × ArH), 7.20–7.09 (m, 2H, 2 × ArH). 13C NMR (126 MHz, d6-DMSO) δ: 143.7 (ArC), 143.1 (ArC), 142.4 (ArC), 141.2 (ArC), 139.4 (ArC), 139.1 (ArC), 135.2 (ArC), 128.8 (ArCH), 125.9 (ArCH), 123.9 (ArCH), 123.4 (ArCH), 121.6 (ArCH), 121.1 (ArCH), 120.5 (ArCH), 119.2 (ArCH). HRMS-ES (m/z): [M+H]+ calculated for C20H16N5O2, 358.1304; found, 358.1305.
5.7.3.5. N,N′-(((6-Nitroquinoxaline-2,3-diyl)bis(azanediyl))bis(4,1-phenylene))diacetamide 17
1H NMR (500 MHz, d6-DMSO) δ: 9.97 (s, 1H, NH), 9.96 (s, 1H, NH), 9.52 (s, 1H, NH), 9.33 (s, 1H, NH), 8.28 (d, J = 2.4 Hz, 1H, ArH), 8.06 (dd, J = 8.9, 2.4 Hz, 1H, ArH), 7.83 (dd, J = 13.6, 8.9 Hz, 4H, 4 × ArH), 7.67–7.57 (m, 5H, 5 × ArH), 2.05 (d, J = 4.9 Hz, 6H, 6 × ArH). 13C NMR (126 MHz, d6-DMSO) δ: 168.1 (Ar-NH-CO-CH3), 168.0 (Ar-NH-CO-CH3), 143.5 (ArC), 143.1 (ArC), 142.3 (ArC), 141.3 (ArC), 135.5 (ArC), 135.3 (ArC), 134.5 (ArC), 134.1 (ArC), 125.7 (ArCH), 122.1 (ArCH), 121.6 (ArCH), 120.4 (ArCH), 119.4 (ArCH), 119.3 (ArCH), 118.9 (ArCH), 23.9 (Ar-NH-CO-CH3). HRMS-ES (m/z): [M+H]+ calculated for C24H22N7O4, 472.1733; found, 472.1750.
5.7.3.6. N,N′-(((6-Nitroquinoxaline-2,3-diyl)bis(azanediyl))bis(3,1-phenylene))diacetamide 18
1H NMR (500 MHz, d6-DMSO) δ 10.02 (s, 2H, 2 × NH), 9.62 (s, 1H, NH), 9.44 (s, 1H, NH), 8.46–8.32 (m, 3H, 3 × ArH), 8.12 (dd, J = 8.8, 2.0 Hz, 1H, ArH), 7.71–7.60 (m, 3H, ArH), 7.36–7.25 (m, 5H, 5 × ArH), 2.09 (s, 3H, CH3), 2.08 (s, 3H, CH3). 13C NMR (126 MHz, d6-DMSO) δ 168.4 (Ar-NHCOCH3), 139.7 (ArC), 135.1 (ArC), 128.8 (ArCH), 125.9 (ArCH), 120.6 (ArC), 119.2 (ArCH), 116.3 (ArCH), 115.8 (ArCH), 114.5 (ArCH), 114.1 (ArCH), 112.2 (ArCH), 111.6 (ArC), 109.1 (ArC), 24.1 (Ar-NHCOCH3). HRMS-ES (m/z): [M+H]+ calculated for C24H22N7O4, 472.1733; found, 472.1733.
5.7.3.7. N2,N3-Bis(4-fluorophenyl)-6-nitroquinoxaline-2,3-diamine 19
1H NMR (500 MHz, d6-DMSO) δ: 9.52 (s, 1H), 9.36 (s, 1H), 8.27 (d, J = 2.4 Hz, 1H), 8.07 (dd, J = 8.9, 2.4 Hz, 1H), 7.92 (ddd, J = 13.8, 8.9, 5.0 Hz, 4H), 7.60 (d, J = 8.9 Hz, 1H), 7.31–7.23 (m, 4H). HRMS-ES (m/z): [M+H] calculated for C20H14N5O3F2, 394.1116; found, 394.1123.
5.7.3.8. 4,4′-(Quinoxaline-2,3-diylbis(azanediyl))diphenol 20
1H NMR (500 MHz, d6-DMSO) δ 9.21 (s, 2H, 2 × OH), 8.71 (s, 2H, 2 × NH), 7.61 (d, J = 8.7 Hz, 4H, 4 × ArH), 7.42 (dt, J = 7.3, 3.4 Hz, 2H, 2 × ArH), 7.23 (dd, J = 6.1, 3.4 Hz, 2H, 2 × ArH), 6.79 (d, J = 8.7 Hz, 4H, 4 × ArH). 13C NMR (126 MHz, d6-DMSO) δ 153.2 (ArC), 141.4 (ArC), 136.2 (ArC), 131.5 (ArC), 125.0 (ArCH), 124.4 (ArCH), 122.8 (ArCH), 115.1 (ArCH). HRMS-ES (m/z): [M+H]+ calculated for C20H17N4O2, 345.1352; found, 345.1347.
5.7.3.9. 4,4′-((6-(Trifluoromethyl)quinoxaline-2,3-diyl)bis(azanediyl))diphenol 21
1H NMR (500 MHz, d6-DMSO) δ: 9.34 (s, 1H, OH), 9.32 (s, 1H, OH), 9.03 (s, 1H, NH), 8.96 (s, 1H, NH), 7.68 (s, 1H, ArH), 7.65–7.59 (m, 4H, 4 × ArH), 7.55 (d, J = 8.5 Hz, 1H, ArH), 7.47 (dd, J = 8.5, 1.8 Hz, 1H, ArH), 6.84–6.78 (m, 4H, 4 × ArH). 13C NMR (126 MHz, d6-DMSO) δ: 170.3 (ArC), 153.8 (ArC), 153.6 (ArC), 142.8 (ArC), 142.3 (ArC), 138.7 (ArC), 135.7 (ArC), 131.0 (ArC), 130.8 (ArC), 125.8 (ArCH), 123.4 (ArCH), 123.2 (ArCH), 121.9 (ArCH), 119.9 (ArCH), 115.2 (ArCH). HRMS-CI (m/z): [M+H]+ calculated for C21H16F3N4O2, 413.12199; found, 413.120133.
5.7.3.10. 3,3′-((6-(Trifluoromethyl)quinoxaline-2,3-diyl)bis(azanediyl))diphenol 22
1H NMR (500 MHz, d6-DMSO) δ: 9.49 (s, 1H, OH), 9.46 (s, 1H, OH), 9.18 (s, 1H, NH), 9.12 (s, 1H, NH), 7.81 (s, 1H, ArH), 7.67 (d, J = 8.4 Hz, 1H, ArH), 7.61–7.56 (m, 2H, 2 × ArH), 7.50 (d, J = 2.5 Hz, 1H, ArH), 7.26 (dd, J = 11.6, 9.0 Hz, 2H, 2 × ArH), 7.18 (td, J = 8.4, 2.5 Hz, 2H, 2 × ArH), 6.57–6.46 (m, 2H, 2 × ArH). 13C NMR (126 MHz, d6-DMSO) δ: 157.6 (ArC), 142.5 (ArC), 142.0 (ArC), 140.7 (ArC), 138.5 (ArC), 135.5 (ArC), 129.3 (ArCH), 126.2 (ArCH), 123.5 (ArC), 122.3 (ArCH), 120.6 (ArC), 112.0 (ArCH), 111.7 (ArCH), 110.5 (ArCH), 110.2 (ArCH), 108.3 (ArCH), 108.0 (ArCH). HRMS-CI (m/z): [M+H]+ calculated for C21H16F3N4O2, 413.12199; found, 413.121732.
5.7.3.11. 2,3-Bis((4-hydroxyphenyl)amino)quinoxaline-6-carbonitrile 23
1H NMR (500 MHz, d6-DMSO) δ: 9.76 (s, 1H, OH), 9.73 (s, 1H, OH), 9.56 (s, 1H, NH), 9.43 (s, 1H, NH), 8.25 (s, 1H, ArH), 8.04 (dt, J = 8.8, 3.2 Hz, 4H, 4 × ArH), 7.93 (ddd, J = 11.1, 8.3, 2.3 Hz, 2H, 2 × ArH), 7.24 (dt, J = 8.8, 3.2 Hz, 4H, 4 × ArH). 13C NMR (126 MHz, d6-DMSO) δ: 153.9 (ArC), 153.7 (ArC), 142.9 (ArC), 142.3 (ArC), 139.6 (ArC), 136.0 (ArC), 130.8 (ArC), 130.5 (ArC), 129.3 (ArCH), 126.5 (ArCH), 125.9 (ArCH), 123.6 (ArCH), 123.2 (ArCH), 119.5 (ArC-CN), 115.19 (ArCH), 115.17 (ArCH), 105.6 (ArC-CN). HRMS-CI (m/z): [M+H]+ calculated for C21H16N5O2, 370.13040; found, 370.129541.
5.7.3.12. 2,3-Bis((3-hydroxyphenyl)amino)quinoxaline-6-carbonitrile 24
1H NMR (500 MHz, d6-DMSO) δ: 9.47 (s, 1H, OH), 9.42 (s, 1H, OH), 9.27 (s, 1H, NH), 9.16 (s, 1H, NH), 7.93 (d, J = 1.7 Hz, 1H, ArH), 7.66–7.57 (m, 2H, 2 × ArH), 7.53 (s, 1H, ArH), 7.49 (s, 1H, ArH), 7.30–7.24 (m, 2H, 2 × ArH), 7.18 (dd, J = 14.3, 8.0 Hz, 2H, 2 × ArH), 6.57–6.47 (m, 2H, 2 × ArH). 13C NMR (126 MHz, d6-DMSO) δ: 157.6 (ArC), 142.6 (ArC), 142.0 (ArC), 140.6 (ArC), 140.3 (ArC), 139.3 (ArC), 135.8 (ArC), 129.7 (ArCH), 129.3 (ArCH), 127.0 (ArCH), 126.3 (ArCH), 119.3 (Ar-C), 112.1 (ArCH), 111.8 (ArCH), 110.8 (ArCH), 110.4 (ArCH), 108.4 (ArCH), 108.1 (ArCH), 106.4 (ArC-CN). HRMS-CI (m/z): [M+H]+ calculated for C21H16N5O2, 370.13040; found, 370.129114.
5.7.3.13. 4,4′-((6-Chloroquinoxaline-2,3-diyl)bis(azanediyl))diphenol 25
1H NMR (500 MHz, d6-DMSO) δ: 9.24 (s, 1H), 9.23 (s, 1H), 8.87 (s, 1H), 8.81 (s, 1H), 7.65–7.55 (m, 4H), 7.45–7.37 (m, 2H), 7.22 (dt, J = 8.6, 2.1 Hz, 1H), 6.83–6.76 (m, 4H). 13C NMR (126 MHz, d6-DMSO) δ: 153.51 (ArC), 153.45 (ArC), 141.97 (ArC), 141.61 (ArC), 137.03 (ArC), 134.97 (ArC), 131.18 (ArC), 131.09 (ArC), 128.04 (ArC), 126.35 (ArCH), 124.25 (ArCH), 123.73 (ArCH), 123.07 (ArCH), 123.04 (ArCH), 115.14 (ArCH). HRMS-ES (m/z): [M+H] calculated for C20H16N4O2Cl, 379.0962; found, 379.0927.
5.7.3.14. 3,3′-((6-Chloroquinoxaline-2,3-diyl)bis(azanediyl))diphenol 26
1H NMR (500 MHz, d6-DMSO) δ: 9.39 (s, 2H), 9.03 (s, 1H), 8.98 (s, 1H), 7.58–7.49 (m, 3H), 7.48 (s, 1H), 7.33 (dd, J = 8.7, 2.3 Hz, 1H), 7.23 (s, 2H), 7.16 (td, J = 8.0, 1.7 Hz, 2H), 6.50 (d, J = 8.0 Hz, 2H). 13C NMR (126 MHz, d6-DMSO) δ: 157.58 (ArC), 141.66 (ArC), 141.25 (ArC), 140.92 (ArC), 140.83 (ArC), 136.82 (ArC), 134.79 (ArC), 129.22 (ArCH), 128.78 (ArC), 126.72 (ArCH), 125.03 (ArCH), 124.05 (ArCH), 111.55 (ArCH), 110.06 (ArCH), 107.83 (ArCH). HRMS-ES (m/z): [M+H] calculated for C20H16N4O2Cl, 379.0962; found, 379.0940.
5.7.3.15. 2,3-Bis((4-hydroxyphenyl)amino)-N-methylquinoxaline-6-carboxamide 27
1H NMR (500 MHz, d6-DMSO) δ 9.26 (s, 1H, OH), 9.25 (s, 1H, OH), 8.89 (s, 1H, Ar-NH), 8.82 (s, 1H, Ar-NH), 8.44 (q, J = 4.5 Hz, 1H, Ar-CONHCH3), 7.94 (d, J = 2.0 Hz, 1H, ArH), 7.71 (dd, J = 8.4, 2.0 Hz, 1H, ArH), 7.63 (d, J = 8.4 Hz, 4H, 4 × ArH), 7.43 (d, J = 8.4 Hz, 1H, ArH), 6.86–6.77 (m, 4H, 4 × ArH), 2.77 (d, J = 4.5 Hz, 3H, Ar-CONHCH3). 13C NMR (126 MHz, d6-DMSO) δ 166.5 (Ar-CONHCH3), 153.5 (ArC), 153.4 (ArC), 142.2 (ArC), 141.9 (ArC), 138.2 (ArC), 137.2 (ArC), 135.4 (ArC), 131.3 (ArC), 131.1 (ArC), 130.0 (ArCH), 124.6 (ArCH), 123.8 (ArCH), 123.1 (ArCH), 115.1 (ArCH), 26.3 (Ar-CONHCH3). HRMS-EI (m/z): [M] calculated for C22H19N5O3, 401.1488; found, 401.1481.
5.7.3.16. 6,7-Bis((3-hydroxyphenyl)amino)-N-methyl-2-naphthamide 28
1H NMR (500 MHz, d6-DMSO) δ 9.42 (s, 1H, OH), 9.41 (s, 1H, OH), 9.04 (s, 1H, NH), 8.98 (s, 1H, NH), 8.51 (d, J = 4.4 Hz, 1H, Ar-CONHCH3), 8.06 (s, 1H, ArCH), 7.80 (d, J = 8.4 Hz, 1H, ArCH), 7.55 (d, J = 8.4 Hz, 1H, ArCH), 7.50 (d, J = 12.0 Hz, 2H, 2 × ArCH), 7.28 (d, J = 8.0 Hz, 2H, 2 × ArCH), 7.17 (t, J = 8.0 Hz, 2H, 2 × ArCH), 6.50 (d, J = 8.0 Hz, 2H, 2 × ArCH), 2.80 (d, J = 4.4 Hz, 3H, Ar-CONHCH3). 13C NMR (126 MHz, d6-DMSO) δ 166.5 (Ar-CONHCH3), 157.6 (ArC), 157.6 (ArC), 141.9 (ArC), 141.6 (ArC), 141.0 (ArC), 140.9 (ArC), 137.9 (ArC), 135.3 (ArC), 130.8 (ArCH), 129.22 (ArCH), 129.19 (ArCH), 124.9 (ArC), 124.3 (ArC), 123.7 (ArC), 111.7 (ArCH), 111.6 (ArCH), 110.2 (ArCH), 110.0 (ArCH), 108.0 (ArCH), 107.9 (ArCH), 26.3 (Ar-CONHCH3). HRMS-EI (m/z): [M] calculated for C22H19N5O3, 401.1488; found, 401.1479.
5.7.4. 4,4′-((6-Aminoquinoxaline-2,3-diyl)bis(azanediyl))diphenol 29
Tin(II) chloride dihydrate (112.5 mg, 0.5 mmol) was added to a solution of 4,4′-((6-nitroquinoxaline-2,3-diyl)bis(azanediyl))diphenol 10 (39 mg, 0.1 mmol) in ethanol (15 mL) and the resulting mixture was brought to boil. A solution of sodium borohydride (18.9 mg, 0.5 mmol) in ethanol (5 mL) was added to the boiling mixture dropwise and refluxed for 1.5 h. Upon cooling, water (10 mL) was added. Sodium hydroxide was added until neutralisation of the solution. The ethanol was evaporated from the mixture and an extraction with ethyl acetate (20 mL) was performed. The organics were washed with brine (3 × 10 mL) and dried over magnesium sulfate, filtered and the solvent evaporated under reduced pressure. The crude material was purified by flash column chromatography using a gradient of 50–80% ethyl acetate in cyclohexane to give the named compound (22.0 mg, 61%). 1H NMR (500 MHz, d6-DMSO) δ: 9.13 (s, 1H, OH), 9.06 (s, 1H, OH), 8.47 (s, 1H, NH), 8.23 (s, 1H, NH), 7.60 (d, J = 8.9 Hz, 2H, 2 × ArH), 7.53 (d, J = 8.9 Hz, 2H, 2 × ArH), 7.15 (d, J = 8.6 Hz, 1H, ArH), 6.75 (dd, J = 10.8, 8.9 Hz, 4H, 4 × ArH), 6.64 (dd, J = 8.6, 2.4 Hz, 1H, ArH), 6.58 (d, J = 2.4 Hz, 1H, ArH), 5.06 (s, 2H, NH2). 13C NMR (126 MHz, d6-DMSO) δ: 152.8 (ArC), 152.4 (ArC), 146.3 (ArC), 141.4 (ArC), 138.5 (ArC), 137.6 (ArC), 132.5 (ArC), 132.0 (ArC), 128.4 (ArC), 125.4 (ArCH), 122.4 (ArCH), 121.8 (ArCH), 115.0 (ArCH), 114.99 (ArCH), 114.8 (ArCH), 106.5 (ArCH). HRMS-CI (m/z): [M+H]+ calculated for C20H18N5O2, 360.1460; found, 360.1451.
Acknowledgement
This research was supported by the Medical Research Council UK with a studentship for F.M.
References and notes
- 1.Derbyshire E., Marletta M. Handb. Exp. Pharmacol. 2009;191:17. doi: 10.1007/978-3-540-68964-5_2. [DOI] [PubMed] [Google Scholar]
- 2.Evgenov O.V., Pacher P., Schmidt P.M., Hasko G., Schmidt H.H.H.W., Stasch J.-P. Nat. Rev. Drug Disc. 2006;5:755. doi: 10.1038/nrd2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moncada S., Higgs E.A. In: Moncada S., Higgs A., editors. Vol. 176. Springer; Berlin, Heidelberger Platz 3, D-14197 Berlin, Germany: 2006. p. 213. (Vascular Endothelium I). [Google Scholar]
- 4.Koesling D., Neitz A., Mittmann T., Mergia E. BMC Pharmacol. 2011;11:O21. [Google Scholar]
- 5.Gómez-Pinedo U., Rodrigo R., Cauli O., Herraiz S., Garcia-Verdugo J.-M., Pellicer B., Pellicer A., Felipo V. Neuroscience. 2010;165:1275. doi: 10.1016/j.neuroscience.2009.11.046. [DOI] [PubMed] [Google Scholar]
- 6.Garthwaite J., Bellamy T.C., Wood J., Goodwin D.A. Proc. Natl. Acad. Sci. U.S.A. 2000;97:2928. doi: 10.1073/pnas.97.6.2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang L., Dawson V.L., Dawson T.M. Pharmacol. Ther. 2006;109:33. doi: 10.1016/j.pharmthera.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 8.Olesen J. Neurotherapeutics. 2010;7:183. doi: 10.1016/j.nurt.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tseng K.Y., Caballero A., Dec A., Cass D.K., Simak N., Sunu E., Park M.J., Blume S.R., Sammut S., Park D.J. PloS One. 2011;6:e27187. doi: 10.1371/journal.pone.0027187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garthwaite J., Southam E., Boulton C.L., Nielsen E.B., Schmidt K., Mayer B. Mol. Pharmacol. 1995;48:184. [PubMed] [Google Scholar]
- 11.Zhao Y.D., Brandish P.E., DiValentin M., Schelvis J.P.M., Babcock G.T., Marletta M.A. Biochemistry. 2000;39:10848. doi: 10.1021/bi9929296. [DOI] [PubMed] [Google Scholar]
- 12.Schrammel A., Behrends S., Schmidt K., Koesling D., Mayer B. Mol. Pharmacol. 1996;50:1. [PubMed] [Google Scholar]
- 13.Babcock G.T., Zhao Y.D., Brandish P.E., DiValentin M., Schelvis J.P.M., Marletta M.A. Biochemistry. 2000;39:10848. doi: 10.1021/bi9929296. [DOI] [PubMed] [Google Scholar]
- 14.Moro M.A., Russel R., Cellek S., Lizasoain I., Su Y., Darley-Usmar V.M., Radomski M.W., Moncada S. Proc. Natl. Acad. Sci. U.S.A. 1996;93:1480. doi: 10.1073/pnas.93.4.1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lies B., Groneberg D., Gambaryan S., Friebe A. Br. J. Pharmacol. 2013;170:317. doi: 10.1111/bph.12275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kumagai Y., Midorikawa K., Nakai Y., Yoshikawa T., Kushida K., Homma-Takeda S., Shimojo N. Eur. J. Pharmacol. 1998;360:213. doi: 10.1016/s0014-2999(98)00666-9. [DOI] [PubMed] [Google Scholar]
- 17.Mittal C.K., Murad F. Proc. Natl. Acad. Sci. U.S.A. 1977;74:4360. doi: 10.1073/pnas.74.10.4360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brune B., Schmidt K.-U., Ullrich V. Eur. J. Biochem. 1990;192:683. doi: 10.1111/j.1432-1033.1990.tb19276.x. [DOI] [PubMed] [Google Scholar]
- 19.Spyridonidou K., Fousteris M., Antonia M., Chatzianastasiou A., Papapetropoulos A., Nikolaropoulos S. Bioorg. Med. Chem. Lett. 2009;19:4810. doi: 10.1016/j.bmcl.2009.06.047. [DOI] [PubMed] [Google Scholar]
- 20.Chang F.-J., Lemme S., Sun Q., Sunahara R.K., Beuve A. J. Biol. Chem. 2005;280:11513. doi: 10.1074/jbc.M412203200. [DOI] [PubMed] [Google Scholar]
- 21.Wood P., Marks V. Ann. Clin. Biochem. 1978;15:25. doi: 10.1177/000456327801500104. [DOI] [PubMed] [Google Scholar]
- 22.Griffiths C., Wykes V., Bellamy T.C., Garthwaite J. Mol. Pharmacol. 2003;64:1349. doi: 10.1124/mol.64.6.1349. [DOI] [PubMed] [Google Scholar]
- 23.Romer D.R. J. Heterocycl. Chem. 2009;46:317. [Google Scholar]
- 24.Mota F., Allerston C.K., Hampden-Smith K., Garthwaite J., Selwood D.L. Bioorg. Med. Chem. Lett. 2014;24:1075. doi: 10.1016/j.bmcl.2014.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meanwell N.A. J. Med. Chem. 2011;54:2529. doi: 10.1021/jm1013693. [DOI] [PubMed] [Google Scholar]
- 26.Martin F., Baskaran P., Ma X., Dunten P.W., Schaefer M., Stasch J.-P., Beuve A., van den Akker F. J. Biol. Chem. 2010;285:22651. doi: 10.1074/jbc.M110.111559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marletta M.A., Stone J.R. Chem. Biol. 1998;5:255. doi: 10.1016/s1074-5521(98)90618-4. [DOI] [PubMed] [Google Scholar]
- 28.Cary S.P.L., Winger J.A., Derbyshire E.R., Marletta M.A. Trends Biochem. Sci. 2006;31:231. doi: 10.1016/j.tibs.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 29.Allerston C.K., von Delft F., Gileadi O. PloS One. 2013;8:e57644. doi: 10.1371/journal.pone.0057644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang G., Liu Y., Qin J., Vo B., Tang W.J., Ruoho A.E., Hurley J.H. Protein Sci. 1997;6:903. doi: 10.1002/pro.5560060417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stasch J.P., Becker E.M., Alonso-Alija C., Apeler H., Dembowsky K., Feurer A., Gerzer R., Minuth T., Perzborn E., Pleiss U., Schroder H., Schroeder W., Stahl E., Steinke W., Straub A., Schramm M. Nature. 2001;410:212. doi: 10.1038/35065611. [DOI] [PubMed] [Google Scholar]
- 32.Hamet P., Tremblay J., Pang S.C., Garcia R., Thibault G., Gutkowska J., Cantin M., Genest J. Biochem. Biophys. Res. Commun. 1984;123:515. doi: 10.1016/0006-291x(84)90260-2. [DOI] [PubMed] [Google Scholar]
- 33.Ahluwalia A., MacAllister R.J., Hobbs A.J. Basic Res. Cardiol. 2004;99:83. doi: 10.1007/s00395-004-0459-6. [DOI] [PubMed] [Google Scholar]
- 34.He X.L., Dukkipati A., Garcia K.C. J. Mol. Biol. 2006;361:698. doi: 10.1016/j.jmb.2006.06.060. [DOI] [PubMed] [Google Scholar]
- 35.Kleppisch T., Feil R. Springer; 2009. cGMP: Generators, Effectors and Therapeutic Implications. p. 549. [Google Scholar]
- 36.Makino R., Yazawa S., Hori H., Shiro Y. Biochemistry. 2012;51:9277. doi: 10.1021/bi3004044. [DOI] [PubMed] [Google Scholar]
- 37.Southam E., Garthwaite J. Neurosci. Lett. 1991;130:107. doi: 10.1016/0304-3940(91)90239-p. [DOI] [PubMed] [Google Scholar]
- 38.Garthwaite G., Hampden-Smith K., Wilson G.W., Goodwin D.A., Garthwaite J. Glia. 2015;63:383. doi: 10.1002/glia.22759. [DOI] [PMC free article] [PubMed] [Google Scholar]